
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Assessing the roles of synthesis method and chemical composition in determining structure–property correlations in alloyed, ultrathin nanowire motifs for the methanol oxidation reaction†
Scott C.
McGuire‡
a,
Nathaniel R.
Hurley‡
a,
Michael G.
Gallagher
a,
Lihua
Zhang
c,
Anatoly I.
Frenkel
 *bd and
Stanislaus S.
Wong
*a
*bd and
Stanislaus S.
Wong
*a
aDepartment of Chemistry, Stony Brook University, Stony Brook, New York 11794-3400, USA. E-mail: stanislaus.wong@stonybrook.edu
bDepartment of Materials Science and Chemical Engineering, Stony Brook University, Stony Brook, New York 11794-2275, USA. E-mail: anatoly.frenkel@stonybrook.edu
cCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, New York 11973, USA
dChemistry Division, Brookhaven National Laboratory, Upton, New York 11973, USA
First published on 29th November 2023
Abstract
In the context of developing novel fuel cell catalysts, we have successfully synthesized in high yields not only ultrathin nanowires with compositions of Pt1Ru1 and Pt3Ru1 but also more complex spoke-like dendritic clusters of Pt1Ru1 and Pt1Ru9 in ambient pressure under relatively straightforward, solution-based reaction conditions, mediated by either cetyltrimethylammonium bromide (CTAB) or oleylamine (OAm), respectively. EXAFS analysis allowed us to determine the homogeneity of the as-prepared samples. Based on this analysis, only the Pt3Ru1 sample was found to be relatively homogeneous. All of the other samples yielded results, suggestive of a tendency for the elements to segregate into clusters of ‘like’ atoms. We have also collected complementary HRTEM EDS mapping data, which support the idea of a segregation of elements consistent with the EXAFS results. We attribute the differences in the observed morphologies and elemental distributions within as-prepared samples to the presence of varying surfactants and heating environments, employed in these reactions. Methanol oxidation reaction (MOR) measurements indicated a correlation of specific activity (SA) values not only with intrinsic chemical composition and degree of alloying but also with the reaction process used to generate the nanoscale motifs in the first place. Specifically, the observed performance of samples tested decreased as a function of chemical composition (surfactant used in their synthesis), as follows: Pt3Ru1 (CTAB) > Pt1Ru1 (CTAB) > Pt1Ru1 (OAm) > Pt1Ru9 (OAm).
1. Introduction
State-of-the-art direct alcohol fuel cell (DAFC) electrocatalysts primarily consist of nanostructured precious metals (i.e., Pt) and metal-based alloys supported onto carbon supports; these metals tend to be both scarce and expensive. Moreover, typical Pt-based electrocatalysts lack the stability and durability required for long-term FC applications, due to a mixture of factors, including CO poisoning, metal dissolution, and surface oxidation.1,2 Hence, approaches towards creating a novel, highly stable, and well-performing catalyst have relied on either maximizing Pt loading, minimizing overall Pt content, or replacing platinum-group metals (PGMs) with more earth-abundant and less expensive alternatives.3One such strategy involves the creation of ultrathin nanowires (NWs), measuring ∼2 nm in average diameter, which are expected to maintain slightly contracted surfaces, the latter of which can weaken the interaction with O2 and prevent their passivation by O2. Specifically, our group has previously noted that ultrathin NWs may be not only more chemically uniform but also more structurally monodisperse with fewer defect sites as compared with commercial bulk and nanoparticulate (NP) analogues.4 In particular, ultrathin Pt NWs maximize the available surface area-to-volume ratio and minimize overall Pt loading, thereby yielding catalytically attractive entities.5
Another parallel strategy involves the tuning of the chemical composition of Pt alone via alloy formation.6,7 Specifically, the precise nature of the metal can modify the d band structure and electron density of Pt through a “ligand effect”. Indeed, both Pt–Pt bond distances and the intrinsic electronic properties of Pt itself are altered by the presence of the underlying, adjoining metal, all of which can manifest themselves as a “lattice strain effect”. These critically relevant ligand and lattice strain effects can reveal themselves as perceptible shifts in the peak positions within the Pt 4f region associated with chemically sensitive X-ray photoelectron spectroscopy and X-ray absorption fine structure (XAFS) spectra. As an example, within PtRu, Pt atoms are expected to present more vacant 5d electronic states as a consequence of the electron withdrawing effect of the adjoining Ru.8
As a matter of combining the desirable approaches of ultrathin NW motifs with alloy generation, we focus on the synthesis of ultrathin, alloyed NWs of Pt with Ru, so as to improve their overall CO tolerance.9–16 Specifically, when alloyed at low-to-moderate Ru concentrations (χRu = 0–0.6), the PtRu alloy forms a solid solution with Ru atoms occupying sites within the Pt's face centered cubic lattice.17 Surface Ru sites can catalyze the oxidation of CO and other C-containing intermediates to CO2 at lower potentials, thereby decreasing the amount of irreversible binding of CO to active Pt sites and promoting stability.18 However, contemporary bimetallic PtRu alloys lack sufficient CO tolerance to be effective, long-lasting catalysts, thereby prompting a strong motivation to probe and understand correlations between the fabrication method and the resulting atomic structure.
Ultrathin PtRu NWs have been previously synthesized using hydrothermal methods.19,20 These reactions resulted in the production of 2–3 nm diameter wires, incorporating different Pt and Ru ratios. With these protocols, surfactants and precursors were dissolved in water, mixed, and subsequently heated within a Teflon-lined steel autoclave. Although hydrothermal methods are certainly reliable, these relatively high-energy-consuming procedures often involve elevated temperatures (upwards of 150–200 °C) and relatively long reaction times (i.e., 24 h or more) with autoclaves run at high pressures.
To mitigate for these potential concerns, herein, we focus on arguably milder solution-based methods to generate our PtRu alloys. As an initial example, in a typical room-temperature, surfactant-mediated synthesis scheme, inspired by previous work by our group and others,12,21–24 the precursors (e.g. H2PtCl6 & RuCl3) are co-reduced within the spatial confines of worm-like micellar pores of cetyltrimethylammonium bromide (CTAB) formed in a water-chloroform microemulsion by the addition of NaBH4. The chemical composition of as-prepared NWs can be reliably and predictably varied (Pt1−xRux, 0.3 ≥ ‘x’ ≥ 0) by changing the ratio of Pt and Ru content in the added precursor solution. Using this soft template approach, we have successfully reported on the use of the CTAB method to produce a multitude of stable ultrathin NWs, characterized by varying compositions including but not limited to Pt, Pd, Pd1−xNix, Pd1−xCux, Pd1−xAux, Pt1−xSnx, Pt1−xFex, Pt1−x−yRuxFey, and Pd1−xPtx.25–38
As a second example, representing a means of comparison with the CTAB method, we have developed a ‘new’ procedure that we are seeking to extend to fabricating RuM (M = arbitrary composition including non-PGMs) NWs. In this context, we have previously used the OAm method to produce ultrathin NW motifs of RuCo alloys.39 In a typical protocol for Ru NWs, RuCl3 was dissolved in oleylamine (OAm) and oleic acid (OA), and heated at 350 °C under an air-sensitive environment (i.e., argon) for 1 h. It is worth noting that oleylamine acted as both a reducing agent and a surfactant, whereas oleic acid behaved as a supplementary surfactant agent. In terms of a specific chemical role in the reaction, it has been previously proposed that both oleylamine and oleic acid acting synergistically in concert can behave as capping ligands to effectively guide and enable the formation of NWs through a process of oriented attachment.40–43
While these two solution-based synthesis protocols are known to be effective at producing ultrathin NWs, what is far less clear is a general understanding of how the structure and chemical composition of as-prepared anisotropic NW products are necessarily contingent upon and vary depending on precisely how they were made. We will therefore probe and address these questions with not only (i) PtRu NWs, synthesized by CTAB versus OAm/OA methods, but also as an added bonus, (ii) within PtRu NWs of varying compositions generated by the CTAB technique. Our analyses will be based on a combination of various characterization techniques, including transmission electron microscopy (TEM), high-resolution TEM (HRTEM), energy dispersive X-ray spectroscopy (EDS), and X-ray absorption fine structure (XAFS). As a subset of XAFS, the extended (EXAFS) signal incorporates crucial information about coordination numbers, interatomic distances, and the nature of disorder within systems (due to both static and dynamic displacements of all atoms from their average positions).44 Not surprisingly, it has been extensively used for studying local structure and composition in bimetallic nanoalloys.29,45–47 In particular, the study of the EXAFS spectra can yield valuable data about the number, type of, and distance to the atoms surrounding the central, X-ray absorbing atom.
In terms of prior EXAFS work, especially with respect to analogous NP systems, the structural evolution and atomic distribution of the “microemulsion lyotropic liquid-crystalline”-templated synthesis of mesoporous PtRu NPs after electroreduction were probed for varied duration times, using X-ray absorption near edge structure (XANES) and EXAFS spectroscopy, complemented by EDS and field-emission TEM.48 Moreover, we have been involved with a comprehensive structural and architectural evaluation based in part on EXAFS and XANES analysis,49 which revealed that 4.4 nm PtRu (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) alloyed NPs actually consisted of crystalline homogeneous random alloys with little twinning in a typical face-centered cubic (fcc) cell; specifically, the Pt atoms were predominantly metallic, whereas the Ru atoms were partially oxidized and were presumably located on the NP surface.
:1) alloyed NPs actually consisted of crystalline homogeneous random alloys with little twinning in a typical face-centered cubic (fcc) cell; specifically, the Pt atoms were predominantly metallic, whereas the Ru atoms were partially oxidized and were presumably located on the NP surface.
In the context of actual PtRu anodes used for operational direct methanol fuel cells (DMFCs),50 the catalyst was found to be essentially metallic with half of the Ru incorporated into a fcc Pt alloy lattice and the remaining half in an amorphous phase. EXAFS analysis suggested that the fcc lattice was not fully disordered and that the phases in which the Ru resided within the catalysts were dissimilar to what would have been expected in conventional oxides. Furthermore, in a separate report,51 catalyst restructuring was noted during in situ DMFC experiments through the observation of increases in the total metal coordination numbers; specifically, EXAFS and XANES showed that the highly oxidized Pt of an as-received commercial Johnson-Matthey PtRu (1:1) catalyst is fully reduced to its metallic state, concomitant with the reduction of a substantial amount of oxidized Ru, upon exposure to an actual operating DMFC environment. Finally, in a third study, XANES and EXAFS data confirmed that a highly alloyed state of PtRu NPs is responsible for their superior electrocatalytic performance as compared with typical commercial electrocatalysts,52 thereby collectively emphasizing the importance of properly assessing chemical composition at the nanoscale.
