DOI:
10.1039/D4TC03220A
(Paper)
J. Mater. Chem. C, 2024,
12, 17913-17924
Computationally guided circularly polarized luminescence simulations in chiral adaptive supramolecular systems†
Received
27th July 2024
, Accepted 3rd September 2024
First published on 5th September 2024
Abstract
Chirality and chiral phenomena associated with electromagnetic waves universally arise across many different forms, which involve transitions between states with opposite symmetries. Recently, adaptive chirality (the post-induced chirality in initial achiral environments by asymmetric guests) has undergone a burgeoning development owing to the multitasking features of tetraphenylethene (TPE) faces restricted in particular chemical buildings. Conceptually, initial achiral hosts bearing TPE commutable helical units (P/M forms) can interface with enantiopure guests to produce supramolecular asymmetric luminophores (CPL-active molecules). In these terms, a robust interpretation of these chiroptical events is yet to be computationally provided. In this study, we provide a solid and accessible computational workflow able to accurately predict the circularly polarized luminescence spectra of TPE-based hosts with stereogenic chiral guests (deoxyribonucleotide monophosphate homo-dimers, dipeptide homo-pairs and organic acids). In virtue of a preliminary DFT benchmark, we decided to use the ωB97XD functional to realize a conformational investigation of H-bonded chiral multicomponents encapsulated within TPE-based architectures in the first excited state by means of Born–Oppenheimer molecular dynamics. Finally, an extensive TD-DFT examination concerning exchange–correlation functionals and basis sets revealed the B3LYP/6-311G(d,p) level as the most precise and transferable method to meticulously model the CPL sign, band shape and peak position in TPE-based chiral reconfigurable host–guest complexes.
1. Introduction
It is not a coincidence that asymmetry is ubiquitous in the universe, from the astronomical level where the Orion Nebula is a source of circularly polarized light1 to the molecular kingdom in which molecules can be enantiomerically resolved.2–7 In this sense, chirality represents the narrative thread of a fascinating story impacting the fundamental phenomena in living matter from its origin.8–11 Notwithstanding the large proliferation of chirality in nature,12–14 its induction, preservation, transfer and, consequently, its complete dominance have caused constant struggle for supramolecular chemists.15–18 Non-covalent interactions, expressed in their different forms (hydrogen bonding,19–23 ion–dipole,24 π–π,25 CH–π,26 electrostatic,27etc.) undoubtedly facilitate the chiral transmission in artificial self-assembly processes.28–33 Benefiting from this biomimicking-oriented approach, various promising applications, such as molecular recognition,34–36 probing,37–39 sensing40–42 and catalysis,43,44 were achieved. Furthermore, unlike covalent chirality, supramolecular chirality can exploit the synergistic assistance of non-covalent interactions and host–guest complexation to produce controllable conformational transformation (flipping, rotating, twisting, etc.) with unique asymmetric packing.45 In recent years, this chiral adaptive strategy has been used to explore the intriguing interaction between the circularly polarized luminescence (CPL) and chiral communication in host–guest chemical architectures.46,47 For this scope, two ingredients shall thus be mixed: (1) an efficient chiral amplification/transfer during host–guest complexation and (2) a strong CPL signal to detect chiral activity. Intentionally omitting notorious hosts such as cyclodextrins,48,49 catenanes,50,51 rotaxanes52,53 and the other macrocycles54,55 that possess intrinsic chiral cavity, adaptive chirality directly interests initial achiral hosts which can exhibit excellent chiral recognition for enantiomeric guests by means post-induced homo-directional movements of internal dynamical units56 (Fig. 1). Novel studies indicate that tetraphenylethene (TPE) scaffolds encapsulated in proper chemical buildings ably combine their high fluorescence emitting efficiency (also favoured by aggregation-induced emission57) with an advantageous conformationally chiral dynamic nature.58 In this pioneering landscape, an in-depth theoretical treatment of CPL signal triggered by adaptive chirality in chiral guest recognition is still missing. Herein, we propose a computational protocol with the aim of understanding the appealing structure-functionality relationship caused by the chiral emission from an induced-asymmetric interior environment. Specifically, three different cases, in which TPE groups assume a dominant role in adaptive chirality, were analysed in order to create a robust DFT guide for their relative CPL spectral simulations in diverse contexts. Our prediction workflow can be ideally split in some preliminary steps: (1) a DFT investigation to catch the more convenient functional to emulate the arrangements (based on single crystal X-ray diffraction data) of TPE components in an octacationic cage. (2) Born–Oppenheimer molecular dynamics to follow the host–guest systems in their excited states (S1) upon the adaptive chirality mechanism. (3) A TD-DFT exchange–correlation functionals and basis sets benchmark with the goal of obtaining a transferable computational model to reproduce skillfully CPL spectral lines in terms of shape and band position.
 |
| | Fig. 1 (top) Chiral recognition through intrinsic chirality. (bottom) Chiral recognition through adaptive chirality. | |
2. Results and discussion
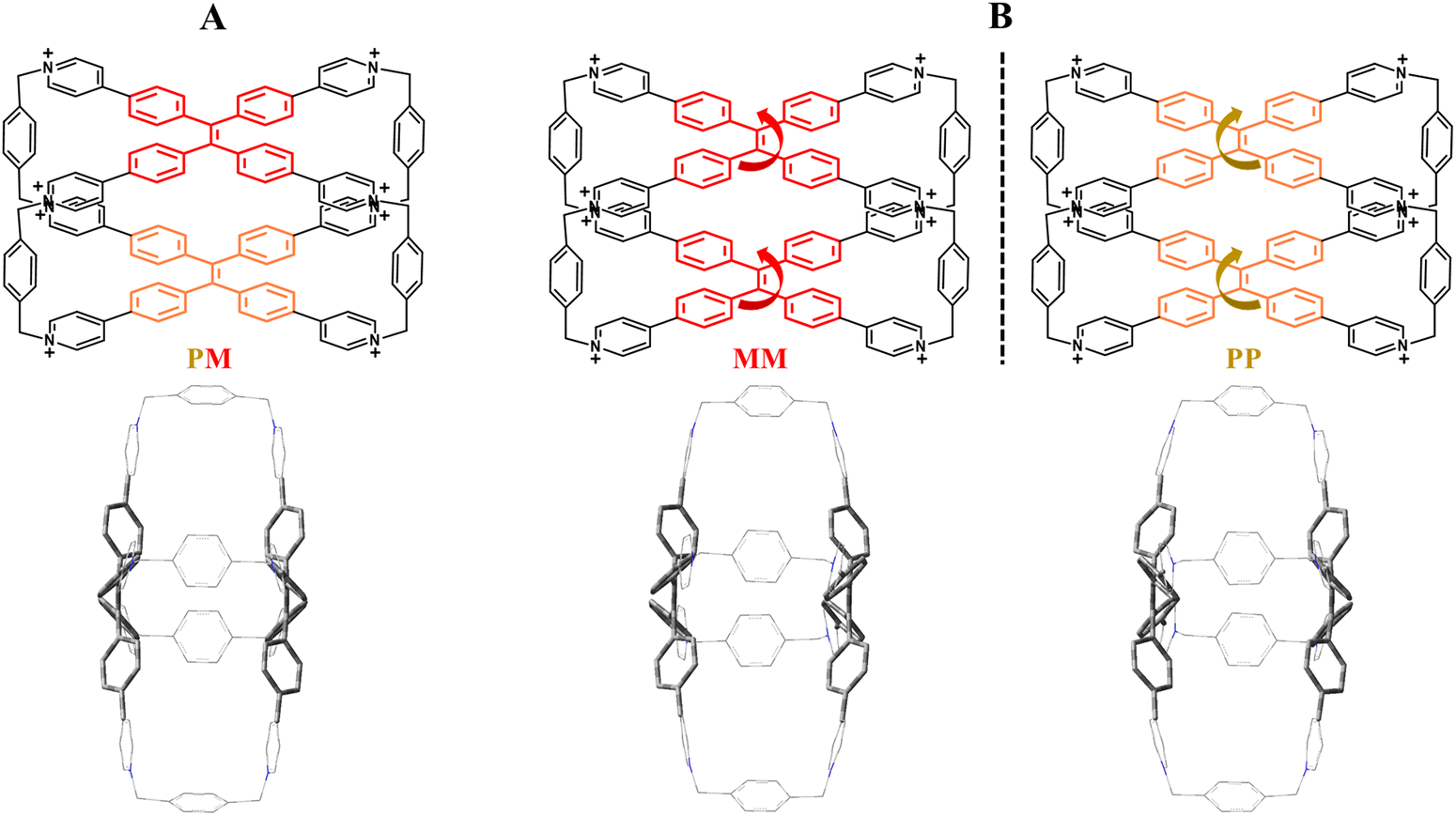
Before immediately accessing the chiroptical behavior of host–guest systems it is worth discussing in detail the structural features of TPE arms to understand their chiral arrangements. In particular, TPE faces are often installed in polycyclic cages with the possibility of having multi-cationic water-soluble compounds.59–63 In such confined pocket TPE fragments can assume a twofold propeller-like dynamic conformation by virtue of the rotation of the four phenyl rings between the two adjacent p-substituted positions. These reversible movements will confer clockwise (P) or anti-clockwise (M) revolutions in the TPE building blocks. However, in the absence of an inner chiral induction mediated by enantiomeric guests, P/M TPE conformations cannot be prevalently homo-directionally restricted (PP or MM) as a consequence of the lack of an energy barrier between the two possible rotations. In a solid state, free TPE-based cages simultaneously adopt P and M configurations resulting in a mesomeric form (P/M) (Fig. 2A) as confirmed by crystallographic data.64
