
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Matrix-dependent high-contrast photochromism in Eu-doped M3MgSi2O8 (M = Ca, Sr, Ba)†
Guna
Krieke
 *ab,
Andris
Antuzevics
ab,
Aleksandr
Kalinko
b,
Alexei
Kuzmin
b,
Tomas
Murauskas
a,
Aivaras
Kareiva
a and
Aleksej
Zarkov
a
*ab,
Andris
Antuzevics
ab,
Aleksandr
Kalinko
b,
Alexei
Kuzmin
b,
Tomas
Murauskas
a,
Aivaras
Kareiva
a and
Aleksej
Zarkov
a
aInstitute of Chemistry, Vilnius University, Naugarduko 24, Vilnius LT-03225, Lithuania
bInstitute of Solid State Physics, University of Latvia, 8 Kengaraga str., LV-1063, Riga, Latvia. E-mail: guna.krieke@cfi.lu.lv; Tel: +371 28260803
First published on 18th September 2024
Abstract
The photochromic properties and charge transfer processes were studied in novel Eu-doped M3MgSi2O8 (M = Ca, Sr, Ba) compounds. These materials exhibit vivid color changes upon irradiation with UV light, resulting in orange, reddish-pink, and green colors. The introduction of europium ions enhances photochromic efficiency and shifts excitation peaks to lower energy ranges. Analysis of diffuse reflectance and electron paramagnetic resonance spectra reveals the formation of both paramagnetic and non-paramagnetic defects, with the dominant signals attributed to electron centers, likely F+-type centers. The Eu2+ → Eu3+ charge transfer during irradiation indicates that Eu2+ acts as a hole center. These findings contribute to a better understanding of the mechanisms underlying photochromism in these materials and highlight their potential for practical applications.
Introduction
Photochromism, or the photochromic effect, is a reversible color change in a material induced by exposure to electromagnetic radiation.1 It was first reported in mineral sodalite, from which some variants exhibited distinctive discoloration of bright pink color to green under sunlight.2 This phenomenon, also known as tenebrescence in minerals,3 was subsequently identified in organic compounds; Fritzsche documented this effect in an orange polycyclic hydrocarbon solution containing tetracene that discolored upon exposure to sunlight and could be restored by heating.4The study of photochromic materials has garnered significant attention due to their diverse applications, including sensors,5,6 displays,7,8 data storage and optical switches,9–11 photochromic lenses,12 smart windows,13,14 textiles,15,16 anti-counterfeiting,17,18 dosimetry,19,20 and others.
Inorganic oxide-based photochromic materials show excellent thermal and chemical stability and long cycling life.1 Examples of such materials include sodalites,21,22 barium silicates,23–25 alkaline earth stannates,26,27 niobates28,29 and transition metal oxides.30
Development of new and efficient photochromic materials requires a deep understanding of the mechanisms underlying photochromism, a task that is both crucial and challenging. In complex oxides, photochromism is typically attributed to charge transfer processes involving point defects such as oxygen1,31 or cation vacancies.26,32 Electrons and holes are trapped in point defects, which exhibit absorbance and can be optically or thermally liberated, resulting in a reversible color change. However, the precise mechanisms behind most inorganic photochromic materials remain elusive.1
The formation of point defects is highly dependent on synthesis conditions and trace impurities.33,34 Variations in synthesis conditions can significantly affect the efficiency of the photochromic effect by altering defect concentration.26 Changes in chemical composition can also shift the positions of absorbance bands, as demonstrated in sodalite.22 Despite all advances, the selection of inorganic materials with good color contrast is limited.1 Consequently, the search for new photochromic materials remains a significant area of research.
In this work, we demonstrate a matrix-dependent photochromism in the Eu2+ doped M3MgSi2O8 (M = Ca–Ba) system. Previous studies have reported a color change from white to orange during irradiation with X-rays or UV light in Ba3MgSi2O8 and (Ba2Ca)MgSi2O8 solid solutions.24,35 However, it was noted that this color change is irreversible.35 By selecting suitable dopant content and optimizing synthesis conditions, we have developed highly efficient photochromic materials, including two novel compounds, Ca3MgSi2O8 and Sr3MgSi2O8. These compounds exhibit reversible color changes and excellent color contrast. The aim of this study is to investigate photochromic properties and the role of europium ions in photochromism. By combining X-ray absorption, optical spectroscopy and electron paramagnetic resonance methods, the dominant defects and charge transfer processes involved were analyzed.
Materials and methods
Samples with the general composition of M3−xMgSi2O8:xEu2+ (x = 0–0.03) were prepared using high-temperature solid-state synthesis. High purity MgO (99.99%, Fisher Scientific), CaCO3 (99.999%, Merck), SrCO3 (99.997%, Fisher Scientific), BaCO3 (99.999%, Merck), SiO2 (99.999%, Fisher Scientific), Eu2O3 (99.999%, Fisher Scientific) and H3BO3 (ACS Merck) were used as precursors. 7.5 mol% H3BO3 was added as a flux by replacing the corresponding SiO2 content. The appropriate amounts of precursors were thoroughly mixed in an agate mortar and pressed in 13 mm pellets using uniaxial hydraulic press from Specacs. The pellets were transferred to graphite plates and placed in tube furnace HTRH 18/100/600 from Carbolite Gero. Ca3MgSi2O8 and Sr3MgSi2O8 samples were heat treated at 1350 °C for 10 h in an H2/Ar (5%/95%) atmosphere with a heating and cooling rate of 5 °C min−1. To prepare single-phase Ba3MgSi2O8, the crystal structure was stabilized by partially replacing Ba2+ with 5 mol% Ca2+. These samples were heat treated at 1250 °C to prevent thermal decomposition.The samples are abbreviated as CMS x%, SMS x%, and BMS x% for Ca3MgSi2O8, Sr3MgSi2O8, and Ba3MgSi2O8 doped with x mol% Eu, respectively.
