
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlling crystallisation and dissolution of biogenic CaCO3via dissolved magnesium cations†
Toby
Morton-Collings
 a,
Minjun
Yang
ab and
Richard G.
Compton
*a
a,
Minjun
Yang
ab and
Richard G.
Compton
*a
aDepartment of Chemistry, Physical and Theoretical Chemistry Laboratory, University of Oxford, South Parks Road, Oxford, UK. E-mail: Richard.Compton@chem.ox.ac.uk
bDepartment of Chemistry, University of Leicester, University Road, Leicester, UK
First published on 20th January 2024
Abstract
The surface of our oceans is teeming with single-cellular ‘plant’ organisms that biomineralise CaCO3 (coccoliths). Globally, an estimate of over 1015 g of atmospheric CO2 per annum is sequestered in the top layers of our ocean. Information of this process is crucial to modelling climate change and achieving net carbon neutrality not least because this rate of CO2 sequestration is comparable to the rate of anthropogenic release of CO2. While the dissolution kinetics of pure calcite (Icelandic Spar, Carrea marble and synthetically grown) have been well-studied in the past decades it remains unclear if biogenic CaCO3 behaves differently, or not, to pure calcite in the marine environment. In this work, we utilise a light microscopy setup to study and compare the precipitation and dissolution of biogenic CaCO3 in both the absence and presence of Mg2+, a known inhibitor, at concentrations similar to seawater. Notably, the time required for a micron-sized calcite particle to dissolve is doubled by approximately doubling the concentration of Mg2+ from 54.6 mM to 100 mM. The work produces two new, key insights. First, there is negligible difference between the rate of mass loss of biogenic and pure, laboratory grown CaCO3 particles when placed in solutions supersaturated and undersaturated with respect to calcite. Second, the mass of the individual micron-sized biogenic coccoliths, ranging from 100–600 picograms, was inferred via image analysis of data from the complete dissolution of coccoliths in aqueous solutions containing seawater levels of Mg2+. This relatively simple light-based approach, allowing the mass of biogenic CaCO3 platelets to be estimated at the single-entity level, shows promise for the development of a proof-of-concept sensor allowing CaCO3 sequestration to be monitored real-time in our oceans.
Environmental significanceMicro-sized photosynthetic plankton which generates “chalk” (CaCO3; coccoliths) have a disproportional impact on the pump of carbon from the atmosphere to the deep ocean. These single-cellular organisms generate >1 billion tonnes (1015 g) of calcite per year matching the rate of CO2 released by mankind. While the crystallisation and dissolution kinetics of pure, synthetic calcite is well-documented in laboratory conditions the fate of biogenic calcite in conditions relevant to seawater remains unclear. In this work, we use dark-field microscopy to reveal that these micron-sized platelets of biogenic CaCO3 behave both chemically and physically as pure, synthetic calcite at seawater levels of dissolved magnesium ions. This information is crucial to understanding and modelling the impact of climate change. Moreover, we show that the mass of individual coccoliths, ranging from 100–600 picograms, can be inferred by simply ‘watching’ their dissolution under dark-field illumination in solutions undersaturated with respect to CaCO3. |
Introduction
The open ocean surface water is oversaturated with respect to calcite as has been likely since the formation of the Earth's oceans.1 Despite the oversaturation, the surface of our ocean is in a meta-stable state because abiotic calcite precipitation is retarded due to the presence of calcite precipitation inhibitors such as Mg2+, Fe2+, SO42− and dissolved organic matter in seawater.2–6 Capitalising on the meta-stable state of our oceans, some marine phytoplankton have evolved to grow platelets of CaCO3 known as coccoliths to give the single-cell an edge in the competition for survival. Globally these calcifying single-cellular photosynthetic plants are estimated to sequester over one billion tonnes (>1015 g) per year of atmospheric CO2 dissolved in the surface waters to form solid CaCO3. Importantly, this rate CO2 sequestration is comparable to the anthropogenic release of CO2!7The intracellular formation of coccoliths starts with a proto-ring baseplate on which the CaCO3 crystals nucleate and grow in the presence of dissolved coccolith polysaccharides. The presence of coccolith polysaccharides is known to elongate the c-axis of the calcite crystalline8 but it remains unclear, however, how polysaccharides, if at all, are incorporated in the structure of coccoliths.9 That said, the surface of the coccolith, once secreted, is known to be coated with a layer polysaccharide which has been thought to act as “organic glue” aiding attachment to the biological cell once excreted10,11 with some authors suggesting an inhibition role in the dissolution of coccoliths.12 The secretion of coccoliths occurs at a rate of one per 1–2 hours to eventually encrust the underlying biological cell with a complete sphere of platelets. The construction of an elaborate calcite shell, despite an estimated cost of one third of its photosynthetic budget,13 has been suggested to provide the single-cell organism with an edge to survival including protection against virus infection, zooplankton grazing, and protection from photo- and/or oxidative damage.13,14 Furthermore, the dense CaCO3 acts as a ballast, sinking the inorganic and organic carbon biomineralized in the sun-lit surface to the deep. Ancient coccoliths found in sediment traps date back as far as ca. 63 million years ago.15 However, due to the pressure-led calcite solubility increase down the water column, as much as 60–80% of the coccoliths formed in the surface water has been estimated to be redissolved in the upper 500–1000 m of the ocean.16 Biomineralized coccoliths thus play a critical role in the marine carbon cycle and hence understanding the precipitation and dissolution kinetics of coccoliths, and how they compare and contrast with pure calcite, provides vital information to modelling the marine carbon cycle and how this may be perturbed in the current climate crisis.