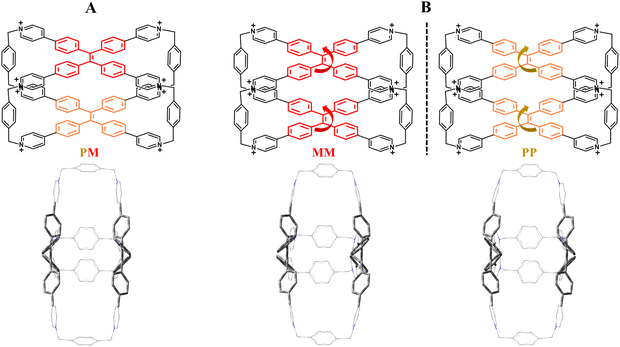 |
| | Fig. 2 (A) X-ray chemical structure of the mesomeric configuration (PM). (B) Enantiomeric forms (PP and MM) related to the TPE-based octacationic cage. Chlorides are the counterions. | |
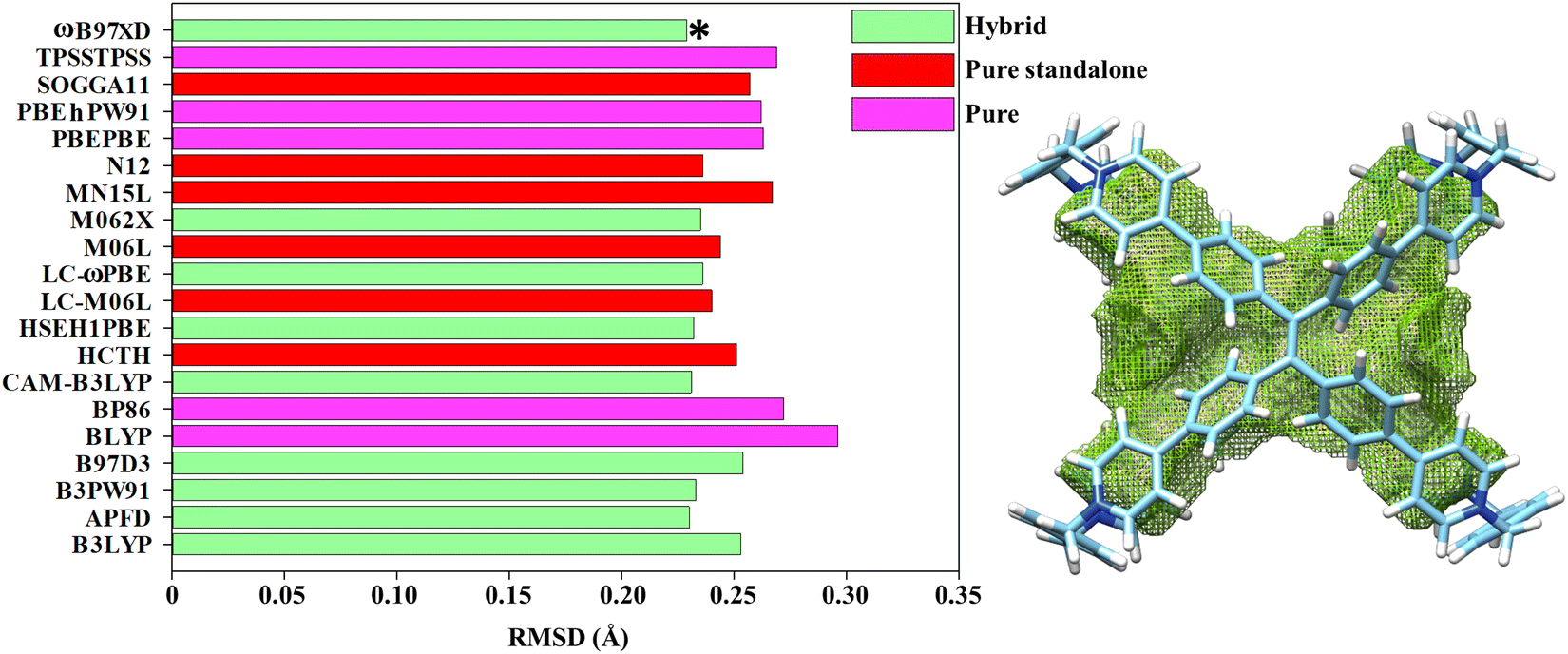
Considering this wealth of conformational effects, it instantly emerges that the chiroptical fingerprints are strongly dependent on the location of the molecular cage within the hypersurface of potential energy.65–69 Computationally speaking, in DFT theory the quality of the energy-minimized conformations is substantially influenced by the choice of the exchange–correlation functionals whose performances are commonly ranked in the virtue of benchmarks targeting the accurate prediction of the atomic positions70,71 defined by X-ray diffraction data. To adequately duplicate the crystallographic coordinates, we decided to define our DFT set conscious that the selected functionals: (1) were already successfully used to model TPE entities,57,72,73 and (2) are convenient for geometrical optimization of organic molecules.74–77 Therefore, APFD, B3LYP, B3PW91, B97D3, BLYP, BP86, CAM-B3LYP, HCTH, HSEH1PBE, LC-M06L, LC-ωPBE, M06L, M062X, MN15L, N12, PBEPBE, PBEhPW91, SOGGA11, TPSSTPSS and ωB97XD were checked in the gas phase to allow valid comparisons with crystal coordinates obtained by single crystal diffraction experiments. As cropped from the previous list, we tried to compose a homogenous box of pure, hybrid and pure-standalone exchange–correlation functionals (also including long-range corrections and dispersion effects) to guarantee a balanced picture in the DFT functionals performances. Finally, the Pople basis set was utilized as it is proven to be quite credible in modelling the second-period elements.78,79 A benchmarking metric, the root-mean-square deviation of all non-hydrogen atoms (RMSD), was elaborated to compare the crystal structures obtained from X-ray diffraction (TPE-based octacationic cage in the PM configuration) with the DFT-optimized gas-phase geometries. The collected statistical data summarized in Fig. 3 attest that averaged skeleton RMSD values are less than 0.3 Å and no excessive fluctuations were recognized. Assisted by Fig. 3 and Table 1, it is easy to intuit that a general DFT trend, in imitating the X-ray structure, can be identified: hybrid > pure-standalone > pure. Within the RMSD values distribution, the hybrid functionals outcomes were exalted by dispersion-embedded ωB97XD (0.229 Å), which appeared as the best candidate to clone the X-ray molecular structure (APFD and CAM-B3LYP still remain valid alternatives, Table 1). Contemplating that a TPE-based octacationic cage will act as a host, we also tried to verify the functional attitudes by calculating the inner cavity volume. It must be emphasized that we did not intend to handle Vocc (Table 1) as a rigorous statistical method (such as RMSD) but as an approximately qualitative tool to estimate the internal accessible topological space. Although it is not obvious to correlate Vocc with the RMSD since the TPE-based octacationic cage is not a closed polygonal structure (like fullerenes), good general DFT performances were detected, corroborating the leading position of ωB97XD (Table 1).
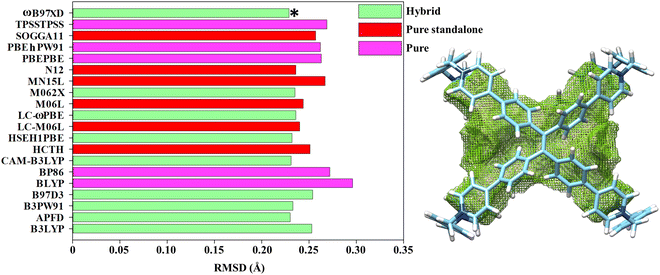 |
| | Fig. 3 (left) Cumulative RMSD histograms (Å) at the functional/6-311G(d,p) level in the gas phase for the PM configuration; the asterisk indicates the best DFT performer. (right) Optimized structure at the ωB97XD/6-311G(d,p) level with its relative mapped cavity volume. | |
Table 1 RMSD values (Å) and Vocc (Å3) related to the whole list of exchange–correlation functionals
| Functional |
RMSD (Å) |
V
occ (Å3) |
| X-Ray |
— |
475.0240 |
| B3LYP |
0.2530 |
519.4560 |
| APFD |
0.2300 |
513.7600 |
| B3PW91 |
0.2330 |
521.6000 |
| B97D3 |
0.2540 |
527.2960 |
| BLYP |
0.2960 |
506.3360 |
| BP86 |
0.2720 |
512.4800 |
| CAM-B3LYP |
0.2310 |
515.8080 |
| HCTH |
0.2510 |
517.7920 |
| HSEH1PBE |
0.2320 |
509.0880 |
| LC-M06L |
0.2400 |
448.6400 |
| LC-ωPBE |
0.2360 |
506.0160 |
| M06L |
0.2440 |
508.4320 |
| M062X |
0.2350 |
503.1360 |
| MN15L |
0.2670 |
530.9120 |
| N12 |
0.2360 |
506.1120 |
| PBEPBE |
0.2630 |
510.8160 |
| PBEhPW91 |
0.2620 |
507.2000 |
| SOGGA11 |
0.2570 |
523.3600 |
| TPSSTPSS |
0.2690 |
509.0560 |
| ωB97XD |
0.2290 |
488.8400 |
2.1. Case 1: TPEcage⊃deoxyribonucleotides monophosphate homo-dimers
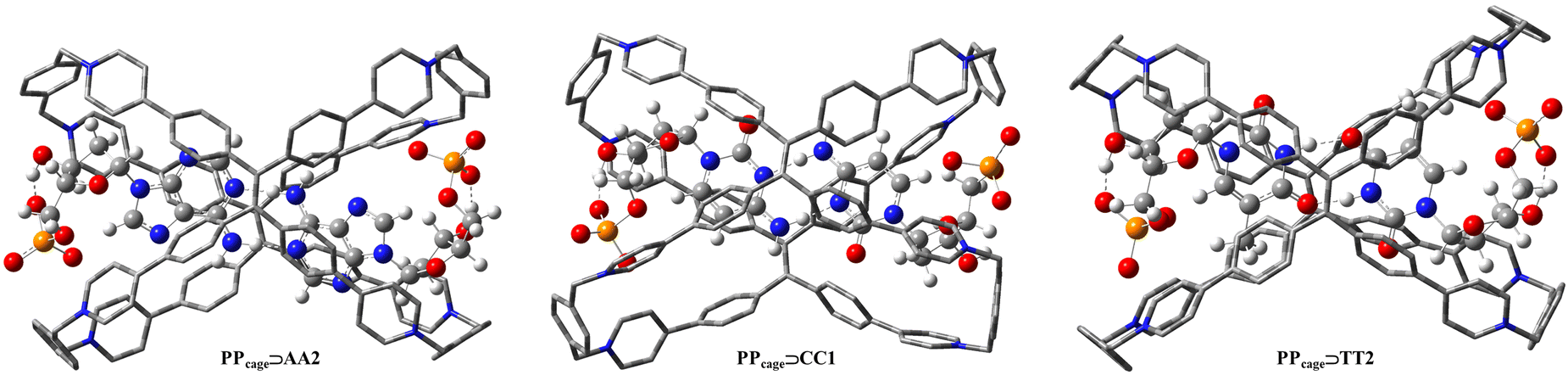
Once the leader functional was identified, we focused on the theoretical analysis of a specific host–guest case with the purpose of using it as “computational probe” in order to achieve a single transferable level of theory for other adaptive chirality examples. Recently, Cao et al. reported a very elegant way of adaptive chirality concerning the association between the TPE-based octacationic cage and enantiomeric deoxyribonucleotide monophosphate guests.80 In this situation, 2′-deoxyadenosine 5′-monophosphate (A), 2′-deoxythymidine 5′-monophosphate (T) and 2′-deoxycytidine 5′-monophosphate (C) induced the PP conformation in the TPE cage through their stereogenic chirality, D-ribodesose, by means the host–guest complexation in aqueous solution. Although water is a hydrogen bonding disruption solvent, stable hydrogen-bonded homo-dimers (AA/TT/CC) were appropriately entrapped in the hydrophobic cavity of the TPE cage. This type of guest recognition was promoted by a through-space chirality propagation in which non-covalent interactions occur between the TPE cage and the homo-dimer, bringing to a multilevel supramolecular adaptive chirality. Before DFT-optimizing the host–guest systems (PPcage⊃TT, PPcage⊃CC and PPcage⊃AA) we inspected the pairing binding energies of the different hydrogen bonding patterns pertinent to the deoxyribonucleotides monophosphate homo-dimers to rank their association stabilities. Among the possible R22(8), R22(9) and R22(10) hydrogen-bonded motifs, TT1, AA2 and CC1 were more stable for deoxyribothymidine, deoxyriboadenosine and deoxyribocytidine monophosphate dimers, respectively (Fig. 4). In spite of the multitude of hydrogen bonding schemes, all the pairs showed an analogous tendency in their chiral groups, in fact, an intramolecular hydrogen bonding between the positive-polarized proton of the hydroxyl function in position 4 and the phosphate pendant was regularly found (Fig. 4 and Fig. S1, ESI†).