The phase composition of the prepared samples was determined by X-ray diffraction (XRD) using Rigaku MiniFlex 600 powder diffractometer (Cu Kα radiation, operated at 40 KV and 15 mA) with Bragg–Brentano θ–2θ geometry.
The boron content in the samples was determined by X-ray photoelectron spectroscopy (XPS) using a Kratos Axis Supra spectrometer with monochromatic Al Kα (25 mA, 15 kV). The instrument was calibrated using metallic gold and copper. The measurements were carried out with a charge neutralization, while the energy scale was charge-corrected to the main line of carbon (C 1s) at 284.8 eV. Spectra were analyzed using CasaXPS software (version 2.3.23rev1.1R). Due to the overlapping of B 1s and Ba 4p1/2 peaks, BMS sample was excluded from the analysis.
X-ray absorption spectra of polycrystalline M3MgSi2O8:Eu (M = Ca, Sr, Ba) were recorded at the Eu L3-edge (6977 eV) at room temperature in fluorescence mode at the DESY PETRA-III P64 Advanced X-ray Absorption Spectroscopy beamline.36 The storage ring operated at E = 6 GeV and current I = 100 mA in top-up 40 bunch mode. A fixed-exit liquid-nitrogen-cooled double-crystal monochromator Si(111) was used to select the required X-ray energy from the undulator photon source. Harmonic rejection was achieved using two silicon mirrors. The X-ray intensity before the sample was monitored with an ionization chamber filled with nitrogen gas, whereas the X-ray fluorescence signal from the sample was measured using a passivated implanted planar silicon (PIPS) detector. The angle between the excitation radiation and the sample was 45 degrees, the same as the angle between the sample and the PIPS detector. The samples were prepared in the form of pellets.
The Eu L3-edge X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra were isolated using the XAESA code,37 following a conventional procedure.38 The qualitative interpretation of the experimental EXAFS spectra χ(k) (where k is the wavenumber of the photoelectron) was performed using theoretical spectra calculated within the multiple-scattering (MS) formalism, as implemented in the FEFF8.50L code.39,40 The MS calculations were performed for clusters with a size of 8 Å, centered at the absorbing europium atom and constructed using the ideal crystallographic structure of monoclinic CMS and SMS with a space group P21/a (14).35,41 No structure relaxation was performed, and the contribution of thermal disorder was neglected. Three structural models were considered, corresponding to europium atoms placed at three different non-equivalent crystallographic sites occupied by strontium (calcium) atoms.35,41 The inelastic losses of a photoelectron were accounted for using the complex exchange–correlation Hedin-Lundqvist potential.42
Diffuse reflectance spectra were measured using a double-beam spectrophotometer Specord 210. The samples were irradiated using a wavelength-tunable pulsed solid-state laser (Ekspla NT342/3UV). For photochromic effect excitation measurements, the laser pulse energy was set to 1.5 ± 0.5 mJ, and the samples were irradiated at a specific wavelength for 30 seconds. Subsequently, the samples were subjected to diffuse reflectance measurements. After each measurement, the samples were bleached by 500 nm excitation for 30 seconds.
Photoluminescence excitation spectra were measured using spectrometer FLS1000 from Edinburgh Instruments with a Xe lamp as an excitation source. The spectra measurements were corrected for the spectral sensitivity of the equipment.
Electron paramagnetic resonance (EPR) spectra were measured using the Bruker ELEXSYS-II E500 CW-EPR spectrometer operated at room temperature, 9.83 GHz microwave frequency, and 100 kHz magnetic field modulation frequency. Other spectra acquisition settings (microwave power, modulation amplitude, magnetic field range, number of points, etc.) were optimized for each sample separately. Before measurements, the samples were irradiated with 250 nm for 5 min.
Results and discussion
Phase composition and crystal structure
XRD analysis was used to investigate the phase composition of the samples. XRD patterns are shown in Fig. 1. All observed peaks matched well with the reference patterns of their respective phases in all samples, with no indications of other phases. | ||
| Fig. 1 XRD patterns of Ca3MgSi2O8 (CMS), Sr3MgSi2O8 (SMS), and Ba3MgSi2O8 (BMS) doped with 0.1% Eu. PDF – powder diffraction file. | ||
The crystal structure of each phase is shown in Fig. S1 (ESI†). The crystal structures of Ca3MgSi2O8 and Sr3MgSi2O8 belong to the monoclinic crystal system with a space group of P21/a, while Ba3MgSi2O8 crystallizes in the trigonal system with a space group of P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) .35,43 In the monoclinic Ca3MgSi2O8 and Sr3MgSi2O8, there are three distinct Ca and Sr positions, one Mg position, two Si positions, and eight O positions. The local symmetry of all positions is C1. In Ba3MgSi2O8 there are three distinct Ba positions with S6, C3 and C1 symmetry, two Mg positions with S6 and C3 symmetry, one Si position with C1 symmetry, and four O positions with C1 symmetry.43 The relatively low symmetry of monoclinic and trigonal phases combined with multiple distinct cationic and anionic positions is desirable for the formation of intrinsic defects, which are crucial for efficient photochromic properties.