While the thermodynamics of carbonate chemical equilibria has been intensively studied over the past century1 the dissolution kinetics of biogenic and abiotic calcite, however, remain obscure and poorly described even under laboratory conditions. A recent review, Batchelor-McAuley et al., explains that the overall rate of (calcite) dissolution is complicated by the interplay between surface kinetics and the rate of mass transport of ions in solution; a lack of appreciation of the mass-transport limitation in different experimental setups leads to an over-simplified, if not erroneous, quantitative interpretation of the surface dissolution kinetics of calcite or any other crystalline solid.17 The ability to measure the rate of single particle dissolutions under well-defined hydrodynamic conditions was highlighted [15] to overcome complications associated with overlapping diffusion layers in the case of measurements made with an ensemble of particles which may lead to a misinterpretation of dissolution kinetics. For example, by gluing individual particles on to the tip of an atomic force microscopy (AFM) cantilever, Hassenkam et al. tracked, in real-time, the weight of pure calcite and biogenic coccoliths immersed in deionised water.18 The rate of mass loss of biogenic coccoliths in deionised water (7.6–9.0 μmol m−2 s−1) was close to that of similar-sized pure calcite particle (10.0 μmol m−2 s−1), hinting that the polysaccharide coating on the surface of the coccolith provides no hindrance to dissolution as compared to pure calcite in deionised water. More recently, Fan et al. demonstrated the use of a simple light microscopy setup allowing the dissolution of pure synthetic calcite single-particles to be tracked in real-time revealing that micron-sized calcite dissolving into deionised water occurs at the mass-transport limit. This result is consistent with the AFM data reported by Hassenkam et al. upon reanalysis of the latter.19 Recent advances in dark-field light scattering microscopy allows sub-micron sized particles to be detected and analysed for environmental monitoring purposes.20
The simplicity of light microscopy setups opens the possibility for the development of optical based sensors allowing the mass of individual coccoliths to be quantified in open surface waters.
In this work we explore the utility of light microscopy to study the precipitation and dissolution of biogenic calcite, in the form of coccoliths, in aqueous solutions relevant to seawater. We question, in physicochemical conditions appropriate to seawater whether biogenic coccoliths behave similarly to pure calcite. We show, using a simple light microscopy setup, that the precipitation and dissolution of biogenic coccoliths can be tracked in real time. Through image analysis, and complementary spectroscopy measurements, we show that biogenic calcite is no different to pure calcite particles in magnesium-containing solutions both over- and undersaturated with respect to calcite in spite of the polysaccharide coating on the coccoliths. Moreover, as demonstrated in previous work,21 we show that the mass of coccolith single entities can be estimated using simple light microscopy regardless of the concentration of calcite dissolution inhibitor (Mg2+) present.
Results and discussion
The work studies the morphological change of an ecologically important species of coccolith, Coccolithus pelagicus subsp. braarudii, when immersed in solutions supersaturated with respect to calcite containing various concentrations of the calcite dissolution inhibitor commonly found in seawater, Mg2+. The composition of seawater is reported in the literature noting that magnesium is present at concentrations as high as 54 mM.22 By using complementary techniques, such as SEM and XRD, we examine the nature of the CaCO3 precipitation and conclude if CaCO3 precipitation on coccolith surfaces is comparable to that of pure calcite. Second, we study the dissolution kinetics of coccolith single-entity using light at the onset of immersion in undersaturated solutions with respect to calcite and we show that the mass of each individual coccolith, after dissolution, regardless of the concentrations of calcite dissolution/precipitation inhibitors, can be estimated by image analysis. The simple inverted microscopy setup used in this work is illustrated in Fig. 1a. It consists of an LED light source, a 20× objective lens and a custom-machined aluminium reaction chamber.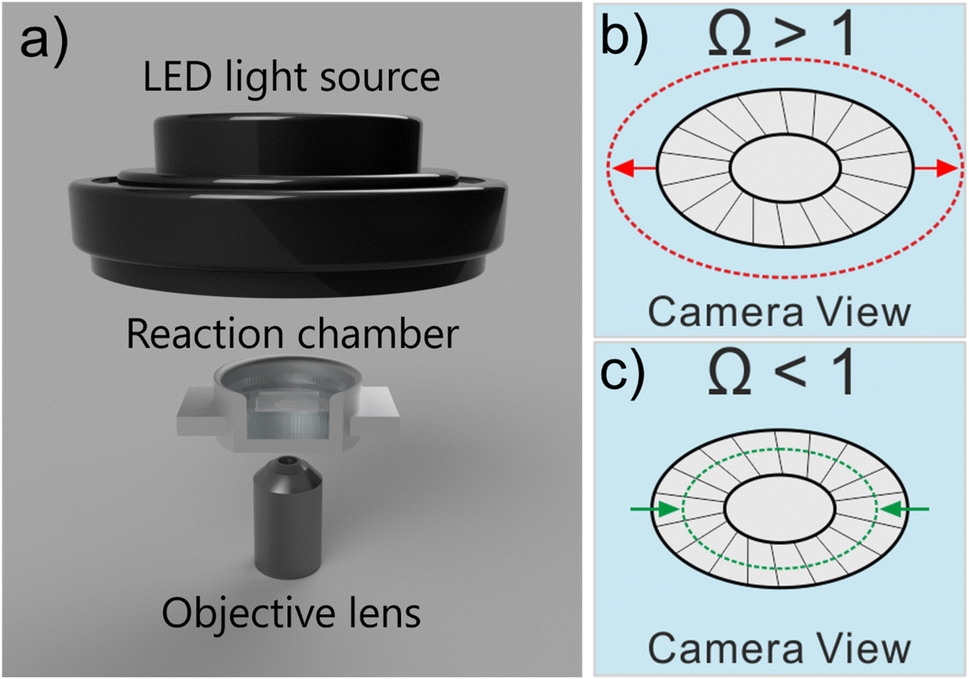 | ||
