
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of lignin–carbohydrate complex (LCC) linkages on cellulose pyrolysis chemistry†
Arul Mozhi Devan
Padmanathan
 a,
Seth
Beck
a,
Khursheed B.
Ansari
bc and
Samir H.
Mushrif
*a
a,
Seth
Beck
a,
Khursheed B.
Ansari
bc and
Samir H.
Mushrif
*a
aDepartment of Chemical and Materials Engineering, University of Alberta, 9211-116 Street Northwest, Edmonton, Alberta T6G 1H9, Canada. E-mail: mushrif@ualberta.ca
bSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, Singapore
cDepartment of Chemical Engineering, College of Engineering, King Khalid University, Abha, 61411, Saudi Arabia
First published on 18th December 2023
Abstract
Understanding the impact of cross-linked cellulose with lignin in lignin–carbohydrate complexes (LCC) on cellulose decomposition reaction kinetics and chemistry is challenging. This study combines first-principles molecular simulations and thin-film experiments to investigate key cellulose decomposition mechanisms, including transglycosylation, ring contraction, and ring opening, which lead to the formation of major bio-oil components (levoglucosan, 5-hydroxymethylfurfural, and glycolaldehyde). Ab initio molecular dynamics and metadynamics are employed to model LCC molecules with β-O-4 benzyl ether linkages at the C2, C3, and C6 carbon positions of cellobiose. Density functional theory (DFT) calculations are used to evaluate the reaction energetics of cellulose activation via these mechanisms. Activation barriers, reaction energies, and frontier molecular orbital interactions are compared between cellobiose with and without LCC, providing insights into the influence of LCC linkages. Experimental product yields from native herbaceous biomass pyrolysis are measured and compared to those from pure cellulose pyrolysis. The results demonstrate that cross-linked cellobiose in LCC exhibits higher activation barriers (2X) and reaction energies (3–4X) compared to pure cellobiose, indicating altered kinetics and thermodynamics. The differences within LCC conformers are minimal, except the blocking of the C6 position due to the LCC linkage. Analysis of HOMO–LUMO interactions reveals a spatial separation of reaction centers in LCC, indicating the favorability of inter-moiety mechanisms over intra-moiety mechanisms. This study underscores the novel role of covalent LCC bonding between lignin and carbohydrates in the reaction kinetics and chemistry of cellulose decomposition in native biomass and in the formation of major bio-oil products.
1 Introduction
Lignocellulosic biomass, the largest renewable natural resource1 for carbon-based liquid fuels, offers a promising solution for sustainable fuel and chemical production. By utilizing fast pyrolysis, a decentralizable processing technology, biopolymers can be thermally cracked without oxygen, yielding renewable crude oil (bio-oil).2 During pyrolysis, biomass is heated in the absence of oxygen to produce a mixture of bio-oil, biochar, and volatile gases. Despite the potential benefits of decentralized biomass processing and lower transportation costs, the commercial viability of pyrolysis technology has been hindered by the instability of bio-oil during storage and transport. Also, the highly oxygenated nature of bio-oil3 necessitates further treatment for integration into existing petroleum infrastructure. Despite extensive attempts over past couple of decades to improve targeted bio-oil production, minimize lignin repolymerization, and enhance the accessibility of cellulose-derived products, the advancement of biomass deconstruction techniques has faced obstacles due to limited understanding of the underlying chemistry. Therefore, understanding the chemistry of biomass decomposition and molecular interactions between cellulose and other biopolymers is crucial for systematic and bottom-up optimization of the pyrolysis process and for improving the yield and quality of the resulting products.The intricate microscopic structure of native biomass in the plant cell wall involves the complex intermingling of cellulose, lignin, and hemicellulose.1 To facilitate a clearer understanding of biomass decomposition, researchers have focused on studying isolated biopolymers chemistry. Among the primary components of biomass, cellulose garnered significant attention due to its rapid decomposition during pyrolysis, leading to the production of substantial quantities of desirable volatile compounds. Multiscale molecular modelling and first principles-based calculations have enabled the discovery of molecular level mechanisms that are inaccessible to experiments alone. First-principles modeling has played a crucial role in advancing our understanding of the intricate mechanisms and kinetics involved in cellulose pyrolysis.4,5 Previous research has mainly concentrated on investigating the initial steps of cellulose pyrolysis, specifically the depolymerization of cellulose chains and the formation of various small molecules. The thermal decomposition of cellulose during pyrolysis produces levoglucosan (LG)6,7 as a dominant product followed by furanic compounds like 5-hydroxymethyl-furfural (5-HMF) and light oxygenates like glycolaldehyde (GA). Moreover, multiple Density Functional Theory (DFT) studies4,8,9 have proven that concerted mechanisms, such as transglycosylation and ring contraction, are more favorable, compared to homolytic or heterolytic cleavage of glycosidic C–O bonds during LG formation. Transglycosylation, specifically, has been proposed as a favorable pathway for glycosidic bond cleavage.10 Concerted transglycosylation mechanism involves simultaneous protonation of the glycosidic bond by the C6-hydroxymethyl group and the formation of a C6–O–C1 bridge (cf.Scheme 1 for atomic nomenclature). Ab initio molecular dynamics (AIMD)-metadynamics simulations have revealed that major volatiles, including furans, can be directly generated from cellulose through ring contraction, without the involvement of small-molecule intermediates like glucose or levoglucosan (LGA).11 Free energy barriers reported for pyranose ring contraction, ring opening, rearrangement, or ring fragmentation during pyrolysis, leading to the formation of pre-LGA and pre-furans revealed ring contraction as the dominant mechanism at 327 °C. Concerted ring contraction mechanism involves the simultaneous protonation of the glycosidic bond and C2–C3 bond fragmentation to form C1–C3 bond, changing to a furanose ring from pyranose. The reorganization of the ring structure leads to the formation of 3,4-(hydroxyl) 5-(hydroxymethyl) furfural and glucose. These mechanisms to form major pyranic and furanic compounds have been previously investigated in both gas-phase10 and in condensed-phase environments.12 Piskorz et al.13 proposed that the two carbon fragments resulting from the cleavage of cellulose monomers during pyrolysis are transformed into glycolaldehyde (GA), a major pyrolysis derived bio-oil product. The mechanisms for small molecular weight products, such as GA, have also been studied using DFT14–17 and a minor fraction of GA has been suggested to be produced from the secondary cleavage of LG.18 However, GA primarily originates from the ring opening of cellulose, particularly through the cleavage of C1–C2, C5–C6, and C3–C4 bonds in the cellulose monomer. DFT calculations showed that the pyran ring, undergoing dehydration, is more inclined to undergo the ring opening reaction.19 1,2-Dehydration mechanism has been found to be the most prevalent, surpassing alcohol dehydration.20,21 Detailed insights into such retro-aldol reactions, which are the primary routes for GA production, have been provided by Assary and Curtiss22 and further supported by Zhang et al.23 Ring opening leads to reactants through a series of steps involving dehydration, cleavage, and isomeric formation of glycolaldehyde (GA). As depicted in Scheme 1, the ring opening process initiates with the 1–2 dehydration of cellobiose, followed by the cleavage of C4′–C5′ and C1′–O5′ bonds, resulting in the opening of the pyran ring. This leads to the formation of two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds (C1′–C2′ and C5′–C6′) and glycolaldehyde. While the aforementioned discussion provides valuable insights into the chemistry of cellulose pyrolysis, with a primary focus on the primary and secondary reactions of cellulose decomposition, there remains a lack of integrated research addressing the mechanism of the initial stage of cellulose pyrolysis in the context of covalent bonding with other plant biopolymers in the plant cell wall.
C double bonds (C1′–C2′ and C5′–C6′) and glycolaldehyde. While the aforementioned discussion provides valuable insights into the chemistry of cellulose pyrolysis, with a primary focus on the primary and secondary reactions of cellulose decomposition, there remains a lack of integrated research addressing the mechanism of the initial stage of cellulose pyrolysis in the context of covalent bonding with other plant biopolymers in the plant cell wall.