As we observed at the time, while the quantitative EXAFS models themselves tend to be mutually consistent in their overall conclusions, they still needed to be improved upon and optimized to acquire a thorough and accurate understanding of local atomic structure within relatively complex systems, such as ultrathin PtRu NW alloys. Hence, the novelty of our current study involves not only (i) demonstrable advances in effective solution-based PtRu NW synthesis using our in-house OAm/OA protocol but also more significantly, (ii) the application of EXAFS and HRTEM EDS in order to compare and contrast the discrete effects of (a) synthesis method and (b) variable chemical composition upon the resulting structure and growth of these alloyed anisotropic PtRu motifs.
As an additional novelty, what we have done herein is to correlate the MOR performance of our novel nanoscale motifs not only with carefully tailored chemical compositions but also ultimately with the synthesis reaction process and associated surfactant used to generate these nanomaterials. This was accomplished primarily through a careful analysis of acquired EXAFS and HRTEM data, which suggested not only that the greater Pt content, the better the catalyst performed but also that even with an identical PtxRuy chemical composition, variable size and morphology do matter significantly for stability under MOR conditions.
2. Experimental
2.1. Materials
All chemicals were used without further purification. Specifically, dihydrogen hexachloroplatinate(IV) hexahydrate (H2PtCl6, 99.9%), ruthenium chloride (RuCl3, 99.9%), oleic acid (OA, 90%), and anhydrous ethanol denatured (99%) were purchased from Beantown Chemical. Hexadecyltrimethylammonium bromide (CTAB, 98%) and oleylamine (OAm, 70%) were obtained through Millipore Sigma. Chloroform (ACS grade) was acquired from VWR. Sodium borohydride (NaBH4, 98%) was bought from Alfa Aesar. Perchloric acid (Optima grade) and methanol (Optima grade) were separately purchased from Fisher Scientific.2.2. Synthesis of nanowires
As an extension of that prior work in our lab for creating sustainable solution-based alternatives for nanoscale synthesis, we employed the following modification. Specifically, for the ultrathin NW synthesis of Pt1Ru1, 0.125 mmol H2PtCl6 and 0.125 mmol RuCl3 were dispersed in 7.5 mL of oleic acid and 7.5 mL of oleylamine. The contents of the flask were degassed under vacuum at 120 °C. The sample was then heated at 300 °C under the flow of Ar gas for 1 h, while stirring. The precipitate was ultimately collected by centrifugation, and washed with hexane and ethanol. For the analogous synthesis of ultrathin NWs of Pt1Ru9, 0.025 mmol of H2PtCl6 and 0.225 mmol of RuCl3 were used.
2.3. Electrochemical measurements
As-prepared samples of PtRu nanowires and dendritic clusters were supported onto Vulcan carbon XC-72R and rendered into catalyst inks by dispersing these dry powders into 200 proof ethanol to create a 1.5 mg mL−1 solution. A glassy carbon rotating disk electrode (GC-RDE, Pine Instruments, 5 mm) was prepared by polishing it using aluminum oxide powder (average particle size of 0.3 μm). Four 5 μL drops of the catalyst ink were then loaded onto the glassy carbon electrode, prior to allowing aliquots of this solution to dry under vacuum. One 5 μL drop of an ethanolic 0.025% Nafion solution was subsequently utilized to seal in the catalyst.A standard three-electrode electrochemical cell was assembled with a Pt counter electrode and an Ag/AgCl3 reference electrode. Electrochemical measurements of PtRu/C were performed in the presence of a 0.1 M perchloric acid (Optima grade) electrolyte, which had been generated from high-purity type 1 water with a measured resistance of 18.2 MΩ cm. Open circuit potentiometry (OCP) was carried out using pure hydrogen gas to assess the potential of the reference electrode. Cyclic voltammogram (CV) curves were collected within an Ar-saturated electrolyte solution, at a scan rate of 20 mV s−1. The measured catalytic activity toward the oxidation of small organic molecules was determined by acquiring linear sweep voltammograms at a scan rate of 20 mV s−1 within a solution mixture, containing de-oxygenated 0.1 M perchloric acid and 0.5 M of methanol. Chronoamperometry measurements indicative of stability were obtained with the same methanol/electrolyte solution by ramping up the voltage from 0.0 V to 0.7 V vs. RHE and then monitoring the current for a period of 3600 s.