.35,43 In the monoclinic Ca3MgSi2O8 and Sr3MgSi2O8, there are three distinct Ca and Sr positions, one Mg position, two Si positions, and eight O positions. The local symmetry of all positions is C1. In Ba3MgSi2O8 there are three distinct Ba positions with S6, C3 and C1 symmetry, two Mg positions with S6 and C3 symmetry, one Si position with C1 symmetry, and four O positions with C1 symmetry.43 The relatively low symmetry of monoclinic and trigonal phases combined with multiple distinct cationic and anionic positions is desirable for the formation of intrinsic defects, which are crucial for efficient photochromic properties.
Despite differences, all three compounds form wide regions of solid solutions.35 This fact was used in the present study to stabilize trigonal Ba3MgSi2O8. The phase stabilization was achieved by introducing Ca2+ into the structure, which prevented its thermal decomposition – a challenging issue in the high-temperature solid-state synthesis of Ba3MgSi2O8.43
The investigated samples contain boron, which was added as a flux. The evaporation of volatile boron-rich compounds could potentially alter the chemical and phase composition of the material. However, the absence of additional phases (Fig. 1) suggests that boron is most likely incorporated into the crystalline lattice. To verify this, boron content was estimated using XPS, as shown in Fig. S2 (ESI†). The average boron content was found to be 7.16 ± 2.72 mol% with respect to Si, indicating that boron losses during the synthesis are negligible. Additionally, when the synthesis was performed with the precursors ratio different from the stoichiometry of M3Mg(Si,B)2O8, some secondary phases were observed in the XRD patterns of the resulting products (not shown). It is important to note that the photochromic effect, discussed in detail later, is also observable in samples that do not contain boron, indicating that it is related to the processes in the M3MgSi2O8 (M = Ca–Ba) phases. A comparison of the diffuse reflectance spectra of Eu2+-doped CMS with and without B is shown in Fig. S3 (ESI†).
XANES and EXAFS analysis
The local environment of Eu in polycrystalline M3MgSi2O8 (M = Ca, Sr, Ba) was investigated using XANES and EXAFS. The Eu L3-edge XANES spectra were collected during the first 16 minutes after the beam shutter opening and are shown in Fig. 2. | ||
| Fig. 2 The Eu L3-edge XANES spectra of polycrystalline CMS, SMS and BMS doped with 0.1% Eu. Dashed lines – spectra measured immediately after opening the beam shutter, solid lines – spectra collected after 16 minutes. | ||
The XANES spectra are dominated by two well-separated peaks, the so-called “white lines”, located at about 6974 eV and 6982 eV. These peaks are attributed to 2p3/2 → 5d bound-state electronic transitions in Eu2+ and Eu3+ ions, respectively.44–46 The ratio of the intensities of these peaks can be used to estimate the Eu2+/Eu3+ ratio in the sample. Following the procedure described in ref. 47, the Eu2+/Eu3+ ratio was evaluated and found to be ≈1.1 in CMS, ≈1.6 in SMS, but changed from ≈1.2 to ≈0.9 after 16 minutes of exposure to X-rays in BMS. Thus, the initial fraction of Eu2+ ions is lower in CMS and BMS than in SMS. X-ray irradiation promotes the recharging of europium ions, resulting in a change in the Eu2+/Eu3+ ratio. A time-dependent variation in the peak ratio was observed for all three samples. However, this effect is insignificant in CMS and SMS, whereas the Eu2+/Eu3+ ratio changes significantly in BMS.
The results of the analysis of the Eu L3-edge EXAFS spectra χ(k)k2 of monoclinic SMS are shown in Fig. S4a and b (ESI†). The ionic radii of Eu2+ and Sr2+ are close,48 therefore the substitution can be easily achieved. Due to the quality of the experimental data, only qualitative comparison can be performed in a rather restricted energy range up to k = 5 Å−1. Nevertheless, the model, corresponding to europium atoms located at the Sr2 crystallographic site, with the eight nearest oxygen atoms and a distribution of Eu–O distances in the range of 2.28–2.89 Å, gives the best agreement with the experiment. The worst agreement is observed for europium atoms located at the Sr1 crystallographic site, with the nine nearest oxygen atoms and a distribution of Eu–O distances in the range of 2.42–2.95 Å: in this case, the frequency of the EXAFS spectrum is larger than in the experimental data and, as a result, the first main peak in the Fourier transform (FT) is shifted to longer distances compared to the experimental one. At the same time, the europium substitution of Sr3 atoms leads to an increase in the amplitude of the EXAFS spectrum due to the ten nearest oxygen atoms located at distances in the range of 2.38–2.89 Å.