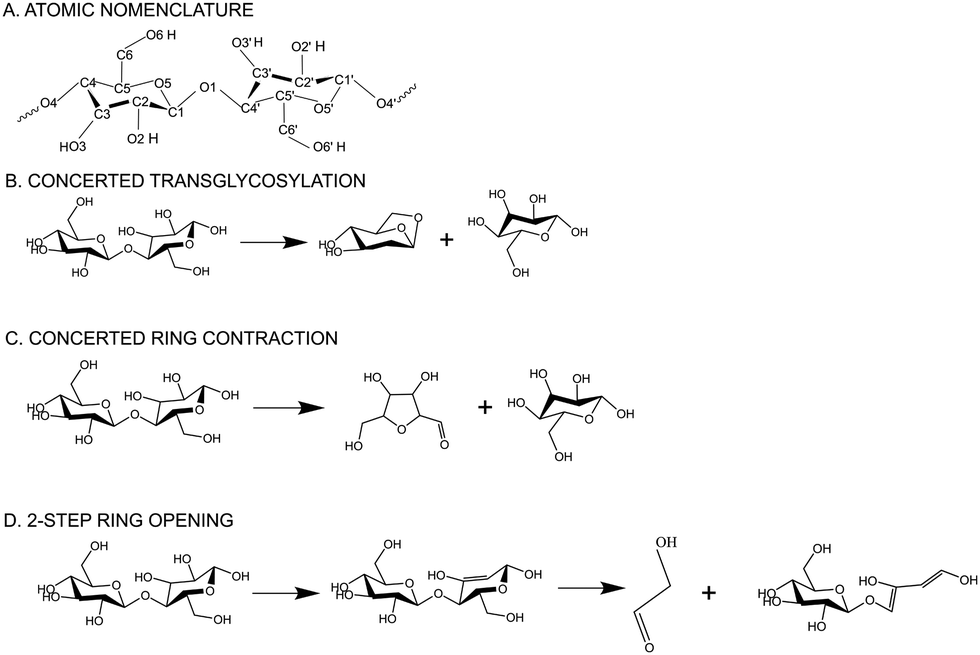 | ||
| Scheme 1 (A) Atomic nomenclature of the monomeric glucose unit (B) transglycosylation reaction mechanism of cellobiose decomposition to form LGA (C) ring contraction mechanism of cellobiose decomposition to form furanic compounds (D) ring opening mechanism of cellobiose decomposition to form glycolaldehyde. | ||
It is well known that the presence of lignin in native biomass contributes to the formation of additional compounds such as phenols (derived primarily from lignin), in addition to furans and levoglucosan (LGA). However, the pyrolysis of native biomass, such as spruce and beech wood, exhibits a significant change in the distribution of cellulose derived components too, as compared to pure cellulose pyrolysis. While the cellulose content in these biomasses can potentially yield up to 48% levoglucosan when pyrolyzed separately, the actual LGA yield is less than 3%.24,25 This phenomenon is also observed in the pyrolysis of synthetic biomass (model biomass prepared by mixing different extracted biopolymers) containing cellulose and lignin mixtures, where the reaction rates slow down, and lignin hinders in cellulose breakdown and volatile formation.26–28 Additionally, the product yields from the pyrolysis of herbaceous biomass (e.g., corn stover, switchgrass) also differ from those of woody biomass (e.g., pine, redwood).29 The herbaceous cellulose–lignin sample results in a 10.28 wt% lower LGA yield which is compensated by 11.38 wt% and 1.45 wt% increase in C1 to C3 products and furans, respectively. However, in the pyrolysis of woody biomass, despite the higher lignin content, the deviation in product distribution from pure cellulose is minimal. This variation between herbaceous and woody biomass is attributed to higher lignin carbohydrate complex (LCC) linkages in herbaceous biomass. While modelling isolated biopolymer molecules has been useful in understanding their individual pyrolysis chemistries, they don’t allow the prediction or even can explain the pyrolysis chemistry of native biomass. It is therefore important to consider the complex linkages between biomass components and their influence on cellulose decomposition. Lignin–carbohydrate complexes (LCC) play a vital role in wood structure, with a considerable portion of lignin (all in coniferous30 and 47–60% in deciduous31) forming covalent bonds with carbohydrates. In softwood, approximately 50% of lignin is bound to cellulose, while in hardwood, this proportion is around 17%.32 The presence of LCCs poses challenges in isolating biomass components with high yield and purity,33,34 mainly due to the reduced accessibility of carbohydrates35,36 and the stability of these covalent bonds37 between lignin and carbohydrates to extraction via alkaline treatment. Traditionally, research on LCCs has focused on cleaving these bonds for extraction purposes, employing indirect wet chemistry methods such as selective acid/alkaline hydrolysis30 followed by FT-IR spectroscopy for analysis. However, for a more comprehensive understanding of the molecular structure, direct techniques like two-dimensional nuclear magnetic resonance (2D NMR), particularly HSQC spectroscopy, are commonly utilized. Among the eight identified types of LCC linkages,36,38–41 benzyl ether, benzyl ester, and phenyl glycosidic linkages are frequently observed,41 exhibiting varying strengths under different conditions. Benzyl ether bonds are dominant in softwood LCCs and are notably more stable and prevalent. Furthermore, benzyl ether bonds hold greater relevance within the context of cellulose and lignin, as the currently postulated mechanisms for phenyl glycoside formation necessitate a reducing end on the carbohydrates42 or monolignol glycoside precursors for lignin.43 Given that cellulose lacks numerous end groups and that monolignol glycoside precursors do not facilitate an LCC linkage between lignin and cellulose, the focus of our studies primarily centres on benzyl ether bonds. Herbaceous biomass, particularly in grass cell walls, displays a higher occurrence of LCC linkages, where benzyl ether linkages crosslink lignin and polysaccharides (Kajikawa et al., 2000). A study conducted by Watanabe et al. in 1989 employed a comprehensive technique involving cellulase digestion, adsorption chromatography, acetylation, DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) oxidation, and methylation to provide direct evidence of binding sites between lignin and carbohydrates. The majority of studies44,45 have reported LCC linkages at the C6 position of the sugar, although acetylation at the C2 and C3 positions, indicating LCC linkages, has been observed in mannose and xylan molecules.45–47 Recent first principles-based calculations48 studying the reaction energies of LCC formation at different bonding sites on glucose demonstrated minimal differences of less than 5 kJ mol−1, indicating thermodynamic preference for LCC formation at any of the three positions (C6, C3, or C2) (cf.Scheme 1(A)).
Cross-linked cellulose in these LCC heteromolecules may exhibit distinct chemical properties compared to pure cellulose. LCC linkages formed at the C6 position in particular can inhibit levoglucosan (LGA) formation, similar to the inhibition observed in polysaccharides with 1,6-glycosidic linkages as opposed to 1,4- or 1,3-glycosidic linkages.49 This inhibition of LGA formation by the benzyl ether LCC linkage at the C6 position aligns with the measured decrease in LGA yield and the subsequent increase in C1–C3 products observed in the pyrolysis of native biomass, particularly herbaceous biomass. This suggests that the LCCs could potentially alter cellulose reaction pathways and energetics because of the covalent cross-linking between lignin and cellulose. However, despite the significance of cross-linked LCC linkages in cellulose decomposition, only one recent study, to the best of our knowledge, has investigated their role in pyrolysis chemistry. The study compared the pyrolysis products of chemically tailored native lignocellulose with selective removal of hemicellulose to a synthetic cellulose–lignin mixture.50 The presence of cross-linked lignin was found to significantly influence the production of small molecules and furan derivatives, increasing their yield by 97%, while hindering the generation of anhydrosugars by up to 47%. Additionally, cross-linked lignin exhibited a more pronounced effect on lignocellulose pyrolysis by promoting glycosyl ring scission and lignin fragmentation compared to free lignin. However, the specific chemistry and energetics underlying these ring scissions in the LCC molecule remain unknown. Studying cellulose pyrolysis chemistry in cross-linked LCCs is limited by the complex nature of conducting first principles calculations for multimolecular systems and the experimental difficulties associated with complex pre-treatments for the isolation of these LCCs from native biomass.
In order to gain understanding of the impact of lignin–carbohydrate linkages on cellulose pyrolysis chemistry, it is crucial to address several significant knowledge gaps – (1) the role of cross-linked LCC linkages in cellulose decomposition energetics, (2) the similarity or dissimilarity in the decomposition chemistry and kinetics between pure cellulose and cross-linked cellulose moieties within LCC, and (3) the influence of the lignin binding site on cellulose decomposition. This study aims to address these gaps by employing a combination of first principles calculations and thin-film pyrolysis experiments. Specifically, the study will utilize ab initio techniques to model different LCC conformations with varying binding sites – C2, C3, and C6 (cf.Scheme 1(A)) and calculate their respective energy barriers and reaction energies using Density Functional Theory (DFT). These calculations will be compared to the energetics calculated for the pyrolysis of pure cellulose. By investigating the decomposition chemistry using model molecules and subsequently validating the findings through experiments, this paper will provide novel insights into the role of LCC linkages and the impact of lignin–cellulose binding sites on cellulose decomposition.
2 Methodology
2.1 Ab initio conformational search
To ensure accurate computation of activation and reaction energies, it is crucial to thoroughly explore the conformational space of the LCC molecule (model cellobiose and lignin compounds covalently bonded via an LCC linkage). In this study, Car–Parrinello molecular dynamics (CPMD) combined with metadynamics (CPMD-metadynamics) was utilized to calculate the free energy surface (FES) as a function of torsional angles within the LCC molecule. The selection of torsion angles aimed to enable extensive sampling of the system, which would not be feasible within reasonable computational timeframes using thermal energy alone. The methods and parameters closely followed the procedures outlined by Beck et al.51 Conformers corresponding to the lowest minima on the FES were subsequently subjected to density functional theory (DFT) optimization and further calculation of the transition state for the cleavage of cellulose moiety in the LCCs through transglycosylation, ring contraction, and ring opening. The computational details are further elaborated in Section S1 and the free energy surfaces are shown in Fig. S1 (in the ESI†).2.2 DFT optimization and transition state calculations