2.4. Characterization
Data analysis was first completed on the EXAFS data of elemental metal foils, wherein the coordination number (N) of the first shell was set to be equal to 12. The passive electron reduction factors (S02) were varied in the fit. For the bimetallic systems, multiple-edge analysis was done to fit the signals, measured from each of the alloying constituent component's absorption edge, simultaneously. Data for the monometallic samples were simulated using FEFF calculations, performed using fcc structures for Pt and the hcp structure for Ru. To calculate FEFF theory for the heterometallic samples incorporating elements A and B, the atoms of type B were put into a first nearest neighbor position within the coordinate list with respect to the atoms of the type A. The S02 parameters for the NW components were subsequently fixed to be equal to those obtained for their respective bulk foil counterparts.
For the bimetallic NWs, the fits were performed for both edges concurrently, and the following constraints were applied.54 In particular, the heterometallic bond lengths were set to be equal (RA–B = RB–A) along with the mean squared bond length disorders (σA–B2 = σB–A2), whereas the homometallic bond lengths (i.e., RA–A and RB–B) and mean squared bond length disorders (i.e., σA–A2 and σB–B2) were varied independently. The coordination numbers were also tuned independently for all samples.
3. Results
3.1. Structural and chemical overview
TEM images and the corresponding size distributions associated with all samples of not only Pt1Ru1 and Pt3Ru1 prepared using the CTAB method but also Pt1Ru1 and Pt1Ru9 generated with the OAm/OA protocol are shown in Fig. 1. Subsequent HRTEM images and lattice spacing for all of the four samples produced have been provided in ESI,† Fig. S1. It is evident that synthesis in the presence of CTAB resulted in a web-like, entangled framework of smaller constituent wire-like subunits, whereas the samples obtained with the OAm/OA fabrication protocol incorporated a more globular mass consisting of localized spoke-like, dendritic clusters. As such, even qualitatively speaking, the isolated NW samples appear to highlight the importance and relevance of the synthesis method in dictating the resulting morphology.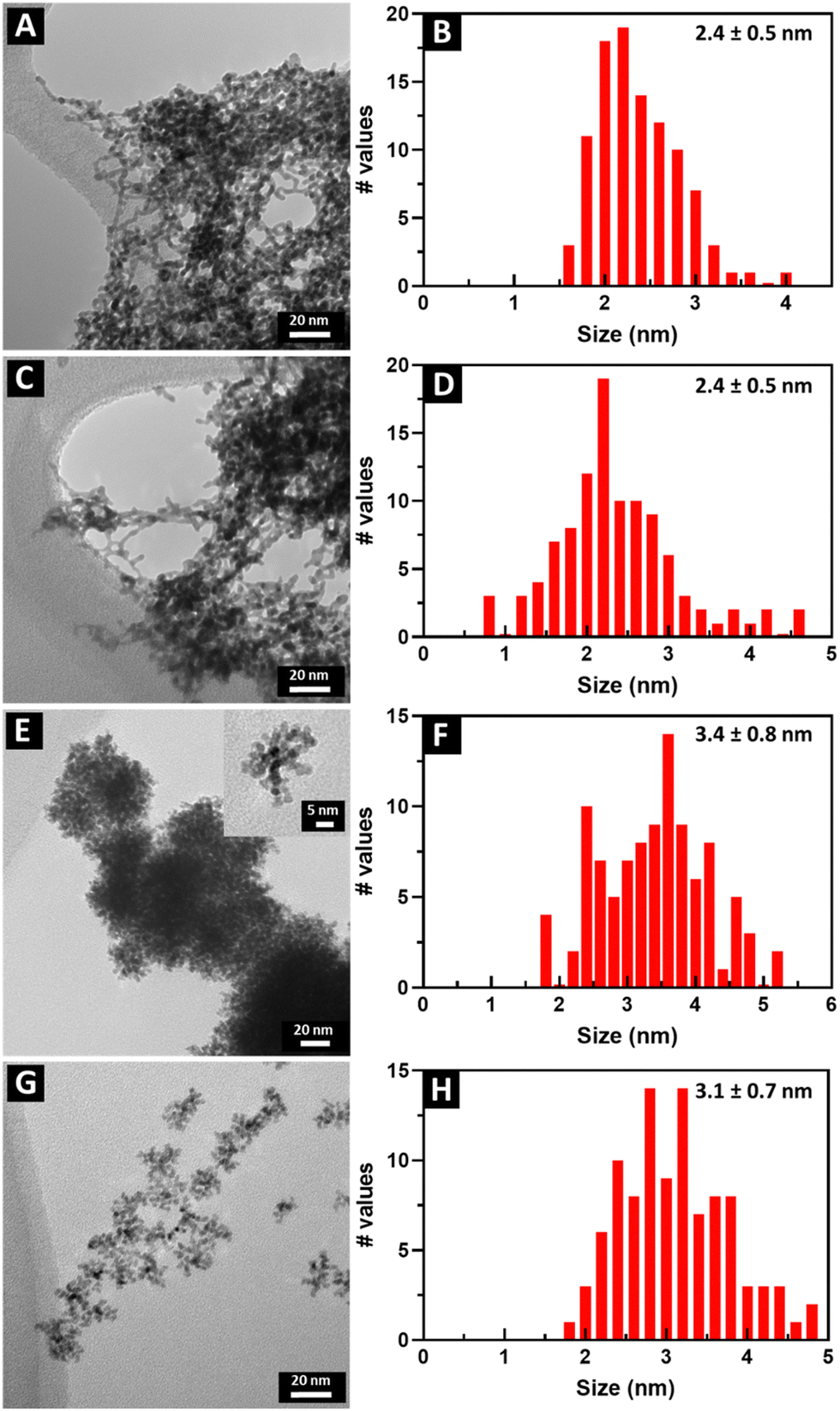 | ||
| Fig. 1 TEM images and size distributions are shown of a range of chemically distinctive ultrathin NWs, prepared by different synthesis methods. (A) and (B) Pt1Ru1 and (C) and (D) Pt3Ru1 were both prepared with CTAB. (E) and (F) Pt1Ru1 with the inset showing an individual cluster as well as (G) and (H) Pt1Ru9 were generated with OAm and OA. | ||
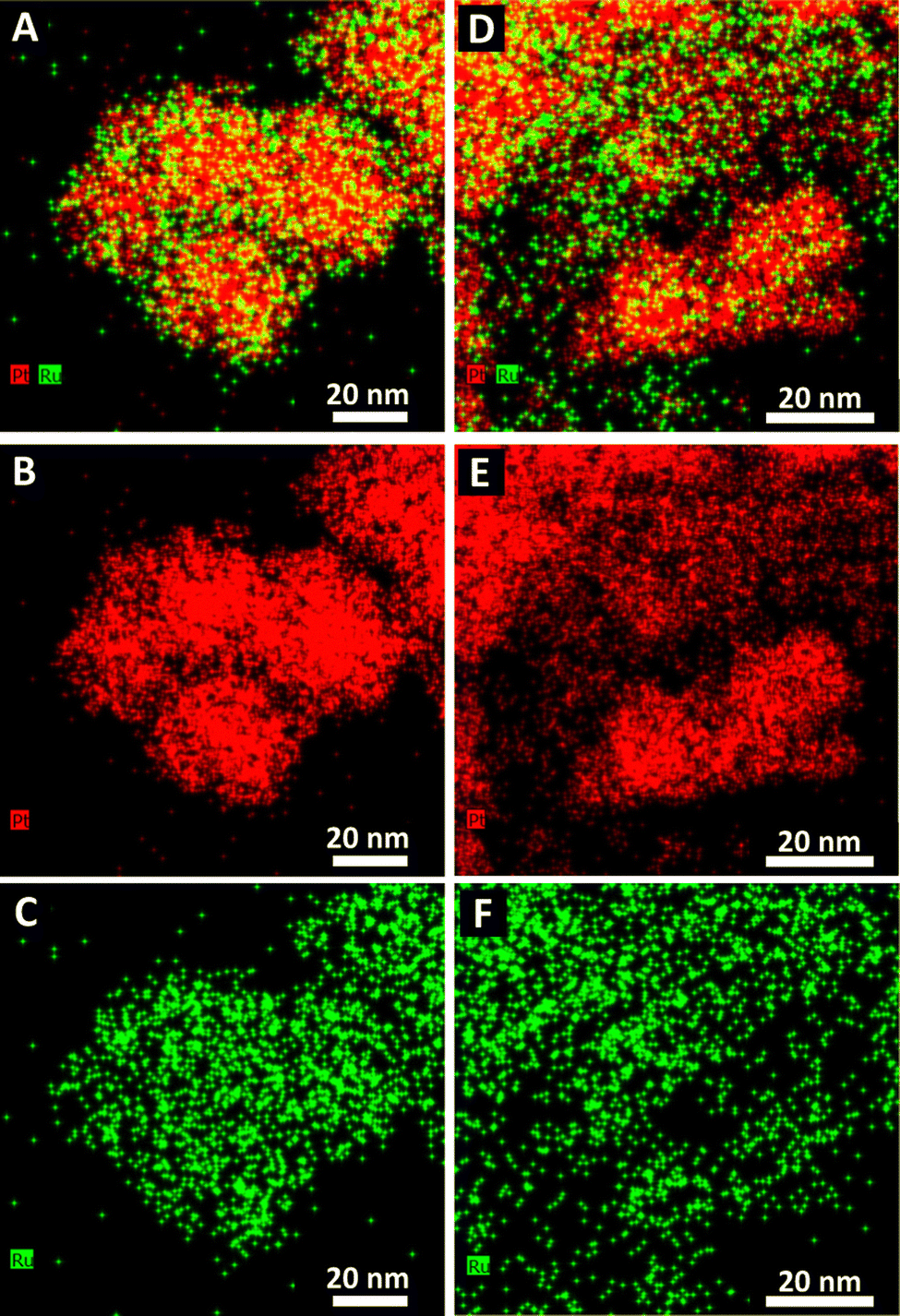
The corresponding EDS elemental maps of the CTAB and OAm/OA-derived samples are provided in Fig. 2 and 3, respectively. They highlight that in all of the samples, the ensembles of the atoms appear to be strongly disordered, forming nanoporous-like features, with the dimensions of the smallest units ranging from ∼1 to 5 nm (Fig. 1). Interestingly, for both of the Pt:Ru 1:1 samples, produced from the CTAB and OAm/OA-derived protocols, as shown in Fig. 2A and 3A, respectively, the elemental maps seem to show a greater propensity of Pt regions to concentrate and localize within the core, whereas the Ru regions give the impression of being more diffuse with a higher concentration at the outer periphery of the structure. In broad strokes, these observations are generally corroborated by the analysis of local compositional motifs via the EXAFS data modeling, as discussed in greater detail below.
 | ||
| Fig. 2 EDS elemental maps of PtRu generated using the CTAB method. Pt1Ru1 maps are provided as follows: (A) the composite of Pt and Ru, (B) Pt, and (C) Ru. Analogous Pt3Ru1 maps are shown, as (D) the composite of Pt and Ru, (E) Pt, and (F) Ru. | ||
 | ||