The results of EXAFS simulations for monoclinic CMS are demonstrated in Fig. S4c and d (ESI†). Here, the observed agreement is worse than for SMS because the ionic radius of Eu2+ is larger than that of Ca2+.48 Therefore, structural relaxation around europium atoms upon substitution is expected. In the ideal structure, the europium atoms at the Ca1 and Ca3 sites are surrounded by eight oxygen atoms located at distances in the ranges of 2.31–2.84 Å and 2.25–2.78 Å, respectively. Meanwhile, there are nine oxygen atoms around the Ca2 site, located at distances in the range of 2.22–2.81 Å. Note that the distribution of oxygen atoms around the Ca1 site has a bi-modal shape, with the four nearest oxygen atoms located at distances in the range of 2.31–2.42 Å and the other four oxygen atoms located at distances in the range of 2.60–2.84 Å. Such a bi-modal distribution is responsible for the shape of the double first peak in the Fourier transform in Fig. S4d (ESI†). The calculated Eu L3-edge EXAFS spectra and their FTs for the europium atoms placed at the Ca2 and Ca3 sites are quite similar. Both models give close frequencies of the EXAFS spectra, reflecting the closeness of the average Eu–O interatomic distances, in qualitative agreement with the experiment. However, they overestimate the amplitude of the EXAFS spectrum, suggesting the presence of structural relaxation upon substitution. Due to the low signal-to-noise ratio of EXAFS spectra, Ba3MgSi2O8 was excluded from analysis, however, a similar substitution to CMS and SMS can be expected.
Photochromic properties
All investigated samples exhibited efficient photochromism in the visible spectral range (Fig. 3a). Before irradiation, Eu-doped samples displayed absorbance bands in the UV to 450 nm range related to 4f7 → 4f65d transitions of Eu2+ ions. During the UV irradiation, the samples underwent a significant color change, and several additional broad absorbance bands appeared. After the irradiation, each sample exhibited at least two distinct absorption bands. | ||
| Fig. 3 (a) Diffuse reflectance spectra of CMS, SMS, and BMS samples doped with 0.1% Eu before (dashed lines) and after (solid lines) excitation with 250 nm for 5 min and (b) photochromic effect excitation spectra of CMS, SMS and BMS samples doped with 0.1% Eu. Inset: Photograph of samples after irradiation of the right side. | ||
In the CMS samples, the dominant absorption bands were centered at 435 nm and 700 nm, and the high intensity of the 435 nm band resulted in a bright orange color. The SMS samples displayed partly overlapping bands with maxima at 530 nm and 770 nm, leading to a reddish-pink color. The BMS samples exhibited the widest absorption bands, with maxima at 400 nm and 720 nm, producing a green color. It should be noted, that the color of BMS differs from the one reported in the literature,24 which indicates the formation of different color centers. This could be due to the use of different fluxes, deviations in chemical composition, or the use of different temperatures for the heat treatment.
All samples showed an excellent color contrast which is the difference in diffuse reflectance intensity of initial and colored states: 61% for CMS, 66% for SMS, and 66% for BMS for the most intense absorption bands. The photochromic effect excitation spectra of the Eu-doped samples were comparable (Fig. 3b), suggesting similar excitation mechanisms for Eu-doped CMS, SMS and BMS. The photochromic effect could be excited over a broad UV range, with the highest intensity achieved with 230–250 nm excitation.
The role of Eu in the photochromic effect was analyzed in the SMS samples. Diffuse reflectance spectra of the samples doped with 0–1% Eu are shown in Fig. 4a. Before the irradiation, Eu-doped samples exhibited only absorption bands related to 4f7 → 4f65d transitions of Eu2+. After irradiation, additional absorption bands were detected in all samples, including undoped SMS, indicating that these absorption bands are associated with intrinsic defects rather than Eu2+ ions. However, the introduction of Eu improved absorption intensity, suggesting that Eu2+ participates in charge compensation processes, most likely acting as a hole trap. These results are in good agreement with XANES analysis which revealed a decrease of the Eu2+/Eu3+ ratio upon X-ray irradiation.
 | ||
| Fig. 4 (a) Diffuse reflectance spectra of SMS samples doped with 0–1% Eu before (dashed lines) and after excitation with 250 nm for 5 min and (b) photochromic effect (dashed lines) excitation spectra of SMS samples doped with 0% and 0.1% Eu and Eu2+ photoluminescence excitation spectrum (solid line) of SMS doped with 0.1% Eu. Inset: Photograph of samples after irradiation of right side. | ||
The highest intensity photochromic effect was observed in samples doped with 0.01% and 0.1% Eu (see Fig. 4a). In the sample doped with 1% Eu, a rapid decrease in intensity was detected, possibly due to changes in charge compensation processes.
The photochromic effect excitation spectra differ considerably between undoped and doped samples (Fig. 4b). In the undoped sample, the maximum excitation efficiency was achieved using 210 nm excitation. The introduction of Eu shifted the maximum to a lower wavelength and increased the overall intensity. Several possible reasons could explain this effect. It may be related to the formation of additional defects in the crystal lattice from the introduction of Eu, due to photoionization or other charge transfer processes of Eu2+ during excitation. If photoionization was the dominant process, the excitation spectra of the photochromic effect should resemble the photoluminescence excitation spectra of Eu2+. These spectra are compared in Fig. 4b. The excitation bands of Eu2+ are located in the 250–350 nm range. Although photoionization may contribute to the photochromic effect, especially in the 250–350 nm range, the formation of other defects likely contributes to the most intense excitation bands for photochromic excitation.