The Gaussian 09 code52 was utilized to perform all-electron DFT calculations, aiming to compare the relative stabilities and to further optimize the lowest energy sample conformers identified through CPMD-metadynamics. Considering the large number of generated starting conformers, a step-wise improvement strategy was adopted for the basis set selection. The output of a less sophisticated basis set served as the input for a more advanced one, providing a systematic approach for conformational screening, with each level of theory screening the lowest energy conformers. Each conformer underwent complete geometry optimization at each level of theory, without imposing constraints on the atoms. Subsequently, frequency calculations were performed to ensure the absence of spurious frequencies in the reactant and product compounds. The hybrid functional RM06-2X was employed in all stages of optimization up to the 6-311+G(d) basis set, as it has been demonstrated to provide sufficient accuracy for modeling cellulose-derived molecules.4,53–55 Transition state (TS) searches were conducted using the Berny algorithm for both unconstrained cellobiose and LCC molecules, focusing on transglycosylation, ring contraction, and ring opening mechanisms (Scheme 1). Following the TS searches, frequency calculations were carried out to differentiate between saddle points and local minima, determined by the presence or absence of an imaginary frequency (corresponding to the reaction coordinate), respectively. Intrinsic reaction coordinates (IRCs) were traced in both directions to verify that the TS corresponded to the correct reactant and product on the potential energy surface. The reported reaction free energies were determined at 1 atm and 500 K. A convergence criterion of 1.00D-06 in energy change was selected for the self-consistent field (SCF) calculations to determine the electronic structure configuration. Similar activation barriers for cellobiose activation via transglycosylation and ring contraction have been previously reported in our earlier work.122.3 Experimental methods
3 Results and discussions
Cellulose undergoes decomposition under pyrolysis conditions through competing reactions involving the cleavage of glycosidic C–O bonds (yielding LGA) and C–C bonds (yielding furan and C1–C3 products). DFT calculations show that concerted transglycosylation and ring contraction (cf.Scheme 1) are the most favorable pathways for the glycosidic bond cleavage,58 while 2-step retro-aldol ring opening has reported be the predominant pathway for the formation of lower molecular weight compounds. The opening of the pyran ring initiates with a dehydration step followed by a ring opening step to form glycolaldehyde (GA), one of the major products in bio-oil19 (cf.Scheme 1). Transglycosylation, ring contraction and 2-step ring opening mechanisms contribute to the formation of major cellulose pyrolysis products, LGA, furans and GA, and are used as representative primary reaction pathways in this study. Also, dimers cellobiose and quinone methide intermediate.513.1 Activation barriers for competing cellulose decomposition reactions
As described in methodology, DFT calculations were performed for an isolated cellobiose molecule. The results, as presented in Fig. 1, illustrate the energy diagram for transglycosylation, ring contraction, and ring opening mechanisms at a temperature of 500 K. The calculated activation barriers for transglycosylation and ring contraction were determined to be 60.98 kcal mol−1 and 68.15 kcal mol−1, respectively. The barriers for the two-step ring opening mechanism were calculated to be significantly lower, 48.96 kcal mol−1 and 39.64 kcal mol−1. Transglycosylation involves the formation of a bridge, while both ring opening and ring contraction entail the reorganization and cleavage of the ring structure. The results depicted in Fig. 1 demonstrate that transglycosylation is favored over ring contraction, which aligns with previous findings from DFT studies.22 Additionally, the stepwise barriers for ring opening are the lowest among the investigated mechanisms.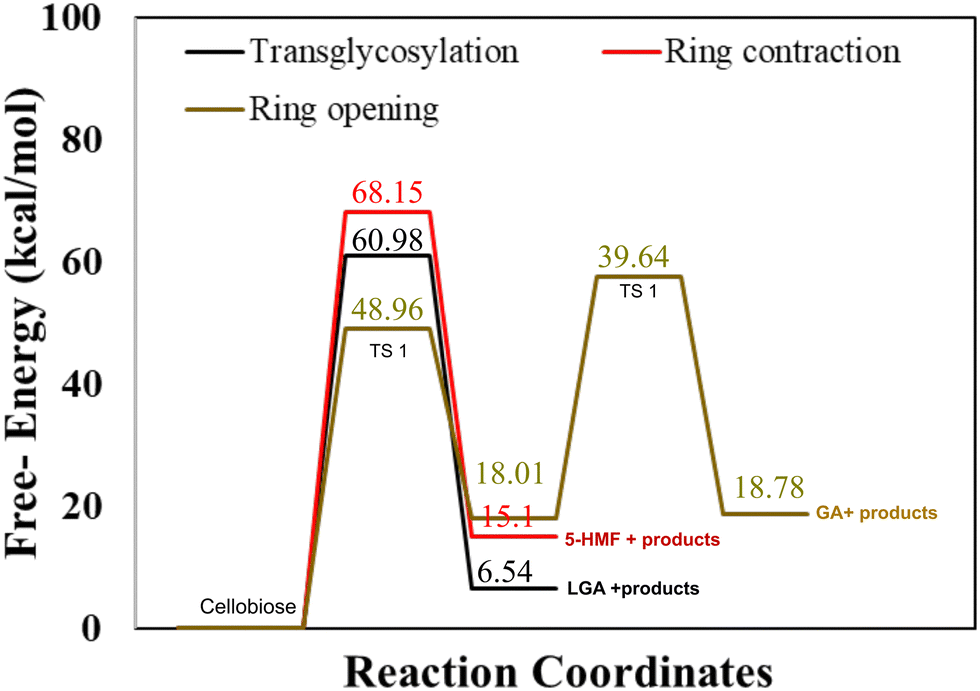 | ||
| Fig. 1 Gas phase activation free energy barriers for cellobiose decomposition calculated using hybrid functional M06-2X with 6-311+G(d) basis set for transglycosylation, ring contraction and ring opening mechanisms. | ||
3.2 Lignin–carbohydrate complex (LCC)
The formation of lignin carbohydrate complex (LCCs) linkages has recently gained attention due to their significant role in the recalcitrant nature of biomass.42,59 LCCs are formed as a result of side reactions during the formation of the predominant β-O-4 linkage in lignin. During the formation of the β-O-4 linkage, a quinone methide (QM) intermediate is generated, which undergoes re-aromatization through nucleophilic addition at the α-carbon. Traditionally, it has been assumed that this intermediate reacts exclusively with water,60,61 leading to only physical interactions between lignin and cellulose in the cell wall. However, Beck et al.48 provided direct evidence of the molecular mechanism behind the formation of benzyl ether and benzyl ester LCC linkages through the speculated lignin–cellobiose polymerization pathway. These LCCs, formed through covalent bonding between cellobiose and lignin, were found to be thermodynamically more stable than the nucleophilic addition of water. Since, among various LCC linkages, benzyl ether LCCs are prevalent and stable,62 the cross-linking between cellobiose and the quinone methide intermediate were made via a benzyl ether bond. While these covalent linkages primarily form at the C6 position of the sugar,44,45 LCCs at C2 and C3 positions have also been shown to be thermodynamically facile. Therefore, this study also aims to investigate whether the site of LCC linkage influences cellulose activation. To address this, covalent linkages were established not only at the C6 position but also at the C2 and C3 positions (refer to Scheme 2).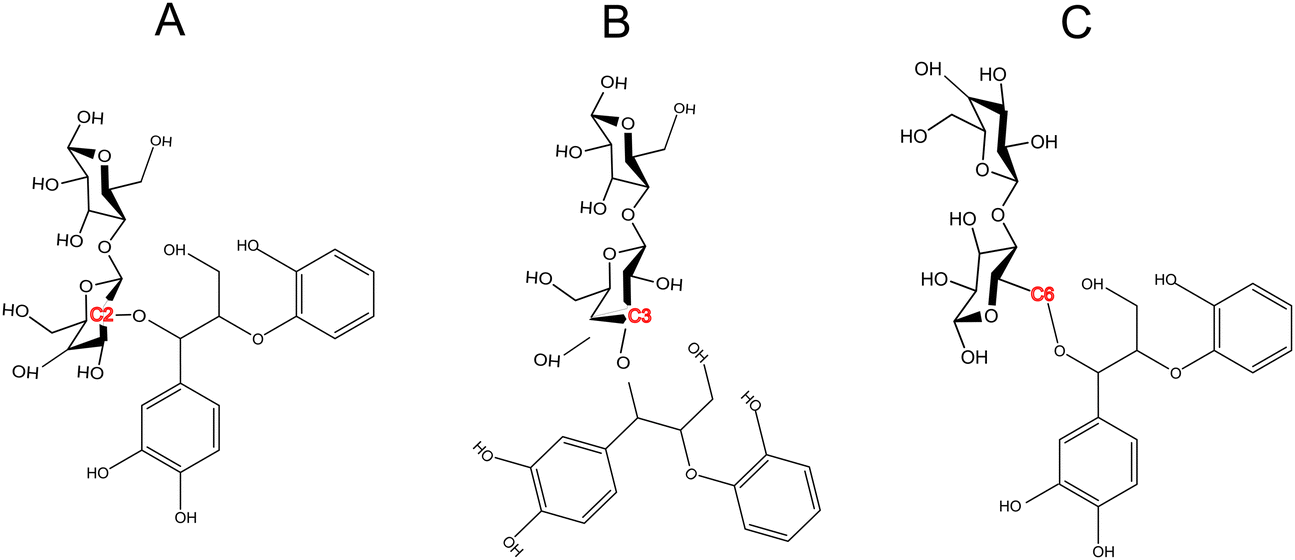 | ||
| Scheme 2 LCC linkage between a lignin moiety (quinone methide intermediate) and the cellobiose dimer bonded at (A) C2 position (B) C3 position (C) C6 position. | ||
However, LCC-C2 can’t undergo ring contraction because the mechanism requires a C2 hydroxyl group that protonates the glycosidic oxygen, which is replaced by the ether linkage in the LCC-C2 molecule. Similarly, LCC-C6 can’t undergo transglycosylation as the mechanism requires the protonation of the glycosidic oxygen by the C6 hydroxyl group; but the C6 oxygen is involved in the LCC ether linkage. These DFT calculations revealed that the activation barrier for transglycosylation in the LCC-C2 and LCC-C3 molecules are 108.04 kcal mol−1 and 109.86 kcal mol−1 (cf.Fig. 2(A)). These barriers calculated in LCC molecules (cross-linked cellobiose) are almost twice of that in a pure cellobiose molecule (60.98 kcal mol−1) for the same mechanism. The activation barrier for ring contraction in the LCC-C3 and LCC-C6 molecules are 117.21 kcal mol−1 and 112.8 kcal mol−1, respectively (cf.Fig. 2(C)). For the 2-step ring opening mechanism in LCC-C2, LCC-C3 and LCC-C6 (cf.Fig. 2(B)), the barriers for the first dehydration step are 104.44 kcal mol−1, 107.7 kcal mol−1 and 104.66 kcal mol−1, respectively. Subsequently, the activation barriers for the second ring opening step in cellobiose cross-linked at the C2, C3 and C6 positions are 37.59 kcal mol−1, 39.38 kcal mol−1 and 37.72 kcal mol−1, respectively. Unlike the large barriers calculated in the decomposition of cross-linked cellobiose in LCC via other mechanisms, the second ring opening step exhibits relatively low activation barriers. This indicates that once the 1,2-dehydration step is complete forming a CC bond on the cellobiose ring, the formation of glycolaldehyde by ring opening is kinetically facile. Comparing the three mechanisms for an LCC conformer cross-linked at a particular site, ring contraction has the highest barrier, followed by transglycosylation, while the first dehydration (rate determining) step in ring opening has the lowest barriers. This lower barrier for dehydration and subsequent formation of lower oxygenates (like GA) is supported by the high yields of GA measured in cellulose pyrolysis experiments.19 For all three mechanisms and in turn all three products (LGA, furans, GA) formation, the barriers in cross-linked cellobiose in LCC are almost twice of those in the isolated cellobiose. Additionally, the decomposition reaction of LCC molecules is also significantly more endergonic (as compared to pure cellobiose). This indicates that the pyrolysis energetics and the kinetics of cellobiose with LCC are different from that of pure cellobiose. Such high activation barriers for the primary decomposition of cellobiose moiety in the presence of LCC has not been reported before. To develop a molecular level understanding and a mechanistic reason for the high barrier to cleave cellulose moiety in the presence of LCC, the reaction energies are compared, and the frontier molecular orbitals are also visualized.