| Fig. 1 (a) Schematic of the dark-field light microscopy setup. The aluminium reaction chamber provides sufficient structural rigidity allowing coccoliths to be studied over timescales in excess of 6 hours. Note that a cross-section view of the reaction chamber is presented. The top of the reaction chamber was covered up by a glass slide (not shown) to minimise evaporation. All experiments were conducted at room temperature (293–295 K). Schematic illustration of coccolith growth (b) and dissolution (c) in oversaturated (Ω >1) and undersaturated (Ω < 1) solutions with respect to calcite, respectively. | ||
The calcite saturation of solutions, Ωcalcite, is referenced to the stoichiometric solubility constant of calcite, Ksp(calcite), which has the value of 3.3 × 10−9 M2 in deionised water at 298K1
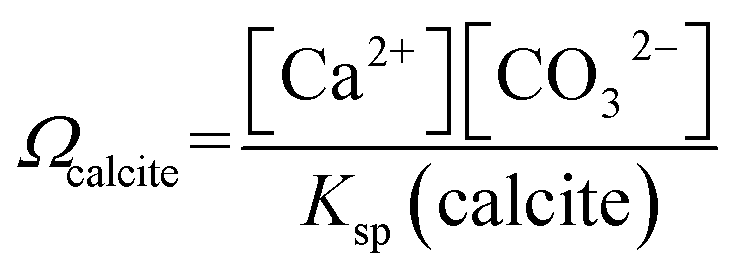 | (1) |
Artificially induced growth: coccolith and pure calcite
The nature of CaCO3 growth on coccoliths and on pure calcite with and without the addition of magnesium ions is examined in this section. Fig. 2a shows the temporal evolution of a representative C. braarudii coccolith immersed in an aqueous solution containing 2.7 mM of Ca2+ and CO32− without Mg2+. Note that both coccoliths and abiotic calcite particles are initially present in the solution prior to the onset of the artificially induced CaCO3 precipitation. The onset of the precipitation was induced by the addition of CO32− (aq) into the reaction chamber where it encounters Ca2+ at suitable concentrations to achieve an overall concentration of 2.7 mM of each ion. Simple calculation of Ωcalcite reveals that the resultant mixture is supersaturated with respect to calcite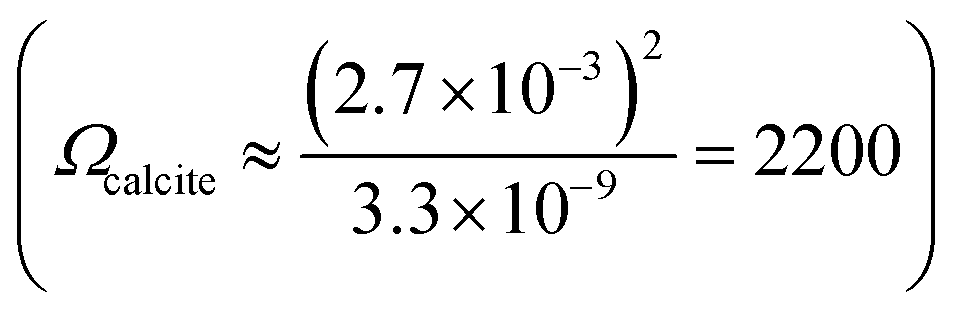 so the precipitation of CaCO3 is thermodynamically favoured. As can be seen in Fig. 2a), the coccolith is seen to increase in size over 12 minutes following the onset of precipitation. A timelapse recording of the growth of coccoliths, along with similarly sized pure synthetic calcite present initially in the same experiment, is shown in video S1.† In the present experiment, biomineralized coccolith and pure calcite particles are both initially present allowing direct comparison of CaCO3 growth on these two different substrates to be made. In both cases, the biogenic and pure, synthetic CaCO3 particles grew over the experimental timescale as shown in Fig. S1a†). It is clear from the optical imaging that the morphology of the pure calcite particles remains rhombohedral which is characteristic of calcite.3 Video S2† presents the above-described experiment in a wide-angle view and it was observed that, in addition to the growth of particles initially present, in situ nucleation of CaCO3(s) occurs concomitantly in the supersaturated solution resulting in an increase of particles sedimented from the solution and onto the supporting substrate, where the initially present coccolith and pure calcite particles are being examined. This results in a significant overlap of the diffusion layers of Ca2+ and CO32− ions to the particles located on the supporting substrate and thus prevents quantitative analysis of the growth of biogenic and abiotic growth kinetics since the diffusion layer thicknesses will be of the order of the size of the particles.24,25 It is noteworthy that the in situ nucleated CaCO3 particles are characteristically rhombohedral in shape as can be seen in the wide-angle images shown in video S2.†
so the precipitation of CaCO3 is thermodynamically favoured. As can be seen in Fig. 2a), the coccolith is seen to increase in size over 12 minutes following the onset of precipitation. A timelapse recording of the growth of coccoliths, along with similarly sized pure synthetic calcite present initially in the same experiment, is shown in video S1.† In the present experiment, biomineralized coccolith and pure calcite particles are both initially present allowing direct comparison of CaCO3 growth on these two different substrates to be made. In both cases, the biogenic and pure, synthetic CaCO3 particles grew over the experimental timescale as shown in Fig. S1a†). It is clear from the optical imaging that the morphology of the pure calcite particles remains rhombohedral which is characteristic of calcite.3 Video S2† presents the above-described experiment in a wide-angle view and it was observed that, in addition to the growth of particles initially present, in situ nucleation of CaCO3(s) occurs concomitantly in the supersaturated solution resulting in an increase of particles sedimented from the solution and onto the supporting substrate, where the initially present coccolith and pure calcite particles are being examined. This results in a significant overlap of the diffusion layers of Ca2+ and CO32− ions to the particles located on the supporting substrate and thus prevents quantitative analysis of the growth of biogenic and abiotic growth kinetics since the diffusion layer thicknesses will be of the order of the size of the particles.24,25 It is noteworthy that the in situ nucleated CaCO3 particles are characteristically rhombohedral in shape as can be seen in the wide-angle images shown in video S2.†