| Fig. 3 EDS elemental maps of PtRu prepared using the OAm/OA method. Pt1Ru1 maps are given as (A) the composite of Pt and Ru, (B) Pt, and (C) Ru. Corresponding Pt1Ru9 elemental maps are shown in (D) the composite of Pt and Ru, (E) Pt, and (F) Ru. | ||
3.2. Visual examination of the raw XAFS data
Pt L3-edge XANES data for Pt1Ru1 and Pt3Ru1 prepared using a CTAB surfactant are shown in Fig. 4A. The k2-weighted EXAFS data in k-space and r-space in which the Fourier transforms were performed for the k2-weighted EXAFS spectra in the k-range from 2 to 15 Å−1 using the Hanning window function and dk = 2 Å−1, are shown in Fig. 4B and C, respectively. The associated Ru K-edge data for the same samples are highlighted in Fig. 5A–C. The Fourier transforms (Fig. 5C) were performed for the k2-weighted EXAFS spectra (Fig. 5B) in the k-range from 2 to 15 Å−1. The corresponding Pt L3 edge SXANES and EXAFS data for Pt1Ru1 and Pt1Ru9 prepared using the OAm/OA surfactant are illustrated in ESI,† Fig. S2. The associated Ru K-edge data are provided in ESI,† Fig. S3.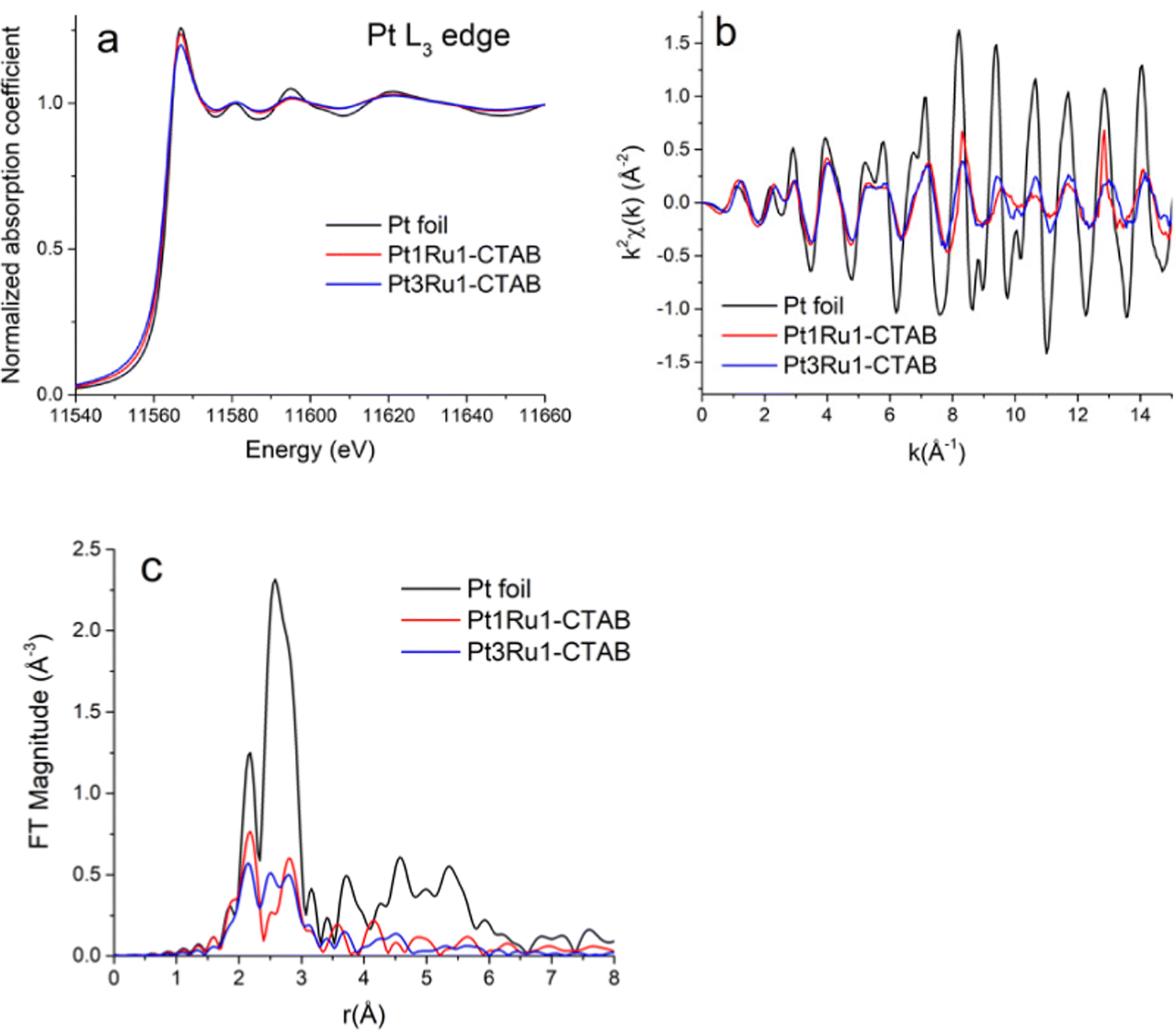 | ||
| Fig. 4 (A) Pt L3-edge XANES data. (B) k2-weighted EXAFS data in k-space. (C) Fourier transform magnitudes of the k2-weighted EXAFS data for Pt foil, Pt1Ru1 (produced with CTAB), and Pt3Ru1 (produced with CTAB). | ||
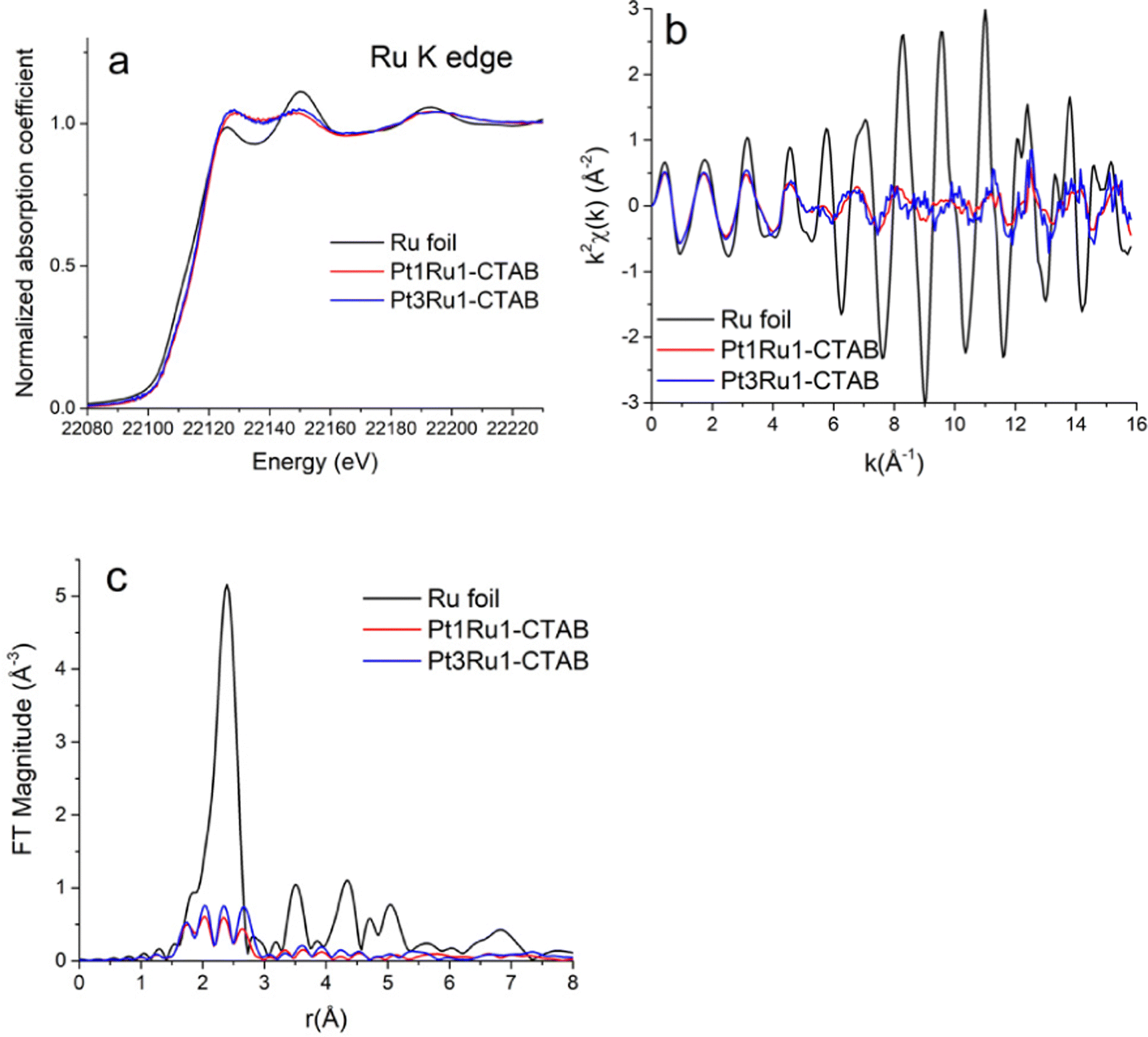 | ||
| Fig. 5 (A) Ru K-edge XANES data. (B) k2-weighted EXAFS data in k-space. (C) Fourier transform magnitudes of the k2-weighted EXAFS data for Ru foil, Pt1Ru1 (produced with CTAB), and Pt3Ru1 (generated with CTAB). | ||
3.3. Results of EXAFS data analysis
Table S1 (ESI†) presents the Fourier transform parameters, fitting ranges, constraints used in the multiple – edge fits, and the resultant r-factors for each analyzed data set. Fourier transform magnitudes of the k2-weighted EXAFS data and fits for the Pt3Ru1 (originating from CTAB) samples at the Pt L3-edge and Ru K-edge are given in Fig. 6A and B, respectively. The corresponding data and the fits for not only the reference Pt and Ru foils but also samples of Pt1Ru1 (derived from CTAB) in addition to Pt1Ru1 and Pt1Ru9 (produced from OAm/OA) are shown in ESI,† Fig. S4A, B, S5, S6 and S7, respectively.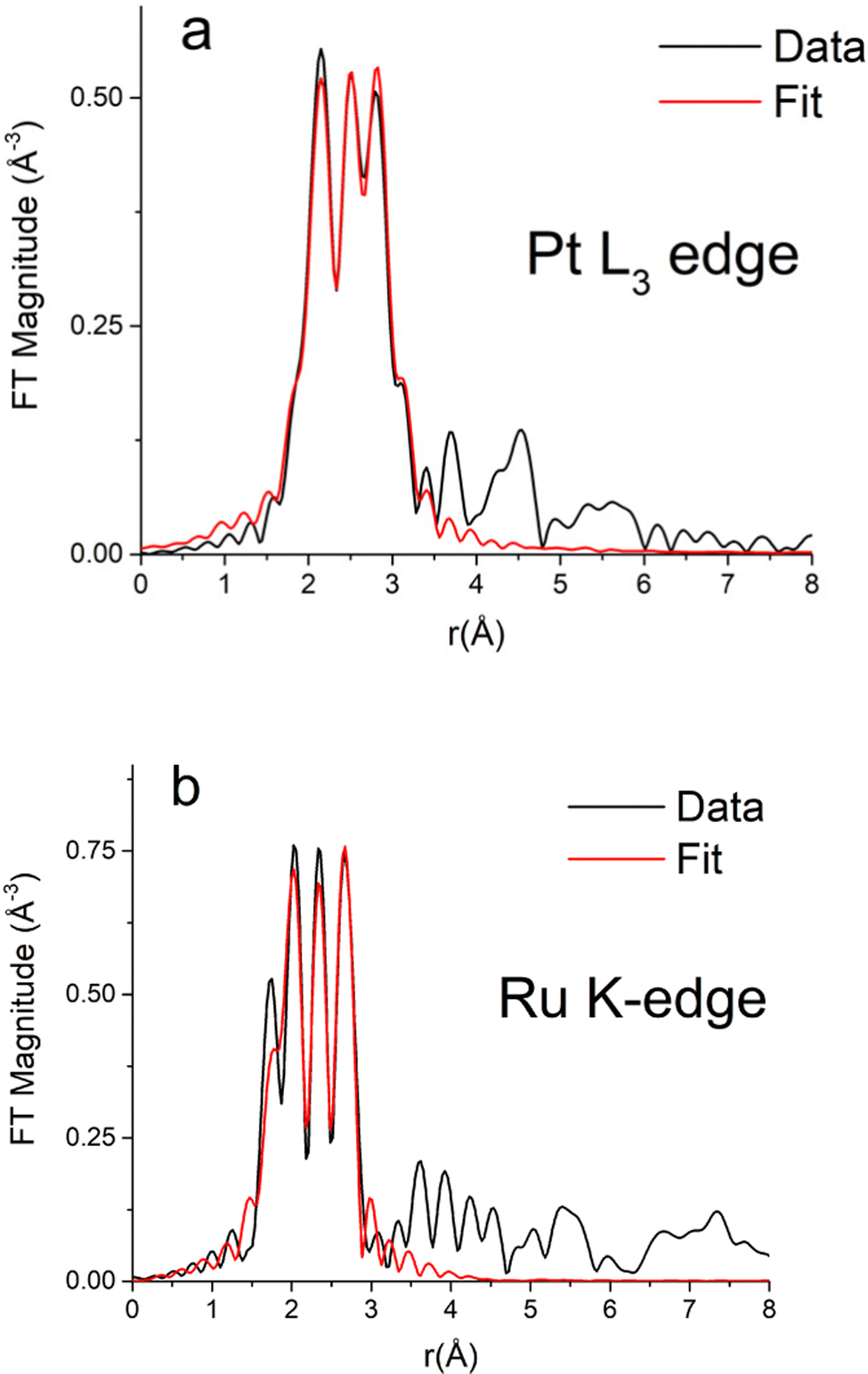 | ||
| Fig. 6 Fourier transform magnitudes of k2-weighted EXAFS data and theoretical fits for the Pt3Ru1 (generated with CTAB) sample at (A) the Pt L3-edge and (B) the Ru K-edge. | ||
The best fit values of the amplitude factor (S02) for Pt and Ru edges were obtained from the analysis of their respective metal foils: 0.84(3) and 0.79(7), respectively. The best fit results of the Pt edge and Ru edge data analyses, together with the results obtained for Pt and Ru foil references, are summarized in ESI,† Table S2.
4. Discussion
4.1. EXAFS
To assess the local compositional motifs of a given bimetallic alloy, Cowley's short range order parameter, α, is often used. It is expressed as:44,47,54,55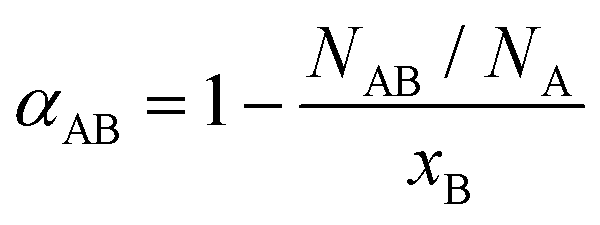 | (1) |
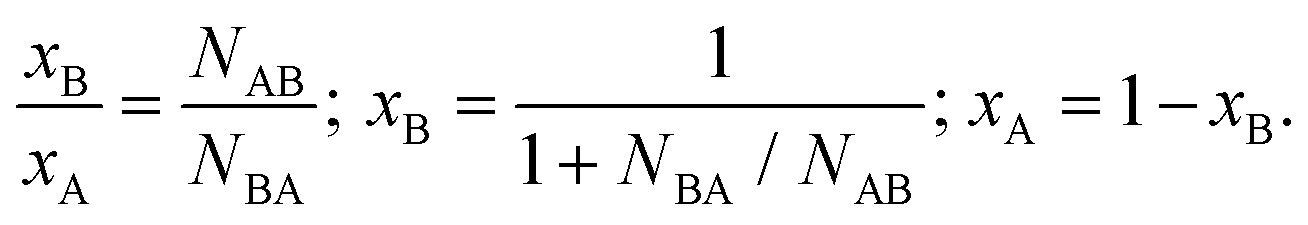 | (2) |
| Ultrathin NW – synthesis method | N Pt | N Ru | x Pt | x Ru | α PtRu | α RuPt |
|---|---|---|---|---|---|---|
| Pt1Ru1-CTAB | 6.9(7) | 4.3(8) | 0.6(4) | 0.4(1) | 0.5(5) | 0.3(3) |
| Pt3Ru1-CTAB | 6.4(4) | 7.1(1.6) | 0.9(6) | 0.12(4) | 0(0) | 0.1(1) |
| Pt1Ru1-OAm/OA | 9.4(1.5) | 8.1(1.4) | 0.5(3) | 0.5(2) | 0.5(5) | 0.4(4) |
| Pt1Ru9-OAm/OA | 9.7(2.0) | 4.6(6) | 0.3(1) | 0.7(5) | 0.7(8) | 0.4(5) |
In our previous work,56 we have highlighted the limits of applicability of the SRO and include a more detailed discussion in the ESI.† That is, its interpretation as an indicator of either positive (when αAB > 0) or negative (when αAB < 0) tendency to clustering of ‘like’ (A–A and B–B) atoms is fundamentally limited to the analysis of relatively homogeneous alloys only, i.e., those bimetallic systems in which the relative number of neighbors of different atom types to each atom is approximately the same, regardless of the atom's location. Using that definition, even in a bimetallic nanoparticle (in which the number of nearest neighbors to atoms on the surface is smaller than in the interior), different components can be homogeneously mixed. That characteristic of homogeneity is convenient, because it can be seamlessly verified in terms of observable EXAFS results.