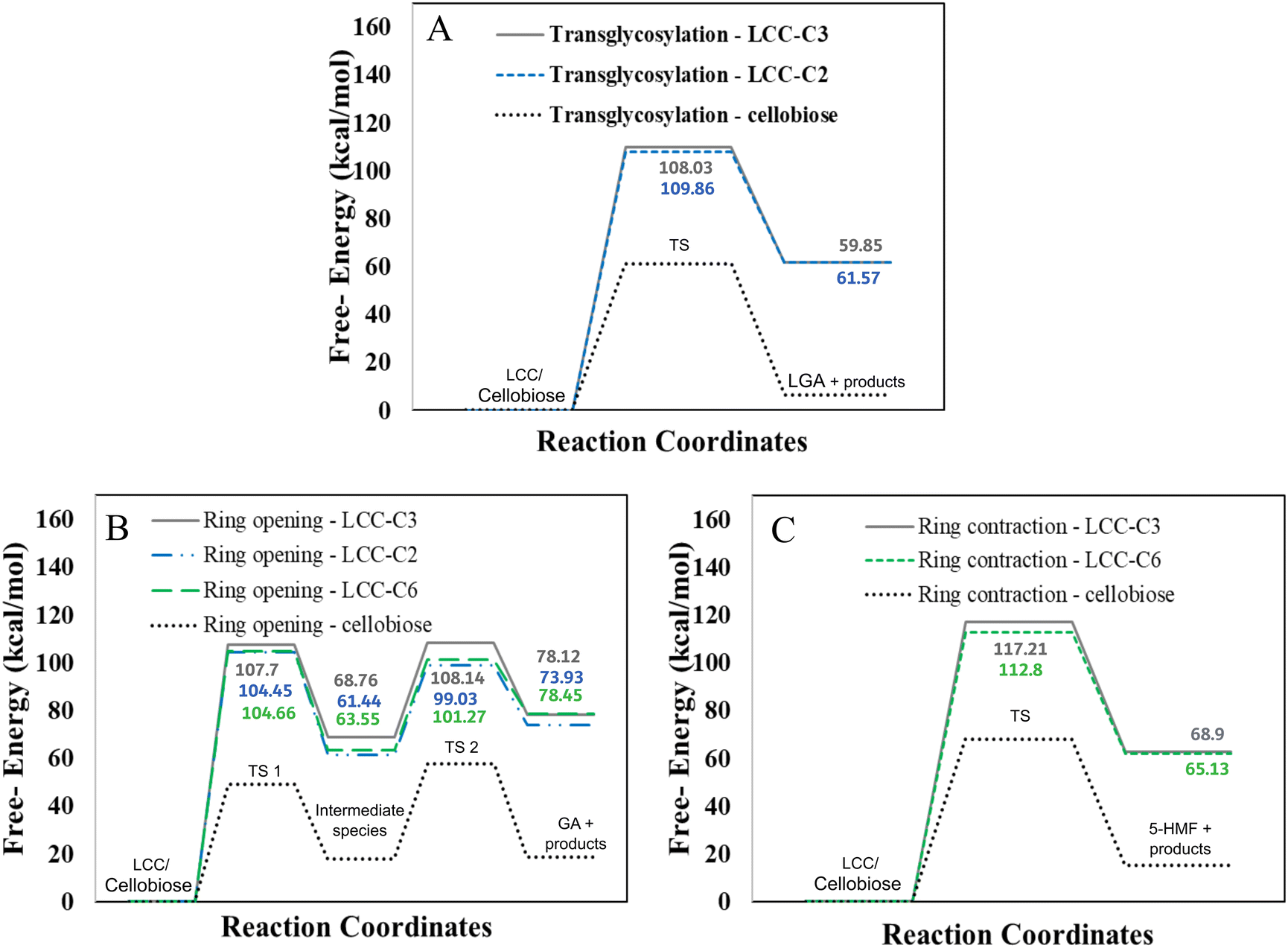 | ||
| Fig. 2 Free energy barriers for LCC decomposition via (A) transglycosylation (B) ring opening and (C) ring contraction mechanisms. | ||
The reaction free energies of all three mechanisms in isolated and cross-linked cellobiose are reported in Table 1. For pure cellobiose the reaction energies for transglycosylation and ring contraction are 6.54 kcal mol−1 and 15.1 kcal mol−1, respectively while that for the 2-step ring opening mechanism are 18.0 kcal mol−1 and 0.79 kcal mol−1. These reaction energies are in excellent agreement with previously published DFT calculations.58 However, the LCC cross-linked cellobiose exhibits significantly higher reaction energies, similar to the large deviation observed in activation energies. The reaction energy for transglycosylation in cellobiose cross-linked at the C2 and C3 positions are 59.85 kcal mol−1 and 61.57 kcal mol−1 while for ring contraction cross-linked at the C3 and C6 positions are 68.9 kcal mol−1 and 65.13 kcal mol−1, respectively. For the ring opening, the reaction energies of the first dehydration step for LCC conformers binding at C2, C3 and C6 positions are 61.45 kcal mol−1, 68.76 kcal mol−1 and 63.56 kcal mol−1 while for the second step it is 12.49 kcal mol−1, 14.3 kcal mol−1 and 14.89 kcal mol−1, respectively. The calculation of conformational changes in LCC molecules revealed a maximum change of ∼15 kcal mol−1. Despite considering a conformational penalty (which we have minimized using the sampling), the reaction energies observed in LCC were found to be more than 40 kcal mol−1 higher compared to those in pure cellobiose. These reaction energies follow the same trend as the activation barriers with only the second ring opening step having lower values compared to the energetics in isolated cellobiose. This indicates that it is not the preferential destabilization of the TS that leads to these high barriers for cellulose decomposition in LCCs. The lignin moiety in LCC seems to shield the cross-linked cellobiose leading to a higher barrier for cellulose cleavage via conventional reaction mechanisms. These representative reaction mechanisms were proposed for the cleavage of cellulose and its oligomers. However, for the cleavage of cross-linked cellobiose in LCC, there could potentially be more energetically favorable mechanisms. The changes in relative stabilities of cross-linked cellobiose (evident from the higher barriers compared to pure cellobiose) can be evaluated using the quantum chemical indicators calculated using the frontier molecular orbitals.63 Electron transfer between/within the reactants is most likely to occur in the frontier orbital and they have been widely used in predicting activity,64 absorption selectivity,63 binding properties65 and so on. In this work, the frontier orbital in the cellobiose and LCC molecules are visualized to investigate the electronic structure's contributions to the higher barriers for the cleavage of cellulose moiety in LCC.
| Reaction mechanism | Cellobiose | LCC | ||
|---|---|---|---|---|
| C2 | C3 | C6 | ||
| Transglycosylation | 6.54 | 59.85 | 61.57 | — |
| Ring contraction | 15.10 | — | 68.90 | 65.13 |
| Ring opening | 18.00 | 61.45 | 68.76 | 63.56 |
| 0.79 | 12.49 | 14.31 | 14.89 | |
Within the framework of frontier molecular orbital theory, the highest energy level occupied orbital is denoted as the highest occupied molecular orbital (HOMO), while the lowest energy level unoccupied orbital is referred to as the lowest unoccupied molecular orbital (LUMO). HOMO reflects the ability to give electrons, and LUMO reflects the ability to accept electrons. The exchange of frontier electrons significantly influences reactions since the interaction between the HOMO and LUMO also correlates with the energy barrier.66,67Fig. 3 shows the frontier orbitals, HOMO and LUMO, for cellobiose and cross-linked cellobiose in LCC. In an isolated cellobiose, both the frontier orbitals are spatially close and are located on the cellobiose molecule. In the LCC molecule, while the LUMO is on the cellobiose moiety, the HOMO is located away on the nucleophilic lignin moiety. The chemical reaction is likely to occur in the position and direction where HOMO and LUMO overlap effectively.68 By examining the spatial extent and localization of the HOMO and LUMO orbitals, specific regions or atoms within a molecule that are more reactive or prone to participate in chemical reactions can be identified. These regions are often associated with higher electron density in the HOMO or regions where the LUMO has significant overlap, indicating favorable sites for electron transfer or bond formation. The sites of HOMO and LUMO orbitals in the cellobiose molecule match with previous calculations investigating adsorption selectivity in anticorrosion coating with biopolymer extracts (specifically cellobiose).63 These favorable sites are active centers that contribute to covalent bonding in cellulose. Unlike the spatial overlap of the HOMO–LUMO orbitals in the isolated cellobiose molecule, in the LCC molecule, they are spatially far. Moreover, the localization of the HOMO orbital on the electron rich lignin and LUMO orbital on the cellobiose moiety suggests that electron transfer between the lignin and cellobiose moiety might be favored over intra-moiety electron transfer. In addition to the position and direction of HOMO and LUMO overlap, the HOMO–LUMO gap has been used to study the molecular stability and activity for cellulose and related systems.64,69,70 The magnitude of the HOMO–LUMO gap correlates with the level of HOMO–LUMO interaction and stability71 in the reaction and a larger HOMO–LUMO gap is indicative of greater kinetic stability and diminished chemical reactivity.63,72 Therefore, the lower HOMO–LUMO gap in cross-linked cellobiose (cf.Fig. 3) as compared to pure cellobiose seems to suggest higher reactivity meaning lower activation barrier. Since the activation barriers are higher for the intra-moiety cellobiose reaction mechanisms reported in Fig. 2, these are possibly not the mechanism through which cross-linked cellulose cleaves. The HOMO–LUMO energy gap is indicative of the inter-moiety reaction but not that of the activity of intra-moiety reaction mechanisms studied here. The HOMO orbital shifting from the cellobiose moiety to lignin moiety in LCC supports this. New mechanisms for cross-linked cellobiose cleavage involving atoms in the lignin and cellobiose moiety could possibly be more favored. Cellobiose decomposition mechanisms involving just the cellobiose moiety investigated here have higher energy penalty as they require the HOMO to be on the cellobiose moiety. However, the highest energy occupied molecular orbital is on lignin moiety which can possibly explain the higher barrier for cross-linked cellulose decomposition calculated in LCCs.
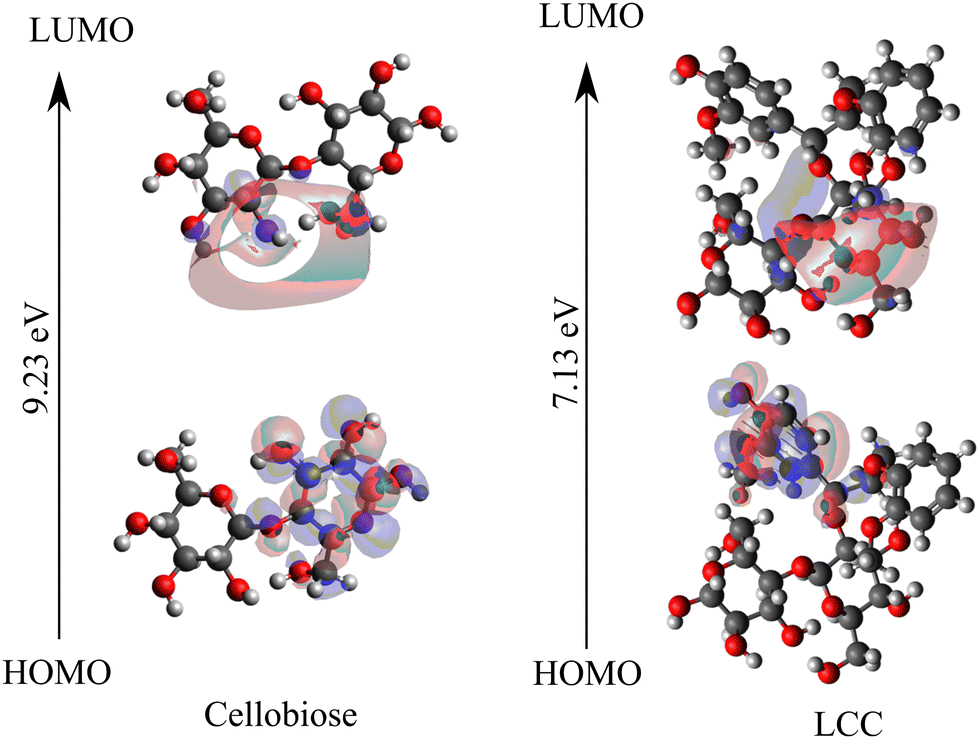 | ||
| Fig. 3 HOMO–LUMO orbitals are visualized using frontier orbital analysis performed on isolated cellobiose and cross-linked cellobiose in LCC. | ||
The reported activation barriers for the three mechanisms produce three major bio-oil components – anhydrosugars (LGA), furans (5-HMF) and lower oxygenates (GA). To validate these first principles calculations and the corresponding activation barriers, thin-film pyrolysis experiments were conducted on bagasse as model biomass for cross-linked cellulose. The product distribution and bio-oil composition are then compared to previously reported thin-film pyrolysis of pure cellulose.73 This enables comparison between the first principles barriers and the experimental yields of the three bio-oil components.
