
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrobially-mediated aerobic oxidation of trace element-bearing pyrite in neutral-pH sandstone aquifer sediments†
Lisa
Haas
ab,
Matthew
Ginder-Vogel
 c,
James J.
Zambito
IV
d,
David
Hart
b and
Eric E.
Roden
*a
c,
James J.
Zambito
IV
d,
David
Hart
b and
Eric E.
Roden
*a
aDepartment of Geoscience, University of Wisconsin, 1215 West Dayton Street, Madison, WI 53706, USA. E-mail: eroden@geology.wisc.edu; ldhaas@wisc.edu
bWisconsin Geological and Natural History Survey, 3817 Mineral Point Rd, Madison, WI 53705, USA. E-mail: david.hart@wisc.edu
cDepartment of Civil and Environmental Engineering, University of Wisconsin, Water Science Engineering Laboratory, 660 N. Park Street, Madison, WI 53706, USA. E-mail: mgindervogel@wisc.edu
dDepartment of Geology, Beloit College, 700 College Street, Beloit, WI 53511, USA. E-mail: zambitoj@beloit.edu
First published on 12th April 2024
Abstract
Pyrite (FeS2) is the most abundant sulfide mineral on Earth and represents a significant reservoir of iron and sulfur in modern and ancient sediments. Oxidation of pyrite in the terrestrial subsurface is commonly associated with lowering of groundwater pH and release of constituent trace elements to solution. Although the central role of microbial activity in pyrite oxidation is well understood in acid mine/rock drainage and other low-pH (e.g. pH < 2) environments, the role of microorganisms in mediating pyrite oxidation under circumneutral pH conditions is not well understood. Here we demonstrate the potential for aerobic microbial metabolism to promote circumneutral pH oxidation of trace element-bearing pyrite in Cambrian-age sandstones from Trempealeau County, WI (USA). Microbial activity accelerated ca. 2–5 fold the rate and extent of sulfate release (a direct measure of pyrite oxidation) from reduced pyrite-bearing sediments. pH values dropped to 3 in biotic microcosms which contained limited carbonate (dolomite) buffering capacity. The overall surface area-specific rate constant for pyrite oxidation inferred from batch reaction modeling of these microcosms (10−7.8 mol m−2 s−1) was ca. 25-fold higher than for the corresponding abiotic reactors (10−9.2 mol m−2 s−1). Calcium and magnesium were proportionally released to solution with sulfate as a result of carbonate and/or Ca-aluminosilicate dissolution by acid generated from pyrite oxidation. When the amount of acid from pyrite oxidation exceeded the system buffering capacity, metals were selectively released from the geological material. No significant release of trace metals took place in abiotic reactors, which showed much lower rates of pyrite oxidation. These findings suggest that groundwaters in contact with pyrite-containing geological formations contain microorganisms capable of accelerating the oxidation of native pyrite in those formations. Analysis of microbial community composition in the microcosms by 16S rRNA gene amplicon sequencing showed enrichment in organisms related to taxa associated with chemolithotrophic metabolism (Candidatus Tenderia electrophaga, Thioprofundum lithophicum, and Thiobacillus thioparus) from background levels (<2%) to up to 40% of total sequence reads. A reactive transport modeling exercise demonstrated how microbial acceleration of pyrite oxidation could have a crucial, near-term (<10 years) impact on pH decline and trace element release in response to influx of oxygenated groundwater into previously reduced geological strata. Our results have key implications for controls on the onset of low-pH conditions and associated changes in groundwater quality in drinking water wells located within pyrite-bearing geological formations.
Environmental significancePyrite is a dominant mineral reservoir for iron and sulfur in sedimentary environments. Reaction of pyrite and other metal sulfides with oxygen results in lowering of pH and the release of iron, sulfate, and trace elements to solution. Microorganisms are known to accelerate pyrite oxidation, particularly in acid mine drainage and other low-pH environments. The potential for microorganisms to mediate pyrite oxidation under circumneutral pH conditions has also recently been recognized. Microbial acceleration of neutral-pH pyrite oxidation has important implications for water quality in aquifers where pyrite-bearing geological strata are exposed to oxygen as a result of human activities. This study demonstrates the ability of aerobic microbial metabolism to promote oxidation of native pyrite phases in circumneutral-pH sandstone aquifer sediments from west-central Wisconsin, USA. Our results have key implications for controls on the onset of low-pH conditions and associated changes in groundwater quality in private and municipal drinking water wells located within pyrite-bearing subsurface environments. |
1. Introduction
Pyrite (FeS2) is the most abundant sulfide mineral on Earth, and its oxidation through biotic and abiotic pathways is a major source of sulfate input to oceans both in modern1,2 and ancient environments.3–5 Oxidative transformations of pyrite and other metal sulfides play a key role in terrestrial element partitioning with broad impacts to contaminant mobility and the formation of acid mine drainage systems.6 Microbial pyrite oxidation has been extensively studied at low pH, mainly in the context of acid mine/rock drainage. Under acidic conditions, microbial catalysis greatly accelerates pyrite oxidation through aerobic oxidation of soluble Fe2+, which produces Fe3+ ions that serve as an effective chemical oxidant for FeS2.7 However, many soil and sediment environments are not acidic but rather have a circumneutral pH, where pyrite oxidation can occur abiotically through reaction with atmospheric oxygen,8 or potentially through biologically catalyzed pathways. While there is a growing body of literature on circumneutral chemolithotrophic microbial oxidation of pyrite coupled to denitrification,9–11 fewer studies have examined the ability of aerobic microorganisms to accelerate pyrite oxidation at circumneutral pH. The growth of such organisms has been previously suggested in hydrothermal habitats12,13 and subglacial ecosystems.14,15 Only recently, however, has explicit evidence for microbially accelerated aerobic pyrite oxidation at neutral pH been obtained.16,17 Percak-Dennett et al.16 demonstrated sustained enhancement of sulfate release in the presence of a live, natural inoculum over multiple generations in cultures grown on synthetic framboidal pyrite. Napieralski et al.17 demonstrated 2–5 fold accelerated oxidation of specimen pyrite by organisms that had previously colonized pyrite incubated in situ in a pyrite-bearing shale formation in central Pennsylvania.Microbial acceleration of aerobic, neutral-pH pyrite oxidation has important implications for low-temperature geochemistry in modern environments, in particular subsurface metal sulfide weathering (e.g. in pyrite-bearing sedimentary rocks) and acid mine/rock drainage systems. In such environments, acidification linked to the formation and accumulation of ferric iron during biotic and/or abiotic pyrite oxidation initiates a propagation cycle that results in massive metal sulfide leaching.6 Depending on the buffering capacity of the local surficial environment, early aerobic and/or nitrate-dependent pyrite oxidation could initiate the process of acidification, thereby accelerating the eventual development of acidic conditions.
Poor water quality conditions exist in many public (and private) groundwater wells in west-central Wisconsin (WI), USA. As illustrated by data from three municipal wells in Fig. 1A, concentrations of iron, manganese, aluminum, various trace metals/metalloids, and sulfate in these subneutral pH (4–4.5) well waters exceed either enforceable or secondary standards by factors of 4 to >1000. West-central WI, similar to the entirety of central WI, is an area where the Cambrian–Ordovician regional aquifer system (see Fig. S1† for map of WI geological units) within sandstone formations (interbedded with shales and carbonate strata) is a primary groundwater resource for municipalities and private well owners. Well construction reports indicate that each of the impacted wells shown in Fig. 1A is open to the Cambrian-aged Tunnel City Group (TCG) and Wonewoc Formation (WF; Elk Mound Group) geologic units. The TCG, which contains variable abundance of carbonate, quartz, feldspar, and the clay mineral glauconite, stratigraphically overlays the quartz-dominated WF. North of the impacted wells where the TCG and WF intersect the land surface, the WF is of economic interest as it has been actively mined for the friable sandstone which consists upwards of 97% sand-sized, rounded quartz grains that is applied as a proppant during hydraulic oil and gas fracturing (frac) operations. Frac sand wash ponds where WF sediments are washed, sieved, and settled out also show poor water quality (Fig. 1B), specifically high iron, manganese, and aluminum. Infiltration of frac sand wash pond fluid into shallow groundwater (e.g. as a result of both normal operations, e.g. unlined ponds, as well as accidental sludge release, e.g. levee failures, to surface waterways) represents a risk to local homeowner well water quality that has received substantial public attention.18
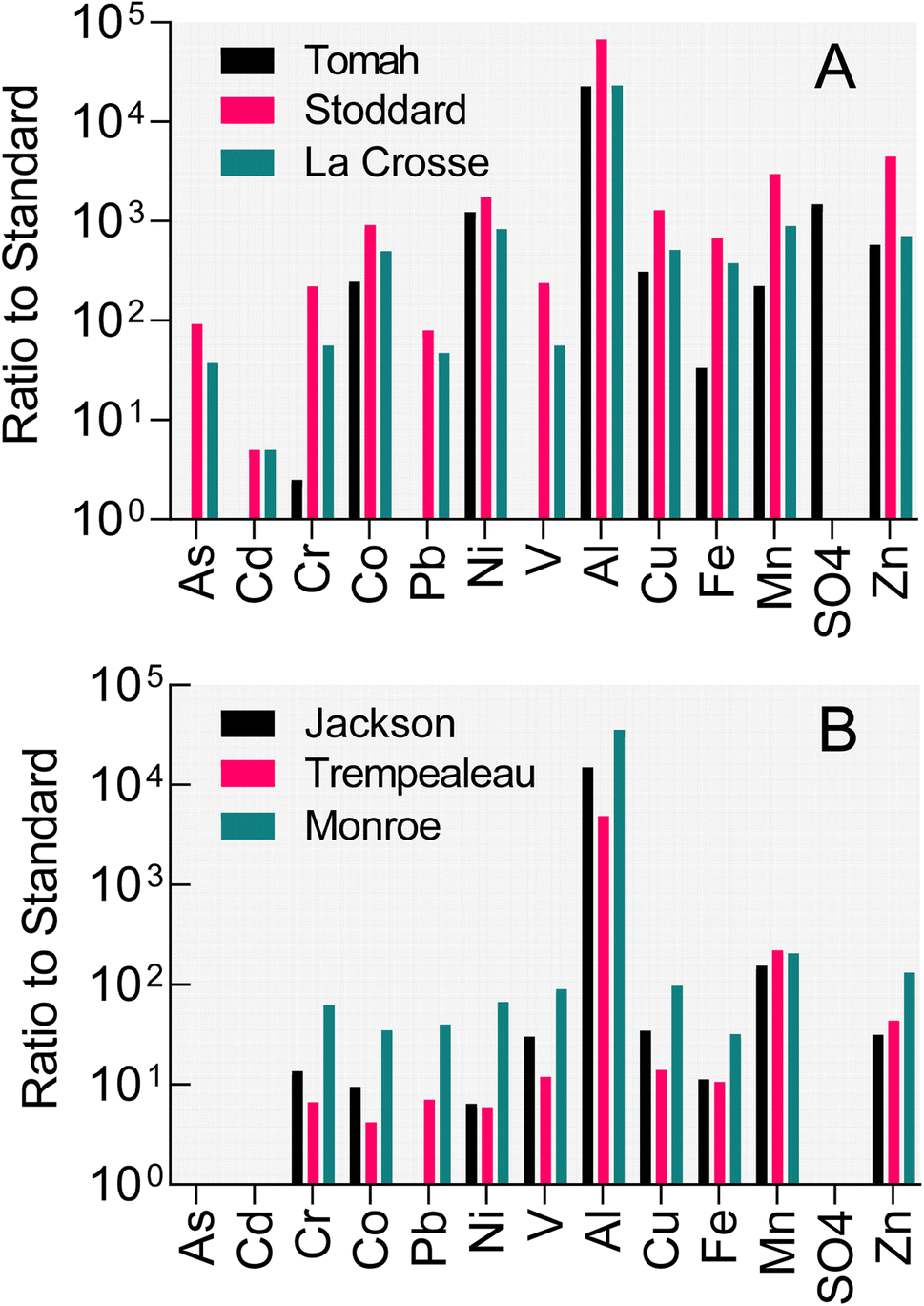 | ||
| Fig. 1 Water quality results from wells open to Tunnel City Group and Wonewoc Formation strata (A) and from frac sand wash ponds in West-Central Wisconsin. (B) Water quality data provided by D. Johnson, Wisconsin Department of Natural Resources (personal communication, 2016). Values plotted represent the ratio of measured concentration to the either the NR140 enforceable standard or the NR809 secondary standard for groundwater and drinking water (Wisconsin-DNR, 2021). | ||
Zambito et al.19 suggested that oxidative dissolution of iron sulfide minerals (mainly pyrite) containing trace abundances of divalent transition metals (Co, Cu, Ni, Pb, Zn) and arsenic (As) could be responsible for generation of the low-pH, elevated dissolved metal conditions present in well water and frac sand pond fluids in west-central WI. This process is responsible for contamination of groundwater in northeastern, WI, where introduction of aerobic groundwater into pyrite-bearing secondary cement horizons in the Sinnipee Group and St. Peter Sandstone (Ancell Group) formations has led to dissolved As concentration far above water quality standards.20,21 As in the case of the St. Peter Sandstone, the pyrite identified in the TCG and WF units is present as sub-cm-sized nodules or disseminated in rock pore spaces as sub-mm-sized crystals and framboids (Fig. 2; see also Fig. S3†). The origin of the pyrite in these units may include early diagenetic authigenic phases (e.g. in Cambrian age marine layers), as well as precipitation from later migrating fluids.22–24
 | ||
| Fig. 2 Rock core images of pre-processed Tunnel City Group (TCG) and Wonewoc Formation (WF) reduced and oxidized that was pulverized into sediment for reactor experiments. Green and red boxes show examples of sub-mm sized disseminated pyrite crystals. Upper redox box shows a magnified image of a Tunnel City Group sample with pyrite lining the inside of a vug, collected at 186 ft depth. Lower red box shows magnified image a Wonewoc formation sample with abundant well-rounded quartz sand and pyrite nodules infilling a ca. 500 million year-old worm burrow, collected at 360 feet depth. See Fig. S2† for larger versions of the images in the red boxes. Images on the right illustrate oxidized version of the same geological strata, in which virtually all pyrite has been replaced with iron oxides present as coatings on quartz sand. | ||
In light of recent studies that demonstrate microbial acceleration of neutral-pH pyrite oxidation16,17 it seems possible that microbial catalysis may be a factor these systems, as well as other similar environments within (i.e. the Sinipee/St. Peter Sandstone) and beyond the state of WI. Indeed, trace metal mobilization from sulfides is a common groundwater quality concern. The sandstone aquifer settings in WI and the associated mechanisms for trace metal mobilization differ from the alluvial sedimentary environments in Asia and southeast Asia where groundwater As contamination is major threat to human health.25 However, oxidation of pyrite and other metal sulfides is ultimately responsible for the generation of iron oxide-associated As in alluvial sediments,26 and may contribute directly to net mobilization of As to groundwaters in Bangladesh27 and other analogous systems.