For example, if the total coordination numbers, NA = NAA + NAB and NB = NBA + NBB are relatively similar, the alloy is homogeneous. Otherwise, there is a macroscopic segregation of components A and B (e.g., cluster-by-cluster or core–shell etc.). Notably, in a core–shell situation, the comparison between the values of NA and NB will communicate which element is more under-coordinated and, hence, preferentially localized on the surface. Only when the alloy is found to be relatively homogeneous can we ponder the next question: What is the short-range order in the alloy?
Therefore, prior to interpreting the SRO parameters, it is important to first investigate the relationship between the total coordination numbers NPt and NRu of the two different elements. Table 1 shows that their values are significantly different for both Pt1Ru1 samples. Namely, they are, first, larger in the CTAB sample as compared with the OAm-derived one, and, second, NPt > NRu in both types of samples. It should also be noted that as the composition of the nanowires changes using both the CTAB and OAm-based methods, the NPt remains constant, but conversely, the NRu does change. For example, with the CTAB protocol, as the relative amount of Ru increases from Pt3Ru1 to Pt1Ru1, the corresponding NRu value decreases from 7.1 to 4.3. Likewise, as the chemical composition of the sample created using the OAm method evolves from Pt1Ru1 to Pt1Ru9, we note that the coordination numbers associated with Ru decrease from 8.1 to 4.6. This observation indicates both a reduction in the metal–metal coordination and a decrease in the homogeneity of the mixture.
These results are in full agreement with the TEM size distributions and the EDS elemental maps (Fig. 1–3) we have previously discussed. In particular, Fig. 1B and F show that the characteristic size of the metal regions in the Pt1Ru1 sample is smaller for the CTAB-derived samples, as compared with their OAm/OA-produced analogues. That finding is in full agreement with the relative decrease of the metal–metal coordination numbers of the former samples as compared with the latter ones (Table 1). In both samples, the elemental maps show much smaller Ru regions on the surfaces of the larger Pt ones (Fig. 2A and 3A), in full agreement with the relationship (NPt > NRu) obtained from EXAFS data analysis (Table 1). The only alloy sample that displays relative homogeneity (NPt ≈ NRu) is Pt3Ru1, derived from CTAB (Table 1). It can also be characterized as quasi-random, because the SRO parameter is consistent with zero (Table 1).
We note that the coordination numbers in all samples are much smaller than those predicted from the average sizes obtained by TEM (Fig. 1). Indeed, for the sizes in the range of 2.4–3.4 nm, the average coordination numbers of metal atoms,57NMM = xANAM + xBNBM, should be of the order of 10 or larger, assuming, as a simple model, an ideal cuboctahedral geometry.58 This estimate is significantly larger than the coordination numbers reported in Table 1. The explanation can likely be ascribed to the strongly disordered, nanoporous-like nature of the metal ensembles (Fig. 2 and 3), which contain much larger numbers of under-coordinated atoms than those predicted from conventional close-packed models typical of the bulk.
4.2. Probing the effects of synthesis methods
To understand the differences between the OAm/OA and CTAB synthetic protocols, as noted, we synthesized ultrathin NWs of Pt1Ru1 using both procedures, with equimolar amounts of the H2PtCl6 and RuCl3 precursors. Many aspects of the synthesis methods can impact the growth of alloyed nanomaterials. Important reaction variables that we can reliably alter include but are not limited to the reaction temperature, surfactant identity, and reduction mechanism. Each of these parameters will be explored in the context of the growth of the wires and particles. As mentioned in Section 3.1, the CTAB method produced a network of tiny nanowires. In this methodology, CTAB forms worm-like micelles at the interface between water and chloroform. The long non-polar tails of CTAB make the surfactant readily soluble in chloroform, thereby enabling the formation of micelles with the hydrophobic tails exposed to chloroform and the more hydrophilic polar heads positioned away from the organic solvent.When water, containing dissolved Pt4+and Ru3+ ions, is introduced to the chloroform, an immiscible emulsion is subsequently formed. With the addition of vigorous stirring between water and chloroform, the micelles themselves are impacted by the presence of the metal ions. Specifically, at the interface of the two solvents, the dissolved metal ions are able to coordinate onto the polar heads of CTAB. When sodium borohydride (NaBH4) is then added into the reaction medium, it reduces Pt4+and Ru3+ to Pt0 and Ru0, respectively. The role of CTAB therefore is to spatially confine the growth of the reduced metals to the localized area contained within the micelles themselves, thereby allowing for the formation of isolated motifs of ultrathin nanowires.37
As mentioned, the OAm/OA procedure yielded a more globular nano-porous agglomerate structure. We postulate that OAm can simultaneously act as a solvent, a reducing agent, and a surfactant, whereas oleic acid functions as an additional “shape-control” ligand.39 The interplay between these two species can be complex and is not fully understood. Nevertheless, it has been shown in the past that both OAm and OA are capping agents that can control the morphology of nanomaterials by binding onto their external surfaces and selectively directing their growth; it is not surprising therefore that a particulate morphology was generated in this case.40,41,59
XRD data are provided in Fig. 7. The relatively broad XRD peaks can be ascribed to the relatively small sizes of the individual particles and wires. For samples of Pt3Ru1 CTAB, Pt1Ru1 CTAB, and Pt1Ru1 OAm, we observed the expected pattern for fcc platinum with no apparent impurity phases of ruthenium. For the Pt1Ru9 sample, we noted the pattern for hcp ruthenium with no platinum phases present. The lack of any obvious impurity phases is a good indication of successful alloying, as it suggests that there was no segregation of crystal phases.
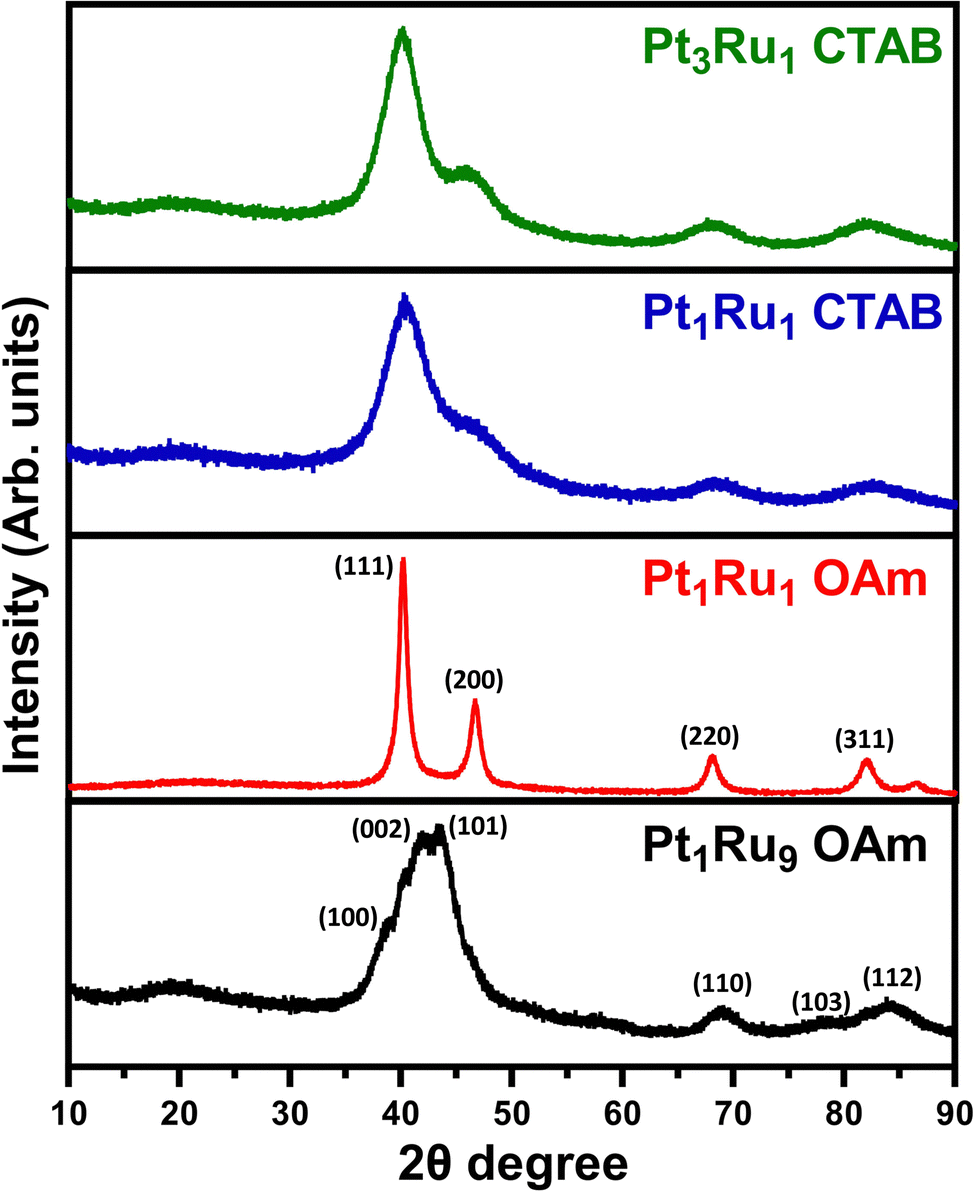 | ||
| Fig. 7 XRD data for all of the alloyed samples generated. | ||
As indicated earlier, both the CTAB and OAm/OA-mediated reactions utilize surfactants to control the growth of as-prepared PtRu nanomaterials. Surfactants can be designated into several broad categories, encompassing ionic (i.e., anionic/cationic), charged non-ionic, non-charged, and polymeric species.60,61 The interactions between the surfactants and the growing particles can fundamentally impact how they form and grow, because of the extent to which these organic molecules will bind (or not) onto exposed facets. In particular, CTAB can be characterized as a cationic surfactant, containing a negatively charged bromide ion coupled with a positively charged organic component. Table S3 (ESI†) gives the chemical structure of the various surfactants used herein.