3.3 Thin-film pyrolysis experiment product yields for pure cellulose and cross-linked cellulose in native biomass (bagasse)
In this section the overall product yield, including the percentages of non-condensable gases, bio-oil, and char, as well as the individual yields of bio-oil components such as anhydrosugars (including LGA), furans, and light oxygenates (including glycolaldehyde) are examined. The effect of cross-linked cellulose on the yields of these bio-oil components, non-condensable gases, and char is highlighted. Further, the experimental data from thin-film pyrolysis provides valuable insights into the thermal decomposition pathways of both cross-linked and pure cellulose. These findings are then compared to first principles calculations to establish parallels between the experimental and theoretical results. The pyrolysis of bagasse thin films in the temperature range of 573–773 K resulted in the production of non-condensable gases (1.3–3.9 wt%), bio-oil (36–69 wt%), and char (17–45 wt%) as major products, as shown in Table 2. The total yield of pyrolysis products, including non-condensable gases, bio-oil, and char, ranged from 86 to 93 wt%, the carbon balance is consistent with previous experiments.56 These pyrolysis yields for bagasse are compared to those reported for cellulose thin-films under similar pyrolysis conditions.73 Increasing temperature resulted in a marginal increase in non-condensable gas production in bagasse (1.3–3.9 wt%) and a prominent increase in pure cellulose (0.12–7.04 wt%). Both materials showed a decreasing trend in char yield with higher pyrolysis temperatures, with bagasse thin-films yielding more char (44.04 to 17.77) than pure cellulose (32.58 to 8.79). The condensable volatile products (boi-oil) during bagasse pyrolysis had higher yields at elevated temperatures, while the char yield decreased, competing with other pyrolysis products.56 However, while the trends in char/bio-oil behavior are similar between cellulose and bagasse pyrolysis, bagasse containing cross-linked cellulose (in LCC) exhibits significantly higher char yields (> +7wt% across all temperatures) compared to pure cellulose. The reduced bio-oil yield in bagasse aligns with the higher activation barriers calculated for decomposition of cellobiose with LCC, compared to pure cellobiose, as discussed in Section 3.2. Further, the major components of bio-oil, including LGA, 5-HMF, and GA, which are products of transglycosylation, ring contraction, and ring opening mechanisms, respectively, were considered for comparison between experimental data and first principles calculations. To facilitate this comparison, the chemical compounds present in bio-oil derived from bagasse were categorized into anhydrosugars, furans, light oxygenates, and phenolic compounds. The formation of anhydrosugars (e.g., levoglucosan), furans (e.g., 5-HMF), and lower oxygenates (e.g., glycolaldehyde) can be attributed to the three mechanisms investigated using first principles calculations. Furthermore, phenolic compounds derived from lignin are also found in bagasse bio-oil.| Temperature (K) | Bagasse product distribution | Cellulose product distribution | ||||
|---|---|---|---|---|---|---|
| Gases (wt%) | Bio-oil (wt%) | Char (wt%) | Gases (wt%) | Bio-oil (wt%) | Char (wt%) | |
| 573 | 1.34 ± 0.08 | 36.74 ± 0.14 | 44.04 ± 0.45 | 0.12 ± 0.01 | 61.16 ± 0.22 | 32.58 ± 0.48 |
| 623 | 1.48 ± 0.05 | 56.68 ± 0.24 | 32.86 ± 0.43 | 2.07 ± 0.013 | 69.47 ± 0.88 | 25.53 ± 0.29 |
| 673 | 2.38 ± 0.06 | 62.36 ± 0.33 | 27.59 ± 0.35 | 5.35 ± 0.07 | 76.23 ± 0.1 | 17.12 ± 0.2 |
| 723 | 2.8 ± 0.05 | 66.23 ± 0.11 | 22.96 ± 0.93 | 6.41 ± 0.04 | 82.81 ± 0.1 | 11.13 ± 0.9 |
| 773 | 3.85 ± 0.29 | 68.44 ± 0.12 | 17.77 ± 0.15 | 7.04 ± 0.07 | 84.99 ± 0.1 | 8.79 ± 0.12 |
The yields of bio-oil components from bagasse and pure cellulose, are measured as a percentage of their carbohydrate content. Biomass pyrolysis research has focused on cellulose-based materials due to their abundance in biomass and their potential impact on bio-oil yield and composition.9,56,74 In this context, comparing the decomposition of bagasse with that of cellulose under similar reaction conditions is valuable. The findings reveal that within the temperature range of 573–773 K, the anhydrosugar yield in bagasse pyrolysis decreased from 16 wt% to 6.85 wt%, while the yields of furans and lower oxygenates increased from 6.57 wt% to 13.22 wt% and from 0 wt% to 34.16 wt%, respectively (cf.Fig. 4). The results also demonstrate that furans derived from bagasse thin-film pyrolysis exhibit a similar temperature-dependent trend as observed in cellulose thin-film pyrolysis. However, the anhydrosugar yields in bagasse were significantly lower compared to cellulose pyrolysis (ranging from 34 wt% to 48 wt%). This study further highlights the influence of cross-linked cellulose with lignin in lignocellulosic complexes (LCCs) on anhydrosugar yields, which are compensated by increased C1–C3 product yields (lower oxygenates). Moreover, the compensation by lower oxygenates is also supported by first-principles calculated reaction energies reported in Table 1. The reaction energy is more endergonic (∼10 kcal mol−1) for the formation of glycolaldehyde compared to LGA or furan, making it more thermodynamically favorable at higher temperatures. A recent publication50 investigating the impact of cross-linking in fast pyrolysis of lignocellulosic biomass presents the only notable comparability to the findings of this study. The selective removal of lignin or hemicellulose from treated biomass led to significant changes in LCC composition, affecting the yields of levoglucosan (LGA) and C1–C3 products, in alignment with the results obtained from pure cellulose.73 The observed trends in product yields from bagasse pyrolysis correspond with the literature for untreated herbaceous biomass, particularly in terms of bio-oil compounds, anhydrosugars, and light oxygenates.
These pyrolysis products originate from cellulose; however, it is noteworthy that bagasse pyrolysis also results in the formation of phenolic compounds in bio-oil. The yields of phenolic compound in the bagasse bio-oil are presented in Table 3. These results indicate that the phenolic yield reduces from 53.9 wt% to 27.87 wt% as temperature increases from 573 to 773 K. Moreover, phenolic compounds are the predominant products at these specified temperature ranges when compared to anhydrosugars and furans, with the exception of lower oxygenates. The yield of lower oxygenates surpasses that of phenolic compounds only beyond 673 K, due to secondary reactions. An important observation was that the weight fraction of phenolic compounds in the bio-oil vastly exceeded the weight fraction of lignin in the original bagasse biomass. This discrepancy is attributed to the presence of carbohydrate derived fragments within the phenolic compounds and aligns with earlier studies indicating a greater production of “lignin-derived” products in native biomass compared to the co-pyrolysis of cellulose and lignin.27 This suggests that the breakdown of lignin in the LCC involves not only lignin itself but also the cleavage of cellulose and hemicellulose molecules.