This study examined the potential for aerobic microbial metabolism to promote circumneutral pH pyrite oxidation in subsurface sediments from the TCG and WF geological units. The geologic materials used were specifically chosen based on their mineral composition representing two subsurface redox endmembers (reduced vs. oxidized) with two pH buffering endmember capacities (TCG with carbonate vs. WF without carbonate). A private well owner provided a source of in situ groundwater (with its native microbial community) from geological strata in proximity to where the geologic materials were collected. A unique aspect of this work is the use of naturally-occurring sedimentary pyrite phases, which to our knowledge have never been examined for their susceptibility microbially-accelerated aerobic oxidation at circumneutral pH. Numerous studies have examined the potential for aerobic microorganisms to obtain energy for growth and/or accelerate pyrite oxidation at both acidic (reviewed in ref. 6) and neutral pH,6,12,16,17,28–32 but virtually all of these studies (and all of the circumneutral pH studies) have been conducted with specimen minerals (arsenopyrite in the case of ref. 32), as opposed to naturally-occurring phases.
We evaluated the hypothesis that microbial activity could increase the rate and extent of sandstone pyrite oxidation by conducting batch incubation experiments with native microbial populations in WF groundwater. The response of the microbial community was assessed through 16S rRNA gene amplicon sequencing, which based on previous work16,17 was expected to show a distinct response to controlled exposure to pyrite-containing sediment. The impact of microbial activity on the surface texture of sediment pyrite grains was assessed through scanning electron microscopy. A one-dimensional, single-cell reactive transport model, employing rate constants derived from the batch experiments, was used to assess the potential for microbially-accelerated pyrite oxidation to promote the onset of low-pH conditions and release to trace metals to solution in poorly-buffered WF aquifer sediments.
2. Materials and methods
2.1 Geological materials
Pyrite-containing reduced (Red) and pyrite-depleted oxidized (Ox) end-member materials from the TCG and WF geologic units were obtained from archived (WGNHS Core repository in Mount Horeb, WI) borehole rock cores drilled by the Wisconsin Geological and Natural History Survey (WGNHS) in 2015 and 2016. Fig. S2† shows the drill core locations in relation to the regional stratigraphy and landscape relief. The sampled intervals include TCG-Red from WGNHS Arcadia Quarry Core 62000166, TCG-Ox from WGNHS Lockington Quarry Core 62000213, WF-Red WGNHS Arcadia Quarry Core 62000166, and WF-Ox Thompson Valley Core 62000218 (see Fig. 2 for photos of sampled core intervals). All boreholes are within 3.5 km of each other. The reduced TCG and WF materials contained ca. 0.7 and 1.6% by weight of pyrite-S, whereas the oxidized representatives of the same lithologies contained 0.001% or lower pyrite-S by weight (Table 1). The TCG lithologies are abundant in pH-buffering carbonate minerals, whereas WF lithologies are not (Table 1). The bulk solid-phase elemental composition of the materials is provided in Tables S1 and S2.† The choice of these materials allowed us examine (1) how exposure of reduced, pyrite-bearing sediments to aerobic conditions could impact groundwater geochemistry, in direct comparison with stratigraphically equivalent oxidized, pyrite-depleted sediments; (2) the influence of sediment carbonate mineral abundance on the geochemical response to native sediment pyrite oxidation.| Material | Total reduced Sa (%) | Total Feb (%) | Ca (%) | TICc (%) | pHd |
|---|---|---|---|---|---|
| a Chromium reduction extraction (Fossing and Jorgensen, 1989) and colorimetric S(−II) analysis (Cline, 1969). b Aqua regia digestion, ICP-OES analysis (ALS Geochemistry Reno, NV). c Total inorganic carbon, high temperature combustion (Shimadzu TOC-VCSH). d After 30 minutes suspension of 1 g dry material in 10 mL of 10 mM CaCl2. e Below detection. | |||||
| TCG-Red | 0.66 | 2.55 | 10.7 | 8.21 | 8.5 |
| TCG-Ox | 0.0012 | 2.44 | 10.4 | 5.14 | 8.2 |
| WF-Red | 1.58 | 0.73 | 0.03 | BDe | 5.7 |
| WF-Ox | 0.0006 | 0.83 | 0.01 | BDe | 5.7 |
Rock core section samples were crushed in a jaw crusher then puck-and-bowl pulverized to produce a homogenized bulk sample. The homogenized material was washed three times with anoxic deionized (DI) water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 solid–liquid ratio) to remove any solid-associated sulfate (e.g. sulfate-bearing phases derived from pyrite oxidation since exposed to the atmosphere during core storage). The suspended material was shaken vigorously and left to settle for 24 hours in an anaerobic chamber. The sample was then centrifuged and the supernatant was removed. This was repeated three times to remove virtually all solid-associated sulfate.
:2 solid–liquid ratio) to remove any solid-associated sulfate (e.g. sulfate-bearing phases derived from pyrite oxidation since exposed to the atmosphere during core storage). The suspended material was shaken vigorously and left to settle for 24 hours in an anaerobic chamber. The sample was then centrifuged and the supernatant was removed. This was repeated three times to remove virtually all solid-associated sulfate.
2.2 Groundwater
Groundwater was collected from a homeowner's outdoor spigot near Arcadia, WI and within 3.5 km of the borehole drill sites. By reviewing the well construction report, the groundwater well sampled for this study was known to be pumping water from 12 m below the stratigraphic contact between the WF and overlying TCG. The well itself was not cased starting at 15 m above the contact in the TCG and ending 18 m below the contact in the WF. The static water level in the well in the year 2000 was 8 m below the WF and TCG stratigraphic contact. The line was flushed for 3 hours at 7.5 L per minute (ca. 1350 L through the 445 L well and 150 L storage tank) prior to sample collection. The well water did not pass through a water softener or other water treatment device or filter before reaching the outdoor spigot where it was collected. pH was measured onsite with a pH test strip and immediately upon returning to the lab. Groundwater was stored in 1 L Nalgene wide-mouth sample bottles filled completely to minimize sample degassing. Samples were transported and chilled in a cooler, then refrigerated at 4 °C before microcosm preparation. Samples for groundwater DNA analysis were obtained by passing ca. 2 L of fluid through in-line 0.2 μm filter cartridges. The cartridges were transported with the groundwater and frozen immediately upon return to the laboratory. The major and trace element composition of the groundwater is provided in Table S3.†2.3 Microcosm reactors
A series of microcosm reactors were constructed with the homogenized geological materials and freshly collected groundwater (see Fig. S4† for experimental set-up). Five grams of solids were suspended in 50 mL of groundwater in 120 mL acid-washed glass serum bottles. The bottles were sealed with an aluminum crimp-capped rubber stopper. For live reactors, 5 mL of groundwater was mixed with the geologic material and the bottles were autoclaved. After cooling to room temperature, 45 mL of fresh groundwater was added to establish a live microbial community. Abiotic controls were autoclaved immediately after mixing 5 g of solids with 50 mL of groundwater.The reactors were sampled initially weekly, then bi-weekly, then monthly over a 580 days period to monitor changes in solid- and aqueous-phase geochemical conditions. Samples were obtained using sterile disposable 18 gauge needles and 3 mL plastic syringes. An equal volume of oxygenated, sterile air was added prior to sample removal to maintain atmospheric pressure in the bottles. A 0.5 mL portion of mixed solid and aqueous phase sample was stored with 0.25 mL 20 mM EDTA for ATP analysis. Two 1 mL samples were centrifuged (13000×g, 5 min) and the supernatant was separated from the solids. The supernatants were combined and stored for aqueous phase analysis (see below). One of the solid phase samples was dried in a fume hood with ethanol for later SEM imaging. Samples for DNA extraction and sequencing were obtained at day 86 and processed as described below. All samples, except those for SEM, were stored in a −20 °C freezer until the assay was carried out.
2.4 Analytical procedures
Total carbon and total inorganic carbon content of the geological materials were determined with a Shimadzu SSM-5000A Solid Sample Combustion Unit equipped with a TOC-V analyzer at the Wisconsin Energy Institute in Madison, WI. Total reduced sulfur (TRS, mainly pyrite) was determined via chromium reduction (Fossing and Jorgensen, 1989). Major and trace elemental composition was determined by ICP-MS analysis performed by ALS Global (ME-MS61L Super Trace Lowest 270 DL 4A package) in Reno, Nevada, U.S.A. This method involves the digestion of sample material by four acids (perchloric, nitric, hydrofluoric, and hydrochloric), and subsequent determination of ion concentration via ICP-MS. SEM imaging of microcosm solid-phase samples was conducted using a Hitachi S-3400 Scanning Electron Microscope (SEM) equipped with electron dispersive spectroscopy (EDS), backscatter electron (BSE), and secondary electron (SE) detectors.pH was measured with a Thermo Scientific Orion Micro-pH Electrode at room temperature. Alkalinity was measured by titrating fresh groundwater samples to a pH of 4.0 with 0.1 M HCl. Sulfate was analyzed by ion chromatography (IC) with suppressed conductivity detection (Dionex ICS-1000). Samples were diluted with 1:5 with deionized water prior to analysis. For aqueous-phase cations (Al, As, Ca, Co, Cu, Fe, Mg, Mn, Ni, Pb, Si, Sr, Zn), supernatant samples (see above) were filtered (0.45 μm), diluted 1:10 or 1:5 in 5% trace-metal HNO3, and stored in a 15 mL Falcon tube. We acknowledge the possibility that some metals associated with colloidal size particles could have passed through the 0.45 μm filter. The samples were analyzed on an Agilent 5110 Inductively-Coupled Plasma Optical Emission Spectrometer (ICP-OES) in the University of Wisconsin Water Sciences and Engineering Laboratory. Sample solutions of microcosm duplicates (1 mL each) were combined prior to the analysis. All elements were analyzed in axial position for about 1 min 40 seconds minutes per sample.