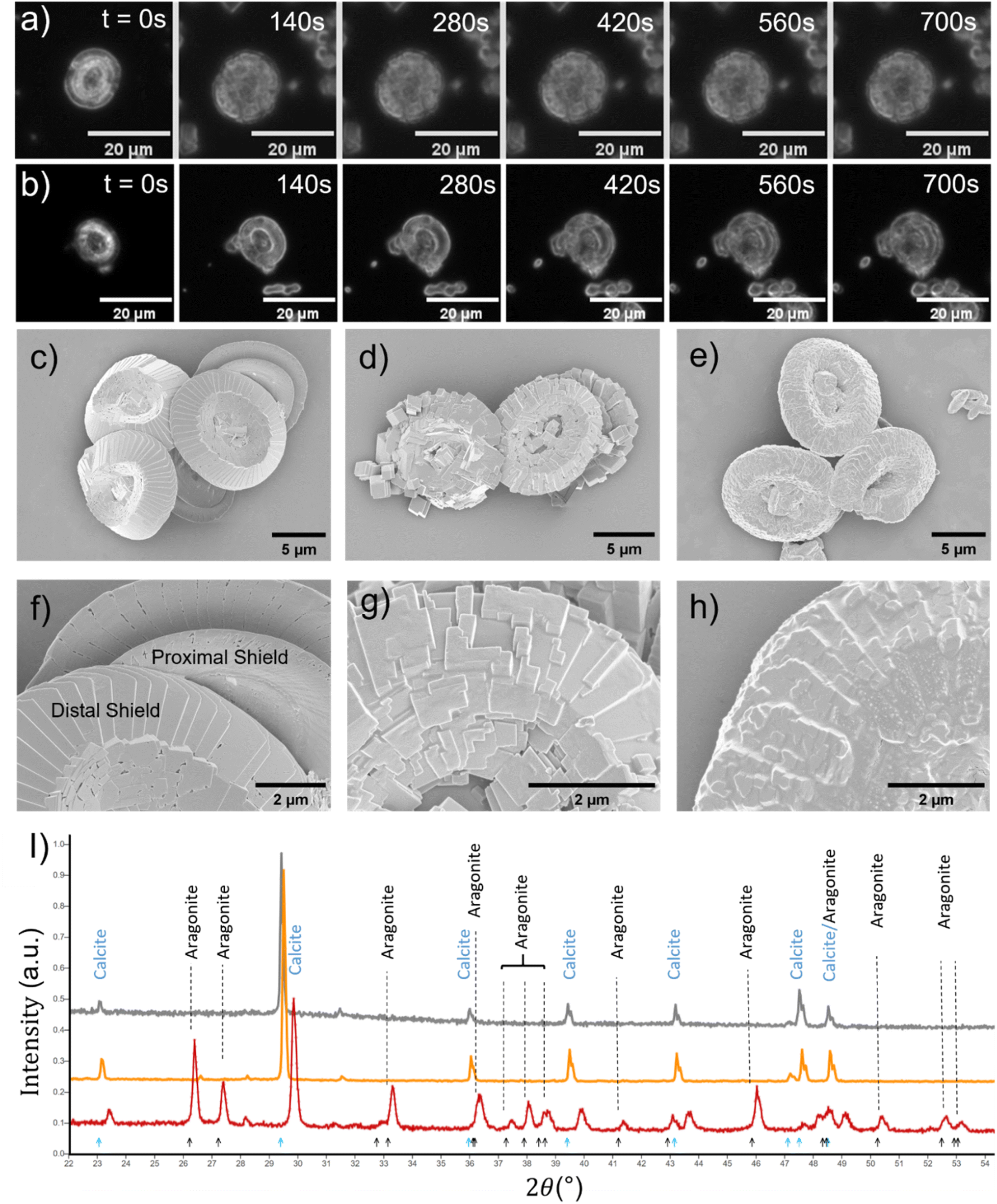 | ||
| Fig. 2 Artificial coccolith growth experiments. (a) Dark-field images of a representative coccolith depicting the temporal evolution of morphology in a solution supersaturated with respect to CaCO3 (Ωcalcite = 2200) in the absence of any added magnesium. The time since initial immersion is overlaid on the images. (b) Temporal evolution of a coccolith in Ωcalcite = 2200 solution with 5.5 mM Mg2+. Scanning electron microscopy images of the coccoliths: (c) and (f) naturally found, unperturbed coccoliths; (d) and (g) coccoliths after 24 hour immersion in ΩCalcite = 2200 solution without any added Mg2+; (e) and (h) coccoliths after 24 hour immersion in ΩCalcite = 2200 solution with 5.5 mM of added Mg2+. (i) Powder X-ray diffraction patterns of natural coccoliths (grey line), CaCO3 precipitates from Ωcalcite = 2200 solution absent of Mg2+ (orange line) and CaCO3 precipitates from Ωcalcite = 2200 with added 5.5 mM of Mg2+ (red line). Overlaid in blue and black arrows are the peak positions of the XRD pattern of calcite and aragonite calculated using the structures in the CrystalMaker library (10.8.2), respectively. | ||
In the case where 5.5 mM of Mg2+ is added to the supersaturated solution, the growth of the pure calcite particles initially present no longer maintains the rhombohedral shape as can be seen in video S3.† This is an important contrast to that seen in the absence of Mg2+ (video S1†). The growth of a coccolith in 5.5 mM of Mg2+ can be seen in Fig. 2b. The change of the projection area of the particles as a function of time is shown in Fig. S1b.† While the nature of the morphology of CaCO3(s) precipitated on coccoliths is unclear via optical imaging alone, the CaCO3(s) that nucleated from the supersaturated solution is different in the case where 5.5 mM Mg2+ is present. This effect can be seen in the wide-angle video shown in the ESI, video S4.† We next turn to examine complementary data from scanning electron microscope (SEM) and X-ray diffraction (XRD) patterns.
The surface morphology of representative C. braarudii coccoliths obtained in different conditions is shown in the SEM images shown in Fig. 2c–h. Fig. 2c and f show naturally formed, unperturbed coccoliths. Fig. 2d and g show coccoliths isolated after 24 hours of immersion in supersaturated solution in the absence of Mg2+, and Fig. 2e and h show those exposed to 5.5 mM Mg2+. First, in the absence of Mg2+, Fig. 2d reveals the surface modification is of calcite-like rhombohedral structure, which is distinctively different to that modified in the presence of Mg2+, as seen in Fig. 2e. Further complimentary SEM images showing the modification made to the proximal shield of the coccolith and coccosphere are reported in Fig. S2 and S3.† The surface modifications seen for biogenic coccoliths are distinctively different with and without Mg2+, and it is clear the surface morphology is akin to particulates that precipitate directly from the solution. This is supported by XRD results shown in Fig. 2i). The biogenic CaCO3 particles in the form of coccoliths are undoubtedly calcite (grey line) as are the particles precipitated abiotically in the absence of Mg2+ (orange line). In the presence of 5.5 mM Mg2+ (red line), a mixture of calcite and aragonite were precipitated.
It can be concluded that the substrate, biomineralized coccoliths or pure calcite, behaves in essentially the same manner under conditions in solution supersaturated with respect to CaCO3. Note that CaCO3 was not observed to precipitate on the central area of coccoliths nor the organic cellular membrane of coccospheres as shown in Fig. S2 and S4.† Likely this was evolved to maintain the biomechanical functionality of the surface. Next, the behaviour of biogenic coccoliths under dissolution is investigated and compared to pure calcite.