The photochromic effect in the investigated materials is reversible. The irradiation of all samples with green light resulted in bleaching of the absorbance bands. The diffuse reflectance spectra of SMS 0.1% sample before irradiation, after irradiation, and after optical bleaching are shown in Fig. S5 (ESI†) as a representative. Excellent reversibility of the photochromic effect was observed after multiple irradiation and bleaching cycles in all samples. The results indicate that these photochromic silicates possess both high efficiency and reversibility, meeting the requirements for practical applications.
EPR analysis
For a better understanding of the photochromic effect mechanism, EPR spectra were analyzed.EPR spectra (Fig. S6, ESI†) demonstrate the incorporation of Eu2+ ions in the investigated samples. The spectra exhibited composition-dependent powder patterns covering a broad field range, with signal intensities scaling proportionally to the europium content. Such signals are typical of Eu2+ ions in various crystalline hosts,49–51 whereas Eu3+ ions usually do not contribute to the EPR spectra. The 8S7/2 ground state of Eu2+ is characterized by an electronic spin of S = 7/2 and a g-factor value close to 2.0. The differences in the zero-field splitting (ZFS) of the ground state for the investigated compositions are exemplified by the variations in resonance positions of the spectra. A gradual increase of signal intensities with europium concentration was observed, which suggests that the same type of Eu2+ centers are created in the investigated concentration range.
EPR spectra were recorded after exposure of the samples to 250 nm laser radiation to elucidate the origin of the photochromic effect in M3MgSi2O8. After the irradiation, a decrease of Eu2+ signal intensity was observed in samples doped with 0.01% and 0.1% Eu, suggesting Eu2+ → Eu3+ conversion during sample excitation. The largest decrease was observed for samples doped with 0.01% Eu. The changes in the intensity before and after irradiation are shown in Fig. S7 (ESI†) and summarized in Table 1. These results indicate that changes in the Eu2+ valence state can be achieved not only by irradiation with X-rays as seen during XANES measurements but also upon irradiation with UV light. Thus, Eu2+ is one of the charge traps present in the investigated materials.
| Eu content, % | Eu2+ EPR signal intensity decrease, % | ||
|---|---|---|---|
| CMS | SMS | BMS | |
| 0.01 | 21 ± 5 | 18 ± 4 | 24 ± 6 |
| 0.10 | 11 ± 2 | 0 ± 1 | 4 ± 2 |
| 1.00 | 0 ± 1 | 0 ± 1 | 2 ± 2 |
In addition to Eu2+, several new signals were observed after sample irradiation; a comparison of the UV-induced EPR signals (the difference between the spectra before and after irradiation) of the investigated samples using identical spectra acquisition parameters is displayed in Fig. 5. Several points can be made from qualitative inspection of the data: (1) distinct paramagnetic centers are generated in each matrix; (2) in CMS, the intensity of UV-generated signals increases with Eu content, whereas for SMS and BMS compositions maximum intensity is observed for 0.01% Eu concentration (see Fig. 5); (3) broadening of EPR spectra at higher doping concentrations occurs, which suggests increased structural disorder induced by the dopant ions.52–54 The 0.01% Eu-doped samples, which showed optimal intensities and signal structure resolution for all compositions, were selected for EPR acquisition parameter optimization and additional measurements.
 | ||
| Fig. 5 EPR spectra of CMS, SMS and BMS samples with different concentrations of Eu2+ ions after irradiation with 250 nm; acquisition settings: 1 mW microwave power, 0.4 mT modulation amplitude. | ||
The time-dependent evolution of EPR spectra after UV irradiation using optimized acquisition parameters for each sample is shown in Fig. 6.