ATP was determined by the BacTiter-Glo™ Microbial Cell Viability Assay from Promega Corporation and analyzed with a TurnerBioSystems 20/20n Luminometer. DNA was extracted from pelletized microcosm samples using Qiagen DNeasy® PowerSoil® DNA Extraction Kits following manufacturer instructions. Groundwater DNA was extracted from filters by slicing the filter into small pieces with a sterile razor, transferring the small pieces to a 2.0 mL screw-top microcentrifuge tube containing ca. 0.25 mL of sterilized 0.1 mm zirconia/silica beads. DNA was extracted from the filters following Napieralski et al.17 16S ribosomal RNA (rRNA) genes in the DNA extracts were amplified and sequenced via Illumina MiSeq 2 × 300 bp at the University of Wisconsin–Madison Biotechnology Center. The demultiplexed, paired-end sequences were imported into QIIME2-2020.6 where primers were removed, and sequences were trimmed, filtered, denoised, and chimeras were removed using R version 3.5.1 (2018-07-02; package Rcpp) DADA2 1.10.0 (Rcpp 1.0.4.6/PcppParallel 5.0.0/), de novo operative taxonomical unit (OTU) clustering with 99% or greater identity similarity with qiime2-vsearch.33,34 Taxonomic matching databases were silva-138-99-nb-classifier.qza and gg-13-8-99-nb-classifier.qza for comparative bacterial identification.35–37 The BLAST search tool38 was used to identify organisms closely related to the most abundant 16S rRNA gene sequences. This information was used to assign tentative physiological properties to the organisms39 as described previously.40
2.5 Reactive transport modeling
The PHREEQC geochemical modeling software41 was used to develop a simplified reactive transport model of microbially-accelerated subsurface pyrite oxidation to assess the potential influence of this process on groundwater geochemistry in pyrite-bearing sandstone aquifers. Although it was beyond the scope of this work to develop a model specific to any particular groundwater environment, we sought to create a framework that could be used for testing purposes. The developed model was restricted to simulation of a single hypothetical representative elementary volume (REV) of pyrite-containing sandstone aquifer material receiving groundwater throughput.PHREEQC was first used to simulate the results of the biotic and abiotic reduced WF Red microcosm experiments (Fig. 9). Surface area-specific pyrite oxidation rate constants were estimated by trial-and-error based on models described in Williamson and Ramstad42 and Manning et al.43 (see ESI Text†). The initial composition of the aqueous phase was defined by the composition of the WF formation groundwater used in the experiments (Table S4†). The abundance of pyrite was defined based on the dry weight TRS content of the WF Red material (1.6%, Table 1) and the mass of sediment added to the microcosms (see Table S4†). The surface area of pyrite rhombs was constrained to a value of 0.02 m2 g−1 based on the SEM imaging results (see Fig. 7). Ni was included as a representative trace element, which was released from pyrite in direct proportion to its dry weight abundance (Table S2†) relative to pyrite. Fe(III) produced during pyrite oxidation was assumed to precipitate to equilibrium as a relatively “high surface area” (150 m2 g−1; ref. 44) goethite phase, consistent with nanoparticulate Fe(III) oxide phases observed in oxidized WF Red sediments (data not shown). Fe(III) oxide phases generated during pyrite oxidation were assumed to engage in surface complexation reactions with aqueous cations and anions according to the diffuse double layer model,45 using the complexation constants included in the MINTEQ thermodynamic database available in the PHREEQC software. Additional modeling details and parameter values are provided in the ESI Text, Tables S5 and S6.†
The pyrite oxidation rate constants estimated for the batch experiments were employed in a series of single-cell, flow-through REV simulations using the 1-D fluid transport capabilities in PHREEQC. The abundance of pyrite (Table S4†) was defined based on the dry weight TRS content of the WF Red material scaled to an assumed in situ porosity of 30%. The simulations were conducted using fluid residence times ranging from 10 to 100 days for the single-cell REV, corresponding to groundwater flow rates of 1 to 10 cm per day (3.65–36.5 m per year) for a 1 m3 REV in the direction of groundwater flow. This range was chosen based on the regional groundwater flow analysis of Hunt et al.,46 in which the upper bedrock aquifer (which is dominated by the Wonewoc Sandstone; see Fig. S2†) has been inferred to have a geometric mean vertical hydraulic conductivity of ca. 70 cm per day (a value well within the range of other studies47,48). A hydraulic head of 0.01 cm cm−1 was calculated using (1) the water level data from the Well Construction Report (WCR) for the homeowner's well from which groundwater was collected, which sits on a ridge ca. 0.4 km from where the core containing the reduced WF material was obtained (Fig. S2†); and (2) the WCR from a home next to a sand mine located in the adjacent valley. Assuming an aquifer porosity of 30%, these values predict a groundwater flow rate on the order of 2 cm per day. The simulations were conducted for a period of 10 years to assess the initial response to influx of aerobic groundwater into pyrite-bearing strata.
3. Results
3.1 Sulfate, pH, and ATP
Experiments with reduced, pyrite-bearing TCG and WF materials showed that microbial activity increased the amount of sulfate release 2.3 (TCG) and 4.5 (WF) fold compared to abiotic controls over the 86 day time period during which both sets of microcosms remained at circumneutral pH (Fig. 3A, D and Table 2). The amount of sulfate released in biotic or abiotic microcosms containing oxidized, pyrite-depleted sediments was 10-fold higher than the bulk reduced S content of the reactors (Table 2), which indicates release from phases other than pyrite (e.g. gypsum). However, the amount of sulfate released from the oxidized sediments was 2–5 fold lower than rates of release in abiotic reactors containing reduced sediments, and were not influenced by biological activity (Table 2). These results indicate that release of sulfate from phases other than pyrite did not confound the use of sulfate release as a measure of pyrite oxidation in the reduced sediment reactors. The fraction of total pyrite S oxidized at circumneutral pH under biotic vs. abiotic conditions was 9.0% vs. 4.6% after 580 days for TCG-Red, and 3.0% vs. 0.9% after 86 days in WF-Red (Table 2).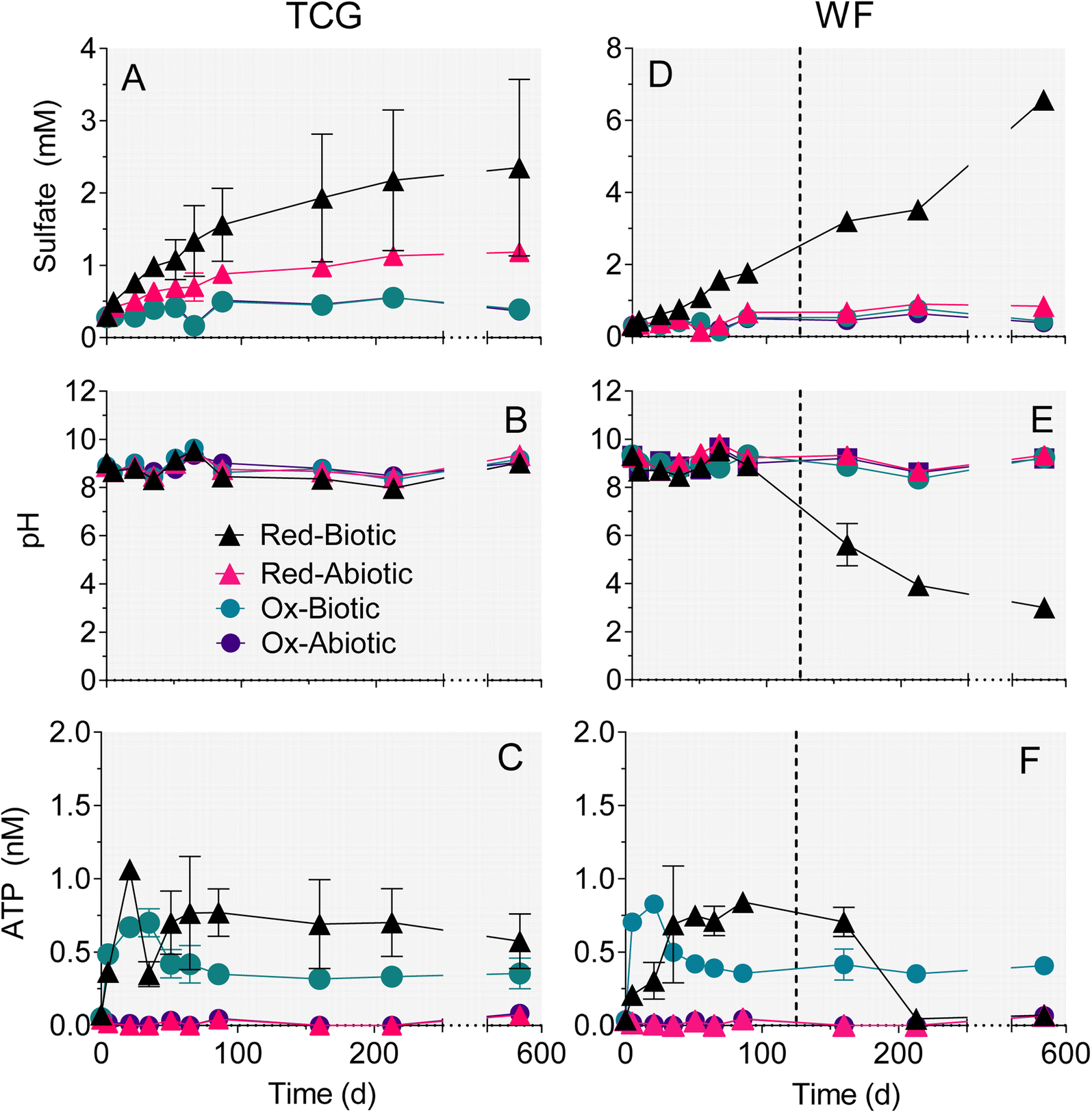 | ||
| Fig. 3 Concentrations of sulfate (A and D), pH (B and E), and ATP (C and F) in the reduced and oxidized TCG and WF sediment incubations. The plotted values represent the average of the duplicate microcosms; error bars show the range. Dotted lines in right-hand panels mark halfway between the two time points when the WF Red live reactors went acidic. | ||
| Treatment | Sulfate released at 86 daysa (mM) | Biotic:abiotic ratio |
Sulfate released at 580 days (mM) | Total reduced Sb (mmol L−1) | % Reduced S oxidizedc |
|---|---|---|---|---|---|
| a All reactors at circumneutral pH. b Bulk concentration calculated from mass of sediment and fluid added to microcosms, and sediment total reduced S content of the sediments (Table 1). c Based on sulfate released after 580 days, except WF-Red biotic based on 86 days. Values are corrected for fluid removed from reactors during sampling over time. d Not determined; amount of sulfate release was greater than the bulk reduced S content of the reactors containing oxidized sediment. | |||||
| TCG-Red biotic | 1.26 ± 1.01 | 2.25 | 2.09 ± 2.37 | 20.8 | 9.02 ± 7.60 |
| TCG-Red abiotic | 0.559 ± 0.005 | 0.792 ± 0.020 | 20.8 | 4.55 ± 0.36 | |
| WF-Red biotic | 1.46 ± 0.054 | 4.51 | 49.5 | 3.05 ± 0.26 | |
| WF-Red abiotic | 0.324 ± 0.003 | 0.508 ± 0.070 | 49.5 | 0.86 ± 0.62 | |
| TCG-Ox biotic | 0.220 ± 0.001 | 1.05 | 0.275 ± 0.017 | 0.038 | NDd |
| TCG-Ox abiotic | 0.210 ± 0.011 | 0.253 ± 0.007 | 0.038 | ND | |
| WF-Ox biotic | 0.231 ± 0.075 | 1.19 | 0.265 ± 0.192 | 0.020 | ND |
| WF-Ox abiotic | 0.194 ± 0.001 | 0.329 ± 0.001 | 0.020 | ND | |
Enough pyrite was oxidized in the WF-Red live microcosms after 86 days to exceed the buffering capacity of the system and they went acidic (Fig. 3E). In contrast, the WF-Red abiotic controls remained circumneutral, as did the biotic and abiotic WF-Ox microcosms. All the TCG microcosms remained circumneutral (Fig. 3B), which was not unexpected given the substantial carbonate mineral content of these materials (Table 1).
ATP content was much higher in the biotic vs. abiotic microcosms and was on average ca. 50% greater in the reduced pyrite-containing microcosms compared to the oxidized pyrite-depleted systems (Fig. 3C and F). These results provide evidence for microbial growth coupled to oxidation of natural pyrite in the reduced sediments.