In a typical CTAB-based method, a ‘soft’ template consisting of worm-like micellar networks is produced in the presence of CTAB within a two-phase water-chloroform system. The charged Pt and Ru precursor ions, added to the mixture, are spatially confined to and subsequently trapped within the ‘micellar pores’, i.e., channels created by the packing and alignment of the highly polar heads of individual CTAB molecules. Hence, porous, high surface area networks of interconnected, ‘worm-like’, and homogeneous metallic nanowires, with average diameters of as small as 1.9 nm, are able to form upon the reduction of these metal precursors with a reducing agent, such as sodium borohydride.62
By contrast, as previously mentioned, with the OAm/OA technique, oleic acid and oleylamine function as both surfactants and solvents. In this procedure, unlike with the CTAB-based method, the surfactants are charged but not necessarily closely coupled with a separate counterion, as is the case with the bromide anion present within CTAB. Hence, for our purposes, these smaller surfactants are considered to be charged but non-ionic. In this mixture, both the OA and OAm molecules will form coordination complexes with the Pt and Ru ions.39 It is likely that the negatively charged oleic acid will coordinate more strongly with the positively charged Pt4+ and Ru3+ ions. Nevertheless, both oleic acid and oleylamine contain similar carbon chains that are 18 carbon atoms long. This renders these surfactants as highly miscible with each other, which likely leads to a more even distribution of precursor elements within the entire solution, thereby enabling the formation of more homogeneous, particulate-like structures as opposed to the worm-like micellar motifs commonly observed with the CTAB method.
With respect to the reducing mechanism, in the CTAB procedure, the precursors are reduced very rapidly, due to the presence of a strong reducing agent, namely NaBH4. By contrast, with the OAm/OA procedure, OAm embraces multiple, simultaneous roles as a solvent, surfactant, and reducing agent. Moreover, because OAm as a reducing agent is a much “weaker” reducing agent than that of NaBH4, it is not surprising that higher temperatures and longer reaction times are needed to facilitate the formation of uniform particles with the OAm/OA process.
From the EXAFS perspective, we observed that the as-synthesized Pt1Ru1 samples possessed an evident degree of heterogeneity with the Pt and Ru atoms preferentially segregated. This level of segregation is consistent with past results for similarly formed types of NWs, such as Ru2Co.39 It is also worth noting that hydrothermally generated PtRu was similarly created preferentially in the form of segregated ultrathin nanowires.
4.3. Probing the effects of varying chemical composition
To confirm the chemical composition of all samples, we looked at both the concentrations calculated from the EXAFS measurements and the atomic % values gathered from the EDS measurements (see Table 2). All of these samples consistently highlight slightly more Pt along with less Ru content than the initial molar ratios mixed during the reactions. For example, the PtRu 1:1 sample generated through the CTAB method yielded a chemical composition closer to that of 6:4 case, based on both the EDS and EXAFS results. Moreover, the PtRu 1:9 OAm sample was characterized by a measured composition of about 3:7. This higher-than-expected amount of Pt may be ascribed to the more positive reduction potential of Pt, which implies a faster reduction process, and consequently, the observed hcp/fcc crystal structure mismatch between Ru and Pt respectively.
| Sample | Chemical concentration derived from XAS | Chemical composition derived from EDS (atomic%) |
|---|---|---|
| Pt1Ru1-CTAB | Pt: 0.6(4) | Pt: 64 ± 8.2% |
| Ru: 0.4(1) | Ru: 36 ± 8.2% | |
| Pt3Ru1-CTAB | Pt: 0.9(6) | Pt: 88 ± 3.0% |
| Ru: 0.12(4) | Ru: 12 ± 3.0% | |
| Pt1Ru1-OAm/OA | Pt: 0.5(3) | Pt: 77 ± 2.9% |
| Ru: 0.5(2) | Ru: 23 ± 2.9% | |
| Pt1Ru9-OAm/OA | Pt: 0.3(1) | Pt: 25 ± 3.4% |
| Ru: 0.7(5) | Ru: 75 ± 3.4% |
As earlier noted, for the CTAB-based method, both Pt3Ru1 and Pt1Ru1 were successfully synthesized as ultrathin NWs. For the sake of completeness, we also attempted to synthesize Pt1Ru9. However, this latter attempt resulted in the formation of NPs with an average size of 1–3 nm and not ultrathin nanowires (see ESI,† Fig. S8A). This observation implies that a sufficiently high enough relative concentration of Pt is necessary to enable wire formation using this particular protocol.
With regards to the OAm/OA method, we successfully generated both Pt1Ru1 and Pt1Ru9 compositions. However, with Pt3Ru1, the resulting product consisted of irregular nanoparticles and aggregates. Fig. S8B (ESI†) highlights TEM images of the Pt3Ru1 OAm sample. The implication was that excess Pt degraded the observed morphology, when using the OAm/OA method. For the CTAB samples, our EXAFS analysis demonstrated that Pt3Ru1 could be described as a random alloy, whereas Pt1Ru1 yielded a degree of segregation between the elements. We note that the Pt4+ → Pt0 reduction process is characterized by a reduction potential of 0.69 V and that the corresponding Ru3+ → Ru0 reduction process has an analogous reduction potential of 0.60 V. Based on these facts, both metals should reduce under similar reaction conditions. However, the preferred crystal structure of Pt0 is fcc, whereas that of Ru0 is hexagonal closed packed (hcp). This structural difference can help to explain why Ru and Pt appear to segregate from one another at higher relative concentrations; that is, the mismatch between the crystal structures of these two elements renders homogeneous alloy formation difficult but not outright impossible. Additionally, we observed that the wire-like morphology is only achievable with large amounts of Pt, whereas Ru tends to preferentially form NPs.
Concerning the OAm method, the differences we observe may be ascribed in part to the differing preferred crystal structures of Pt and Ru, previously discussed, and the corresponding slow reduction process. Although the reduction potentials of Pt4+ and Ru3+ are similar and as such, both species should reduce essentially simultaneously, that of Pt is slightly more positive. In practice, in a sample with a comparatively higher concentration of Pt, relatively more Pt may form first and the Ru may either generate more slowly or not alloy as effectively at all. Therefore, as higher and higher amounts of Pt are introduced, these will preferentially form Pt particles first and will not necessarily alloy with the slower-forming Ru0 species. EXAFS analysis appeared to indicate that both compositions created from the OAm process gave rise to perceptible elemental segregation.
5. MOR
Our ultimate goal was to understand the effects of not only the synthesis method but also the inherent chemical composition of these samples on the observed electrochemical performance of the PtRu alloys. To do this, in separate runs, we deposited the ultrathin wires and spoke-like, dendritic clusters onto Vulcan carbon XC-72R with a nominal loading of about 20%. Since drying the samples can lead to significant aggregation, we confirmed the actual loading quantities using TGA, after the alloyed samples had been combined with the carbon support. Fig. S9 (ESI†) provides for TGA mass loss curves for the loading amounts of either Pt or PtRu onto the carbon support. These values were subsequently used to calculate the specific surface area (SSA) for each sample, as shown in ESI,† Table S4.Representative CV curves for the samples are given in ESI,† Fig. S10. All show the expected hydrogen adsorption and desorption peaks situated at around 0–0.2 V, which is characteristic of platinum. We detected little to no Pt–O reduction peaks present, likely due to their being suppressed by the incorporation of Ru. MOR LSVs and the associated specific activities (SA) are shown in Fig. 8.
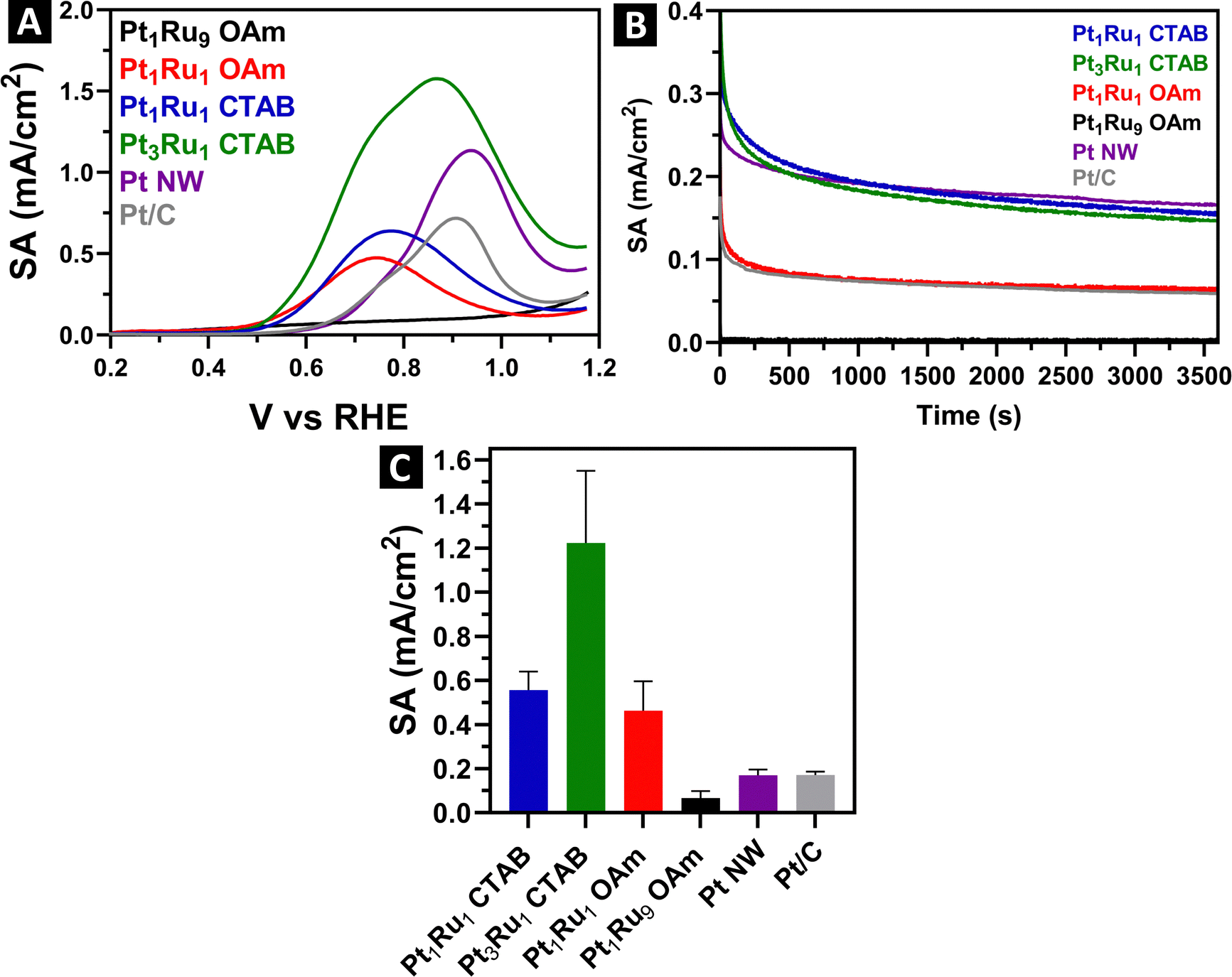 | ||
| Fig. 8 MOR results for all PtRu alloyed ultrathin wires as well as particulate clusters, in addition to the Pt nanowire and Pt/C commercial control samples, with their corresponding synthesis methods and surfactants. Data shown herein include (A) MOR LSVs, (B) chronoamperometry plots, and (C) bar graphs of the specific activity measurements of the various nanoscale motifs. | ||
All of the SA values were acquired at 0.7 V vs. RHE. Specifically, Pt3Ru1 and Pt1Ru1 derived from the CTAB method yielded specific activities of 1.22 mA cm−2 and 0.55 mA cm−2, respectively. By contrast, Pt1Ru1 and Pt1Ru9 created from the OAm/OA protocol gave rise to corresponding activity values of 0.46 mA cm−2 and 0.07 mA cm−2, respectively. We compared these results with those of pure Pt wires generated using the CTAB method. In effect, as a control sample, pure Pt NWs were characterized by an SA of 0.17 mA cm−2, suggesting that the best performing PtRu alloys exhibited an almost 7-fold increase in activity as compared with Pt alone. Approximately the same increase in SA was observed when comparing the behavior of as-synthesized PtRu alloys with those of commercial Pt/C nanoparticles.