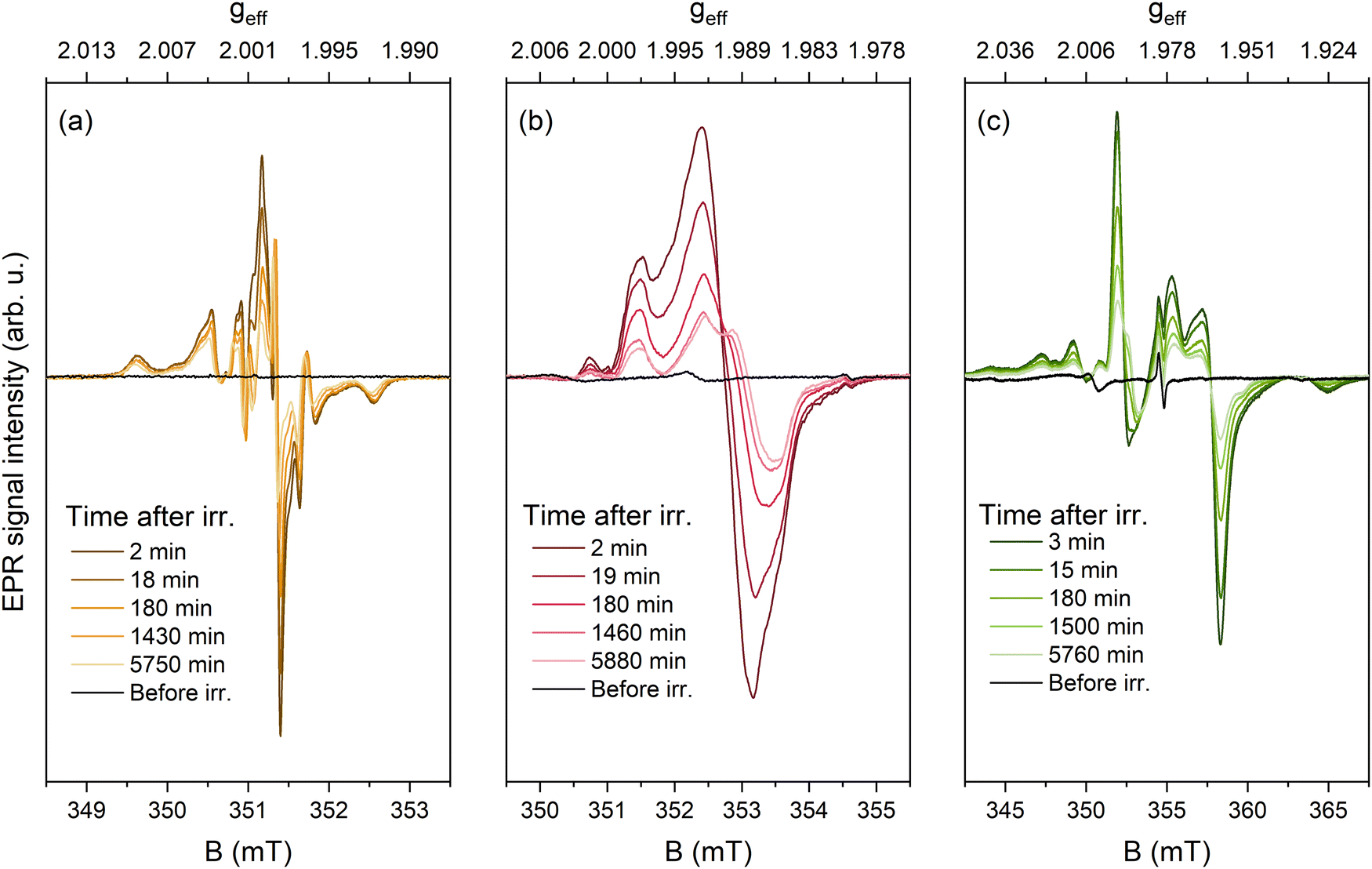 | ||
| Fig. 6 EPR spectra after different time intervals following 250 nm irradiation of 0.01% Eu. (a) CMS (acquisition settings: 0.01 mW microwave power, 0.05 mT modulation amplitude); (b) SMS (0.10 mW, 0.10 mT), (c) BMS (1.00 mW, 0.40 mT) samples. | ||
A gradual decrease in signal intensity was observed in all samples. As a result of minimizing power-saturation and excessive modulation amplitude broadening effects, it was possible to achieve better resolution of the signal structure. To discuss the nature of UV-induced paramagnetic centers in the investigated silicates, the effective g-factor axis can be introduced by using the simple relation geff = hν/μBB, where h – the Planck's constant, ν – microwave frequency, and μB – the Bohr magneton. The highest intensity peaks at the center of the spectrum, likely representing the real g-factor of the paramagnetic centers, can be evaluated as follows: CMS – geff ≈ 1.999–2.000; SMS – geff ≈ 1.989–1.991; BMS – geff ≈ 1.956–1.994. The estimated geff values are negatively shifted from the free electron value of ge = 2.0023, which implies that the signals originate from trapped electrons.55 One of the simplest electron centers in oxides is a single electron localized in an anion vacancy, for which comparable g-factor values have been reported in a series of alkaline earth metal oxides, e.g., g = 2.000 in CaO, g = 1.9845 in SrO, g = 1.9355 in BaO.56 Due to the availability of several anion sites with lower point symmetry in M3MgSi2O8, a superposition of several signals with anisotropic g-factors could be expected for similar defects. This hypothesis is corroborated by the evolution of spectra with time as the signal decay is not monotonous, i.e., EPR signal intensity decrease is accompanied by a change of signal shape providing evidence for the coexistence of several paramagnetic centers in each composition. Additional measurements and simulations are required to get the full picture of paramagnetic UV-induced centers in M3MgSi2O8.
Comparison of photochromic properties with EPR data
Similarly to EPR, diffuse reflectance measurements showed a gradual decrease in the intensity of absorption bands after storage in darkness (Fig. 7a). In SMS samples, different stabilities were detected for 530 nm and 770 nm bands. The decay of the 530 nm band was considerably faster, resulting in a 39% decrease in 5 hours after irradiation. In contrast, the 770 nm band decreased by only 7%, indicating higher stability of the defects responsible for this band. The decay rate of the 530 nm band correlated well with changes in EPR intensity, indicating the involvement of paramagnetic defects in the formation of this band. In contrast, the slower decay rate of the 770 nm band compared to the EPR signal suggests the presence of more stable, non-paramagnetic defects. These findings indicate that the photochromic effect in the investigated materials involves the formation of both paramagnetic and non-paramagnetic defects. However, the most intense photochromic absorbance band is associated with electron centers, likely electrons trapped in oxygen vacancies (i.e., F+-type centers). | ||
| Fig. 7 (a) Diffuse reflectance spectra detected after different time intervals following 250 nm irradiation of SMS doped with 0.1 Eu3+ and (b) comparison of reflectance and EPR signal decay rate. | ||
The thermal stability of paramagnetic defects was compared with the photochromic effect in SMS samples doped with 0.1% Eu. A gradual decrease in intensity was observed for both the EPR signal and all absorption bands (Fig. 8a and b). However, the decay of the 770 nm band was detected at a higher temperature, indicating greater thermal stability. These results are in good agreement with the time-dependent decay of the photochromic effect shown in Fig. 7b, which revealed a slower decay for the 770 nm band.