3.2 Major cations
The reduced TCG and WF sediment reactors showed a steady increase in aqueous-phase Ca (Fig. 4A and C) and Mg (Fig. 4B andD). The enhanced release of Ca and Mg from the biotic WF-Red and TCG-Red treatments is consistent with the greater generation of sulfate compared to abiotic controls, indicating carbonate mineral dissolution coupled to microbially-accelerated pyrite oxidation and acid generation. In contrast to the reduced sediments, Ca and Mg concentrations remained more-or-less constant in both the live and abiotic oxidized sediments. The reason for the higher Ca concentrations in the WF-Ox reactors is unknown, but perhaps related to microbial respiratory activity as indicated by the increase in ATP content in the reactors (Fig. 3F).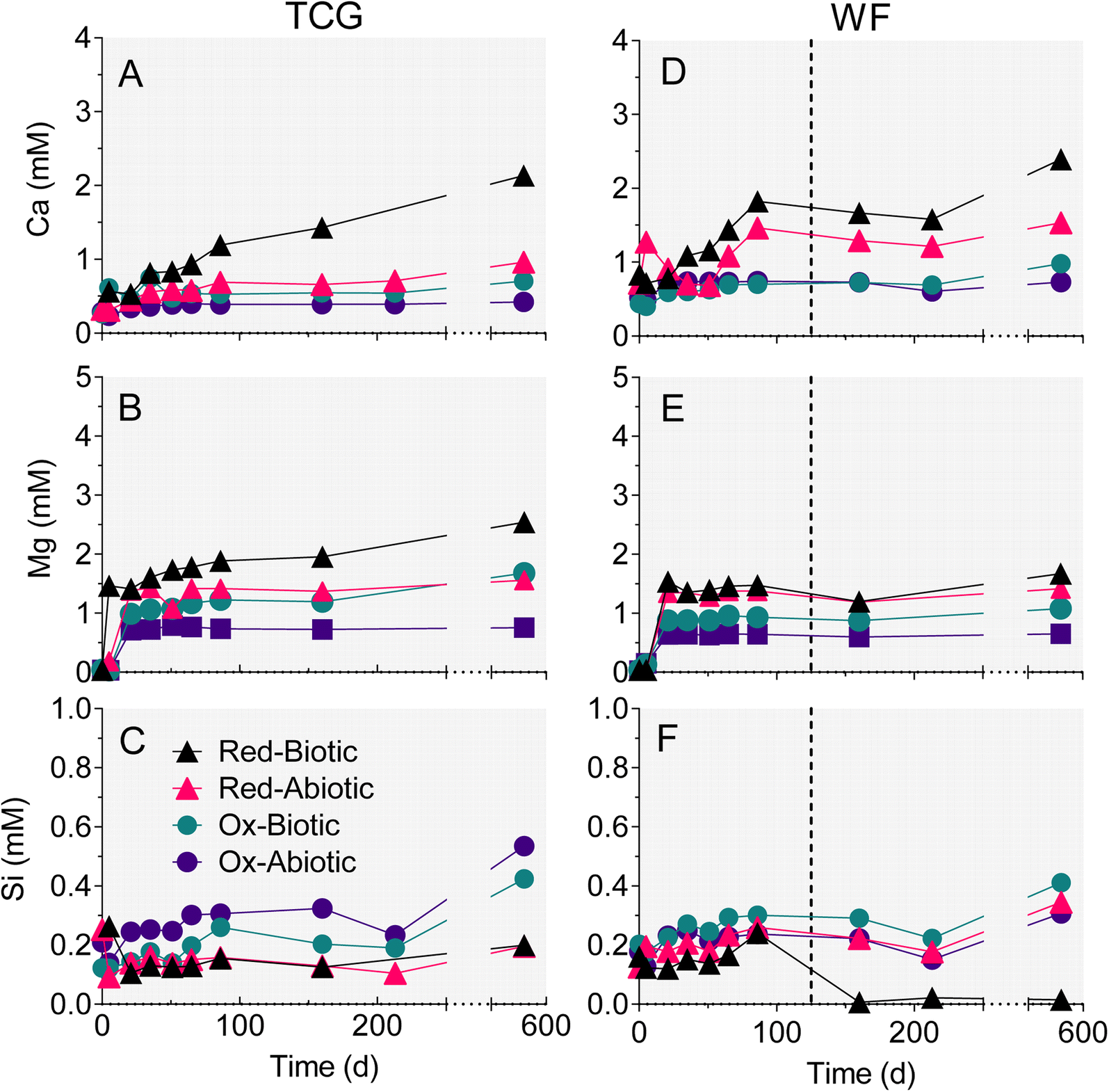 | ||
| Fig. 4 Changes in aqueous-phase Ca (A and D), Mg (B and E), and Si (C and F) in the reduced and oxidized TCG and WF sediment incubations. The plotted values for biotic TCG-Red represent the average of the duplicate microcosms. All other data points are one sample of combined aqueous-phase duplicates. Dotted lines in right-hand panels mark halfway between the two time points when the biotic WF-Red reactors went acidic. | ||
There was no change in aqueous-phase Fe, Al, and Mn, and minor Si generation in reactors that remained circumneutral pH for all four treatments (Fig. 4 and 5). However, Fe, Al, and Mn went into solution (Fig. 5D–F), and Si precipitated out of solution (Fig. 4F), when pH dropped to below 5 after day 86 in the biotic WF-Red reactors. Approximately 12% of the Fe content of the sedimentary pyrite was released to solution by the end of the biotic WF-Red incubations; corresponding values for Al and Mn were 2.5% and 60% respectively.
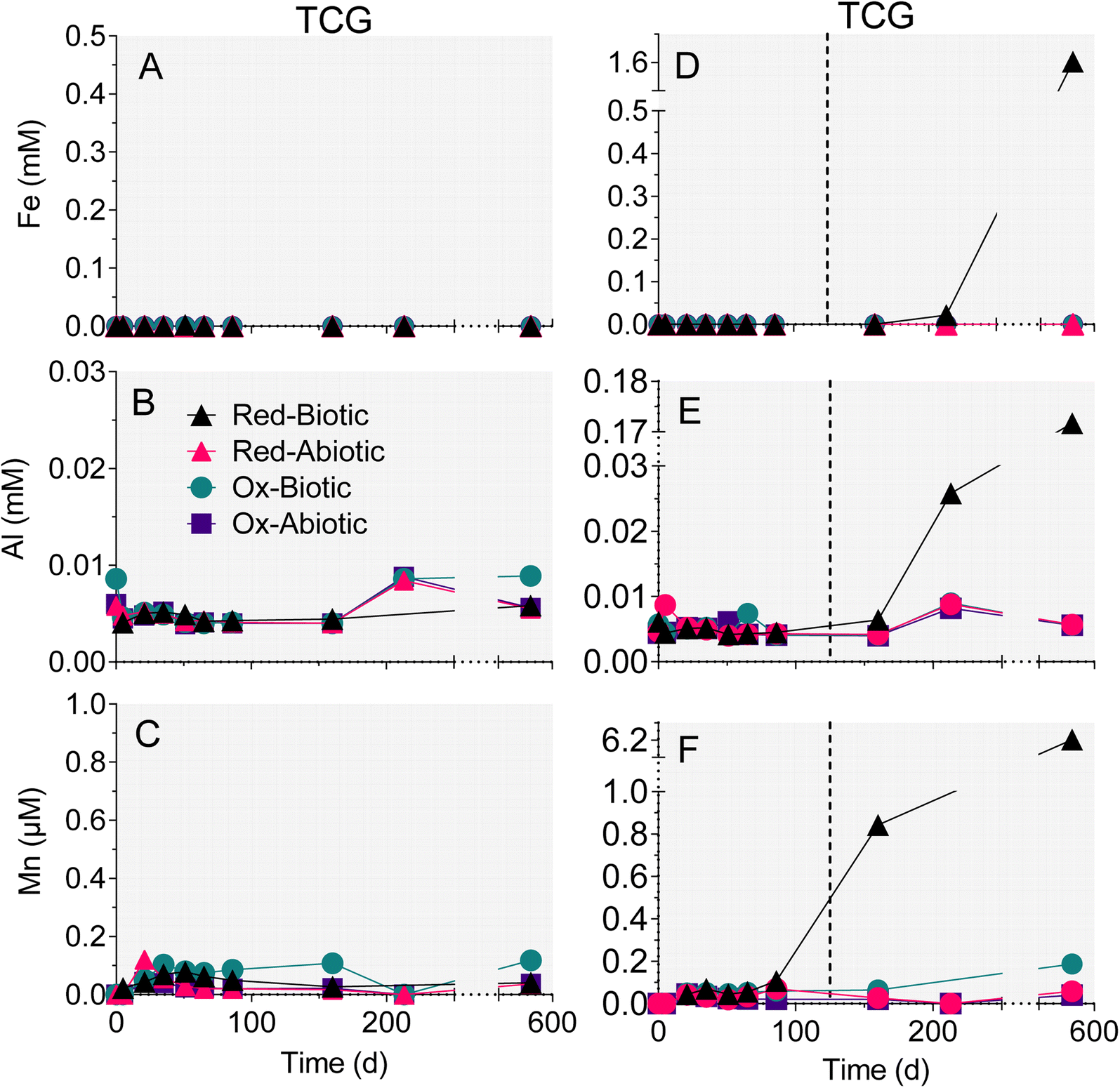 | ||
| Fig. 5 Changes in aqueous-phase Fe (A and D), Al (B and E), and Mn (C and F) in the reduced and oxidized TCG and WF sediment incubations. The plotted values for biotic TCG-Red represent the average of the duplicate microcosms. All other data points are one sample of combined aqueous-phase duplicates. Dotted lines in right-hand panels mark halfway between the two time points when the biotic WF-Red reactors went acidic. | ||
3.3 Trace metals
Trends in dissolved trace metal concentration were generally similar to those for Fe, Al and Mn: Ni, Co, and Cu (Fig. 6D–F) went into solution after the biotic WF-Red reactors went acidic (pH 3). The rest of the treatments, which remained circumneutral, demonstrated no increase in Ni, Co or Cu concentration (Fig. 6A–C). Aqueous-phase As and Pb remained low and fairly constant throughout the experiments, including the biotic WF-Red reactors (Fig. S5†). No significant trends in Sr or Zn were observed in any of the reactors (data not shown). Based on the bulk trace metal content of the geological materials (Table S2†), the extent of mobilization for those metals that showed release in the biotic WF-Red reactors was 54% for Ni, 41% for Co, and 35% for Cu.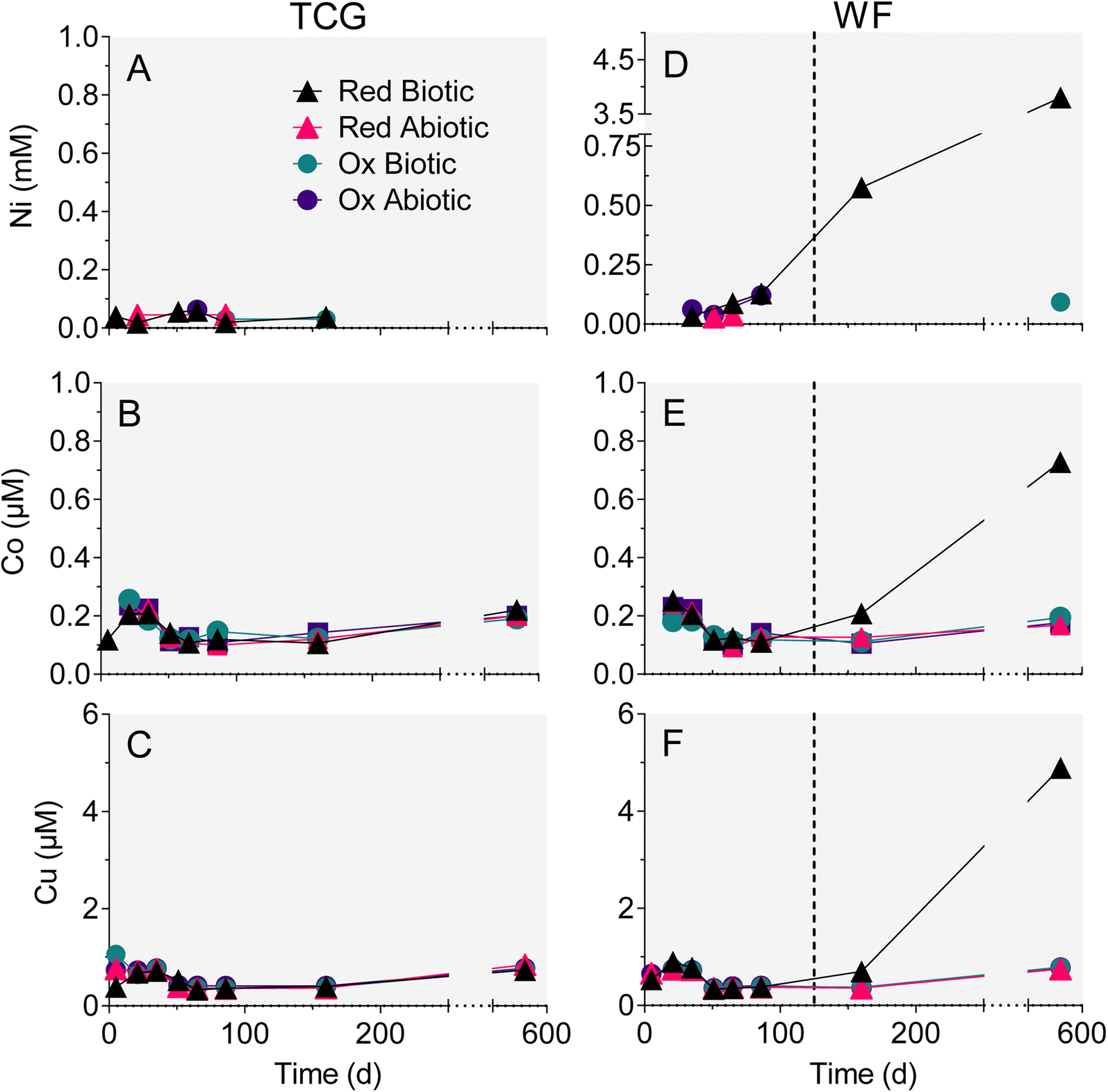 | ||
| Fig. 6 Changes in aqueous-phase nickel Ni (A and D), Co (B and E), and Cu (C and F) in the reduced and oxidized TCG and WF sediment incubations. The values for biotic TCG-Red represent the average of the duplicate microcosms. All other data points are one sample of combined aqueous-phase treatment duplicates. Values below limit of detection of the analytical assay are not plotted. Dotted lines in right-hand panels mark halfway between the two time points when the biotic WF-Red reactors went acidic. | ||
3.4 SEM imaging
SEM imaging was carried out on natural pyrite grains from the incubated biotic and abiotic WF-Red sediments (Fig. 7). Octahedral pyrite crystals about 20–200 μm in length were observed. Euhedral etching features with three-fold axes symmetry in the pit centers were common and observed before the start of incubation (Fig. 7A). A visual comparison of BSE-SEM images of pyrite sampled after 86 days of incubation shows that the size and shape irregularity of the pits was greater in the biotic than the abiotic treatments (Fig. 7B and C). The SE-SEM image of day 86 pyrite in the biotic treatment captures the irregularity of the surfaces, including a coating of fluff or debris (Fig. 7D). The debris was interpreted as biofilm coating the pyrite surface formed during microbial colonization of the pyrite grain surface. | ||
| Fig. 7 SEM images of natural octahedral pyrite from the WF Red reactors. (A) BSE image of day 0 pyrite, (B) BSE image day 86 pyrite from abiotic reactor, (C and D) BSE and SE image of day 86 pyrite from the biotic reactor. All scale bars indicate a length of 20 μm. | ||
3.5 Microbial community analysis
The biotic WF Red microcosms contained 16S rRNA gene sequences similar to those of three different groups of chemolithotrophic bacteria: candidates Tenderia electrophaga (22% sample abundance, 96% identity), uncultured Acidiferrobacteracea (NCBI Blast match Thioprofundum lithophicum) (20% sample abundance, 93% identity), and Thiobacillus thioparus (14% sample abundance, 97% identity) (Fig. 8). Candidatus Tenderia electrophaga-like sequences were also identified in passively filtered (>0.45 μm) groundwater from the same well. In contrast to the similarity to chemolithoautotrophic sequences found in the WF Red-live microcosms, no such similarity was found for the WF Ox-live microcosms. Both the groundwater and WF Ox-live microbial communities were dominated by a variety of heterotrophic taxa from the Gammaproteobacteria, Alphaproteobacteria, and Actinobacteria.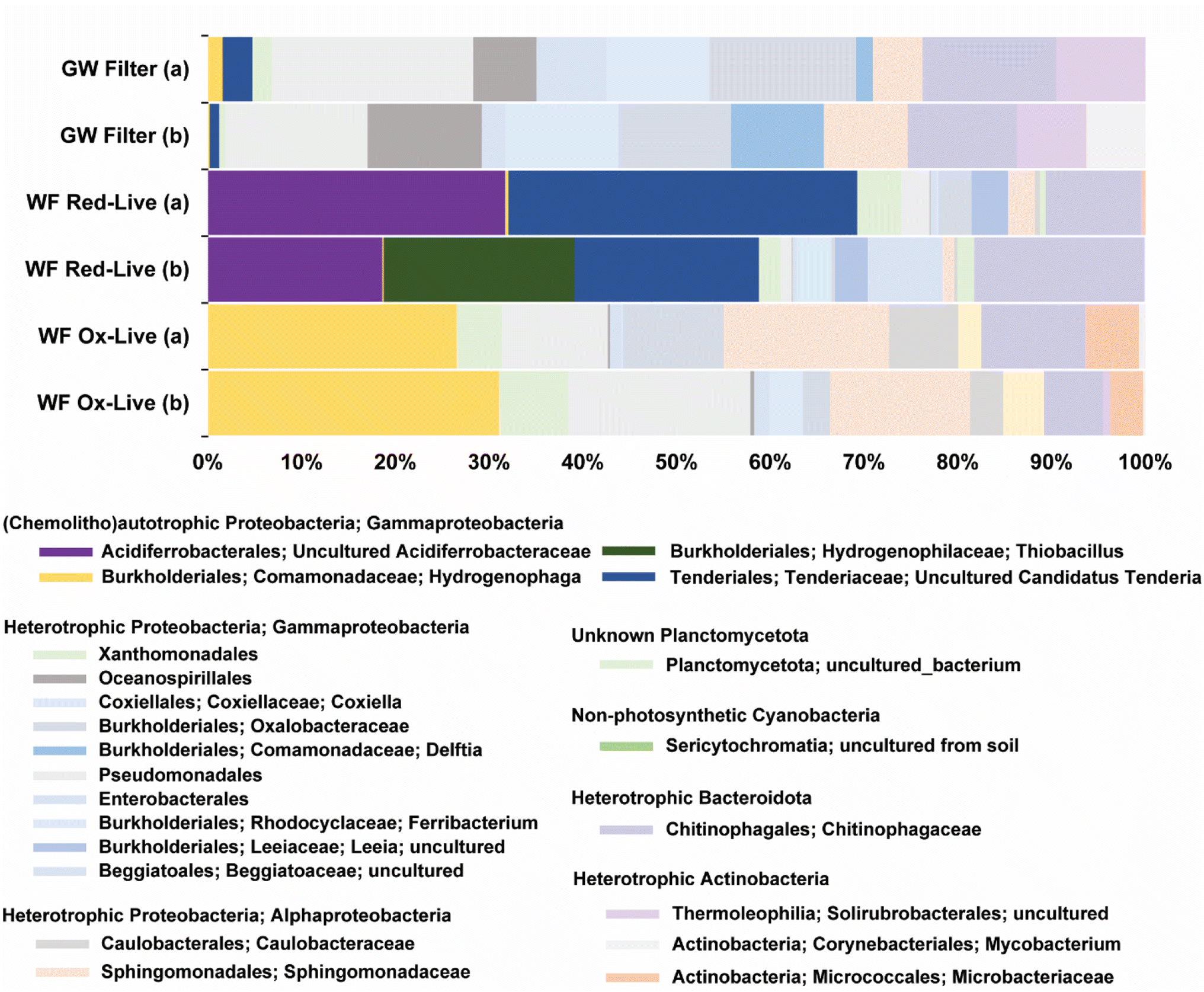 | ||
| Fig. 8 Operational taxonomic unit matching of the most abundant (to 10% of taxa) amplified 16S rRNA sequences based on 99% similarity to sequences hosted in Silva-139-99 database. Chemolithoautotrophs include uncultured Acidiferrobacteraceae (purple), Thiobacillus from the order Burkholderiales (dark green), and uncultured Candidatus Tenderia from the order Tenderiales (dark blue). | ||
3.6 Pyrite oxidation model