Auto-dissolution of biomineralized CaCO3
The work now turns to auto-dissolution of biomineralized coccoliths in solutions containing various concentrations of Mg2+, ranging from 0 mM to 100 mM, with a constant ionic strength at 0.3 M maintained using varying amounts of NaCl. Fig. 3a shows snapshots of a representative coccolith undergoing dissolution in an aqueous solution containing 2.5 mM of Mg2+, in the absence of any added Ca2+ or CO32−. The coccolith can be seen to completely dissolve within 30 minutes from the initial immersion. The top row shows the raw images and the bottom row shows the binary images after auto-thresholding. Fig. 3b) plots the temporal evolution of the coccolith area measured from the thresholded images for different concentrations of Mg2+. In the presence of a sea-water concentration of Mg2+ (54.6 mM, blue line) the time required to fully dissolve the biogenic coccolith is approximately 120 minutes, a 4-fold increase in comparison to the solution containing no magnesium. Fig. S5 and S6† shows the raw coccolith dissolution transient data recorded for various concentrations of Mg2+ ranging from 0 mM to 100 mM. The initial rate of the coccolith dissolution was measured between time zero and five minutes of the onset of dissolution as illustrated in Fig. S7.† Interestingly, when comparing the initial rate of coccolith dissolution (m2 s−1) to that of pure, similarly sized calcite particles23 the rate of coccolith dissolution is found to be approximately doubled across all concentrations of Mg2+ (Fig. S8†).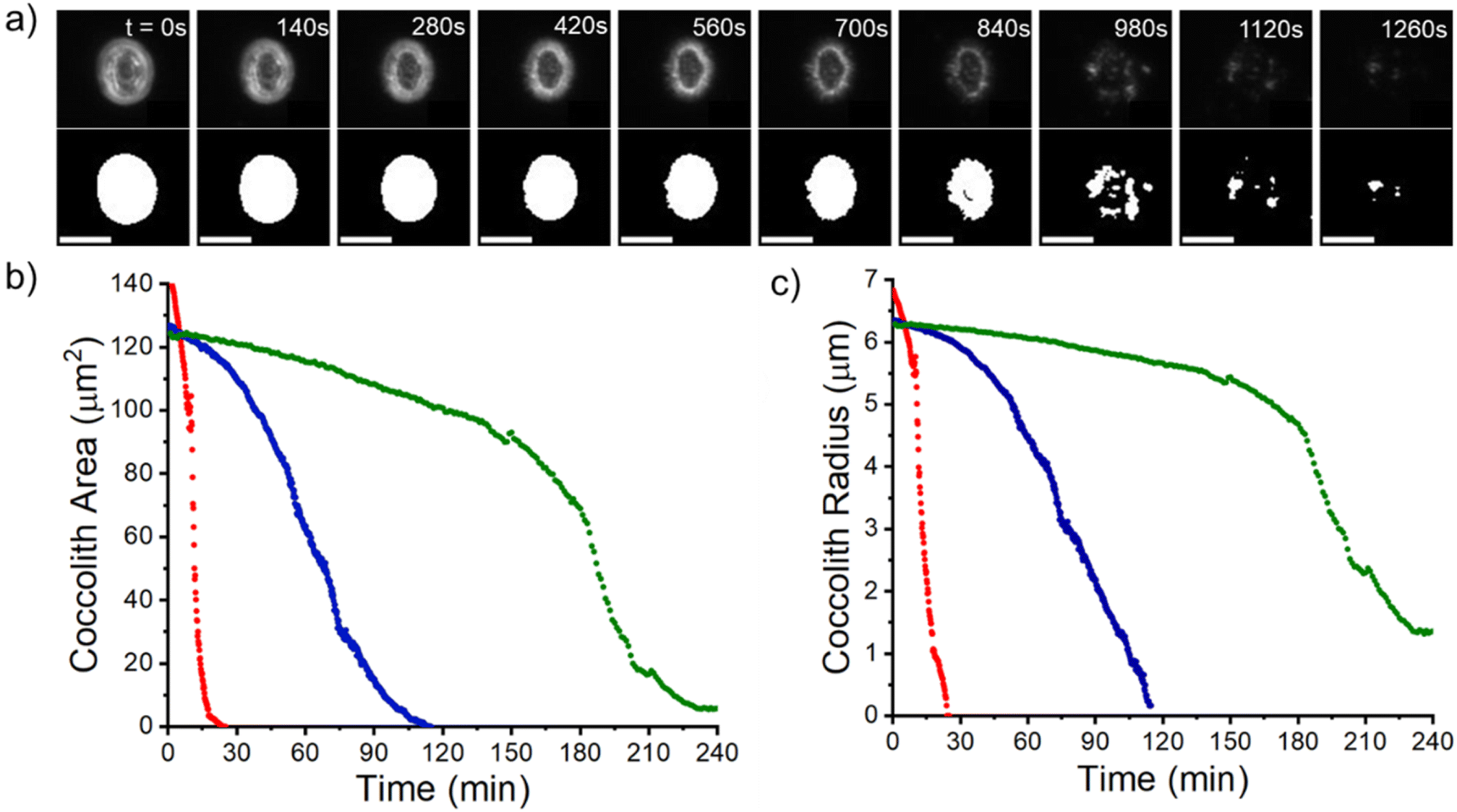 | ||
| Fig. 3 Coccolith dissolution experiments. (a) Temporal evolution of a representative C. braarudii coccolith dissolving in an aqueous solution containing 2.5 mM of Mg2+. The top row shows the raw images and the bottom row shows the binary images after auto-thresholding. Scale bar = 10 μm. (b) Shows the measured coccolith area as a function of time since immersion in an aqueous solution containing 0 mM (red), 54.6 mM (blue) and 100 mM concentrations of Mg2+(green). (c) A plot of the effective coccolith radius equivalent to the area of a perfect circle (area = πr2). The non-zero end point seen in the green data is likely an artifact of a small, non-soluble detritus. | ||
This difference is most likely due to the fact that the structure of coccoliths (Fig. 2c and f) is an intricate arrangement of biomineralized platelets which have likely evolved with the function to interlock other coccoliths to enclose the underlying bio-organism but with sufficient porosity to allow for nutrient uptake.26 We therefore consider a correction factor associated with the complex shape of the coccolith which results in the entity dissolving with the dissolution of less calcite compared to a hypothetical pure calcite particle with a similar geometric size but composed of a uniform solid. We use the literature reported coccolith ‘shape factor’ (ks) which relates the length of the coccolith (L) cubed to the estimated volume of a coccolith (Vcoccolith)27
| Vcoccolith = ksL3 | (2) |
The shape factor, ks, is species-specific and the recommended value for C. braarudii coccoliths is 0.06. The physical interpretation of eqn (2) is shown in Fig. 4a where the true volume of the coccolith is the volume of a cubic with side-length L multiplied by the shape factor. We next convert the shape factor, representing the correction for volume, to the projected coccolith area. We assume that the coccolith thickness (T) is one tenth of its side-length (L) as suggested by literature.28
| Vcoccolith = 10ksTL2 | (3) |
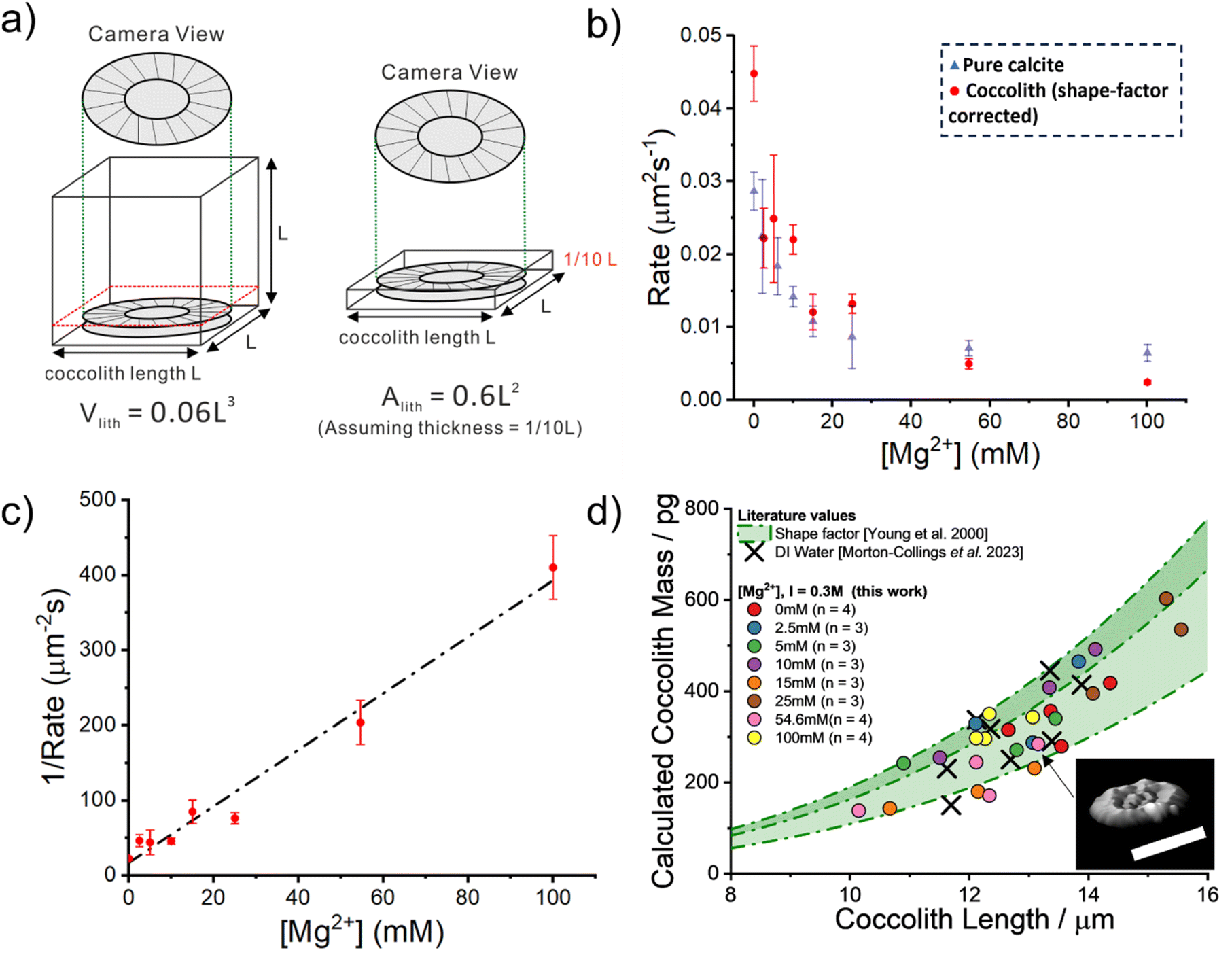 | ||