 | ||
| Fig. 8 (a) EPR and (b) diffuse reflectance spectra of SMS samples doped with 0.1% Eu after irradiation with 250 nm for 5 min. (c) Comparison of diffuse reflection and EPR signal intensity after heat treatment for 10 min at different temperatures. | ||
During thermal treatment, the maximum of the 530 nm band shifted to higher energies (470 nm), suggesting the presence of several overlapping bands. The changes in the EPR signal intensity followed a similar trend to the 530 nm band, though the intensity decrease of the EPR signal was slightly faster (Fig. 8c). These results suggest the presence of at least three distinct trap centers in the material, with only the least thermally stable defects being paramagnetic. The other non-paramagnetic defects could be F-centers (two electrons trapped in oxygen vacancies); however, further investigation is needed to accurately identify these non-paramagnetic defects in the material.
Mechanism of photochromism
Although precise identification of the specific defects responsible for the photochromic effect requires further investigation, the general mechanism in Eu-doped Ca3MgSi2O8, Sr3MgSi2O8, and Ba3MgSi2O8 is schematically depicted in Fig. S8 (ESI†).The defects responsible for the photochromic effect are intrinsic and exist even in undoped samples. The introduction of Eu improves photochromic efficiency and shifts the excitation peak to a lower energy range. The similarities in the excitation spectra and EPR signals of the CMS, SMS, and BMS samples suggest that the dominant defects in these materials are of the same type. The color variations between Ca3MgSi2O8, Sr3MgSi2O8, and Ba3MgSi2O8 can be attributed to the incorporation of these defects into different local environments, leading to shifts in the spectral positions of the absorbance bands and resulting in different colors of each material.
Experimental evidence of Eu2+ to Eu3+ charge transfer during irradiation indicates that Eu2+ acts as a hole center. During the irradiation, intrinsic defects and Eu2+ are excited and the energy is transferred to intrinsic defects through a conduction band or tunneling. The irradiation leads to the formation of several paramagnetic electron-type defects, which likely consist of single electrons trapped at oxygen vacancies or F+-type centers. However, not all defects responsible for the color change are paramagnetic, suggesting the presence of other defects such as F centers. Optical or thermal stimulation liberates these trapped charges, resulting in the decoloration of the samples and a reversible photochromic effect.
The unusually high efficiency of the photochromic effect of M3MgSi2O8 makes them ideal for practical applications. As an example, a self-erasing “flower” design is shown in Fig. 9.
 | ||
| Fig. 9 Photochromic self-erasing design of CMS, SMS and BMS samples doped with 0.1% Eu before irradiation and after 20 s–15 min storage in ambient light. | ||
The design is created by irradiation of samples through a photomask and it is gradually erased by ambient light. This effect can be used for displays, rewritable media, and anti-counterfeiting applications.
Conclusions
Novel orange, reddish-pink and green photochromic M3MgSi2O8 (M = Ca, Sr, Ba) based materials were developed. Excellent photochromic properties with good reversibility and color contrast exceeding 60% were obtained. Although the photochromic effect can be detected in undoped samples, it is greatly improved by the incorporation of europium ions into the lattice. The XANES and EPR analysis indicate that during irradiation, Eu2+ acts as a hole trap by forming Eu3+. The comparison of thermal stability and time evaluation of EPR signals and diffuse reflectance spectra indicate that the dominant absorbance bands are related to electron centers, which are likely F+-type centers; however, not all of the detected color centers are paramagnetic. These materials show promising photochromic properties for practical applications.Data availability
The data supporting this study have been included within the article and the corresponding ESI.† The code XAESA for analysis of extended X-ray absorption fine structure (EXAFS) spectra can be found at https://gitlab.desy.de/aleksandr.kalinko/xaesa.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the Research Council of Lithuania (LMTLT), agreement no. S-PD-24-67. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Part of this research was carried out at the PETRA III synchrotron within proposal no. I-20230821 EC. Institute of Solid State Physics, University of Latvia as the Center of Excellence has received funding from the European Union's Horizon 2020 Framework Programme H2020-WIDESPREAD-01-2016-2017-TeamingPhase2 under grant agreement no. 739508, project CAMART2.References
- J. Du, Z. Yang, H. Lin and D. Poelman, Responsive Mater., 2024, 2, e20240004 CrossRef.
- R. Allan, A manual of mineralogy comprehending the more recent discoveries in the mineral kingdom, A. and C. Black, London, 1834 Search PubMed.
- D. B. Medved, Am. Mineral., 1954, 39, 615–629 CAS.
- J. Fritzsche, C. R. Acad. Sci., 1867, 1035–1037 Search PubMed.
- M. Qin, Y. Huang, F. Li and Y. Song, J. Mater. Chem. C, 2015, 3, 9265–9275 RSC.
- J. Zhang, J. Wang and H. Tian, Mater. Horiz., 2014, 1, 169–184 RSC.
- Z. Yang, J. Du, L. I. D. J. Martin, D. Van der Heggen and D. Poelman, Laser Photonics Rev., 2021, 15, 2000525 CrossRef CAS.