The surface area-specific pyrite oxidation rate constants estimated from WF sediment batch incubation experiments (see Table S5†) were within the range of those reported in studies of specimen pyrite oxidation.42 The rate constant for the biotic system (10−7.8 mol m−2 s−1) was ca. 25-fold higher than for the abiotic (10−9.2 mol m−2 s−1). The batch simulations reproduced the overall time course of pH and sulfate concentration in the biotic and abiotic reactor experiments (Fig. 9). The simulations also captured the trend in Ni release to solution following the initial downshift in pH in the biotic reactors. Incorporation of these rate constants into single-cell REV reactive transport simulations (Fig. 10) yielded results which suggest that microbially-accelerated pyrite oxidation could have a significant impact on pH and dissolved metal concentration with groundwater fluid residence times on the order of 10–100 years, i.e. values within the range of those expected given groundwater flow rates in the Wonewoc sandstone aquifer in western WI.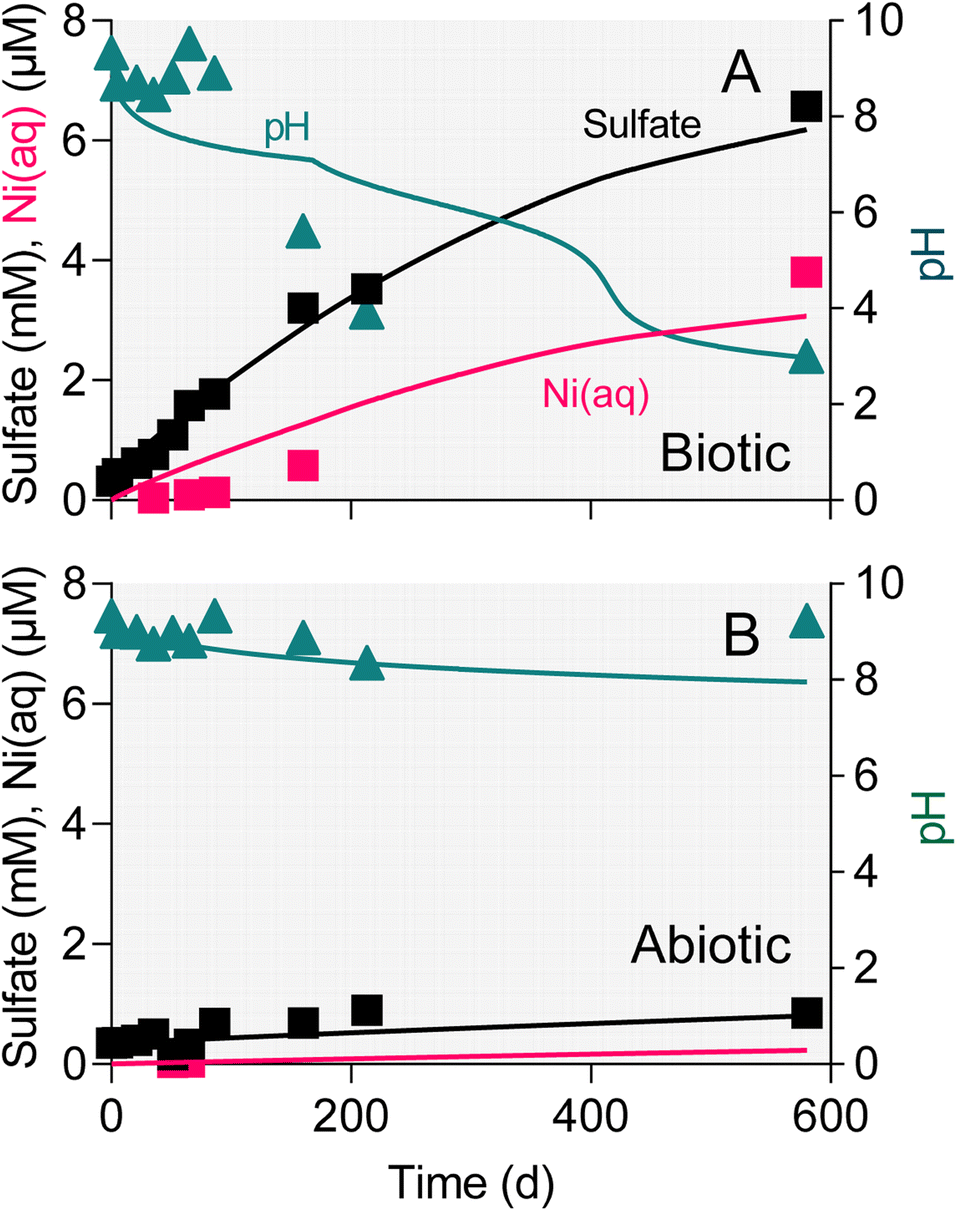 | ||
| Fig. 9 Results of PHREEQC batch simulation of pyrite oxidation in the reduced WF biotic (a) and abiotic (a) microcosm experiments. Symbols show the average of duplicate reactors; solid lines show model results. | ||
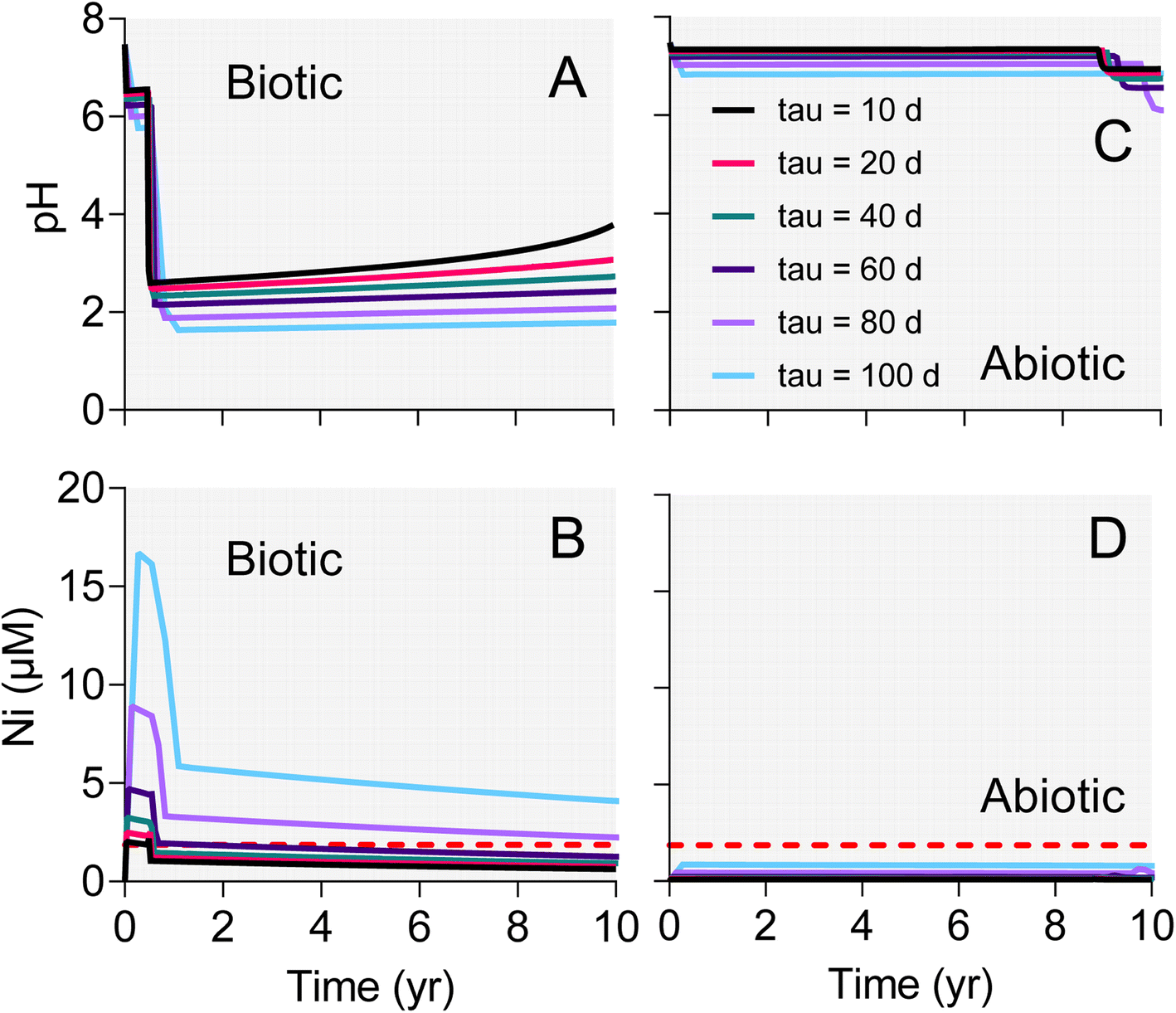 | ||
| Fig. 10 Results of single-cell REV reactive transport simulations employing biotic (A and B) and abiotic (C and D) pyrite oxidation rate constants derived from the WF Red microcosm experiments shown in Fig. 9. The different lines represent results for various fluid residence times (tau values) in the hypothetical REV of pyrite-bearing aquifer sediment. | ||
4. Discussion
4.1 Microbial acceleration of natural pyrite oxidation
To our knowledge this is the first study to demonstrate microbially-mediated aerobic oxidation of natural subsurface pyrite. The sulfate release observed in the microcosm experiments showed that microbial activity accelerated oxidation of naturally-occurring pyrite phases compared to abiotic controls (Fig. 3). In the case of the reduced WF material, there was a 4.5 fold increase in the amount of sulfate released over the first 86 days at circumneutral pH (Table 2). Microbial activity led to a 2–3-fold increase in pyrite oxidation in the reduced TCG microcosms. Much smaller amounts of sulfate were released in microcosms containing oxidized end-member WF and TCG materials, which directly ties sulfate release to oxidation of pyrite in the reduced sediments. Similar results were obtained in a repeat experiment with the WF-Red sediment and freshly collected groundwater (data not shown). Though limited in scope and detail, SEM observations on microbially-altered pyrite phases from the WF-Red microcosms (Fig. 7) provide additional evidence for the ability of microorganisms to promote pyrite dissolution. Percak-Dennett et al.16 argued that microbial acceleration during the early stages of pyrite oxidation may play a key role in the consumption of buffering capacity, which when depleted leads to conditions conducive to low-pH oxidation processes akin to those well-known in acid mine drainage and related environments. The results for the WF-Red material shown in Fig. 3E illustrate this phenomenon, where extensive oxidation of natural pyrite and eventual acidification of the system occurred only in the presence of microbial activity.4.2 Potential pyrite-oxidizing microorganisms in WF reactors
The most abundant (22% of total sequences) amplified 16S rRNA gene sequence identified in the WF Red-live reactors (Fig. 8) was a match (95% identity) to Candidatus Tenderia electrophaga. Ca. T. electrophaga has been documented in biocathode (i.e. negatively poised graphite electrodes) microbial communities, and proposed to gain energy via acquisition of electrons from the electrode through “extracellular electron transfer” (EET) processes coupled to aerobic respiration.49 Although Ca. T. electrophaga has not been successfully cultivated in isolation from its community, these organisms have been shown to express EET-associated pathways,50 including Cyc2, an outer membrane-associated cytochrome known to be involved in extracellular Fe(II) oxidation.51,52 Percak-Dennett et al.16 identified organisms containing Cyc2 in synthetic pyrite-oxidizing cultures, and suggested their possible involvement in circumneutral pH pyrite oxidation through a sorbed Fe redox cycling mechanism akin to that proposed for neutral-pH abiotic pyrite oxidation.8 Relatives of Ca. T. electrophaga may be able to mediate electron transfer from a solid-phase mineral (i.e. pyrite) to oxygen, and it seems possible that the biofilm coating on the pyrite crystal sampled from the WF Red – live reactor at day 86 (Fig. 5D) could have included Ca. T. electrophaga cells.The second most abundant sequence (20% of total sequences) was a 93% match to Thioprofundum lithotrophicum strain 106. Strain 106 grows chemolithoautotrophically with elemental sulfur, thiosulfate, tetrathionate or sulfite as a sole electron donor and with nitrate or oxygen as an electron acceptor.53 Organic compounds served as neither an energy nor carbon sources, and this organism is therefore a strictly chemolithoautotrophic. The third most abundant sequence (14% of total sequences) was closely related (97% identity) to Thiobacillus thioparus strain White,54 which is also an inorganic sulfur-oxidizing bacterium. However, the amplified T. thioparus-like sequences was only found in one of the WF Red-live reactors (Fig. 8). It is unlikely that the T. thioparus identified in WF Red-live provided significant contribution to pyrite oxidation because the generation of sulfate in the two WF Red-live reactors were so similar.
The microbial community analyses provide limited but convincing evidence for the presence of organisms in the WF-Red reactors that were capable of gaining energy from oxidation of inorganic sulfur compounds. More extensive metagenomic investigation of the aquifer microbial community is required to gain insight into which organisms and pathways are involved in circumneutral pH pyrite oxidation. Whether or not these organisms are active in the Trempealeau County (and other analogous) sandstones remains unknown, but our studies clearly provide motivation and microbiological targets for further investigation of groundwater microbial communities that may mediate in situ sulfide mineral oxidation.
4.3 Fate of trace metals during pyrite oxidation