| Fig. 4 Quantitative analysis of the coccolith dissolution observed using a light microscope. (a) Shows an illustration of how the volume of a coccolith can be estimated by using the measured coccolith length (l) and the literature reported shape-factor,27Vlith = ksl3. The recommended C. braarudii shape-factor is 0.06. The equivalent absolute coccolith area are derived (discussed in the main text) with the assumption that the coccolith thickness is approximately one-tenth its length. (b) A plot of the measured rate of coccolith dissolution, in units of area per second, against the concentration of Mg2+ in an aqueous solution. Note that the rates obtained in this study, shown as red circles, are corrected by a factor of 0.6. The ionic strength of all the solutions is 0.3 M. Overlaid as blue triangles are the equivalent rate of dissolution for pure calcite. (c) A plot of the reciprocal rate of coccolith dissolution as a function of Mg2+ concentration. Note that the rate has again been corrected by a factor of 0.6, derived from the reported shape factor. The error bars show 1 standard deviation. The slope of the line of best fit is 3.76 μm−2 s mM−1 with a y-intercept at 16.8 μm−2 s. (d) shows a plot of the calculated mass of each coccolith individual obtained via 3-D reconstruction using information obtained from the opto-dissolution experiment. The concentration of Mg2+ is shown in the legend, along with the sample size (n). The method used to 3-D reconstruct the coccolith volume and thus an estimation of the coccolith mass is discussed in detail in the main text and elsewhere.28 Inset: 3D image of a representative coccolith after complete dissolution. Scale bar 10 μm. Shaded in green are the estimated coccolith mass using the literature-reported coccolith shape factor. The green dotted lines represent the mass estimated using the reported shape factor of 0.04, 0.06 and 0.07. These three values are the lower, recommended and upper values of the C. braaudii shape-factor.27 | ||
Dividing eqn (3) by the coccolith thickness (T) gives
| Acoccolith = 10ksL2 = 0.6L2 | (4) |
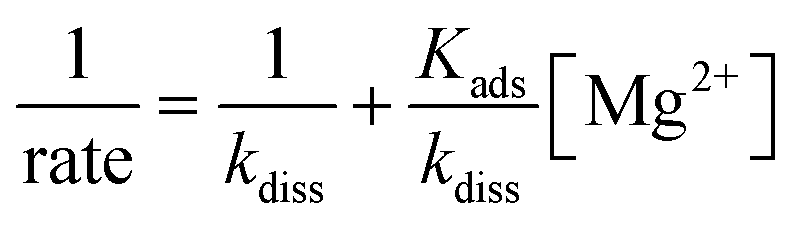 | (5) |
Having shown that biomineralised coccoliths behave similarly to pure calcite, we next show that the CaCO3 mass of coccoliths can be inferred via analysis of the optically acquired dissolution images regardless of the level of magnesium present in the solution. This requires quantifying the initial rate of dissolution of the coccolith length (μm s−1). The true volume of the coccolith can be reconstructed on a per coccolith basis using thresholded optical time-series images such as those shown in Fig. 3b.21,28 The coccolith mass is calculated by upscaling the calculated volume by the density of calcite.27Fig. 4d plots the individual coccolith mass calculated after complete dissolution in aqueous solutions undersaturated with respect to calcite with varying Mg2+ concentrations from zero to 100 mM. Note that the ionic strength of the media used in this work was fixed at 0.3 M which allows the rate of coccolith dissolution (μm2 s−1) to be directly compared to the rate of dissolution of pure calcite particles reported in previous work.23 Overlaid in green shade are corresponding values calculated using literature-reported C. braarudii coccolith volume shape factors (ks, as shown in eqn (2)) reported by Young et al., which range from 0.04 to 0.07 and a value of 0.06 was recommended.27 The black crosses are independent results of the mass of C. braarudii coccoliths reconstructed after complete dissolution in deionised water.21 The excellent agreement between current work and literature values provides validation of the mass reported herein.