It has been postulated that the MOR enhancement of PtRu alloys is due to not only (i) their enabling of the direct conversion of methanol to CO2 (without any intermediate CO through a non-CO pathway) but also (ii) the stimulation of CH3OH adsorption and concomitant formation of active OH species from H2O which can oxidize intermediate (and poisoning)63 CO bound to Pt sites.64 Moreover, the strain effect65 resulting from the lattice mismatch between Pt and Ru can decrease the binding energy of reaction intermediates, thereby favoring high CO tolerance.
To confirm that these apparent differences could not be ascribed to the degradation of either the wires or dendritic particulate clusters, we obtained TEM images of the samples, both before and after the MOR electrochemical process. These microscopy data are provided in ESI,† Fig. S11 for the sample synthesized by the OAm/OA method and in ESI,† Fig. S12 for that generated from the corresponding CTAB protocol. The corresponding TEM images of the analogous pure Pt/C NW system are given in ESI,† Fig. S13. We found little to no sample degradation, and in fact, there was no obvious change in the degree of sample aggregation after running MOR for our PtRu alloys. By contrast, in the case of the pure Pt NW controls, we observed the presence of an obvious anisotropic wire morphology prior to MOR but after MOR, there was noticeably more sample aggregation. All of these results collectively indicate that the incorporation of Ru into our samples yielded tangibly better structural and operational stability towards MOR in an acidic environment as compared with pure monometallic Pt NWs.
In terms of chemical composition, we noted a direct correlation between the MOR performance and the amount of Pt incorporated within the alloyed samples. Not surprisingly, Pt3Ru1 containing the highest relative Pt content gave rise to the best performance detected, whereas Pt1Ru9 evinced almost no measurable MOR activity. Structurally, as we have seen from EXAFS, the Pt3Ru1 sample maintains the greatest amount of alloying, as indicated by its SRO parameter of about 0.1. Hence, we postulate that Pt3Ru1 behaves the best for MOR, in part because of a higher degree of coordination between the Pt and Ru atoms, and in fact, per ESI,† Table S2, the Ru–Pt coordination number (NRu–Pt) of 5.6 is at least 2–3 times higher than of the other samples tested.
We note that the NRu–Pt of Pt1Ru1-OAm sample was 2.7, which is a higher figure than that of the 1.9 value computed for Pt1Ru1-CTAB. However, the MOR values associated with these samples were very similar, with the NWs created by the CTAB method yielding a slighter better performance metric. This observation indicates that as a tunable parameter, the degree of alloying, in the samples possessing the same apparent composition, in and of itself, is not sufficient to dramatically affect the MOR performance. Instead, the electrocatalytic performance of these samples would appear to be impacted more significantly by morphological and size differences. In particular, the smaller diameters of the NWs generated using the CTAB method gave rise to better performance than their larger-diameter analogues.
Moreover, both of the Pt1Ru1 samples created with OAm/OA and CTAB procedures possessed very similar SA values. This finding indicates that MOR performance is much more connected to and can be viewed as a function of the actual Pt–Ru composition, which to a large extent is independent of the synthesis method employed. It is worth highlighting that the EXAFS data for both OAm/OA and CTAB Pt1Ru1 samples are also relatively comparable to each other, in that both materials possess almost identical SRO values.
To further probe and understand the differences in performance between the samples, we acquired chronoamperometry measurements, provided in Fig. 8B. We ran these measurements at a steady state voltage of 0.7 V vs. RHE for 3600 s. Pt is easily poisoned by CO, so we can attribute the initial rapid drop to the de-hydrogenation of methanol followed by the poisoning of accessible Pt active sites with CO. Steady state SA measurements indicate that both samples generated using the CTAB method yielded the highest durability and stability, followed by the Pt1Ru1 sample produced using the OAm/OA method. As expected, the Pt1Ru9 sample gave rise to almost no steady state performance, in line with its very low MOR SA. We also observed that the steady state SA values of pure Pt are similar to that of Pt7Ru3 and Pt1Ru1 generated using the CTAB method, thereby indicating that our PtRu alloys and monometallic Pt controls maintain comparable levels of tolerance to CO.
Interestingly, the chronoamperometry results imply a significant discrepancy between synthesis protocols, that had not been apparent with MOR performance data alone. Specifically, the Pt1Ru1 produced with CTAB gave rise to a steady state SA that was more than 2× higher than that of the identical composition fabricated with the OAm/OA procedure. We assert that this observation is associated with the size and morphology of the ultrathin nanowires, made using the CTAB method, versus that of the discrete clusters, produced with the analogous OAm/OA protocol. It is worth noting that we have observed similar analogous improvements in the electrochemical performance of our ultrathin motifs versus that of their corresponding particulate morphologies in a number of our prior studies.25–38
In general, ultrathin anisotropic nanowires not only are smaller in diameter but also possess lower total coordination numbers than their more isotropic particulate cluster counterparts. Hence, considering that our EXAFS analysis was suggestive of a core–shell-like distribution of elements with Pt preferentially localized in the core and Ru confined to the shell, it is reasonable to hypothesize a greater preponderance and proportion of Pt atoms situated on the outer surfaces of the nanowires, mixed in with the adjacent Ru atoms, thereby leading to potentially higher numbers of available active surface sites, which are conducive factors to enabling an overall improvement in measured MOR activity especially as compared with pure Pt alone.
6. Conclusions
We have successfully synthesized not only ultrathin Pt1Ru1 and Pt3Ru1 nanowires using the CTAB method but also more organized ensembles, consisting of localized spoke-like, dendritic clusters of Pt1Ru1 and Pt1Ru9, utilizing an effective, in-house, solution-based OAm/OA protocol that we have generalized from mono-metallic to more complex bi-metallic alloy species. HRTEM EDS elemental mapping data indicated that all of the samples gave rise to relatively disordered groupings of atoms to varying degrees, depending on the precise composition. As an example, Pt:Ru 1:1 samples, generated through either the CTAB or OAm/OA-derived protocols, appear to maintain a Pt core/Ru shell structure. This finding was corroborated by our XAS results which were overall suggestive of elemental segregation. Interestingly, Table 1 shows that the NPt and NRu values are significantly different for both Pt1Ru1 samples. Specifically, (i) both values are larger in the CTAB sample as compared with the OAm/OA-derived one and (ii) NPt > NRu in both types of samples, suggesting that Pt is localized within the core, while Ru is preferentially arranged at the surface.
Therefore, highlighting the key role of the synthesis methodology in dictating the observed morphology, we postulate that the reducing environment within the CTAB method enables the Ru and Pt precursors to be reduced relatively quickly at room temperature with NaBH4. By contrast, since OAm is a weaker reducing agent than NaBH4, more highly elevated temperatures are required to enable the reduction process, thereby leading to greater elemental segregation and architectural complexity. With respect to the equally important role of the viability of generating products with specific chemical compositions, we determined that the mismatch in the crystal structures of the fcc Pt vs. the hcp Ru likely leads to the presence of increased elemental segregation at higher loadings of Ru, irrespective of the synthesis method employed.
Finally, MOR measurements indicate a direct correlation and connection between both chemical composition and the degree of alloying versus the electrochemical performance. In particular, our Pt3Ru1 sample yielded an almost 2× greater SA value as compared with analogous samples tested. We explain these observations, based on the greater relative Pt content coupled with the high degree of alloying associated with our Pt3Ru1 sample, as evinced by its small, positive SRO parameter of 0.1. Moreover, the CTAB protocol also produced ultrathin alloyed nanowires that not only appeared to be more tolerant to CO poisoning than nanoscale motifs generated using the OAm/OA fabrication method based on our chronoamperometry data but also gave rise to a 7× increase in performance as compared with pure Pt NWs alone. A comparison of our PtRu results versus the corresponding MOR data of various PtRu alloys previously reported in the literature emphasizes that the findings discussed herein are certainly within the expected range of prior values previously noted with PtRu (Table S5, ESI†).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The synthesis protocols and associated structural characterization reported herein are based on work performed in SSW's laboratory, supported by the U.S. National Science Foundation under Grant No. CHE-1807640. Certain characterization experiments (i.e., TEM, HRTEM, EDS, and SEM) for this manuscript were performed in part at the Center for Functional Nanomaterials, located at Brookhaven National Laboratory, which is supported by the U.S. Department of Energy under Contract No. DE-SC-00112704. Moreover, the collaborative studies were also supported in part by a seed grant from the Stony Brook University Office of the Vice President for Research. AIF acknowledges the support of his work by the National Science Foundation under grant CHE 2203858. This research used beamline 7-BM (QAS) of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. This research used resources from the Center for Functional Nanomaterials, which is a U.S. DOE Office of Science Facility at Brookhaven National Laboratory under Contract No. DE-SC0012704. We thank Drs Lu Ma, Steven Ehrlich, and Nebojsa Marinkovic, in addition to Dr Haodong Wang for their collective expert help during the synchrotron measurements at QAS beamline. Beamline operations were supported in part by the Synchrotron Catalysis Consortium (U.S. DOE, Office of Basic Energy Sciences, Grant No. DE-SC0012335).References
- Y. Shao, G. Yin and Y. Gao, J. Power Sources, 2007, 171, 558–566 CrossRef CAS.
- J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 315, 220–222 CrossRef CAS PubMed.
- L. Swette and N. Kackley, J. Power Sources, 1990, 29, 423–436 CrossRef CAS.
- L. Li and S. S. Wong, ACS Omega, 2018, 3, 3294–3313 CrossRef CAS PubMed.