The results presented here demonstrate that pH had a crucial impact on aqueous/solid-phase trace metal speciation during pyrite oxidation. In general, divalent trace metal cations are expected to sorb to mineral surfaces at circumneutral pH, including hydroxyl sites on Fe(III) oxides produced during pyrite oxidation.55 Sorption is greatly reduced at acidic pH, due to competition between H+ and metal cations on oxide surfaces. Substantial mobilization of trace metals took place after exhaustion of buffering capacity and decline in pH in the biotic WF-Red reactors, where Ni, Co, and Cu (Fig. 6D–F), as well as Fe, Al, and Mn (Fig. 5D–F) accumulated to levels several-fold higher than background. In contrast, virtually no solubilization of trace metals took place in the TCG-Red reactors (Fig. 5A–C and 6A–C), which remained at circumneutral pH (Fig. 3B) despite extensive biotic and abiotic oxidation (Fig. 3A). The major cations Ca2+ and Mg2+, which do not sorb strongly to hydroxyl surface groups, accumulated in solution in both the TCG-Red and WF-Red reactors (Fig. 4A–D) during dissolution of carbonate phases (i.e. dolomite) driven by oxidative pyrite dissolution. The extent of Co, Cu, Mn, and Ni mobilization approached 50% of the bulk metal content of the solids. Prior studies have shown that these metals are hosted in disseminated sedimentary pyrite and Fe(III) oxides, as well pyrite-bearing nodules.19,56 Release of metals from all these sources is expected during groundwater acidification linked to pyrite oxidation.Taken together our results provide experimental verification of the hypothesized connection between sulfide mineral oxidation (as reflected by sulfate accumulation), groundwater acidification, and trace metal contamination in wells open to reduced, pyrite-bearing WF strata in West-Central Wisconsin (Fig. 1A). Our results suggest that microorganisms are likely to be readily capable of accelerating pyrite oxidation in both groundwater aquifer and frac sand wash pond environments. Depending on aqueous and solid-phase pH buffering and the metal sorption properties of the geologic materials, groundwater acidification and metal contamination are expected to proceed much faster in the presence of microbial activity. A preliminary analysis of TCG and WF materials collected from a sand mine in Trempealeau County showed only very low abundance (<0.02%) of sulfide minerals. It remains to be determined whether other sand mine refuse and overburden materials contain sufficient sulfide mineral content to account (vis-à-vis biotic and/or abiotic pyrite oxidation) for the elevated sulfate, acidic pH, and elevated trace metal content of the frac sand wash pond fluids documented in Fig. 1B.
4.4 Implications for in situ microbially-mediated pyrite oxidation in Trempealeau sandstones and other WI groundwaters
Our experiments with the reduced WF and TCG materials suggests that natural subsurface pyrite oxidation, particularly when accelerated by microbial activity, is likely to occur over a time scale of months to years under fully aerobic aqueous conditions. Although translation of results from laboratory mineral dissolution studies to natural systems is notoriously difficult,57 the high reactivity of pyrite with dissolved oxygen is such that pyrite would not be expected to persist over geological time scales (e.g. thousands to millions of years) in aquifer sediments subjected to aerobic groundwater throughput. In light of this reality, active, ongoing pyrite oxidation and associated groundwater acidification and trace metal/metalloid contamination is likely to be limited to situations where aerobic fluids have only recently impinged on reduced, sulfide-bearing sediments. Such situations have been identified in several settings in Wisconsin, with important implications for groundwater quality. In the west-central WI systems studied by Zambito et al.19,56 and examined in this project, both natural water table fluctuations as well as faulty well construction have been identified as causes for sulfide oxidation-induced groundwater contamination. Similarly, release of As during oxidation of sulfides in the St. Peter Sandstone in north-eastern WI has been linked to natural vertical hydraulic leakage and groundwater recharge, as well as borehole installation and water table fluctuations associated with pumping.20,21 In addition, Stewart et al.58 suggested a linkage between sulfide mineral oxidation and elevated groundwater As detection in the vicinity of bedrock folds in Beaver Dam anticline in southeast Wisconsin. In this setting, elevated hydraulic conductivity near the fold axis (e.g. reflecting a higher concentration of vertical fractures) may have led to systematic changes in the distribution and concentration of arsenic-bearing mineral hosts, whose oxidation in turn is responsible for the observed pattern of groundwater As detection. All of these environments represent situations where microbial activity might be expected to enhance rates of pyrite oxidation with attendant impacts on groundwater chemistry. Microbial acceleration of pyrite oxidation is likely to be most important in relation to the short-term response to aerobic groundwater influx due to well installation or groundwater pumping. In this case, our results suggest that groundwater destined for homeowner or municipal use could potentially become contaminated on a time frame substantially shorter than would occur in the absence of microbial activity. Data obtained through University of Wisconsin-Stevens Point Well Water Quality Viewer (https://www.uwsp.edu/cnr-ap/watershed/Pages/WellWaterViewer.aspx), which contains data from private wells across WI, shows elevated groundwater sulfate within certain areas of Trempealeau County. Our experimental and modeling (see below) suggest that microbial activity could play a role in generating these elevated sulfate concentrations.A recent compilation of the temporal distribution of pyrite in sedimentary rocks59 suggests that the majority (83%) of pyrite-bearing geological strata are siliciclastic and dominantly fine-grained, i.e. the types of materials that would typically represent confining units in intermediate and regional groundwater flow systems.60 However, pyrite bearing rocks were also shown to include pure sandstones and conglomerates (ca. 6%), limestones, dolomites and cherts (ca. 3%), and interbedded variants (ca. 8%). These represent units in which groundwater is used for private and municipal water supplies, and are thus at risk for contamination linked to pyrite oxidation and associated geochemical impacts.