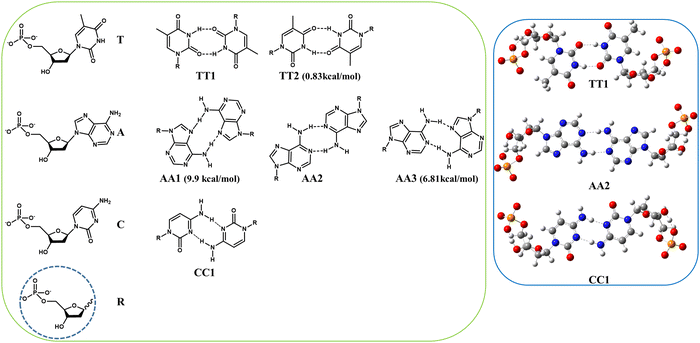 |
| | Fig. 4 (green) Neutral hydrogen-bonded homo-dimers accessible for deoxyribonucleotide monophosphate. (blue) Geometrical arrangements of the more stable homo-dimers (for each nucleobase) optimized at the ωB97XD/6-311G(d,p) level in the gas phase. | |
Despite experimental proton resonances certifying the occupation of the H-bonded nucleobase moieties at the center of the cavity, and NMR titration, isothermal titration calorimetry and mass spectra supporting the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry in the PPcage⊃deoxyribonucleotides host–guest complexes,80 their relative hydrogen bonding templates still remain unknown. With this in mind, we optimized all the achievable complexed forms (PPcage⊃TT1, PPcage⊃TT2, PPcage⊃AA1, PPcage⊃AA2, PPcage⊃AA3 and PPcage⊃CC1) to establish if the energetic rankings individuated for the uncomplexed homo-dimers (Fig. 4 and Fig. S1, ESI†) were also maintained in the host–guest systems (Fig. S2, ESI†). Firstly, the energy-minimized conformations of all the complexes preserved the original homo-dimers hydrogen bonding schemes (Fig. S2, ESI†) in the center of the cage. Beyond the very articulated CH–π/π–π interactions between nucleobases and the TPE faces, in each complex the anionic monophosphate chain heads to the electron-deficient pyridinium rings to realize the multiple hydrogen bonds (Fig. 5 and Fig. S3, ESI†). For deoxyriboadenosine complexes (PPcage⊃AA1, PPcage⊃AA2 and PPcage⊃AA3), their stability energies reasonably match with the ones of isolated homo-dimers, de facto the energetic order is retained (PPcage⊃AA2 > PPcage⊃AA3 > PPcage⊃AA1). Conversely, a marked inversion in the stability sequence was revealed for the deoxyribothymidine complexes compared to the isolated homo-dimers (TT1 > TT2 but PPcage⊃TT2 > PPcage⊃TT1, see Fig. S1 and S2, ESI†) as a result of a more diagonal disposition of the ribo-monophosphate pendants that makes spatially difficult hydrogen bonds with the terminal phenyl and pyridinium rings of the cage. Neglecting the myriad of non-covalent interactions in the low/middle electronic-density regime (yellow/green surfaces), NCI (non-covalent index) mapped surface spotlighted how monophosphate oxygen atoms of TT2 in PPcage managed to form two more strong hydrogen bonds (blue/violet surfaces) than TT1 in PPcage (Fig. S4 and S5, ESI†), justifying the energy gap inversion (Δ = 8.37 kcal mol−1). Additionally, the presence of the monophosphate fragments actively influences the H-bonded package of the nucleobases, which tend to curve in the supramolecular frontier to guarantee a stable accommodation of –PO42− at the edges of the cage. This phenomenon was authenticated by re-optimizing PPcage⊃AA2, PPcage⊃TT2 and PPcage⊃CC1 in which their monophosphate units were truncated (only nucleobase–deoxyribose was considered, Fig. S6, ESI†), their acquired minima depicted flat H-bonded regions emphasizing how the –PO42− pendants simultaneously determine both the host–guest association energies and the chiral propagation via hydrogen bonding. On the other hand, if we focus on the H-bonding lengths between the nucleobases, it is possible to note an asymmetric shortening in the homo-dimer intermolecular distances as an effect of the multiple interactions with the cage (Fig. S7, ESI†).
:2 stoichiometry in the PPcage⊃deoxyribonucleotides host–guest complexes,80 their relative hydrogen bonding templates still remain unknown. With this in mind, we optimized all the achievable complexed forms (PPcage⊃TT1, PPcage⊃TT2, PPcage⊃AA1, PPcage⊃AA2, PPcage⊃AA3 and PPcage⊃CC1) to establish if the energetic rankings individuated for the uncomplexed homo-dimers (Fig. 4 and Fig. S1, ESI†) were also maintained in the host–guest systems (Fig. S2, ESI†). Firstly, the energy-minimized conformations of all the complexes preserved the original homo-dimers hydrogen bonding schemes (Fig. S2, ESI†) in the center of the cage. Beyond the very articulated CH–π/π–π interactions between nucleobases and the TPE faces, in each complex the anionic monophosphate chain heads to the electron-deficient pyridinium rings to realize the multiple hydrogen bonds (Fig. 5 and Fig. S3, ESI†). For deoxyriboadenosine complexes (PPcage⊃AA1, PPcage⊃AA2 and PPcage⊃AA3), their stability energies reasonably match with the ones of isolated homo-dimers, de facto the energetic order is retained (PPcage⊃AA2 > PPcage⊃AA3 > PPcage⊃AA1). Conversely, a marked inversion in the stability sequence was revealed for the deoxyribothymidine complexes compared to the isolated homo-dimers (TT1 > TT2 but PPcage⊃TT2 > PPcage⊃TT1, see Fig. S1 and S2, ESI†) as a result of a more diagonal disposition of the ribo-monophosphate pendants that makes spatially difficult hydrogen bonds with the terminal phenyl and pyridinium rings of the cage. Neglecting the myriad of non-covalent interactions in the low/middle electronic-density regime (yellow/green surfaces), NCI (non-covalent index) mapped surface spotlighted how monophosphate oxygen atoms of TT2 in PPcage managed to form two more strong hydrogen bonds (blue/violet surfaces) than TT1 in PPcage (Fig. S4 and S5, ESI†), justifying the energy gap inversion (Δ = 8.37 kcal mol−1). Additionally, the presence of the monophosphate fragments actively influences the H-bonded package of the nucleobases, which tend to curve in the supramolecular frontier to guarantee a stable accommodation of –PO42− at the edges of the cage. This phenomenon was authenticated by re-optimizing PPcage⊃AA2, PPcage⊃TT2 and PPcage⊃CC1 in which their monophosphate units were truncated (only nucleobase–deoxyribose was considered, Fig. S6, ESI†), their acquired minima depicted flat H-bonded regions emphasizing how the –PO42− pendants simultaneously determine both the host–guest association energies and the chiral propagation via hydrogen bonding. On the other hand, if we focus on the H-bonding lengths between the nucleobases, it is possible to note an asymmetric shortening in the homo-dimer intermolecular distances as an effect of the multiple interactions with the cage (Fig. S7, ESI†).