| Temperature (K) | Phenolic compounds (wt%) |
|---|---|
| 573 | 53.9 |
| 623 | 39.3 |
| 673 | 32.17 |
| 723 | 32.17 |
| 773 | 27.87 |
3.4 Reaction kinetics and thermochemistry for cross-linked cellulose (in LCC) decomposition
We have reported a significant increase in the activation barrier (∼2X) for the formation of three major pyrolysis products in cross-linked cellobiose (LCC) as compared to pure cellobiose (cf.Fig. 2). Additionally, cross-linking between lignin and cellulose also seems to alter the thermodynamics of the reaction. The reaction energies (cf.Table 1) also increase significantly (∼3X–4X). Probing primary decomposition reaction kinetics experimentally is limited by the timescale difference between product evolution and analysis. However, the apparent activation energies of product formations calculated from millisecond scale kinetics can be used as reactivity criteria for comparing different biomass feedstocks and relative rates of product formations. The only millisecond scale data available is for woody biomass (loblolly)75 which has been suggested to have significantly low number of LCC linkages as compared to herbaceous biomass. Consequently, the overall kinetics between pure cellulose and such woody biomass exhibits minimum difference as it's the LCC linkage that alters product yields and not the mere presence of lignin.50 Such millisecond scale experimental kinetics is currently unavailable for herbaceous biomass hindering the investigation of the direct role of cross-linked cellulose on its decomposition kinetics and thermochemistry. However, as mentioned in the previous section, product yields from the pyrolysis of chemically treated and untreated native biomass reveals a drastic shift in LGA and C1–C3 product yields. The difference in product yields was suggested to be because of the LCC linkage being made at the glycosyl C6 position that hinders the formation of C6–O–C1 bridge resulting in less efficient release of LGA end-groups.29 However, other thermodynamically feasible LCCs48 made at 3 linkage sites (LCC-C2, LCC-C3, LCC-C6) exhibited comparable activation barriers (cf.Fig. 2) for cellobiose decomposition with LCC-C3 having marginally higher barriers. Further details about these minimal deviations among different LCC sites can be found in the ESI.†The increased experimental yield of light oxygenates compared to LGA could be a result of altered kinetics, thermochemistry and/or reaction pathway. The higher reaction energy (∼10–15 kcal mol−1) and the endergonic nature of GA (light oxygenate) compared to LGA indicate that the formation of GA will be relatively more favoured at higher temperatures. Higher temperatures also facilitate the overcoming of kinetic barriers, making the more endergonic formation of light oxygenates favoured. Moreover, in addition to the enhancement of kinetic or thermochemistry, the breakdown of cellulose in LCC molecules could involve alternate reaction pathways, such as ring opening and dehydration rearrangement, leading to the formation of C1–C3 small molecules.76 The lower HOMO–LUMO energy gap and the position of the HOMO orbital on electron-rich phenyl groups suggest that inter-moiety mechanisms could be more favourable. These findings indicate that the difference in product distribution during the pyrolysis of cross-linked cellobiose, compared to pure cellobiose, not only reflects a change in kinetics but also potentially a difference in reaction pathway. To gain further insights into alternate chemistry and kinetics, one can draw parallels between the experimental yields measured in this study and first principles calculated activation barriers. The presence of LCC linkages can alter kinetic and thermodynamic parameters for cellulose cleavage, such as reaction energy and activation barriers for the same reaction mechanism. Alternatively, the LCC linkage can promote alternate reaction pathways (other than cleavage of glycosidic linkage in cellulose as the first step in cellulose decomposition), thereby changing the ultimate product yields. Comparing the relative differences in product yields between cellulose and lignocellulosic complexes (LCCs), such as bagasse, provides qualitative insights to distinguish between the effects of altered kinetics or altered reaction pathway and reaction pathways. Analysis of Fig. 4 shows a consistent gap in the yields of anhydrosugars between bagasse and cellulose (with a constant relative yield), indicating kinetic inhibition in LCCs while maintaining the reaction mechanism for LGA formation. For C1–C3 products, both relative and absolute yields change, indicating changes in both kinetics and reaction pathways. On the other hand, the absolute and relative yields of furans are comparable, suggesting that the reaction pathway and kinetics of furan formation during cellulose decomposition remain unchanged in both bagasse and cellulose. This also indicates that the LCC linkage only affects the primary decomposition of cellulose, as a significant portion of furans is formed through secondary reactions during cellulose pyrolysis,19 which remain unaffected. The increased presence of phenolic compounds in the bio-oil resulting from bagasse pyrolysis supports the notion of inter-moiety cleavage. The higher proportion of phenolics compared to the original lignin content in the biomass suggests that the interaction between lignin and carbohydrate within the LCC molecule leads to remnants of cellulose and hemicellulose molecules in the formed phenolic products. Hosoya et al. previously introduced a mechanistic model to clarify the elevated presence of lignin-derived products, including phenols, guaiacols, and syringols, in cellulose–lignin mixtures.27 In this model, volatile compounds originating from cellulose serve as hydrogen donors, while volatiles derived from lignin, in their radical form, function as hydrogen acceptors.77 This model provides a compelling illustration of inter-moiety interactions in which carbohydrates function as hydrogen donors, leaving behind segments of cellulose/hemicellulose chains in phenolic products. This explanation accounts for the relatively lower yields of anhydrosugars and furans compared to the generation of lignin-derived phenolic products in synthetic cellulose–lignin mixtures. It's worth noting that this phenomenon may be further accentuated in native biomass due to the covalent LCC linkage between cellulose and lignin, with depolymerization involving the cleavage of lignin alongside cellulose molecules through inter-moiety mechanisms leaving cellulose/hemicellulose fragments in phenolic products. Furthermore, the modification of lignin functional groups has been observed to facilitate lignin depolymerization.78 Similarly, the interaction between lignin and carbohydrates in LCCs could induce novel chemical reactions, such as inter-moiety mechanisms, leading to cellulose cleavage involving lignin depolymerization.
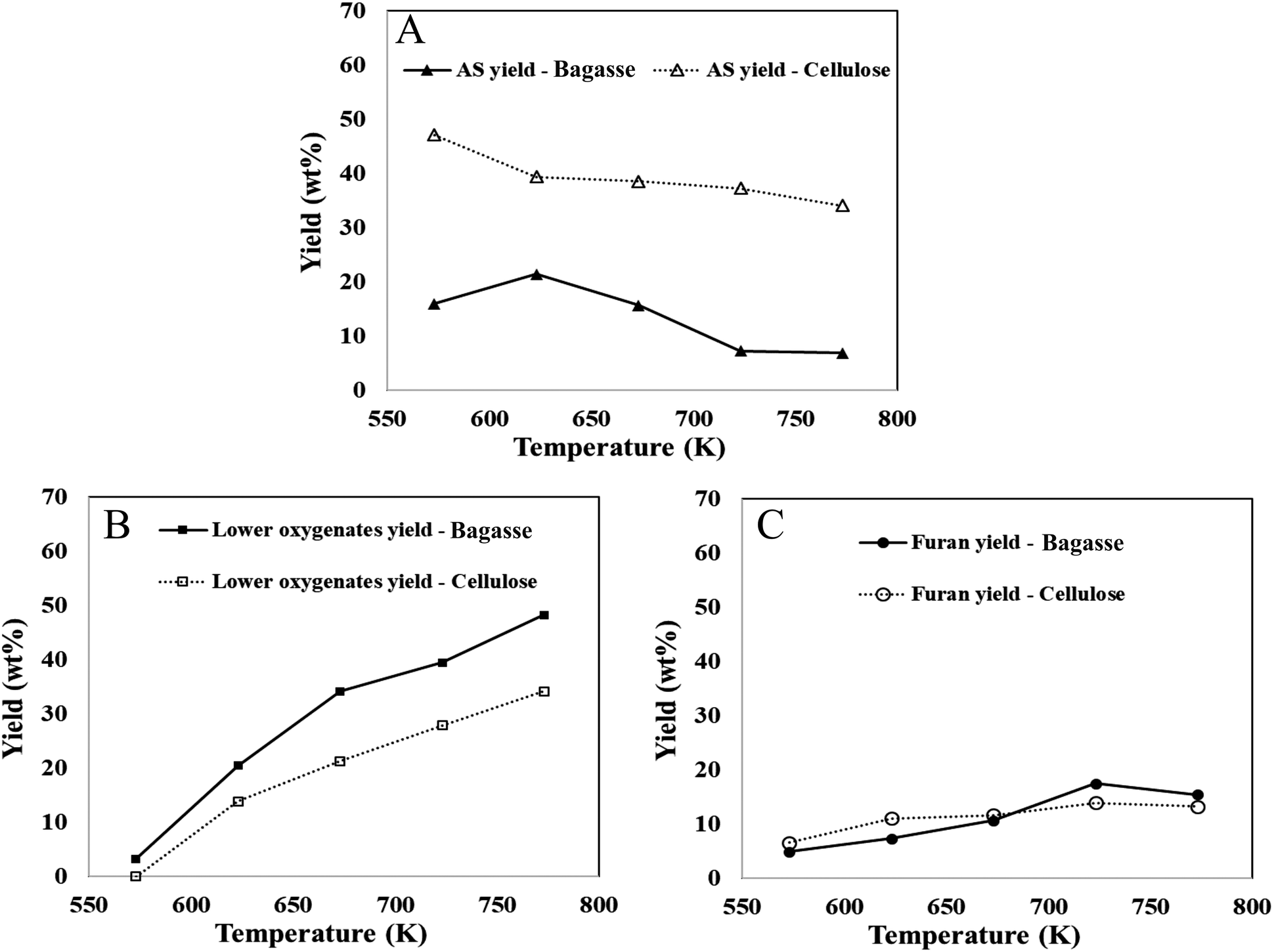 | ||
| Fig. 4 Yields (wt%) of bio-oil components ((A) anhydrosugars, (B) lower oxygenates, (C) furans) in the thin-film pyrolysis of native biomass, bagasse (solid line) and that of pure cellulose (dash line). | ||
4 Conclusions
This study investigates the role of cross-linked cellulose with lignin in lignin–carbohydrate complexes (LCC) during cellulose decomposition, focusing on its effects on reaction kinetics, thermochemistry, and reaction pathways. Ab initio molecular dynamics and metadynamics simulations are employed to model LCC molecules with β-O-4 benzyl ether linkages, connecting cellulose and lignin dimers (cellobiose and quinone methide intermediate) at thermodynamically feasible positions in different conformations. First principles density functional theory (DFT) calculations are then conducted to screen for the lowest energy conformers and to determine the transition states for three major reaction mechanisms (transglycosylation, ring contraction, ring opening) producing bio-oil components (levoglucosan, 5-hydroxymethylfurfural, glycolaldehyde) at 500 K. Activation barriers, reaction energies, and frontier molecular orbital interactions are analyzed to gain insights into the role of LCC linkages in cellulose decomposition. Experimental measurements of anhydrosugars, furans, and lower oxygenates in native herbaceous biomass (bagasse) pyrolysis are compared with yields from pure cellulose pyrolysis. The calculated activation barriers and experimental product yields provide evidence of different kinetics, thermochemistry and potentially reaction pathways induced by cross-linked lignin in LCC. Notably, higher activation barriers and reaction energies are observed for cross-linked cellobiose cleavage in LCC compared to pure cellobiose, indicating altered kinetics/thermochemistry. The higher endergonic nature (reaction energy) of GA formation in comparison to LGA suggests that cellulose decomposition can be effectively promoted at higher temperatures, favoring the production of lighter oxygenates, particularly GA. This is in strong agreement with the increased relative yields of lower oxygenates over anhydrosugars in bagasse. In addition, the preference for inter-moiety mechanisms over intra-moiety cellulose decomposition is indicated by the high activation barrier for the intra-moiety mechanism and the low HOMO–LUMO energy gap. Comparison of relative differences in product yields between bagasse and cellulose provides additional evidence supporting the presence of alternative reaction pathways. This combined computational and experimental study sheds light on the distinct role played by cross-linked lignin–carbohydrate bonding in influencing reaction kinetics, thermochemistry and mechanisms of cellulose decomposition.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Arul Mozhi Devan Padmanathan was responsible for the conceptualization, methodology, validation, performing simulations, investigation, data analysis, writing, review, and editing. Seth Beck performed CPMD-metadynamics simulations for conformation search. Dr Khursheed B. Ansari was responsible for experimental methodology and performing thin-film pyrolysis experiments. Dr S. H. Mushrif was the supervisory author in the conceptualization, investigation, review, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by funding from the Canada First Research Excellence Fund as part of the University of Alberta's Future Energy Systems Research Initiative, the Natural Sciences and Engineering Research Council of Canada (NSERC) via their Discovery Grants program and the Digital Research Alliance of Canada provided computational resources.References
- S. Kudo, X. Huang, S. Asano and J. Hayashi, Catalytic Strategies for Levoglucosenone Production by Pyrolysis of Cellulose and Lignocellulosic Biomass, Energy Fuels, 2021, 35, 9809–9824, DOI:10.1021/acs.energyfuels.1c01062.