Our results raise the question of whether or not microbial activity could accelerate pyrite oxidation in more fine-grained geological units. Ongoing studies at the Susquehanna Shale Hills Critical Zone Observatory in Pennsylvania, USA have identified the presence of a pyrite oxidation front that is hypothesized to play a key role in the dissolution of other primary minerals (e.g. illite, chlorite, and feldspar) in the shales.61 These findings led Napieraski et al.17 to examine the potential for microbial pyrite oxidation by microorganisms in groundwater in the vicinity of the oxidation front. Although organisms capable of accelerating pyrite were identified, the study ultimately concluded the nm-size pore throats that dominate the lithology at Shale Hills are likely too small to allow for bacteria to contact the main in situ reservoir of pyrite. Rather, pyrite oxidation in the shale lithologies at this and in other similar subsurface environments is likely to be dominated by abiotic processes,62 with microorganisms playing a role only on surfaces of larger fractures in rock matrix.17 This is in contrast to the relatively porous and poorly-cemented WF and variably-cemented TCG strata examined in this study,63,64 where the fundamental pore size length scale is on the order of tens of μm.65 Based on previous studies in Cretaceous-age Atlantic Coastal Plain aquifer sediments,66 bacteria would be expected to be able to directly access disseminated and nodular pyrite in pores of this size range.
4.5 Reactive transport modeling
The single-cell REV approach adopted for the reactive transport modeling reflects a conceptual model where oxygenated groundwater impinges on a previously reduced, pyrite-bearing geological stratum. In this model, the “impingement” can be viewed as being essentially instantaneous on a geological time scale, i.e. representative of a decadal time-scale occurrence associated with either an abrupt change in a natural flow paths, or (more likely) some kind of human-induced change in fluid flow (e.g. associated with groundwater pumping, poorly constructed multiaquifer wells, or frac sand mining operations as discussed above). The paper by Manning et al.43 provided a guiding impetus for this exercise. Manning et al.43 employed the reactive transport code TOUGHREACT to analyze the oxidation of pyrite during climate-induced changes in redox conditions in a pyrite-bearing aquifer in the Upper Snake River region of Colorado. Of central interest in that study was the short-term response of a pyrite-bearing subsurface system to increased input of oxygenated fluid (in both saturated and unsaturated zones) during lowering of the water table. Our system is different in that we consider fully saturated sediments, where changes in fluid flow path may be responsible for enhanced pyrite oxidation. However, the idea is the same in that we are interested in a transport-reaction system where the time scale of reaction is on the order of years to decades.The simulation results provide important insight into how a several-fold increase in pyrite oxidation rate constant due to microbial activity (rate constants for biotic and abiotic oxidation of 10−7.8 and 10−9.2 mol m−2 s−1, respectively) could have a major impact on the short-term response to influx of oxygenated groundwater into previously reduced geological strata which (like the WF) are low in carbonate mineral buffering capacity. Despite the presence of 5 mM alkalinity in the intruding groundwater, accelerated pyrite in the biotic case caused pH values to fall below 2 within 1–2 years depending on fluid residence time (Fig. 10A). Increased pyrite oxidation and decreased pH resulted in several-fold increase in dissolved As (data not shown) and Ni concentration (Fig. 10B) compared to the abiotic case (Fig. 10D), in which pH values (Fig. 10C) remained circumneutral throughout the simulated period. It should be noted that there was virtually no decrease (<1%) in dissolved oxygen in both the abiotic and biotic simulations, which indicates that oxygen availability is not likely to play a role in influencing pyrite oxidation in these sandstones. Although preliminary, the modeling results provide the starting point for development of reactive transport simulations for a specific well or series of wells in pyrite oxidation-impacted groundwaters in WI, e.g. akin to the modeling analysis of Manning et al.43 Of particular interest would be to develop multi-dimensional reactive transport simulations of the “borehole” and “dewatering” scenarios described in Schreiber et al.21 In these scenarios, oxygen intrusion into the sulfide cement layer leads to pyrite oxidation and release of dissolved As to groundwater, where the “borehole” scenario is directly analogous to the proposed well-drilling associated mechanism for sulfuric acid and trace element contamination of the west-central WI sandstone aquifers.
5. Conclusions
The results of this study shed light on factors that may control the onset of low-pH conditions and associated changes in groundwater quality in private and municipal drinking water wells located within pyrite-bearing formations in WI. Specifically, our findings provide experimental verification of the potential linkage between sulfide mineral oxidation (i.e. as reflected by sulfate accumulation), groundwater acidification, and trace metal contamination in pyrite-bearing sandstone units subject to intrusion of oxygenated groundwater. It seems likely that microbial activity is involved in promoting the generation of low-pH, metal-contaminated (above advisory levels for aluminum, arsenic, cadmium, chromium, cobalt, copper, iron, lead, manganese, nickel, vanadium) aquifers on the highlands east of La Crosse, WI, where some private wells draw water from WF-TCG contact interval rocks.19,56 Collectively our findings have important implications for controls on rates of pyrite oxidation in a wide variety of undisturbed (e.g. natural water table fluctuation) and disturbed (e.g. well drilling, pumping, overburden removal, and frac sand mining) sandstone aquifers, where naturally- and anthropogenically-induced changes in subsurface groundwater flow paths can introduce oxygen into previous reduced geologic strata. Further studies of microbial communities (and their potential pyrite oxidation activity) in a variety of sulfide oxidation-impacted groundwater environments (e.g. in comparison with hydrogeologically analogous, unimpacted systems) could reveal taxonomic indicators of situations where exposure of reduced sediments to oxygenated groundwater is triggering microbially-accelerated pyrite oxidation and associated drinking water quality degradation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Groundwater Research Project WR19R001 (awarded to E. E. Roden and M. Ginder-Vogel) from the University of Wisconsin System, Wisconsin Groundwater Research and Monitoring Program. L. Haas acknowledges the support of a Geological Society of America Graduate Student Research Grant, and a graduate student research grant from the University of Wisconsin, Department of Geoscience. We thank James Lazarcik, University of Wisconsin, Water Science and Engineering Laboratory for expert assistance with the OCP-OES analyses, and Dr Stephanie Napieralski (PhD graduate from University of Wisconsin–Madison, Department of Geoscience) for assistance with the 16S rRNA gene amplicon analysis.References
- E. K. Berner and R. A. Berner, Global Environment: Water, Air, and Geochemical Cycles, Prentice Hall, Upper Saddle River, NJ, 1996 Search PubMed.
- P. A. Raymond and N. H. Oh, Long term changes of chemical weathering products in rivers heavily impacted from acid mine drainage: Insights on the impact of coal mining on regional and global carbon and sulfur budgets, Earth Planet. Sci. Lett., 2009, 284, 50–56 CrossRef CAS.
- S. A. Crowe, L. N. Døssing, N. J. Beukes, M. Bau, S. J. Kruger, R. Frei and D. E. Canfield, Atmospheric oxygenation three billion years ago, Nature, 2013, 501, 535–538 CrossRef CAS PubMed.
- K. O. Konhauser, S. V. Lalonde, N. J. Planavsky, E. Pecoits, T. W. Lyons, S. J. Mojzsis, O. J. Rouxel, M. E. Barley, C. Rosiere, P. W. Fralick, L. R. Kump and A. Bekker, Aerobic bacterial pyrite oxidation and acid rock drainage during the Great Oxidation Event, Nature, 2011, 478, 369–374 CrossRef CAS PubMed.
- E. E. Stueken, D. C. Catling and R. Buick, Contributions to late Archaean sulphur cycling by life on land, Nat. Geosci., 2012, 5, 722–725 CrossRef.
- A. Schippers, Biogeochemistry of metal sulfide oxidation in mining environments, sediments, and soils, in Sulfur Biogeochemistry – Past and Present GSA Special Papers 379, ed. J. P. Amend, K. J. Edwards and T. W. Lyons, Geological Society of America, Boulder, CO, 2004, pp. 49–62 Search PubMed.
- C. O. Moses, D. K. Nordstrom, J. S. Herman and A. L. Mills, Aqueous pyrite oxidation by dissolved oxygen and by ferric iron, Geochim. Cosmochim. Acta, 1987, 51, 1561–1571 CrossRef CAS.
- C. O. Moses and J. S. Herman, Pyrite oxidation at circumneutral pH, Geochim. Cosmochim. Acta, 1991, 55, 471–482 CrossRef CAS.
- J. Bosch and R. U. Meckenstock, Rates and potential mechanism of anaerobic nitrate-dependent microbial pyrite oxidation, Biochem. Soc. Trans., 2012, 40, 1280–1283 CrossRef CAS PubMed.
- S. C. M. Haaijer, L. P. M. Lamers, A. J. P. Smolders, M. S. M. Jetten and H. J. M. Op den Camp, Iron sulfide and pyrite as potential electron donors for microbial nitrate reduction in freshwater wetlands, Geomicrobiol. J., 2007, 24, 391–401 CrossRef CAS.
- C. J. Jorgensen, O. S. Jacobsen, B. Elberling and J. Aamand, Microbial oxidation of pyrite coupled to nitrate reduction in anoxic groundwater sediment, Environ. Sci. Technol., 2009, 43, 4851–4857 CrossRef PubMed.
- C. O. Wirsen, H. W. Jannasch and S. J. Molyneaux, Chemosynthetic microbial activity at Mid-Atlantic ridge hydrothermal vent sites, J. Geophys. Res.: Solid Earth, 1993, 98, 9693–9703 CrossRef CAS.
- K. J. Edwards, W. Bach and D. R. Rogers, Geomicrobiology of the ocean crust: A role for chemoautotrophic Fe-bacteria, Biol. Bull., 2003, 204, 180–185 CrossRef CAS PubMed.
- E. S. Boyd, T. L. Hamilton, J. R. Havig, M. L. Skidmore and E. L. Shock, Chemolithotrophic primary production in a subglacial ecosystem, Appl. Environ. Microbiol., 2014, 80, 6146–6153 CrossRef PubMed.
- Z. R. Harrold, M. L. Skidmore, T. L. Hamilton, L. Desch, K. Amada, W. van Gelder, K. Glover, E. E. Roden and E. S. Boyd, Aerobic and anaerobic thiosulfate oxidation by a cold-adapted, subglacial chemoautotroph, Appl. Environ. Microbiol., 2016, 82, 1486–1495 CrossRef CAS PubMed.
- E. M. Percak-Dennett, S. He, B. J. Converse, H. Konishi, H. Xu, C. S. Chan, A. Bhayyacharyya, T. Borch and E. E. Roden, Microbial acceleration of aerobic microbial pyrite oxidation at circumneutral pH, Geobiology, 2017, 15, 690–703 CrossRef CAS PubMed.
- S. A. Napieralski, Y. H. Fang, V. Marcon, B. Forsythe, S. L. Brantley, H. F. Xu and E. E. Roden, Microbial chemolithotrophic oxidation of pyrite in a subsurface shale weathering environment: Geologic considerations and potential mechanisms, Geobiology, 2022, 20, 271–291 CrossRef CAS PubMed.
- Associated-Press, Frac sand mine spill sends sludge into Wisconsin waterways, Milwaukee Journal Sentinel, May 25, 2018 Search PubMed.
- J. J. Zambito, L. D. Haas, M. J. Parsen and P. I. McLaughlin, Geochemistry and mineralogy of the Wonewoc – Tunnel City contact interval strata in western Wisconsin, Wisconsin Geological and Natural History Survey, Wisconsin Open-File Report 2019-01, 2019 Search PubMed.