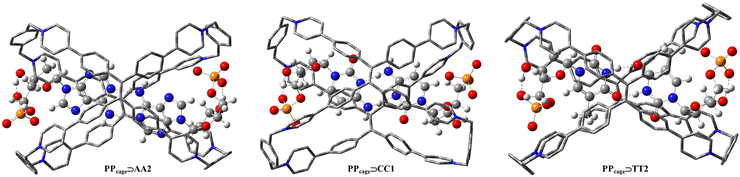 |
| | Fig. 5 DFT optimization of the more stable PPcage⊃homo-dimers (for each nucleobase) calculated at the ωB97XD/6-311G(d,p) level in the gas phase. Hydrogen atoms of the cages are omitted for clarity. | |
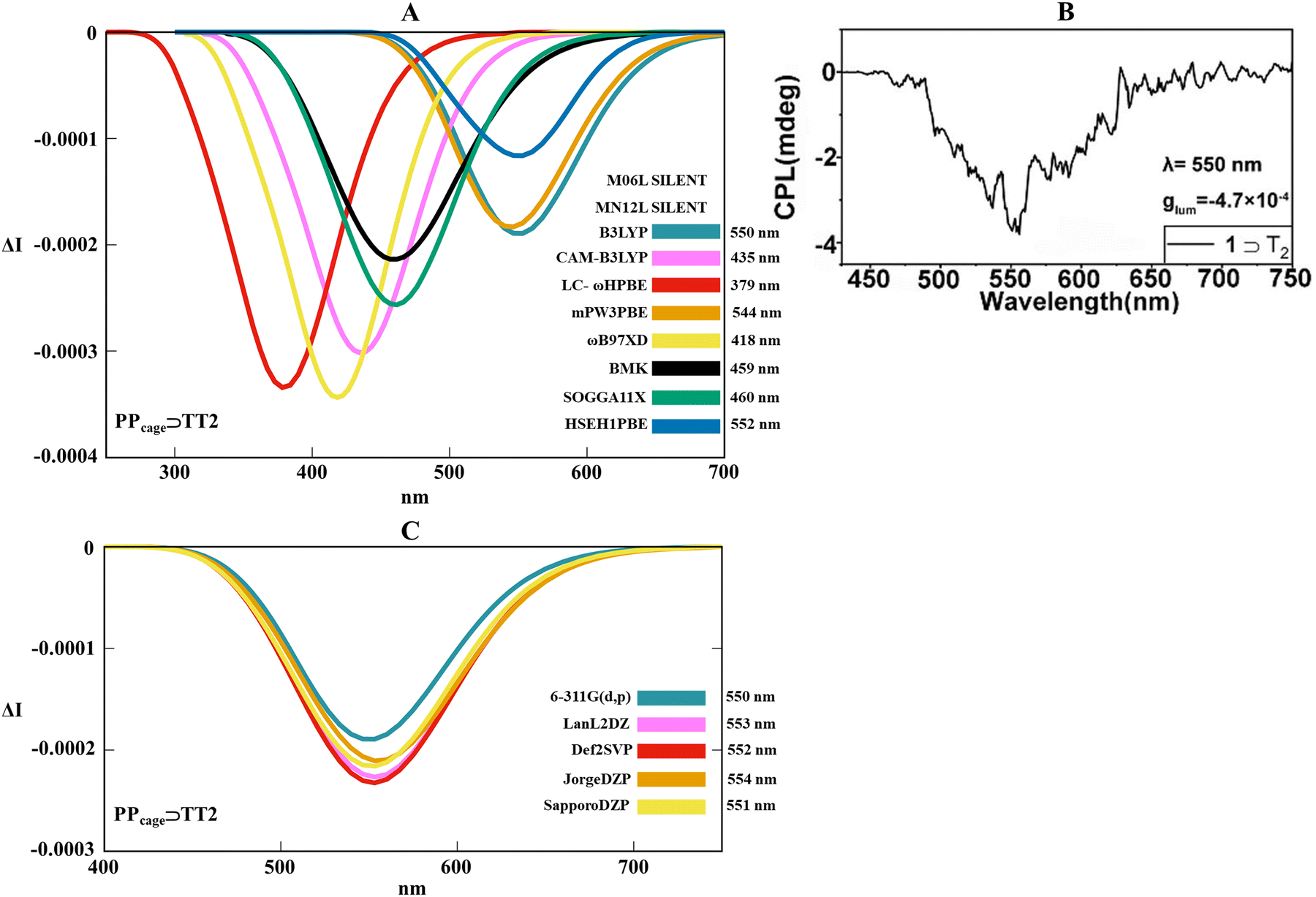
By condensing the previous conformational findings, it evidently comes up that thermal movements between the three supramolecular-interconnected components (the two monomers and the cage) become relevant (especially in the solvent phase) and certainly impacts the final spectral signatures of the CPL outputs. Given this framework, the asymmetry pocket associated with the supramolecular host–guest adduct was explored by combining thermal-dependent fluctuations with light irradiation along the excited photochemical pathway. For such scope, the ab initio excited molecular dynamics (Born–Oppenheimer molecular dynamic, BOMD) for PPcage⊃homo-dimers in the solvent phase allowed us to carry out an appropriate phase space conformational sampling of the first excited state, permitting the collection of an ensemble of statistically relevant conformers. The final CPL spectra were thus reconstructed as a weighted sum of the spectra performed at the TD-DFT level on temporally interspersed molecular conformation snapshots recovered from BOMD trajectories. The time-dependent formalism becomes fundamental to comprehend how the emission wavelengths (band position) and the rotatory strengths (intensity and sign of the signal) react when the mechanism of adaptive chirality acts. Convinced by our initial DFT benchmark, we adopted the ωB97XD/6-311G(d,p) level for a satisfactory description of the excited potential energy surfaces over a picosecond scale. At first glance, the study of the excited state trajectories pointed to a general trend regarding the H-bonding supramolecular distances for PPcage⊃AA2, PPcage⊃TT2 and PPcage⊃CC1 between ground and the first excited states (S0 and S1, respectively). As a point of fact, in the normalized distributions for the three host–guest systems, spatial proximity between the nucleobases moieties (expressed as NH–N and NH–O hydrogen bondings) is on average slightly more extended in S1 than in S0 (Fig. S11–S13, ESI†). Furthermore, no proton transfer was detected between donor and acceptor hydrogen bonding groups during the S1 potential energy surface (PES) scanning. Once the three S1 trajectories were evaluated, a conspicuous list of exchange–correlation functionals at the TD-DFT level was tested to detect the coexistence of the luminescent macrocycle with the chiral homo-dimer guest in the chiral adaptive mechanism. Hence, M06L, MN12L, B3LYP, CAM-B3LYP, LC-ωhPBE, mPW3PBE, ωB97XD, BMK, SOGGA11X and HSEH1PBE were trialed for this aim. For technical reasons, we first decided to apply our TD-DFT benchmark on the deoxyribothymidine host–guest system as an upshot of its more regular and less fragmented CPL spectral line. At this point, it should be stressed that our primary goal centrepieces on the realistic quantification of the host–guest chiroptical responses in terms of the sign, intensity, shape profile and band positions of the CPL spectra. Following this background, Fig. 6A gives a complete depiction of the heterogeneous functional performances in reproducing the CPL spectral outline. Remembering that experimental CPL maxima is placed at 550 nm (Fig. 6B), it is possible to see that a consistent number of functionals (CAM-B3LYP, LC-ωhPBE, ωB97XD, BMK, SOGGA11X) underestimates the CPL peak position, showing blue-shifted spectra between 380 and 460 nm. Diverging from CAM-B3LYP, LC-ωhPBE, ωB97XD, and SOGGA11X which present a too-sharp and excessively intense chiral emission, M06L and MN12L suffered a silent signal due to the almost zero rotatory strengths during the radiative process (Table S1, ESI†). Undoubtedly, the central interval around 550 nm represents a salient spectral range, with HSEH1PBE, mPW3PBE and B3LYP, which excellently capture both the asymmetrical pseudo-Gaussian lineshape and the CPL maximum (552, 544 and 550 nm, respectively). This collection of spectra straightly testimonies that the diagnostic negative sign (which univocally identifies the PP TPE conformation) was adroitly calculated using all the functionals that conferred an active CPL output. At this point of such computational searching, it easily arrives that just two functionals (mPW3PBE and B3LYP) managed to combine theoretically the features of the optical emission with the chiral feedback (HSEH1PBE moderately belittles the band intensity). Taking into account B3LYP as our gold standard, we attempted to verify if different basis sets can influence the CPL spectral pattern, hence 6-311G(d,p), LanLDZ, Def2SVP, JorgeDZP and SapporoDZP were applied. Fig. 6C revealed that the basis set has a negligible effect on the 0 ← 1 energy transition (peak position changes very subtly) and a moderate impact on the CPL magnitude, which reflects how a different number of primitive functions can lead to appreciable differences in the rotatory strengths. Coherently with a perfect band position (550 nm), we maintained 6-311G(d,p) coupled with B3LYP.
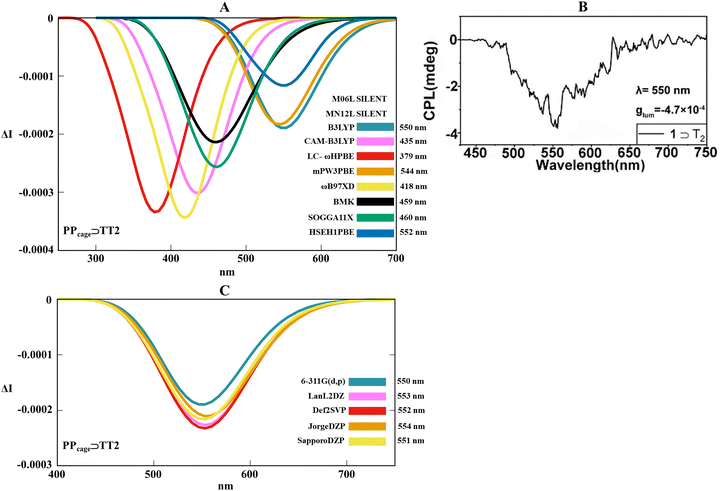 |