- K. Sanderson, A field in ferment, Nature, 2006, 444, 673–676 CrossRef CAS PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak and C. L. Liotta, The path forward for biofuels and biomaterials, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- H. B. Mayes and L. J. Broadbelt, Unraveling the Reactions that Unravel Cellulose, J. Phys. Chem. A, 2012, 116, 7098–7106, DOI:10.1021/jp300405x.
- S. Mushrif, Multiscale molecular modeling can be an effective tool to aid the development of biomass conversion technology: A perspective, Chem. Eng. Sci., 2015, 121, 217–235, DOI:10.1016/j.ces.2014.08.019.
- D. F. Arseneau, Competitive Reactions in the Thermal Decomposition of Cellulose, Can. J. Chem., 1971, 49, 632–638, DOI:10.1139/v71-101.
- I. Milosavljevic, V. Oja and E. M. Suuberg, Thermal Effects in Cellulose Pyrolysis: Relationship to Char Formation Processes, Ind. Eng. Chem. Res., 1996, 35, 653–662, DOI:10.1021/ie950438l.
- R. S. Assary and L. A. Curtiss, Thermochemistry and reaction barriers for the formation of levoglucosenone from cellobiose, ChemCatChem, 2012, 4, 200–205 CrossRef CAS.
- M. Mettler, S. Mushrif, A. Paulsen, A. Javadekar, D. Vlachos and P. Dauenhauer, Revealing pyrolysis chemistry for biofuels production: Conversion of cellulose to furans and small oxygenates, Energy Environ. Sci., 2012, 5, 5414–5424, 10.1039/C1EE02743C.
- J. S. Arora, J. W. Chew and S. H. Mushrif, Influence of Alkali and Alkaline-Earth Metals on the Cleavage of Glycosidic Bond in Biomass Pyrolysis: A DFT Study Using Cellobiose as a Model Compound, J. Phys. Chem. A, 2018, 122, 7646–7658, DOI:10.1021/acs.jpca.8b06083.
- V. Agarwal, P. J. Dauenhauer, G. W. Huber and S. M. Auerbach, Ab Initio Dynamics of Cellulose Pyrolysis: Nascent Decomposition Pathways at 327 and 600 °C, J. Am. Chem. Soc., 2012, 134, 14958–14972, DOI:10.1021/ja305135u.
- A. M. D. Padmanathan and S. H. Mushrif, Pyrolytic activation of cellulose: energetics and condensed phase effects, React. Chem. Eng., 2022, 7, 1136–1149, 10.1039/D1RE00492A.
- J. Piskorz, D. Radlein and D. S. Scott, On the mechanism of the rapid pyrolysis of cellulose, J. Anal. Appl. Pyrolysis, 1986, 9, 121–137 CrossRef CAS.
- T. Hosoya, Y. Nakao, H. Sato, H. Kawamoto and S. Sakaki, Thermal degradation of methyl beta-D-glucoside. a theoretical study of plausible reaction mechanisms, J. Org. Chem., 2009, 74, 6891–6894, DOI:10.1021/jo900457k.
- J. Huang, D. Wu, H. Tong and L. Ren, Density functional theory studies on pyrolysis mechanism of β-O-4 type lignin dimer model compound, J. Anal. Appl. Pyrolysis, 2014, 109, 98–108, DOI:10.1016/j.jaap.2014.07.007.
- M. J. Climent, A. Corma and S. Iborra, Converting carbohydrates to bulk chemicals and fine chemicals over heterogeneous catalysts, Green Chem., 2011, 13, 520–540, 10.1039/C0GC00639D.
- F. Momany, M. Appell, G. Strati and J. Willett, B3LYP/6-311++G** study of monohydrates of α- and β-D-glucopyranose: Hydrogen bonding, stress energies, and effect of hydration on internal coordinates, Carbohydr. Res., 2004, 339, 553–567, DOI:10.1016/j.carres.2003.10.013.
- J. Banyasz, S. Li, J. Lyons-Hart and K. Shafer, Cellulose pyrolysis: The kinetics of hydroxyacetaldehyde evolution, J. Anal. Appl. Pyrolysis, 2001, 57, 223–248, DOI:10.1016/S0165-2370(00)00135-2.
- Q. Wang, H. Song, S. Pan, N. Dong, X. Wang and S. Sun, Initial pyrolysis mechanism and product formation of cellulose: An Experimental and Density functional theory(DFT) study, Sci. Rep., 2020, 10, 3626, DOI:10.1038/s41598-020-60095-2.
- M. R. Nimlos, S. J. Blanksby, X. Qian, M. E. Himmel and D. K. Johnson, Mechanisms of glycerol dehydration, J. Phys. Chem. A, 2006, 110, 6145–6156, DOI:10.1021/jp060597q.
- S. Mishra, P. K. Singh, S. Dash and R. Pattnaik, Microbial pretreatment of lignocellulosic biomass for enhanced biomethanation and waste management, 3 Biotech., 2018, 8, 458, DOI:10.1007/s13205-018-1480-z.
- R. S. Assary and L. A. Curtiss, Comparison of Sugar Molecule Decomposition through Glucose and Fructose: A High-Level Quantum Chemical Study, Energy Fuels, 2012, 26, 1344–1352, DOI:10.1021/ef201654s.
- Y. Zhang, C. Liu and X. Chen, Unveiling the initial pyrolytic mechanisms of cellulose by DFT study, J. Anal. Appl. Pyrolysis, 2015, 113, 621–629, DOI:10.1016/j.jaap.2015.04.010.
- A. Demirbas, The influence of temperature on the yields of compounds existing in bio-oils obtained from biomass samples via pyrolysis, Fuel Process. Technol., 2007, 88, 591–597, DOI:10.1016/j.fuproc.2007.01.010.
- J. Yu, N. Paterson, J. Blamey and M. Millan, Cellulose, xylan and lignin interactions during pyrolysis of lignocellulosic biomass, Fuel, 2017, 191, 140–149, DOI:10.1016/j.fuel.2016.11.057.
- S. Wang, X. Guo, K. Wang and Z. Luo, Influence of the interaction of components on the pyrolysis behavior of biomass, J. Anal. Appl. Pyrolysis, 2011, 91, 183–189, DOI:10.1016/j.jaap.2011.02.006.
- S. Wu, D. Shen, J. Hu, H. Zhang and R. Xiao, Cellulose–lignin interactions during fast pyrolysis with different temperatures and mixing methods, Biomass Bioenergy, 2016, 90, 209–217, DOI:10.1016/j.biombioe.2016.04.012.
- L. Zhu and Z. Zhong, Effects of cellulose, hemicellulose and lignin on biomass pyrolysis kinetics, Korean J. Chem. Eng., 2020, 37, 1660–1668 CrossRef CAS.
- J. Zhang, Y. S. Choi, C. G. Yoo, T. H. Kim, R. C. Brown and B. H. Shanks, Cellulose–Hemicellulose and Cellulose–Lignin Interactions during Fast Pyrolysis, ACS Sustainable Chem. Eng., 2015, 3, 293–301, DOI:10.1021/sc500664h.
- M. Lawoko, G. Henriksson and G. Gellerstedt, Structural differences between the lignin-carbohydrate complexes present in wood and in chemical pulps, Biomacromolecules, 2005, 6, 3467–3473, DOI:10.1021/bm058014q.
- G. Henriksson, M. Lawoko, M. Eugenio and G. Gellerstedt, Lignin-carbohydrate network in wood and pulps: A determinant for reactivity, Holzforschung, 2007, 61, 668–674, DOI:10.1515/HF.2007.097.
- Z. Jin, K. S. Katsumata, T. B. T. Lam and K. Iiyama, Covalent linkages between cellulose and lignin in cell walls of coniferous and nonconiferous woods, Biopolym. Orig. Res. Biomol., 2006, 83, 103–110 CAS.
- M. Y. Balakshin, E. A. Capanema and H. Chang, MWL fraction with a high concentration of lignin-carbohydrate linkages: Isolation and 2D NMR spectroscopic analysis, Holzforschung, 2007, 61, 1–7, DOI:10.1515/HF.2007.001.
- J. S. Kim, Y. Y. Lee and T. H. Kim, A review on alkaline pretreatment technology for bioconversion of lignocellulosic biomass, Bioresour. Technol., 2016, 199, 42–48, DOI:10.1016/j.biortech.2015.08.085.
- T. B.-T. Lam, K. Iiyama and B. A. Stone, Hot alkali-labile linkages in the walls of the forage grass Phalaris aquatica and Lolium perenne and their relation to in vitro wall digestibility, Phytochemistry, 2003, 64, 603–607, DOI:10.1016/s0031-9422(03)00301-7.
- X. Zhao, F. Qi and D. Liu, Hierarchy Nano- and Ultrastructure of Lignocellulose and Its Impact on the Bioconversion of Cellulose, in Nanotechnology for Bioenergy and Biofuel Production, ed. M. Rai and S. da Silva, Green Chemistry and Sustainable Technology, Springer, Cham, 2017, pp. 117–151 DOI:10.1007/978-3-319-45459-7_6.
- V. Silva, H. Jameel, F. Gomes, L. Batalha, M. Coura and J. Colodette, Effect of Lignin Carbohydrate Complexes of Hardwood Hybrids on the Kraft Pulping Process, J. Wood Chem. Technol., 2016, 37, 1–10, DOI:10.1080/02773813.2016.1235584.
- Ö. Eriksson, D. A. I. Goring and B. O. Lindgren, Structural studies on the chemical bonds between lignins and carbohydrates in spruce wood, Wood Sci. Technol., 1980, 14, 267–279, DOI:10.1007/BF00383454.
- B. Košíková and A. Ebringerová, Lignin-carbohydrate bonds in a residual soda spruce pulp lignin, Wood Sci. Technol., 1994, 28, 291–296, DOI:10.1007/BF00204215.
- M. Lawoko, Lignin polysaccharide networks in softwood and chemical pulps: characterisation, structure and reactivity, PhD thesis, KTH, 2005, https://kth.diva-portal.org/smash/get/diva2:12959/FULLTEXT01.pdf Search PubMed.
- S. C. Fry, Plant cell walls. From chemistry to biology, Ann. Bot., 2011, 108, viii–ix, DOI:10.1093/aob/mcr128.
- N. Giummarella, Y. Pu, A. J. Ragauskas and M. Lawoko, A critical review on the analysis of lignin carbohydrate bonds, Green Chem., 2019, 21, 1573–1595, 10.1039/C8GC03606C.
- Y. Miyagawa, Y. Tobimatsu, P. Y. Lam, T. Mizukami, S. Sakurai, H. Kamitakahara and T. Takano, Possible mechanisms for the generation of phenyl glycoside-type lignin–carbohydrate linkages in lignification with monolignol glucosides, Plant J., 2020, 104, 156–170, DOI:10.1111/tpj.14913.