- C. Koenigsmann, M. E. Scofield, H. Liu and S. S. Wong, J. Phys. Chem. Lett., 2012, 3, 3385–3398 CrossRef CAS.
- J. L. Fernandez, D. A. Walsh and A. J. Bard, J. Am. Chem. Soc., 2005, 127, 357–365 CrossRef CAS PubMed.
- J. Greeley and J. K. Nørskov, J. Phys. Chem. C, 2009, 113, 4932–4939 CrossRef CAS.
- G. A. Camara, M. J. Giz, V. A. Paganin and E. A. Ticianelli, J. Electroanal. Chem., 2002, 537, 21–29 CrossRef CAS.
- H. Liu, C. Song, L. Zhang, J. Zhang, H. Wang and D. P. Wilkinson, J. Power Sources, 2006, 155, 95–110 CrossRef CAS.
- J.-H. Wee and K.-Y. Lee, J. Power Sources, 2006, 157, 128–135 CrossRef CAS.
- X. Zhang and K.-Y. Chan, Chem. Mater., 2002, 15, 451–459 CrossRef.
- R. Basnayake, Z. Li, S. Katar, W. Zhou, H. Rivera, E. S. Smotkin, D. J. Casadonte and C. Korzeniewski, Langmuir, 2006, 22, 10446–10450 CrossRef CAS PubMed.
- S. Sun, H. Xu, S. Tang, J. Guo, H. Li, L. Cao, B. Zhou, Q. Xin and G. Sun, Chin. J. Catal., 2006, 27, 932–936 CrossRef CAS.
- G.-Y. Zhao, C.-L. Xu, D.-J. Guo, H. Li and H.-L. Li, J. Power Sources, 2006, 162, 492–496 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS PubMed.
- S. Garbarino, A. Ponrouch, S. Pronovost and D. Guay, Electrochem. Commun., 2009, 11, 1449–1452 CrossRef CAS.
- H. A. Gasteiger, N. Markovic, P. N. Ross and E. J. Cairns, J. Phys. Chem., 1993, 97, 12020–12029 CrossRef CAS.
- P. Liu and J. K. Nørskov, Fuel Cells, 2001, 1, 192–201 CrossRef CAS.
- W. Zhao, D. Huang, Q. Yuan and X. Wang, Nano Res., 2016, 9, 3066–3074 CrossRef CAS.
- W. Zhao, B. Ni, Q. Yuan, Y. Wang, Q. Zhang and X. Wang, Langmuir, 2017, 33, 8070–8075 CrossRef CAS PubMed.
- Z. Shen, M. Yamada and M. Miyake, Chem. Commun., 2007, 245–247 RSC.
- N. Jha, A. L. M. Reddy, M. M. Shaijumon, N. Rajalakshmi and S. Ramaprabhu, Int. J. Hydrogen Energy, 2008, 33, 427–433 CrossRef CAS.
- S. Yang, F. Hong, L. Wang, S. Guo, X. Song, B. Ding and Z. Yang, J. Phys. Chem. C, 2009, 114, 203–207 CrossRef.
- H. Liu, C. Koenigsmann, R. R. Adzic and S. S. Wong, ACS Catal., 2014, 4, 2544–2555 CrossRef CAS.
- C. Koenigsmann, W.-p Zhou, R. R. Adzic, E. Sutter and S. S. Wong, Nano Lett., 2010, 10, 2806–2811 CrossRef CAS PubMed.
- C. Koenigsmann and S. S. Wong, Energy Environ. Sci., 2011, 4, 1161–1176 RSC.
- C. Koenigsmann, E. Sutter, R. R. Adzic and S. S. Wong, J. Phys. Chem. C, 2012, 116, 15297–15306 CrossRef CAS.
- N. P. Tarasova, Y. V. Smetannikov and A. A. Zanin, Russ. Chem. Rev., 2010, 79, 463–477 CrossRef CAS.
- X. Teng, M. Feygenson, Q. Wang, J. He, W. Du, A. I. Frenkel, W. Han and M. Aronson, Nano Lett., 2009, 9, 3177–3184 CrossRef CAS PubMed.
- H. Zhou, W.-p Zhou, R. R. Adzic and S. S. Wong, J. Phys. Chem. C, 2009, 113, 5460–5466 CrossRef CAS.
- W.-P. Zhou, M. Li, C. Koenigsmann, C. Ma, S. S. Wong and R. R. Adzic, Electrochim. Acta, 2011, 56, 9824–9830 CrossRef CAS.
- M. E. Scofield, Y. Zhou, S. Yue, L. Wang, D. Su, X. Tong, M. B. Vukmirovic, R. R. Adzic and S. S. Wong, ACS Catal., 2016, 6, 3895–3908 CrossRef CAS.
- M. E. Scofield, C. Koenigsmann, D. Bobb-Semple, J. Tao, X. Tong, L. Wang, C. S. Lewis, M. B. Vukmirovic, Y. Zhu, R. R. Adzic and S. S. Wong, Catal.: Sci. Technol., 2016, 6, 2435–2450 RSC.
- H. Liu, R. R. Adzic and S. S. Wong, ACS Appl. Mater. Interfaces, 2015, 7, 26145–26157 CrossRef CAS PubMed.
- C. Koenigsmann, A. C. Santulli, K. Gong, M. B. Vukmirovic, W.-P. Zhou, E. Sutter, S. S. Wong and R. R. Adzic, J. Am. Chem. Soc., 2011, 133, 9783–9795 CrossRef CAS PubMed.
- C. Koenigsmann, D. B. Semple, E. Sutter, S. E. Tobierre and S. S. Wong, ACS Appl. Mater. Interfaces, 2013, 5, 5518–5530 CrossRef CAS PubMed.
- M. E. Scofield, C. Koenigsmann, L. Wang, H. Liu and S. S. Wong, Energy Environ. Sci., 2015, 8, 350–363 RSC.
- L. Li, H. Liu, C. Qin, Z. Liang, A. Scida, S. Yue, X. Tong, R. R. Adzic and S. S. Wong, ACS Appl. Nano Mater., 2018, 1, 1104–1115 CrossRef CAS.
- S. C. McGuire, A. M. Ebrahim, N. Hurley, L. Zhang, A. I. Frenkel and S. S. Wong, Chem. Sci., 2021, 12, 7158–7173 RSC.
- A. Halder and N. Ravishankar, Adv. Mater., 2007, 19, 1854–1858 CrossRef CAS.
- S. Mourdikoudis and L. M. Liz-Marzan, Chem. Mater., 2013, 25, 1465–1476 CrossRef CAS.
- Z. Peng, H. You and H. Yang, ACS Nano, 2010, 4, 1501–1510 CrossRef CAS PubMed.
- N. Poudyal, G. S. Chaubey, V. Nandwana, C.-B. Rong, K. Yano and J. P. Liu, Nanotechnology, 2008, 19, 355601 CrossRef PubMed.
- A. I. Frenkel, Chem. Soc. Rev., 2012, 41, 8163–8178 RSC.
- R. M. Anderson, L. Zhang, J. A. Loussaert, A. I. Frenkel, G. Henkelman and R. M. Crooks, ACS Nano, 2013, 7, 9345–9353 CrossRef CAS PubMed.
- J. Timoshenko, C. J. Wrasman, M. Luneau, T. Shirman, M. Cargnello, S. R. Bare, J. Aizenberg, C. M. Friend and A. I. Frenkel, Nano Lett., 2019, 19, 520–529 CrossRef CAS PubMed.
- A. I. Frenkel, V. S. Machavariani, A. Rubshtein, Y. Rosenberg, A. Voronel and E. A. Stern, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 9364–9371 CrossRef CAS.
- S.-A. Chen, Y.-C. Liang, K.-T. Lu, C.-W. Pao, J.-F. Lee, T.-L. Lin and J.-M. Chen, Phys. Chem. Chem. Phys., 2014, 16, 3939–3945 RSC.
- S. Alayoglu, P. Zavalij, B. Eichhorn, Q. Wang, A. I. Frenkel and P. Chupas, ACS Nano, 2009, 3, 3127–3137 CrossRef CAS PubMed.
- S. Stoupin, E.-H. Chung, S. Chattopadhyay, C. U. Segre and E. S. Smotkin, J. Phys. Chem. B, 2006, 110, 9932–9938 CrossRef CAS PubMed.
- S. Stoupin, H. Rivera, Z. Li, C. U. Segre, C. Korzeniewski, D. J. Casadonte Jr., H. Inoue and E. S. Smotkin, Phys. Chem. Chem. Phys., 2008, 10, 6430–6437 RSC.
- S.-H. Liu, W.-Y. Yu, C.-H. Chen, A.-Y. Lo, B.-J. Hwang, S.-H. Chien and S.-B. Liu, Chem. Mater., 2008, 20, 1622–1628 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. I. Frenkel, Q. Wang, S. I. Sanchez, M. W. Small and R. G. Nuzzo, J. Chem. Phys., 2013, 138, 064202 CrossRef PubMed.
- J. M. Cowley, Phys. Rev., 1965, 138, 1384–1389 CrossRef CAS.
- S. C. McGuire, A. M. Ebrahim, N. Hurley, L. Zhang, A. I. Frenkel and S. S. Wong, Chem. Sci., 2021, 12, 7158–7173 RSC.
- A. Frenkel, Z. Kristallogr. – Cryst. Mater., 2007, 222, 605–611 CrossRef CAS.
- D. Glasner and A. I. Frenkel, AIP Conf. Proc., 2007, 882, 746–748 CrossRef CAS.
- N. Poudyal, G. S. Chaubey, V. Nandwana, C.-B. Rong, K. Yano and J. P. Liu, Nanotechnology, 2008, 19, 355601 CrossRef PubMed.
- T. F. Tadros, An Introduction to Surfactants, De Gruyter, 2014 Search PubMed.
- T. Song, F. Gao, S. Guo, Y. Zhang, S. Li, H. You and Y. Du, Nanoscale, 2021, 13, 3895–3910 RSC.
- Y. Song, R. M. Garcia, R. M. Dorin, H. Wang, Y. Qiu, E. N. Coker, W. A. Steen, J. E. Miller and J. A. Shelnutt, Nano Lett., 2007, 7, 3650–3655 CrossRef CAS PubMed.
- Y. Hu, A. Zhu, Q. Zhang and Q. Liu, Int. J. Hydrogen Energy, 2016, 41, 11359–11368 CrossRef CAS.
- L. Zhao, S. Wang, Q. Ding, W. Xu, P. Sang, Y. Chi, X. Lu and W. Guo, J. Phys. Chem. C, 2015, 119, 20389–20400 CrossRef CAS.
- S. Lu, K. Eid, D. Ge, J. Guo, L. Wang, H. Wang and H. Gu, Nanoscale, 2017, 9, 1033–1039 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ya00278k |
| ‡ These authors contributed equally to this manuscript. |
| This journal is © The Royal Society of Chemistry 2024 |