| | Fig. 6 (A) Collection of TD-DFT calculated CPL spectra at the functional/6-311G(d,p) level along the MD trajectory for PPcage⊃TT2 in H2O. (B) Experimental CPL spectrum of the PPcage⊃TT in water. Adapted with permission from ref. 80 © 2020 Chinese Chemical Society. (C) Overview of TD-DFT calculated CPL spectra at the B3LYP/basis set level along the MD trajectory for PPcage⊃TT2 in H2O. | |
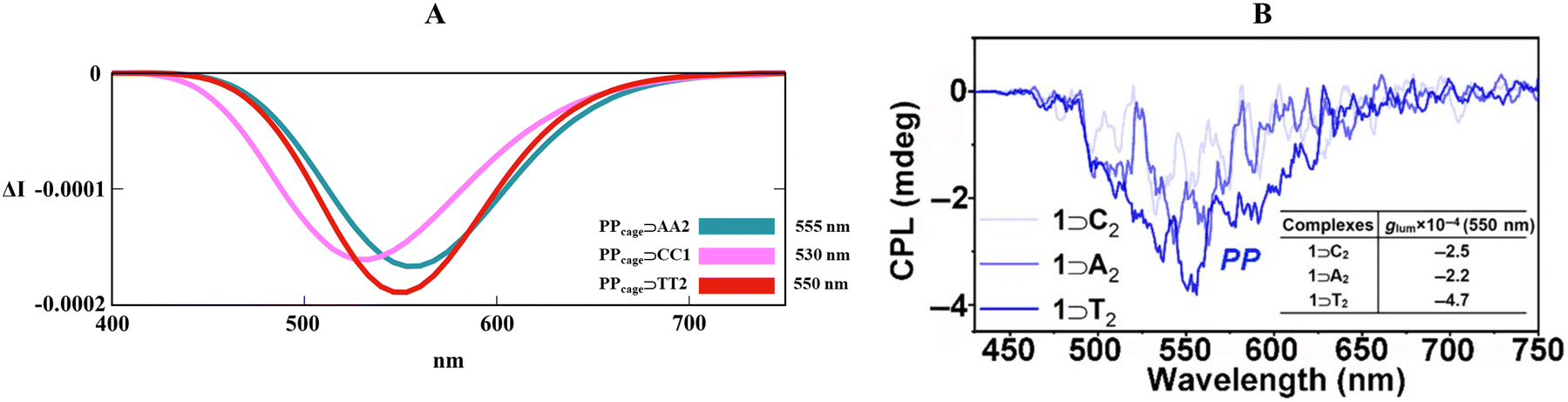
Motivated by the previous outcomes, we also employed our customized B3LYP/6-311G(d,p) level for PPcage⊃AA2 and PPcage⊃CC1. Fig. 7 exemplified that experimental right-handed (isoenergetic) circularly polarized luminescence (Fig. 7B) was correctly imitated by our theoretical protocol (Fig. 7A), with an excellent match for the PPcage⊃AA2 CPL maximum (555 nm) and modest blue shift for the PPcage⊃CC1 CPL band (530 nm). Surprisingly, the selected TD-DFT level elegantly caught the right order in the band intensity (PPcage⊃TT2 > PPcage⊃AA2 > PPcage⊃CC1). Lastly, it must be underlined that guanosine-based (G) host–guest was not the computationally treated cause of the photoinduced-electron transfer (PET) mechanism (usually induced by the electron-donating tendency of guanine heterocycle), which quenches the TPE luminogen activity.80
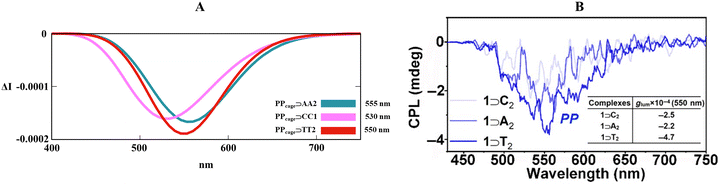 |
| | Fig. 7 (A) Collection of TD-DFT calculated CPL spectra at the B3LYP/6-311G(d,p) level along the MD trajectory for PPcage⊃TT2, PPcage⊃AA2 and PPcage⊃CC1 in H2O. (B) Experimental CPL spectra of the PPcage⊃TT, PPcage⊃AA and PPcage⊃CC in water. Adapted with permission from ref. 80 © 2020 Chinese Chemical Society. | |
2.2. Case 2: TPEcage⊃dipeptide homo-pairs
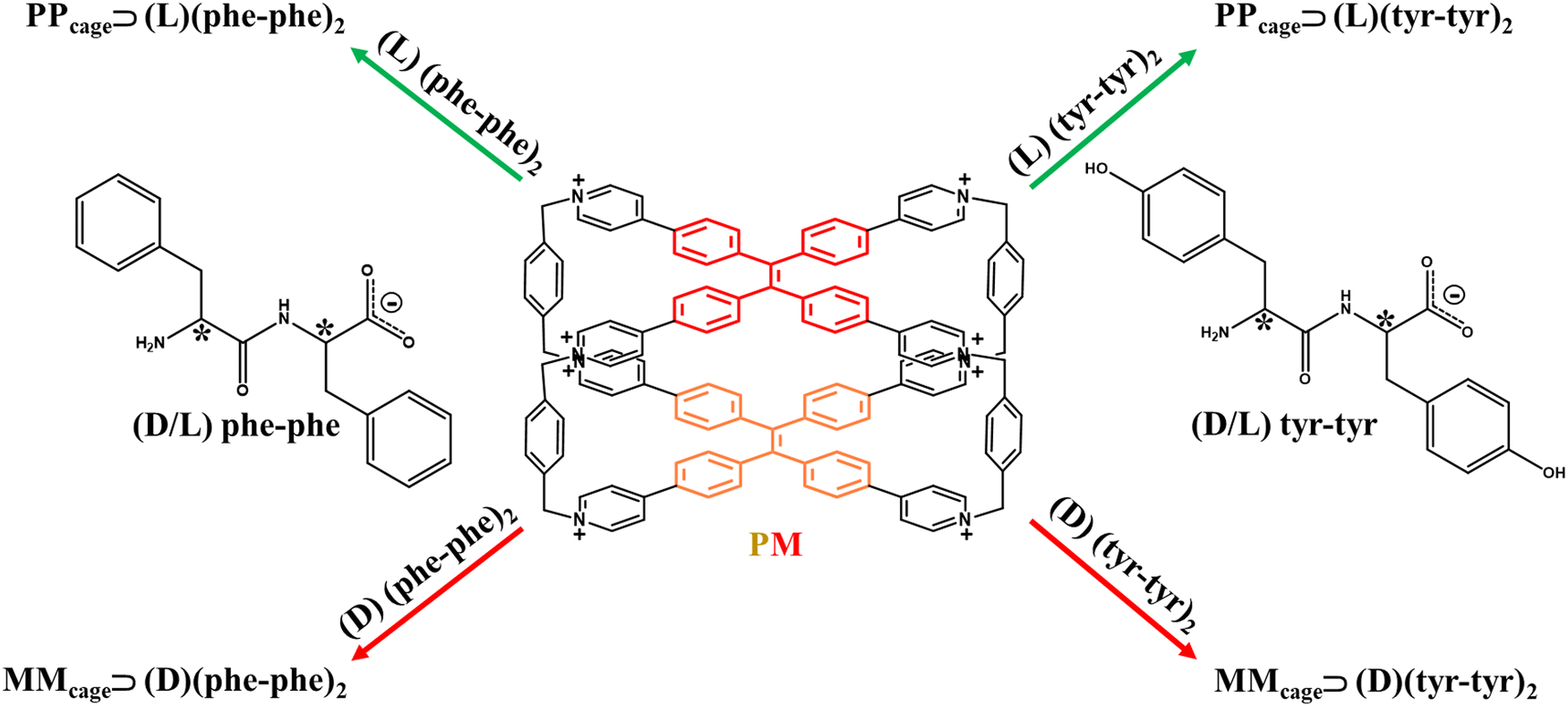
The efficiency of TPE-based octacationic cages in biocompatible media was also proficiently exploited for the sequence-specific recognition of peptides with chiroptical responses in an aqueous solution. Newly, Cheng et al. published an unprecedented case of chiral adaptive recognition with sequence specificity of aromatic dipeptides reporting mirror-symmetrical CPL spectra.64 In such an example, the two direction-selective rotational TPE entities were utilized to accomplish chiral discrimination for supramolecular binding and dimerization of aromatic dipeptides with a 1:2 stoichiometry in the TPE-based octacationic cage, hence the enantiopurity of the L/D-dipeptide mixture can be determined. Precisely, dipeptides pair with a specific sequence of amino acids and transmit their chiral information to the cage resulting in a PP conformation for L-dipeptides and an MM one for D-dipeptides. Despite the negligible CPL signal of most biomolecules such as peptides, phenylalanine–phenylalanine (phe–phe) and tyrosine–tyrosine (tyr–tyr) dipeptides homo-pairs were ingeniously encapsulated in the hydrophobic cavity of the cage, combining their stereogenic chirality (L and D) with the cage luminescent activity to attain a chiral emission (Fig. 8).
 |
| | Fig. 8 Possible combinations of (D/L) phe–phe and (D/L) tyr–tyr homo-pairs with the TPE-based octacationic cage in 1:2 host–guest systems. Asterisks highlight stereogenic carbons. Anionic forms for the guests were chosen coherently with the basic conditions (sodium phosphate, pH = 7.4) in the experimental work.64 | |
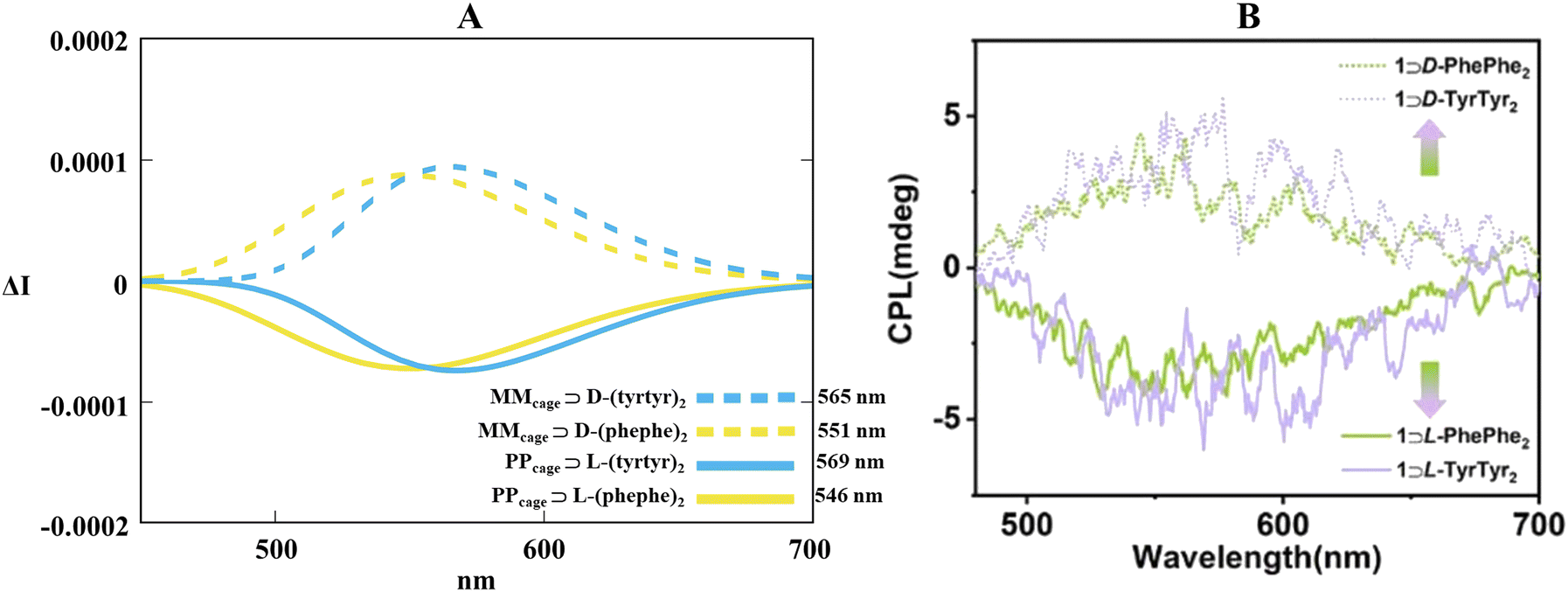
Geometrical optimizations of PPcage⊃(L)(phe–phe)2, PPcage⊃(L)(tyr–tyr)2, MMcage⊃(D)(phe–phe)2 and MMcage⊃(D)(tyr–tyr)2 (ωB97XD/6-311G(d,p) in the gas phase) delineated a diagonal arrangement of the guests along the x-shaped molecular skeleton of the cage (Fig. S14, ESI†). This kind of spatial disposition may be promoted by two key factors: (1) a proper relaxation of the ternary steric hindrance between the dipeptides and the cage. (2) A sum of supramolecular cooperations (electrostatic interactions, hydrogen bonds and ion–dipole interactions) between the carboxyl and amido functions with pyridinium rings (a similarity already perceived in Section 2.1 for the monophosphate action). Curiously, an energetic analysis turned up that PPcage⊃(L)(dipeptides)2 are more stable than their relative MMcage⊃(D)(dipeptides)2 counterparts (about 7.8 kcal for phe–phe and 3.4 kcal mol−1 for tyr–tyr, Fig. S15, ESI†). Confident in our theoretical model we executed an excited state molecular dynamic (ωB97XD/6-311G(d,p)) and subsequently a TD-DFT treatment (B3LYP/6-311G(d,p)) for the four supramolecular species. Notwithstanding the non-perfectly symmetrical CPL bands, our computational guide located the theoretical peaks very close to the experimental ones (560 nm) in terms of spectral position (Fig. 9). Fig. 9A displayed how the simulated right-handed emission (produced by the PP conformation) and the left-handed emission (produced by the MM conformation) are coherent with the experimental part in a matter of sign (negative for PP, positive for MM). Centering the attention, it is also possible to observe that calculated spectra of host–guest systems with phe–phe dipeptides homo-pairs are subjected to a blue-shift (546 nm and 551 nm for PPcage⊃(L)(phe–phe)2 and MMcage⊃(D)(phe–phe)2, respectively) while the tyr–tyr ones are characterized by a red-shift (569 nm and 565 nm for PPcage⊃(L)(tyr–tyr)2 and MMcage⊃(D)(tyr–tyr)2, respectively). This type of chiroptical behavior may be due to not-exactly specular packages of (phe–phe)2 and (tyr–tyr)2 inside the cage.