- M. E. Schreiber, M. B. Gotkowitz, J. A. Simo and P. G. Freiberg, Mechanisms of arsenic release to ground water from naturally occurring sources, Eastern Wisconsin, in Arsenic in Ground Water: Geochemistry and Occurrence, ed. A. H. Welch and K. G. Stollenwerk, Springer US, Boston, MA, 2003, pp. 259–280 Search PubMed.
- M. E. Schreiber, J. A. Simo and P. G. Freiberg, Stratigraphic and geochemical controls on naturally occurring arsenic in groundwater, eastern Wisconsin, USA, Hydrogeol. J., 2000, 8, 161–176 CrossRef CAS.
- J. P. Girard and D. A. Barnes, Illitilization and paleothermal regimes in the middle Ordovician St. Peter Sandstone, Central Michigan Basin - K-Ar, oxygen isotope, and fluid inclusion data, AAPG Bull., 1995, 79, 49–69 CAS.
- B. L. Winter, C. M. Johnson, J. A. Simo and J. W. Valley, Paleozoic fluid history of the Michigan Basin - Evidence from dolomite geochemistry in the Middle Ordovician St. Peter Sandstone, J. Sediment. Res., Sect. A, 1995, 65, 306–320 Search PubMed.
- D. T. A. Symons, K. Kawasaki and S. J. Pannalal, Paleomagnetic mapping of the regional fluid flow event that mineralized the Upper Mississippi Valley Zn-Pb ore district, Wisconsin, USA, J. Geochem. Explor., 2010, 106, 188–196 CrossRef CAS.
- J. Podgorski and M. Berg, Global threat of arsenic in groundwater, Science, 2020, 368, 845–850 CrossRef CAS PubMed.
- H. Masuda, Arsenic cycling in the Earth's crust and hydrosphere: interaction between naturally occurring arsenic and human activities, Prog. Earth Planet. Sci., 2018, 5, 68 CrossRef.
- M. L. Polizzotto, C. F. Harvey, S. R. Sutton and S. Fendorf, Processes conducive to the release and transport of arsenic into aquifers of Bangladesh, Proc. Natl. Acad. Sci. U.S.A., 2005, 102, 18819–18823 CrossRef CAS PubMed.
- C. O. Wirsen, T. Brinkhoff, J. Kuever, G. Muyzer, S. Molyneaux and H. W. Jannasch, Comparison of a new Thiomicrospira strain from the Mid-Atlantic Ridge with known hydrothermal vent isolates, Appl. Environ. Microbiol., 1998, 64, 4057–4059 CrossRef CAS PubMed.
- A. Schippers and B. B. Jorgensen, Biogeochemistry of pyrite and iron sulfide oxidation in marine sediments, Geochim. Cosmochim. Acta, 2001, 65, 915–922 CrossRef CAS.
- R. E. Mielke, D. L. Pace, T. Porter and G. Southam, A critical stage in the formation of acid mine drainage: Colonization of pyrite by Acidithiobacillus ferrooxidans under pH-neutral conditions, Geobiology, 2003, 1, 81–90 CrossRef CAS.
- K. J. Edwards, D. R. Rogers, C. O. Wirsen and T. M. McCollom, Isolation and characterization of novel psychrophilic, neutrophilic, Fe-oxidizing chemolithoautotrophic α- and γ-Proteobacteria from the deep sea, Environ. Microbiol., 2003, 69, 2906–2913 CrossRef CAS PubMed.
- E. D. Rhine, K. M. Onesios, M. E. Serfes, J. R. Reinfelder and L. Y. Young, Arsenic transformation and mobilization from minerals by the arsenite oxidizing strain WAO, Environ. Sci. Technol., 2008, 42, 1423–1429 CrossRef CAS PubMed.
- E. Bolyen, et al., Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2, Nat. Biotechnol., 2019, 37, 852 CrossRef CAS PubMed.
- B. J. Callahan, P. J. McMurdie, M. J. Rosen, A. W. Han, A. J. A. Johnson and S. P. Holmes, DADA2: High-resolution sample inference from Illumina amplicon data, Nat. Methods., 2016, 13, 581–583 CrossRef CAS PubMed.
- N. A. Bokulich, B. D. Kaehler, J. R. Rideout, M. Dillon, E. Bolyen, R. Knight, G. A. Huttley and J. G. Caporaso, Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin, Microbiome, 2018, 6, 90 CrossRef PubMed.
- D. McDonald, M. N. Price, J. Goodrich, E. P. Nawrocki, T. Z. DeSantis, A. Probst, G. L. Andersen, R. Knight and P. Hugenholtz, An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea, ISME J., 2012, 6, 610–618 CrossRef CAS PubMed.
- C. Quast, E. Pruesse, P. Yilmaz, J. Gerken, T. Schweer, P. Yarza, J. Peplies and F. O. Glockner, The SILVA ribosomal RNA gene database project: improved data processing and web-based tools, Nucleic Acids Res., 2013, 41, D590–D600 CrossRef CAS PubMed.
- S. F. Altschul, T. L. Madden, A. A. Schaffer, J. H. Zhang, Z. Zhang, W. Miller and D. J. Lipman, Gapped BLAST and PSI-BLAST: a new generation of protein database search programs, Nucleic Acids Res., 1997, 25, 3389–3402 CrossRef CAS PubMed.
- N. R. Pace, A molecular view of microbial diversity and the biosphere, Science, 1997, 276, 734–740 CrossRef CAS PubMed.
- E. Roden, J. M. McBeth, M. Blothe, E. M. Percak-Dennett, E. J. Fleming, R. R. Holyoke, G. W. Luther and D. Emerson, The microbial ferrous wheel in a neutral pH groundwater seep, Front. Microbiol., 2012, 3, 172 Search PubMed.
- D. A. Parkhurst and C. A. Appelo, User's Guide to PHREEQC (Version 2), Water-Resources Investigation Report 99-4259, 1999 Search PubMed.
- M. A. Williamson and J. D. Rimstidt, The kinetics and electrochemical rate-determining step of aqueous pyrite oxidation, Geochim. Cosmochim. Acta, 1994, 58, 5443–5454 CrossRef CAS.
- A. H. Manning, P. L. Verplanck, J. S. Caine and A. S. Todd, Links between climate change, water-table depth, and water chemistry in a mineralized mountain watershed, Appl. Geochem., 2013, 37, 64–78 CrossRef CAS.
- E. E. Roden, Microbiological controls on geochemical kinetics 1: Fundamentals and case study on microbial Fe(III) reduction, in Kinetics of Water-Rock Interactions, ed. S. L. Brantley, J. Kubicki and A. F. White, Springer, New York, 2008, pp. 335–415 Search PubMed.
- D. A. Dzombak and F. M. M. Morel, Surface Complexation Modeling: Hydrous Ferric Oxide, John Wiley & Sons, New York, 1990 Search PubMed.
- R. J. Hunt, D. A. Saad and D. M. Chapel, Numerical simulation of ground-water flow in La Crosse County, Wisconsin, and into nearby pools of the Mississippi River, Water-Resources Investigations Report 03–4154, 2003, https://pubs.usgs.gov/wri/wri034154 Search PubMed.
- A. C. Runkel, R. G. Tipping, E. C. Alexander and S. C. Alexander, Hydrostratigraphic characterization of intergranular and secondary porosity in part of the Cambrian sandstone aquifer system of the cratonic interior of North America: Improving predictability of hydrogeologic properties, Sediment. Geol., 2006, 184, 281–304 CrossRef.
- M. J. Parsen, P. F. Juckem, M. B. Gotkowitz and M. N. Fienen, Groundwater Flow Model for Western Chippewa County, Wisconsin, Wisconsin Geological and Natural History Survey, Bulletin, vol. 112, 2019 Search PubMed.
- A. P. Malanoski, B. C. Lin, B. J. Eddie, Z. Wang, W. J. Hervey and S. M. Glaven, Relative abundance of “Candidatus Tenderia electrophaga” is linked to cathodic current in an aerobic biocathode community, Microb. Biotechnol., 2018, 11, 98–111 CrossRef CAS PubMed.
- B. J. Eddie, Z. Wang, W. J. Hervey, D. H. Leary, A. P. Malanoski, L. M. Tender, B. C. Lin and S. M. Strycharz-Glaven, Metatranscriptomics Supports the Mechanism for Biocathode Electroautotrophy by “Candidatus Tenderia electrophaga”, mSystems, 2017, 2, e00002 CrossRef CAS PubMed.
- S. He, R. A. Barco, D. Emerson and E. E. Roden, Comparative genomic analysis of neutrophilic iron(II) oxidizer genomes for candidate genes in extracellular electron transfer, Front. Microbiol., 2017, 8, 1584 CrossRef PubMed.
- J. L. Keffer, S. M. McAllister, A. I. Garber, B. J. Hallahan, M. C. Sutherland, S. Rozovsky and C. S. Chan, Iron oxidation by a fused cytochrome-porin common to diverse iron-oxidizing bacteria, mBio, 2021, 12, e01074 CrossRef CAS PubMed.
- K. Takai, M. Miyazaki, H. Hirayama, S. Nakagawa, J. Querellou and A. Godfroy, Isolation and physiological characterization of two novel, piezophilic, thermophilic chemolithoautotrophs from a deep-sea hydrothermal vent chimney, Environ. Microbiol., 2009, 11, 1983–1997 CrossRef PubMed.
- R. Boden, D. Cleland, P. N. Green, Y. Katayama, Y. Uchino, J. C. Murrell and D. P. Kelly, Phylogenetic assessment of culture collection strains of Thiobacillus thioparus, and definitive 16S rRNA gene sequences for T. thioparus, T. denitrificans, and Halothiobacillus neapolitanus, Arch. Microbiol., 2012, 194, 187–195 CrossRef CAS PubMed.
- W. Stumm and J. J. Morgan, Aquatic Chemistry, John Wiley & Sons, Inc., New York, 1996 Search PubMed.
- J. J. Zambito, L. D. Haas and M. J. Parsen, Identifying the source of groundwater contaminants in West-Central Wisconsin, U.S.A.: Geochemical and mineralogical characterization of the Cambrian sandstone aquifer, J. Contam. Hydrol., 2022, 247, 103966 CrossRef CAS PubMed.
- A. F. White and S. L. Brantley, The effect of time on the weathering of silicate minerals: why do weathering rates differ in the laboratory and field?, Chem. Geol., 2003, 202, 479–506 CrossRef CAS.
- E. D. Stewart, E. K. Stewart, K. R. Bradbury and W. Fitzpatrick, Correlating Bedrock Folds to Higher Rates of Arsenic Detection in Groundwater, Southeast Wisconsin, USA, Groundwater, 2021, 59, 829–838 CrossRef CAS PubMed.
- J. F. Emmings, S. W. Poulton, J. Walsh, K. A. Leeming, I. Ross and S. E. Peters, Pyrite mega-analysis reveals modes of anoxia through geological time, Sci. Adv., 2022, 8, eabj5687 CrossRef CAS PubMed.
- F. H. Chapelle, Ground-Water Microbiology and Geochemistry, John Wiley & Sons, Inc., New York, 2001 Search PubMed.
- S. L. Brantley, M. E. Holleran, L. X. Jin and E. Bazilevskaya, Probing deep weathering in the Shale Hills Critical Zone Observatory, Pennsylvania (USA): the hypothesis of nested chemical reaction fronts in the subsurface, Earth Surf. Processes Landforms, 2013, 38, 1280–1298 CrossRef CAS.
- X. Gu, P. J. Heaney, F. Reis and S. L. Brantley, Deep abiotic weathering of pyrite, Science, 2020, 370, eabb8092 CrossRef CAS PubMed.
- A. C. Runkel, R. G. Tipping, E. C. Alexander and S. C. Alexander, Hydrostratigraphic characterization of intergranular and secondary porosity in part of the Cambrian sandstone aquifer system of the cratonic interior of North America: Improving predictability of hydrogeologic properties, Sediment. Geol., 2006, 184, 281–304 CrossRef.
- S. M. Sellwood, D. J. Hart and J. M. Bahr, An in-well heat-tracer-test method for evaluating borehole flow conditions, Hydrogeol. J., 2015, 23, 1817–1830 CrossRef.
- E. S. Amirtharaj, M. A. Ioannidis, B. Parker and C. D. Tsakiroglou, Statistical synthesis of imaging and porosimetry data for the characterization of microstructure and transport properties of sandstones, Transp. Porous Media, 2011, 86, 135–154 CrossRef.
- F. H. Chapelle and D. R. Lovley, Rates of microbial metabolism in deep coastal plain aquifers, Appl. Environ. Microbiol., 1990, 56, 1865–1874 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3va00399j |
| This journal is © The Royal Society of Chemistry 2024 |