Conclusions
The precipitation and dissolution of biogenic coccoliths at seawater levels of magnesium are similar to pure synthetic calcite particles grown in laboratory conditions despite the presence of a layer of coccolith-associated-polysaccharide. This is significant as it suggests biogenic calcite, sequestered in the sun-lit layer of the ocean in the form of coccoliths, behaves chemically like pure calcite. This is key to understanding and modelling the marine carbon cycle as biomineralized calcite dissolves at depth, resulting in a dynamic cycle. Moreover, the simple and non-esoteric light microscope setup employed allows the mass of biomineralized braarudii coccoliths, ranging from 100–600 picograms, to be inferred by simply observing their dissolution in undersaturated solutions regardless of the concentration of magnesium present. We observed that the time required to fully dissolve a micron-sized coccolith is doubled by approximately doubling the concentration of Mg2+ from 54.6 mM to 100 mM. The results may hold for other seawater calcite dissolution inhibitors as long as the rate of dissolution is not completely retarded. Importantly the measurement directly monitors the net dissolution/growth of the solid unlike alternative methods where the appearance or disappearance of ions in bulks solution are used for indirectly inferences and hence are strongly model-dependent. The simple microscopy setup offers promise for development of a proof-of-concept CaCO3 sensor allowing the rate of CaCO3 dissolution as a function of depth to be monitored.Materials and methods
Chemicals
Ultrapure deionised water (MilliQ, resistivity 18.2 MΩ cm−1 at 25 °C) was used to produce all solutions. CaCl2·2H2O and MgCl2 were sourced from Sigma-Aldrich, Germany. Na2CO3 was purchased from Acros Organics, Germany.Cultures
Cultures of Coccolithus pelagicus subsp. braarudii (RCC1198) were supplied by the Roscoff Culture Collection (RCC), France. The cultures were maintained on a K/2 enriched growth medium modified from the recipe for K medium by Keller et al. with f/2 vitamins.30 Aquil synthetic ocean water was used instead of natural seawater.31 The final molarity of each of the medium components in the K/2 recipe is summarized elsewhere.28,32Optical microscopy
Timelapse dissolution images were acquired on an openFrame inverted microscope (supplied by Cairn Research, UK in 2020) using an AuraPro Phase-Contrast illuminator (Cairn Research, UK), fitted with a 20× objective lens (NA = 0.5. Olympus, Japan). Images were captured using an ORCA-Flash 4.0 V3 C13440 CMOS camera (Hamamatsu Photonics, Japan) to provide 16-bit, 4-megapixel images. Images were analysed using ImageJ freeware (Fiji distribution). The calcite growth and dissolution experiments were performed in a custom machined aluminium (6082-T6 alloy) ‘Petri dish’ (STL file available in the online ESI†). The aluminium dish was found to provide superior rigidity as compared to the previously used, plastic-based 3-D printed dish.21 This was essential to maintain optical focus for experiments exceeding 6 hours in duration.X-Ray powder diffraction (XRD) and scanning electron microscopy
XRD diffractograms were collected on a Bruker D8 Advance diffractometer with a LynxEye detector and Cu Kα1 radiation (λ = 1.5406 Å), operating at 40 kV and 25 mA (step size at 0.019°, time per step at 0.10 s, total number of steps at 4368). Samples in powder form were pressed onto a glass preparative slide which was then attached to a sample holder. All measurements were scanned at 2θ of 5–90°.Prior to the scanning electron microscopy imaging, the coccolith and coccolithophore samples were washed with ethanol and dried overnight before a thin layer of gold (10 nm) was coated over the sample using a rotary pumped coater (Q150RES, Quorum, UK). The Scanning Electron Microscopy (SEM) images were obtained using a Sigma 300 FEG-SEM from Zeiss with an accelerating voltage of 2.0 kV.
Synthesis of abiotic calcite particles
Pure, synthetic calcite particles were formed by mixing 5.4 mM Ca2+(aq) and CO32−(aq) solutions in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio. The CaCO3 supersaturated solution was left to react for 15 minutes before the formed particles were resuspended, via centrifugation, in an aqueous solution saturated with respect of CaCO3 (Ωcalcite ≈ 3). The micron-sized rhombohedral shape CaCO3 particles were stable in the solution for 3–4 days before significant agglomeration was seen.
:1 volume ratio. The CaCO3 supersaturated solution was left to react for 15 minutes before the formed particles were resuspended, via centrifugation, in an aqueous solution saturated with respect of CaCO3 (Ωcalcite ≈ 3). The micron-sized rhombohedral shape CaCO3 particles were stable in the solution for 3–4 days before significant agglomeration was seen.