 |
| | Fig. 9 (A) Collection of TD-DFT calculated CPL spectra at B3LYP/6-311G(d,p) level along the MD trajectories for PPcage⊃(L)(phe–phe)2, PPcage⊃(L)(tyr–tyr)2, MMcage⊃(D)(phe–phe)2 and MMcage⊃(D)(tyr–tyr)2 in H2O. (B) Experimental CPL spectra of PPcage⊃(L)(phe–phe)2, PPcage⊃(L)(tyr–tyr)2, MMcage⊃(D)(phe–phe)2 and MMcage⊃(D)(tyr–tyr)2 in water. Adapted with permission from ref. 64 © 2023 Royal Society of Chemistry. | |
2.3. Case 3: TPEplatform⊃organic acids
The last case we present intends to be a validation test of the transferability of our theoretical protocol when TPE groups are not contained in the previously examined octacationic cages (see Sections 2.1 and 2.2). Recently, Zheng et al. divulged a very interesting paper in which TPE units were segregated in a triangular macrocycle bearing three crown ethers to accommodate external guests.81 Practically, three TPE groups were interconnected through three ethynylene bridges forming a tri-modular platform which thanks to the torsion between the phenyl rings and double bond plane proclaimed the typical propeller-like structure. In solution, the rapid interconversion between the right-(P) and left-handed (M) helical fashion mediated by a dynamic equilibrium does not trigger a preponderant single-handed helical conformation. At the same time, in solid, the restricted internal rotation (RIR effect) increased the fluorescence and univocally defined a crystal racemic mixture (MMP/PPM). The interference of the trimeric platform with chiral guests (S/R-mandelic acid and L/D camphorsulfonic acid), through hydrogen bonds with the polyethylenglycol rings, activated prevailing single-handed propeller-like conformations (PPP/MMM, Fig. 10), which became CPL sources in the solid state. It must be noted that it is not unmistakably possible to assign the chiroptical event to PPP/MMM conformations only, since, also, an excess of single helical configuration in the presence of TPE units with opposite propeller-like helical conformations can emit chiral luminescence82 (MMP/PPM in this specific case). Considering the TPE hetero-directional rotation (MMP/PPM) a case limit and aware of the criticalities in separating the multiple contributions, we decided to focus exclusively on the TPE homo-directional rotation (PPP/MMM).
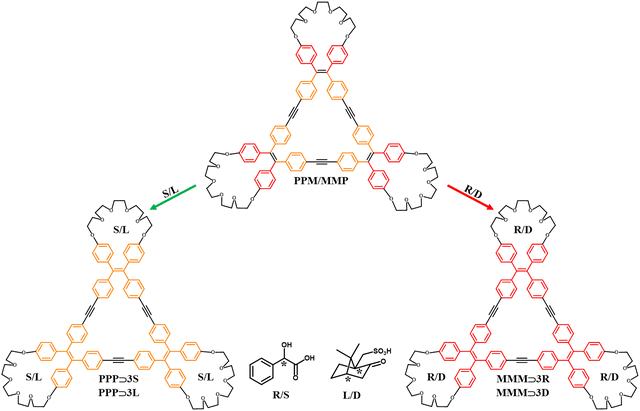 |
| | Fig. 10 Schematic routes to obtain chiral luminophores starting from the racemic crystal. | |
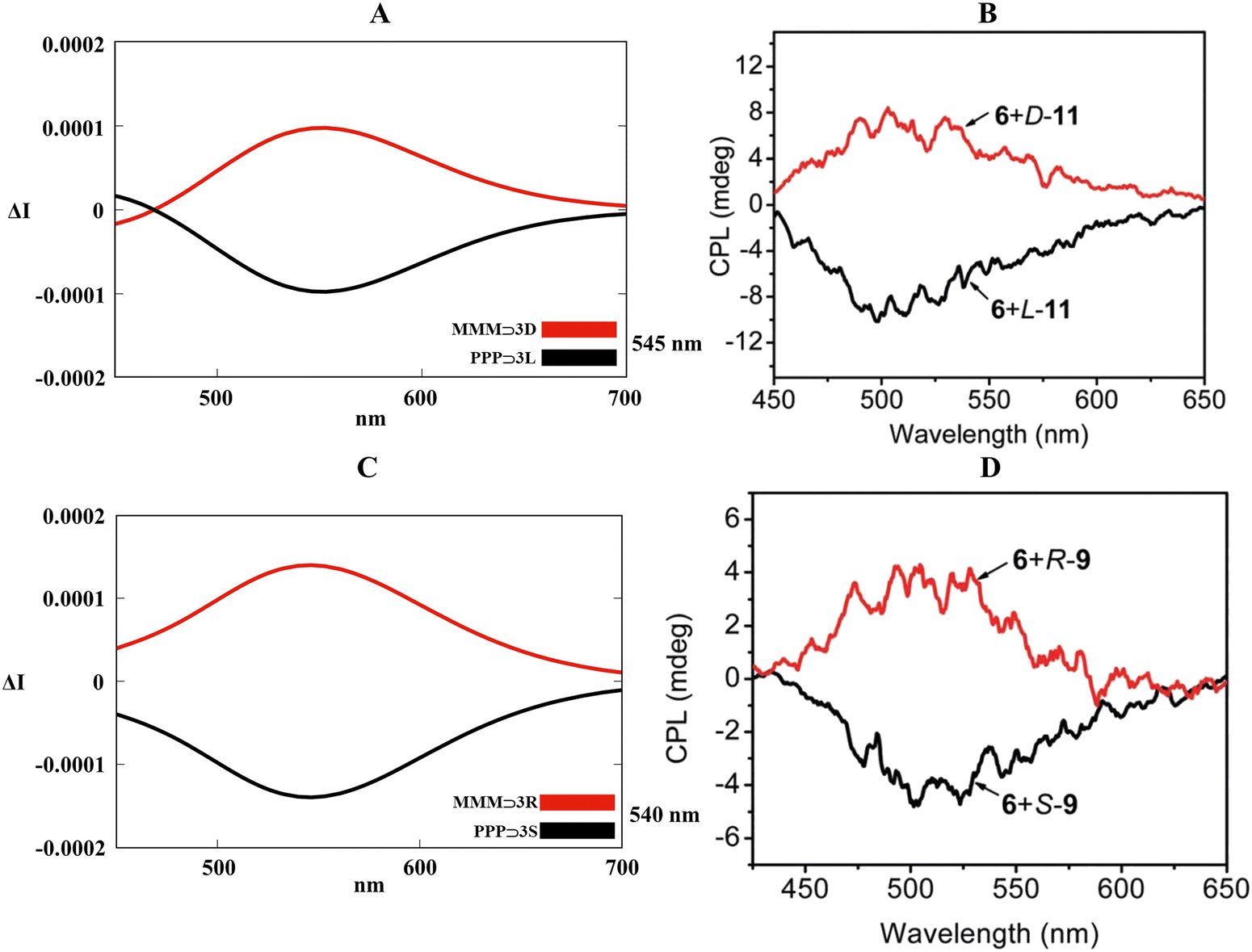
Optimization in the gas phase for PPP⊃3S, PPP⊃3L, MMM⊃3R and MMM⊃3D (B3LYP/6-311G(d,p)) confirmed the embedding of the enantiopure guests inside the hexaethylene glycol tethers in which carboxyl and sulfonic fragments maximized the supramolecular interactions with the crown ethers (Fig. S16 and S17, ESI†). Although not as precise as the previous cases (see Sections 2.1 and 2.2), excited molecular dynamic (ωB97XD/6-311G(d,p) in the gas phase) and the relative TD-DFT methodology (B3LYP/6-311G(d,p) in the gas phase) brought to satisfactory CPL spectra for the four host–guest systems. Fig. 11A and C described an acceptable red shift for the simulated CPL spectral lines compared to the experimental maxima (520 nm, Fig. 11B and D), symmetrical mirroring CPL bands were affected by errors of 25 nm and 20 nm for PPP⊃3L/MMM⊃3D and PPP⊃3S/MMM⊃3R, respectively. As also announced for the previous cases (Sections 2.1 and 2.2), our theoretical model competently recognized that the predominance of P conformations triggered the right-handed light emission (negative sign in the spectra), while M conformations activated the left-handed light emission (positive sign in the spectra).