- Y. Zhou, H. Stuart-Williams, G. D. Farquhar and C. H. Hocart, The use of natural abundance stable isotopic ratios to indicate the presence of oxygen-containing chemical linkages between cellulose and lignin in plant cell walls, Phytochemistry, 2010, 71, 982–993, DOI:10.1016/j.phytochem.2010.03.001.
- J. W. Choi, D.-H. Choi and O. Faix, Characterization of lignin-carbohydrate linkages in the residual lignins isolated from chemical pulps of spruce (Picea abies) and beech wood (Fagus sylvatica), J. Wood Sci., 2007, 53, 309–313, DOI:10.1007/s10086-006-0860-x.
- N. Giummarella, L. Zhang, G. Henriksson and M. Lawoko, Structural features of mildly fractionated lignin carbohydrate complexes (LCC) from spruce, RSC Adv., 2016, 6, 42120–42131, 10.1039/C6RA02399A.
- N. Giummarella and M. Lawoko, Structural Insights on Recalcitrance during Hydrothermal Hemicellulose Extraction from Wood, ACS Sustainable Chem. Eng., 2017, 5, 5156–5165, DOI:10.1021/acssuschemeng.7b00511.
- J. Hu, B. Jiang, J. Liu, Y. Sun and X. Jiang, Influence of interactions between biomass components on physicochemical characteristics of char, J. Anal. Appl. Pyrolysis, 2019, 144, 104704 CrossRef CAS.
- P. R. Patwardhan, J. A. Satrio, R. C. Brown and B. H. Shanks, Product distribution from fast pyrolysis of glucose-based carbohydrates, J. Anal. Appl. Pyrolysis, 2009, 86, 323–330, DOI:10.1016/j.jaap.2009.08.007.
- Y. Zhang, W. Xu, N. Ma, Y. Shen, F. Xu, Y. Wang, N. Wu, Z. Guo and L. Jiang, Revealing the key role of structural cross-link between lignin and polysaccharides during fast pyrolysis of lignocellulose, Bioresour. Technol., 2022, 361, 127714, DOI:10.1016/j.biortech.2022.127714.
- S. Beck, P. Choi and S. H. Mushrif, Origins of Covalent Linkages within the Lignin Carbohydrate Network of Biomass, Phys. Chem. Chem. Phys., 2022, 24, 20480–20490, 10.1039/D2CP01683D.
- R. Car and M. Parrinello, Unified Approach for Molecular Dynamics and Density-Functional Theory, Phys. Rev. Lett., 1985, 55, 2471–2474, DOI:10.1103/PhysRevLett.55.2471.
- G. I. Csonka, A. D. French, G. P. Johnson and C. A. Stortz, Evaluation of Density Functionals and Basis Sets for Carbohydrates, J. Chem. Theory Comput., 2009, 5, 679–692, DOI:10.1021/ct8004479.
- M. D. Wodrich, C. Corminboeuf, P. R. Schreiner, A. A. Fokin, P. von and R. Schleyer, How Accurate Are DFT Treatments of Organic Energies?, Org. Lett., 2007, 9, 1851–1854, DOI:10.1021/ol070354w.
- J. S. Arora, K. B. Ansari, J. W. Chew, P. J. Dauenhauer and S. H. Mushrif, Unravelling the catalytic influence of naturally occurring salts on biomass pyrolysis chemistry using glucose as a model compound: a combined experimental and DFT study, Catal. Sci. Technol., 2019, 9, 3504–3524, 10.1039/C9CY00005D.
- A. Paulsen, M. Mettler and P. Dauenhauer, The Role of Sample Dimension and Temperature in Cellulose Pyrolysis, Energy Fuels, 2013, 27, 2126–2134, DOI:10.1021/ef302117j.
- K. B. Ansari, J. S. Arora, J. W. Chew, P. J. Dauenhauer and S. H. Mushrif, Effect of Temperature and Transport on the Yield and Composition of Pyrolysis-Derived Bio-Oil from Glucose, Energy Fuels, 2018, 32, 6008–6021, DOI:10.1021/acs.energyfuels.8b00852.
- J. S. Arora, J. W. Chew and S. H. Mushrif, Influence of Alkali and Alkaline-Earth Metals on the Cleavage of Glycosidic Bond in Biomass Pyrolysis: A DFT Study Using Cellobiose as a Model Compound, J. Phys. Chem. A, 2018, 122, 7646–7658, DOI:10.1021/acs.jpca.8b06083.
- C. Couhert, J.-M. Commandre and S. Salvador, Is it possible to predict gas yields of any biomass after rapid pyrolysis at high temperature from its composition in cellulose, hemicellulose and lignin, Fuel, 2009, 88, 408–417, DOI:10.1016/j.fuel.2008.09.019.
- Y.-H. P. Zhang, S.-Y. Ding, J. R. Mielenz, J.-B. Cui, R. T. Elander, M. Laser, M. E. Himmel, J. R. McMillan and L. R. Lynd, Fractionating recalcitrant lignocellulose at modest reaction conditions, Biotechnol. Bioeng., 2007, 97, 214–223, DOI:10.1002/bit.21386.
- J. Ralph, J. H. Grabber and R. D. Hatfield, Lignin-ferulate cross-links in grasses: active incorporation of ferulate polysaccharide esters into ryegrass lignins, Carbohydr. Res., 1995, 275, 167–178, DOI:10.1016/0008-6215(95)00237-N.
- Y. Zeng, S. Zhao, S. Yang and S.-Y. Ding, Lignin plays a negative role in the biochemical process for producing lignocellulosic biofuels, Curr. Opin. Biotechnol, 2014, 27, 38–45, DOI:10.1016/j.copbio.2013.09.008.
- W. Daoudi, A. El Aatiaoui, O. Dagdag, K. Zaidi, R. Haldhar, S.-C. Kim, A. Oussaid, A. Aouinti, A. Berisha, F. Benhiba, E. E. Ebenso and A. Oussaid, Anti-Corrosion Coating Formation by a Biopolymeric Extract of Artemisia herba-alba Plant: Experimental and Theoretical Investigations, Coatings, 2023, 13, 611, DOI:10.3390/coatings13030611.
- J. Fang, W. Zheng, K. Liu, H. Li and C. Li, Molecular design and experimental study on the synergistic catalysis of cellulose into 5-hydroxymethylfurfural with Brønsted–Lewis acidic ionic liquids, Chem. Eng. J., 2020, 385, 123796, DOI:10.1016/j.cej.2019.123796.
- K. Singh, A. Kumar, S. K. Pandey, S. Awasthi, S. P. Gupta and P. Mishra, Interpretation of Adsorption Behavior of Carboxymethyl Cellulose onto Functionalized Accurel Polymeric Surface, Ind. Eng. Chem. Res., 2020, 59, 19102–19116, DOI:10.1021/acs.iecr.0c03894.
- J.-D. Bradley and G.-C. Gerrans, Frontier molecular orbitals. A link between kinetics and bonding theory, J. Chem. Educ., 1973, 50, 463, DOI:10.1021/ed050p463.
- K. Fukui, Role of frontier orbitals in chemical reactions, Science, 1982, 218, 747–754, DOI:10.1126/science.218.4574.747.
- K. Fukui, The Role of Frontier Orbitals in Chemical Reactions (Nobel Lecture), Angew. Chem., Int. Ed. Engl., 1982, 21, 801–809, DOI:10.1002/anie.198208013.
- J. Cho, J. M. Davis and G. W. Huber, The intrinsic kinetics and heats of reactions for cellulose pyrolysis and char formation, ChemSusChem, 2010, 3, 1162–1165 CrossRef CAS PubMed.
- T. Vispute and G. Huber, Breaking the chemical and engineering barriers to lignocellulosic biofuels, Int. Sugar J., 2008, 110, 138–149 CAS.
- U. Azeem, R. A. Khera, A. Naveed, M. Imran, M. A. Assiri, M. Khalid and J. Iqbal, Tuning of a A–A–D–A–A-Type Small Molecule with Benzodithiophene as a Central Core with Efficient Photovoltaic Properties for Organic Solar Cells, ACS Omega, 2021, 6, 28923–28935, DOI:10.1021/acsomega.1c03975.
- R. Pontoh, V. E. Rarisavitri, C. Yang, M. F. Putra and D. S. B. Anugrah, Density Functional Theory Study of Intermolecular Interactions between Amylum and Cellulose, Indones. J. Chem., 2022, 22, 253, DOI:10.22146/ijc.69241.
- K. B. Ansari, J. S. Arora, J. W. Chew, P. J. Dauenhauer and S. H. Mushrif, Fast Pyrolysis of Cellulose, Hemicellulose, and Lignin: Effect of Operating Temperature on Bio-oil Yield and Composition and Insights into the Intrinsic Pyrolysis Chemistry, Ind. Eng. Chem. Res., 2019, 58, 15838–15852, DOI:10.1021/acs.iecr.9b00920.
- C. Hutchinson and Y. Jin Lee, Evaluation of Primary Reaction Pathways in Thin-Film Pyrolysis of Glucose Using 13C Labeling and Real-Time Monitoring, ACS Sustainable Chem. Eng., 2017, 5, 8796–8803, DOI:10.1021/acssuschemeng.7b01601.
- S. Maduskar, G. G. Facas, C. Papageorgiou, C. L. Williams and P. J. Dauenhauer, Five Rules for Measuring Biomass Pyrolysis Rates: Pulse-Heated Analysis of Solid Reaction Kinetics of Lignocellulosic Biomass, ACS Sustainable Chem. Eng., 2018, 6, 1387–1399, DOI:10.1021/acssuschemeng.7b03785.
- P. R. Patwardhan, Understanding the product distribution from biomass fast pyrolysis, PhD thesis, Iowa State University, 2010.
- T. Hosoya, H. Kawamoto and S. Saka, Solid/liquid- and vapor-phase interactions between cellulose- and lignin-derived pyrolysis products, J. Anal. Appl. Pyrolysis, 2009, 85, 237–246, DOI:10.1016/j.jaap.2008.11.028.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Solar energy-driven lignin-first approach to full utilization of lignocellulosic biomass under mild conditions, Nat. Catal., 2018, 1, 772–780, DOI:10.1038/s41929- 018-0148-8.
Footnote |
| † Electronic supplementary information (ESI) available: First principles computational methods, images of thin-films preparation used in experiments, comparison of activation barriers in LCC conformers, images of TS configurations, first principles barriers calculated using DFT and atomic coordinates of reaction intermediates. See DOI: https://doi.org/10.1039/d3ya00427a |
| This journal is © The Royal Society of Chemistry 2024 |