Biogenic coccolith growth and dissolution experiments
Prior to the dissolution experiment performed under the light microscope, the sample of biogenic calcite was resuspended from the culture medium into an aqueous solution via centrifugation saturated with respect to calcium carbonate (Ωcalcite ≈ 3). This was to minimise the possibility of contaminating the electrolyte of interest with calcite growth/dissolution inhibitors present in the culture medium (e.g. Mg2+) upon transfer. A small aliquot of the resuspended coccolith sample (20 μL) was transferred into the custom aluminium ‘Petri dish’ containing a large excess of the electrolyte of interest (15 mL) to initiate coccolith growth or dissolution. The time between the addition of the coccolith to setting up optical focus and image acquisition was approximately 20 seconds. The image acquisition was 1 frame per 10 seconds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out with the support of the Oxford Martin School Programme on Monitoring Ocean Ecosystems. We thank Dr Samuel Barton, Earth Science department, University of Oxford, for supplying samples of coccoliths. T Morton-Collings thanks St John's College, Oxford, for their generous support in his summer project. We thank the Mechanical Workshop in the Department of Chemistry, Oxford, for their help in machining the aluminium ‘Petri dish’ used in this work. We thank Dr Yiyang (Bruce) Li, Chemistry Department, University of Oxford, for obtaining XRD measurements.References
- F. J. Millero, Chem. Rev., 2007, 107, 308–341 CrossRef CAS.
- J. W. Morse, R. S. Arvidson and A. Lüttge, Chem. Rev., 2007, 107, 342–381 CrossRef CAS PubMed.
- Y. Pan, Y. Li, Q. Ma, H. He, S. Wang, Z. Sun, W.-J. Cai, B. Dong, Y. Di and W. Fu, Mar. Chem., 2021, 237, 104036 CrossRef CAS.
- J. L. Katz, M. R. Reick, R. E. Herzog and K. I. Parsiegla, Langmuir, 1993, 9, 1423–1430 CrossRef CAS.
- J. W. Morse and R. S. Arvidson, Earth-Sci. Rev., 2002, 58, 51–84 CrossRef CAS.
- T. Sabbides, E. Giannimaras and P. G. Koutsoukos, Environ. Biotechnol., 1992, 13, 73–80 CAS.
- J. D. Milliman, Global Biogeochem. Cycles, 1993, 7, 927–957 CrossRef CAS.
- K. Kayano, K. Saruwatari, T. Kogure and Y. Shiraiwa, Mar. Biotechnol., 2011, 13, 83–92 CrossRef CAS.
- J. Walker and G. Langer, Acta Biomater., 2021, 125, 83–89 CrossRef CAS.
- C. E. Walker, S. Heath, D. L. Salmon, N. Smirnoff, G. Langer, A. R. Taylor, C. Brownlee and G. L. Wheeler, Front. Mar. Sci., 2018, 5, 306 CrossRef.
- R. H. M. Godoi, K. Aerts, J. Harlay, R. Kaegi, C.-U. Ro, L. Chou and R. Van Grieken, Microchem. J., 2009, 91, 266–271 CrossRef CAS.
- K. Henriksen, J. Young, P. Bown and S. Stipp, Palaeontology, 2004, 47, 725–743 CrossRef.
- F. M. Monteiro, L. T. Bach, C. Brownlee, P. Bown, R. E. Rickaby, A. J. Poulton, T. Tyrrell, L. Beaufort, S. Dutkiewicz and S. Gibbs, Sci. Adv., 2016, 2, e1501822 CrossRef PubMed.
- M. Yang, C. Batchelor-McAuley, S. Barton, R. E. Rickaby, H. A. Bouman and R. G. Compton, Small, 2023, 2300346 CrossRef CAS PubMed.
- B. U. Haq and G. Lohmann, Mar. Micropaleontol., 1976, 1, 119–194 CrossRef.
- J. Milliman, P. Troy, W. Balch, A. Adams, Y.-H. Li and F. Mackenzie, Deep Sea Res., Part I, 1999, 46, 1653–1669 CrossRef CAS.
- C. Batchelor-McAuley, M. Yang, R. E. Rickaby and R. G. Compton, Chem.–Eur. J., 2022, 28, e202202290 CrossRef CAS.
- T. Hassenkam, A. Johnsson, K. Bechgaard and S. Stipp, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8571–8576 CrossRef CAS.
- X. Fan, C. Batchelor-McAuley, M. Yang and R. G. Compton, ACS Meas. Sci. Au, 2022, 2, 422–429 CrossRef CAS PubMed.
- R. Fakhrullin, L. Nigamatzyanova and G. Fakhrullina, Sci. Total Environ., 2021, 772, 145478 CrossRef CAS PubMed.
- T. Morton-Collings, M. Yang, C. Batchelor-McAuley, S. Barton, R. E. M. Rickaby, H. A. Bouman and R. G. Compton, Environ. Sci.: Adv., 2023, 2, 645–651 CAS.
- F. J. Millero, R. Feistel, D. G. Wright and T. J. McDougall, Deep Sea Res., Part I, 2008, 55, 50–72 CrossRef.
- M. Yang,L. Tan, C. Batchelor-McAuley and R. G. Compton, The solubility product controls the rate of calcite dissolution in pure water and seawater, 10.1039/d3sc04063a.
- R. Wong, C. Batchelor-McAuley, M. Yang and R. G. Compton, J. Electroanal. Chem., 2021, 903, 115818 CrossRef CAS.
- R. Wong, C. Batchelor-McAuley, M. Yang and R. G. Compton, J. Phys. Chem. Lett., 2022, 13, 7689–7693 CrossRef CAS PubMed.
- I. Jakob, M. A. Chairopoulou, M. Vučak, C. Posten and U. Teipel, Eng. Life Sci., 2017, 17, 605–612 CrossRef CAS PubMed.
- J. R. Young and P. Ziveri, Deep Sea Res., Part II, 2000, 47, 1679–1700 CrossRef.
- M. Yang, C. Batchelor-McAuley, S. Barton, R. E. Rickaby, H. A. Bouman and R. G. Compton, Angew. Chem., 2021, 133, 21167–21174 CrossRef.
- R. G. Compton and C. A. Brown, J. Colloid Interface Sci., 1994, 165, 445–449 CrossRef CAS.
- M. D. Keller, R. C. Selvin, W. Claus and R. R. Guillard, J. Phycol., 1987, 23, 633–638 CrossRef.
- F. M. Morel, J. Rueter, D. M. Anderson and R. Guillard, J. Phycol., 1979, 15, 135–141 CrossRef CAS.
- M. Yang, C. Batchelor-McAuley, S. Barton, R. E. Rickaby, H. A. Bouman and R. G. Compton, Environ. Sci.: Adv., 2022, 1, 156–163 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3va00362k |
| This journal is © The Royal Society of Chemistry 2024 |