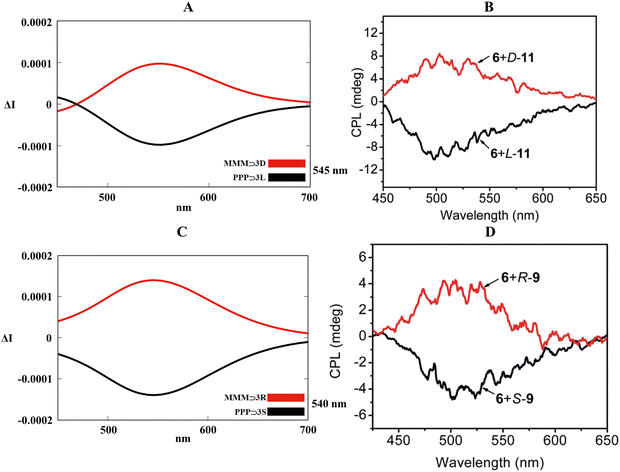 |
| | Fig. 11 (A) and (C) Collection of TD-DFT calculated CPL spectra at the B3LYP/6-311G(d,p) level along the MD trajectory for PPP⊃3L, PPP⊃3S, MMM⊃3D and MMM⊃3R in the gas phase. (B) and (D) Experimental CPL spectra of PPP/PPM⊃3L, PPP/PPM⊃3S, MMM/MMP⊃3D and MMM/MMP⊃3R in the solid phase. Adapted with permission from ref. 81 © 2018 Royal Society of Chemistry. | |
3. Computational details
The X-ray crystallographic structure of the TPE-based cage (CCDC: 2100784†) was energy-optimized in the gas phase employing the following DFT functionals benchmark: APFD, B3LYP, B3PW91, B97D3, BLYP, BP86, CAM-B3LYP, HCTH, HSEH1PBE, LC-M06L, LC-ωPBE, M06L, M062X, MN15L, N12, PBEPBE, PBEhPW91, SOGGA11, TPSSTPSS and ωB97XD combined with 6-311G(d,p) basis set, using an ultrafine grid and a tight criterion for energy and geometry optimization convergence. As a direct consequence of the previous DFT benchmark, the minima of all the presented systems in the manuscript were calculated at the ωB97XD/6-311G(d,p) level in the gas phase. For PPcage⊃TT2, PPcage⊃AA2, PPcage⊃CC1, PPcage⊃(L)(phe–phe)2, PPcage⊃(L)(tyr–tyr)2, MMcage⊃(D)(phe–phe)2, MMcage⊃(D)(tyr–tyr)2, PPP⊃3L, PPP⊃3S, MMM⊃3D and MMM⊃3R, Born–Oppenheimer molecular dynamic (BOMD) runs were performed at 298 K in the first excited electronic state (S1) at the ωB97XD/6-311G(d,p) level in the solvent phase (water, Sections 2.1 and 2.2) or gas phase (Section 2.3) with a total simulation time of 1 ps and a step size of 2 fs. The solvation box was defined by using the integral equation formalism for the polarizable continuum model (IEF-PCM), the default parameters of Gaussian16 were used for the construction of the cavity, built as the envelope of interlocked spheres centered on each atom of the solute (water: ε = 78.3553). Preliminarily, for PPcage⊃TT2, 25 equi-temporally interspersed snapshots (every 40 fs) were extracted from the BOMD and TD-DFT subjected to recover rotatory strengths, excitation energies and oscillator strengths at M06L, MN12L, B3LYP, CAM-B3LYP, LC-ωhPBE, mPW3PBE, ωB97XD, BMK, SOGGA11X and HSEH1PBE/6-311G(d,p) levels considering the first excited state (solvent phase: water, Sections 2.1, 2.2 and the gas phase for Section 2.3). Subsequently, the previous TD-DFT benchmark was also extended for the basis sets, hence 6-311G(d,p), LanLDZ, Def2SVP, JorgeDZP and SapporoDZP were coupled with the leader functional B3LYP. As an effect of the two precedent TD-DFT benchmarks, the B3LYP/6-311G(d,p) level was applied for all the CPL-active molecules. The CPL intensities ΔI were computed as follow:83| |  | (1) |
where ħ represents the reduced Planck's constant, c is the speed of light, ρ(Eemi) is the Gaussian band shape centered in the Eemi energy and R0m are the rotational strengths associated with the transition 0 ← 1 (expressed as R length). Eqn (1) is expressed in cgs units and the band shape was assumed as Gaussian with a bandwidth of 850 cm−1. All the computational jobs were carried out with Gaussian16 package.84 Every time energy comparisons between supramolecular architectures were evaluated, the relative computational jobs were corrected with the counterpoise correction to limit the basis set superposition error (BSSE). Cavity volumes (Vocc) were calculated by means of two-probes mode with a small probe radius of 1.2 Å, a large probe radius of 3.0 Å, and a grid resolution of 0.1 Å, implemented in the Molovol code.85 NCI surfaces were generated by the Multiwfn code86 with a high-quality grid for electron density and visualized using VMD.87
4. Conclusions
In the current work, we introduced a computational guide to study the mechanism of adaptive chirality in supramolecular systems through circularly polarized luminescence. The novel emerging TPE-based cages and platforms encouraged us to investigate theoretically the connection between stereogenic chiral-transmitter guests and the luminescent propeller-like (P/M) tetraphenylethene faces. Mindful of this, three different cases were argued: (1) TPE cage with deoxyribonucleotides. (2) TPE cage with dipeptides pairs. (3) TPE trimeric macrocycle with chiral organic acids. An opening DFT benchmark set on the TPE-based octacationic cage helped us to individuate the correct exchange–correlation functional to outline accurately the conformational preferences of TPE moieties when confined in complex chemical architectures. ωB97XD, in the role of best DFT-performer, was therefore applied for the Born–Oppenheimer molecular dynamics in the first excited state to collect information about spatial interactions between host and guests when chirality induction takes place. The abundance of collected supramolecular features and a decent experimental CPL spectral line persuaded us to use PPcage⊃deoxyribonucleotides monophosphate (specifically PPcage⊃TT2) as a computational probe for a TD-DFT evaluation of exchange–correlation functionals and basis sets in the CPL spectral simulation. B3LYP (mPW3PBE is also an excellent alternative) joint to 6-311G(d,p) proved to be a very precise and transferable method for the chiral emission prediction of all the PPcage⊃deoxyribonucleotides monophosphate complexes. The predictive efficacy of our customized level of theory was then challenged with the TPEcage⊃dipeptides homo-pairs and TPEplatform⊃acids. This theoretical survey certified how our computational treatment was able to authenticate the coexistence of the P/M preponderant conformations with the right/left-handed luminescence (respectively) through the exact computation of the CPL spectral sign (negative for P dominance, positive for M one). In each case, the CPL peak and band shape were deftly emulated with very small deviations compared to the experimental values. Although debates about the chirality transfer at the supramolecular level are continuous, our guide constitutes the first attempt to access computationally the chirality adaptive mechanism through the circularly polarized luminescence of TPE-engineered molecules.
Data availability
Crystallographic data for the TPE-based cage can be found in the Cambridge Structure Database (CCDC: 2100784†). Experimental CPL spectra were adapted with permission from (1) https://doi.org/10.31635/ccschem.020.202000509 (Fig. 6a in the main article and Fig. S21b in the ESI†), (2) https://doi.org/10.1039/D2SC05854E (Fig. 3j in the main article) and (3) https://doi.org/10.1039/C7TC05759H (Fig. 5 in the main article). The datasets supporting this article and the relative code/software are detailed in the “Computational details” section and in the ESI.†
Author contributions
Giovanni Bella: conceptualization, methodology, software, validation, investigation, project administration, writing – original draft. Giuseppe Bruno: resources, supervision, writing – review and editing. Antonio Santoro: supervision, writing – review and editing visualization, resources, data curation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the CINECA award under the ISCRA initiative, for the availability of high-performance computing resources and support.
Notes and references
- J. Bailey, A. Chrysostomou, J. H. Hough, T. M. Gledhill, A. McCall, S. Clark, F. Ménard and M. Tamura, Science, 1998, 281, 672–674 CrossRef CAS.
- M. Ciriani, R. Oliveira and C. A. M. Afonso, Green Chem., 2022, 24, 4328–4362 RSC.
- Y. Teng, C. Gu, Z. Chen, H. Jiang, Y. Xiong, D. Liu and D. Xiao, Chirality, 2022, 34, 1094–1119 CrossRef CAS PubMed.
- T. J. Ward and K. D. Ward, Anal. Chem., 2012, 84, 626–635 CrossRef CAS PubMed.
- Y. Jeong, H. W. Kim, J. Ku and J. Seo, Sci. Rep., 2020, 10, 16166 CrossRef CAS PubMed.
- K. Das, H. Balaram and K. Sanyal, ACS Omega, 2024, 9, 5084–5099 CrossRef CAS PubMed.
- C. Lee, J. M. Weber, L. E. Rodriguez, R. Y. Sheppard, L. M. Barge, E. L. Berger and A. S. Burton, Symmetry, 2022, 14 Search PubMed.
- J. A. Cowan and R. J. Furnstahl, ACS Earth Space Chem., 2022, 6, 2575–2581 CrossRef CAS.
- S. F. Ozturk and D. D. Sasselov, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2204765119 CrossRef CAS PubMed.
- F. Devínsky, Symmetry, 2021, 13(12), 2277 CrossRef.
- V. Valković and J. Obhođaš, Astron. J., 2022, 163, 270 CrossRef.
- T. Taniguchi, T. Hongen and K. Monde, Polym. J., 2016, 48, 925–931 CrossRef CAS.
- J. R. Enders and J. A. McLean, Chirality, 2009, 21, E253–E264 CrossRef CAS PubMed.
- L. Guo, Y. Guo, R. Wang, J. Feng, N. Shao, X. Zhou and Y. Zhou, Molecules, 2023, 28 Search PubMed.
- N. H. Cho, A. Guerrero-Martínez, J. Ma, S. Bals, N. A. Kotov, L. M. Liz-Marzán and K. T. Nam, Nat. Rev. Bioeng., 2023, 1, 88–106 CrossRef.
- Y. Wen, M.-Q. He, Y.-L. Yu and J.-H. Wang, Adv. Colloid Interface Sci., 2021, 289, 102376 CrossRef CAS PubMed.
- M. Sun, X. Wang, X. Guo, L. Xu, H. Kuang and C. Xu, Chem. Sci., 2022, 13, 3069–3081 RSC.
- M. Deng, J. Yu and D. G. Blackmond, Nature, 2024, 626, 1019–1024 CrossRef CAS PubMed.
- D. Herschlag and M. M. Pinney, Biochemistry, 2018, 57, 3338–3352 CrossRef CAS PubMed.
- S. C. C. van der Lubbe and C. Fonseca Guerra, Chem. – Asian J., 2019, 14, 2760–2769 CrossRef CAS PubMed.
- G. Bella, A. Santoro, F. Nicolò, G. Bruno and M. Cordaro, ChemPhysChem, 2021, 22, 593–605 CrossRef CAS PubMed.
- A. Giannetto, F. Nastasi, F. Puntoriero, G. Bella, S. Campagna and S. Lanza, Dalton Trans., 2021, 50, 1422–1433 RSC.
- R. Sylvain, L. Vendier, C. Bijani, A. Santoro, F. Puntoriero, S. Campagna, P. Sutra and A. Igau, New J. Chem., 2013, 37, 3543–3548 RSC.
- K. H. Sippel and F. A. Quiocho, Protein Sci., 2015, 24, 1040–1046 CrossRef CAS PubMed.
- R. Thakuria, N. K. Nath and B. K. Saha, Cryst. Growth Des., 2019, 19, 523–528 CrossRef CAS.
- S. Tsuzuki and A. Fujii, Phys. Chem. Chem. Phys., 2008, 10, 2584–2594 RSC.
- H.-X. Zhou and X. Pang, Chem. Rev., 2018, 118, 1691–1741 CrossRef CAS PubMed.
- H. Yang, W.-L. Xu, X.-Y. Zeng, J. Chen, L. Yu and L. Zhou, Org. Lett., 2021, 23, 9315–9320 CrossRef CAS PubMed.
- A. Shchyrba, M.-T. Nguyen, C. Wäckerlin, S. Martens, S. Nowakowska, T. Ivas, J. Roose, T. Nijs, S. Boz, M. Schär, M. Stöhr, C. A. Pignedoli, C. Thilgen, F. Diederich, D. Passerone and T. A. Jung, J. Am. Chem. Soc., 2013, 135, 15270–15273 CrossRef CAS PubMed.
- J. Guo, Y. Duan, Y. Jia, Z. Zhao, X. Gao, P. Liu, F. Li, H. Chen, Y. Ye, Y. Liu, M. Zhao, Z. Tang and Y. Liu, Nat. Commun., 2024, 15, 139 CrossRef CAS.
- S. Sevim, A. Sorrenti, J. P. Vale, Z. El-Hachemi, S. Pané, A. D. Flouris, T. S. Mayor and J. Puigmartí-Luis, Nat. Commun., 2022, 13, 1766 CrossRef CAS PubMed.
- G. Yang and Y. Xu, J. Chem. Phys., 2009, 130, 084105 CrossRef PubMed.
- S. Pandey, S. Mandal, M. B. Danielsen, A. Brown, C. Hu, N. J. Christensen, A. V. Kulakova, S. Song, T. Brown, K. J. Jensen, J. Wengel, C. Lou and H. Mao, Nat. Commun., 2022, 13, 76 CrossRef CAS PubMed.
- T. J. Mooibroek, J. M. Casas-Solvas, R. L. Harniman, C. M. Renney, T. S. Carter, M. P. Crump and A. P. Davis, Nat. Chem., 2016, 8, 69–74 CrossRef CAS PubMed.
- L.-X. Wang, J.-T. Zhang, X. Sun, D.-W. Yang and Y.-L. Tang, Dyes Pigm., 2021, 185, 108882 CrossRef CAS.
- P. Peluso and B. Chankvetadze, Chem. Rev., 2022, 122, 13235–13400 CrossRef CAS PubMed.
- X.-F. Wu, Q.-M. Ge, N. Jiang, W.-F. Zhao, M. Liu, H. Cong and J.-L. Zhao, Chemosensors, 2023, 11, 269 CrossRef CAS.
- C. Chappaz-Gillot, G. Canard, F. Andreoli, N. Vanthuyne, M. Giorgi, J.-V. Naubron, V. Monnier, R. Rosas, C. Roussel and T. S. Balaban, Eur. J. Org. Chem., 2012, 6526–6536 CrossRef.
- Y. Zhang, C. Yu, L. Han, M. Liu, Y. Guo and Z. Zhang, J. Phys. Chem. Lett., 2022, 13, 3523–3528 CrossRef CAS PubMed.
- L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef CAS PubMed.
- T. Hirao, S. Kishino and T. Haino, Chem. Commun., 2023, 59, 2421–2424 RSC.
- G. Magna, M. Šakarašvili, M. Stefanelli, G. Giancane, S. Bettini, L. Valli, L. Ustrnul, V. Borovkov, R. Aav, D. Monti, C. Di Natale and R. Paolesse, ACS Appl. Mater. Interfaces, 2023, 15, 30674–30683 CrossRef CAS PubMed.
- J. Dong, Y. Liu and Y. Cui, Acc. Chem. Res., 2021, 54, 194–206 CrossRef CAS PubMed.
- G. Liu, M. G. Humphrey, C. Zhang and Y. Zhao, Chem. Soc. Rev., 2023, 52, 4443–4487 RSC.
- H. Nian, L. Cheng, L. Wang, H. Zhang, P. Wang, Y. Li and L. Cao, Angew. Chem., Int. Ed., 2021, 60, 15354–15358 CrossRef CAS PubMed.
- T. Li, X. Zhu, G. Ouyang and M. Liu, Mater. Chem. Front., 2023, 7, 3879–3903 RSC.
- K. Takaishi, C. Maeda and T. Ema, Chirality, 2023, 35, 92–103 CrossRef CAS PubMed.
- J. Guo, J. Hou, J. Hu, Y. Geng, M. Li, H. Wang, J. Wang and Q. Luo, Chem. Commun., 2023, 59, 9157–9166 RSC.
- G. Hancu, L. A. Papp, G. Tóth and H. Kelemen, Molecules, 2021, 26, 2261 CrossRef CAS PubMed.
- Y. Wang, J. Gong, X. Wang, W.-J. Li, X.-Q. Wang, X. He, W. Wang and H.-B. Yang, Angew. Chem., Int. Ed., 2022, 61, e202210542 CrossRef CAS PubMed.
- G. Bella, M. Milone, G. Bruno and A. Santoro, J. Mater. Chem. C, 2024, 12, 3005–3012 RSC.
- J. Puigcerver, M. Marin-Luna, J. Iglesias-Sigüenza, M. Alajarin, A. Martinez-Cuezva and J. Berna, J. Am. Chem. Soc., 2024, 146, 2882–2887 CrossRef CAS PubMed.
- E. M. G. Jamieson, F. Modicom and S. M. Goldup, Chem. Soc. Rev., 2018, 47, 5266–5311 RSC.
- Q.-Y. Feng, Y.-Q. Mao, M.-X. Wang and S. Tong, Org. Lett., 2022, 24, 7107–7112 CrossRef CAS PubMed.
- E. Bang, J.-W. Jung, W. Lee, D. W. Lee and W. Lee, J. Chem. Soc., Perkin Trans. 2, 2001, 1685–1692, 10.1039/B102026I.
- Y. Li, Q. Li, X. Miao, C. Qin, D. Chu and L. Cao, Angew. Chem., Int. Ed., 2021, 60, 6744–6751 CrossRef CAS PubMed.
- M. Qayyum, T. Bushra, Z. A. Khan, H. Gul, S. Majeed, C. Yu, U. Farooq, A. J. Shaikh and S. A. Shahzad, ACS Omega, 2021, 6, 25447–25460 CrossRef CAS PubMed.
- N. B. Shustova, T.-C. Ong, A. F. Cozzolino, V. K. Michaelis, R. G. Griffin and M. Dincă, J. Am. Chem. Soc., 2012, 134, 15061–15070 CrossRef CAS PubMed.
- H. Duan, Y. Li, Q. Li, P. Wang, X. Liu, L. Cheng, Y. Yu and L. Cao, Angew. Chem., Int. Ed., 2020, 59, 10101–10110 CrossRef CAS PubMed.
- Y. Duan, J. Wang, L. Cheng, H. Duan, P. Tian, Y. Zhang and L. Cao, Org. Biomol. Chem., 2022, 20, 3998–4005 RSC.
- H. Duan, F. Cao, M. Zhang, M. Gao and L. Cao, Chin. Chem. Lett., 2022, 33, 2459–2463 CrossRef CAS.
- H. Duan, T. Yang, Q. Li, F. Cao, P. Wang and L. Cao, Chin. Chem. Lett., 2024, 35, 108878 CrossRef CAS.
- Y. Dong, L. Cheng, Y. Duan, H. Xu, R. Dong, B. Guo and L. Cao, Synlett, 2023, 109–112 Search PubMed.
- L. Cheng, P. Tian, H. Duan, Q. Li, X. Song, A. Li and L. Cao, Chem. Sci., 2023, 14, 833–842 RSC.
- Q. Yang, M. Fusè and J. Bloino, Front. Chem., 2020, 8, 801 CrossRef CAS PubMed.
- N. Lin, F. Santoro, X. Zhao, A. Rizzo and V. Barone, J. Phys. Chem. A, 2008, 112, 12401–12411 CrossRef CAS PubMed.
- Q. Xu, L. Chi, Y. Liu, Q. Li, S. Chen and M. Wang, J. Lumin., 2021, 239, 118374 CrossRef CAS.
- G. Bella, G. Bruno and A. Santoro, J. Mol. Liq., 2023, 391, 123268 CrossRef CAS.
- G. Bella, G. Bruno and A. Santoro, FlatChem, 2023, 40, 100509 CrossRef CAS.
- É. Brémond, M. Savarese, N. Q. Su, Á. J. Pérez-Jiménez, X. Xu, J. C. Sancho-García and C. Adamo, J. Chem. Theory Comput., 2016, 12, 459–465 CrossRef PubMed.
- G. Bella, M. Milone, G. Bruno and A. Santoro, Phys. Chem. Chem. Phys., 2022, 24, 26642–26658 RSC.
- M. Roger, Y. Bretonnière, Y. Trolez, A. Vacher, I. Arbouch, J. Cornil, G. Félix, J. De Winter, S. Richeter, S. Clément and P. Gerbier, Int. J. Mol. Sci., 2023, 24, 8715 CrossRef CAS PubMed.
- P. Bhandari, S. Ahmed, R. Saha and P. S. Mukherjee, Chem. – Eur. J., 2024, 30, e202303101 CrossRef CAS PubMed.
- M. Bursch, J.-M. Mewes, A. Hansen and S. Grimme, Angew. Chem., Int. Ed., 2022, 61, e202205735 CrossRef CAS PubMed.
- N. Mardirossian and M. Head-Gordon, Mol. Phys., 2017, 115, 2315–2372 CrossRef CAS.
- G. Bella and A. Rotondo, Chem. Phys. Lipids, 2020, 232, 104973 CrossRef CAS PubMed.
- C. Liang, J. Yang, G. Luo and Y. Luo, Comput. Theor. Chem., 2020, 1187, 112942 CrossRef CAS.
- B. C. Ferrari and C. J. Bennett, J. Phys. Conf. Ser., 2019, 1290, 012013 CrossRef CAS.
- G. Bella, A. Santoro, M. Cordaro, F. Nicolò and G. Bruno, Chin. J. Chem., 2020, 38, 163–168 CrossRef CAS.
- L. Cheng, K. Liu, Y. Duan, H. Duan, Y. Li, M. Gao and L. Cao, CCS Chem., 2020, 3, 2749–2763 CrossRef.
- W.-G. Qiao, J.-B. Xiong, Y.-X. Yuan, H.-C. Zhang, D. Yang, M. Liu and Y.-S. Zheng, J. Mater. Chem. C, 2018, 6, 3427–3434 RSC.
- H. Qu, Y. Wang, Z. Li, X. Wang, H. Fang, Z. Tian and X. Cao, J. Am. Chem. Soc., 2017, 139, 18142–18145 CrossRef CAS PubMed.
- G. Longhi, E. Castiglioni, S. Abbate, F. Lebon and D. A. Lightner, Chirality, 2013, 25, 589–599 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J. B. Maglic and R. Lavendomme, J. Appl. Cryst., 2022, 55, 1033–1044 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Giuseppe
Bruno
and
Antonio
Santoro
*,
Giuseppe
Bruno
and
Antonio
Santoro