- R. Hirayama, A. Shiraki, M. Naruse, S. Nakamura, H. Nakayama, T. Kakue, T. Shimobaba and T. Ito, Sci. Rep., 2016, 6, 31543 CrossRef CAS PubMed.
- S. Kawata and Y. Kawata, Chem. Rev., 2000, 100, 1777–1788 CrossRef CAS PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- J. Andréasson and U. Pischel, Isr. J. Chem., 2013, 53, 236–246 CrossRef.
- A. V. Dotsenko, L. B. Glebov and V. A. Tsekhomsky, Physics and Chemistry of Photochromic Glasses, CRC Press, 2020 Search PubMed.
- Y. J. Hwang, S. B. Pyun, M. J. Choi, J. H. Kim and E. C. Cho, Chem. Nanomater., 2022, 8, e202200005 CAS.
- Y. Ke, J. Chen, G. Lin, S. Wang, Y. Zhou, J. Yin, P. S. Lee and Y. Long, Adv. Energy Mater., 2019, 9, 1902066 CrossRef CAS.
- M. A. Chowdhury, M. Joshi and B. S. Butola, J. Eng. Fibers Fabr., 2014, 9, 107–123 Search PubMed.
- A. P. Periyasamy, M. Vikova and M. Vik, Text. Prog., 2017, 49, 53–136 CrossRef.
- L. Song, Y. Dong, B. Zhao, Y. Wu and J. Deng, Adv. Opt. Mater., 2024, 12, 2400215 CrossRef CAS.
- Y. Shen, X. Le, Y. Wu and T. Chen, Chem. Soc. Rev., 2024, 53, 606–623 RSC.
- Z. Yang, J. Hu, D. Van der Heggen, M. Jiao, A. Feng, H. Vrielinck, P. F. Smet and D. Poelman, Laser Photonics Rev., 2023, 17, 2200809 CrossRef CAS.
- S. Vuori, P. Colinet, I. Norrbo, R. Steininger, T. Saarinen, H. Palonen, P. Paturi, L. C. V. Rodrigues, J. Göttlicher, T. Le Bahers and M. Lastusaari, Adv. Opt. Mater., 2021, 9, 2100762 CrossRef CAS.
- I. Norrbo, P. Gluchowski, I. Hyppänen, T. Laihinen, P. Laukkanen, J. Mäkelä, F. Mamedov, H. S. Santos, J. Sinkkonen, M. Tuomisto, A. Viinikanoja and M. Lastusaari, ACS Appl. Mater. Interfaces, 2016, 8, 11592–11602 CrossRef CAS PubMed.
- H. C. Byron, C. Swain, P. Paturi, P. Colinet, R. Rullan, V. Halava, T. Le Bahers and M. Lastusaari, Adv. Funct. Mater., 2023, 33, 2303398 CrossRef CAS.
- M. Akiyama, Appl. Phys. Lett., 2010, 97, 181905 CrossRef.
- Y. Yonezaki and S. Takei, J. Lumin., 2016, 173, 237–242 CrossRef CAS.
- M. Akiyama, H. Yamada and K. Sakai, J. Ceram. Soc. Jpn., 2011, 119, 338–341 CrossRef CAS.
- S. Kamimura, H. Yamada and C. N. Xu, Appl. Phys. Lett., 2013, 102(3), 031110 CrossRef.
- H. Chen, Z. Xi, F. Guo, W. Long, X. Zhang and A. He, J. Alloys Compd., 2023, 949, 169812 CrossRef CAS.
- Q. Jia, Y. Li, L. Guan, H. Sun, Q. Zhang and X. Hao, J. Mater. Sci.: Mater. Electron., 2020, 31, 19277–19292 CrossRef.
- H. Sun, J. Liu, X. Wang, Q. Zhang, X. Hao and S. An, J. Mater. Chem. C, 2017, 5, 9080–9087 RSC.
- A. B. A. Kayani, S. Kuriakose, M. Monshipouri, F. A. Khalid, S. Walia, S. Sriram and M. Bhaskaran, Small, 2021, 17, 2100621 CrossRef CAS PubMed.
- P. Colinet, H. Byron, S. Vuori, J.-P. Lehtiö, P. Laukkanen, L. Van Goethem, M. Lastusaari and T. Le Bahers, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202487119 CrossRef CAS PubMed.
- K. Iakoubovskii, G. J. Adriaenssens and M. Nesladek, J. Phys.: Condens. Matter, 2000, 12, 189–199 CrossRef CAS.
- H. L. Tuller and S. R. Bishop, Annu. Rev. Mater. Res., 2011, 41, 369–398 CrossRef CAS.
- C. Freysoldt, B. Grabowski, T. Hickel, J. Neugebauer, G. Kresse, A. Janotti and C. G. Van de Walle, Rev. Mod. Phys., 2014, 86, 253–305 CrossRef.
- Y. Yonesaki, T. Takei, N. Kumada and N. Kinomura, J. Solid State Chem., 2009, 182, 547–554 CrossRef CAS.
- W. A. Caliebe, V. Murzin, A. Kalinko and M. Görlitz, AIP Conf. Proc., 2019, 2054, 060031 CrossRef.
- A. Kalinko, ( 2023). https://gitlab.desy.de/aleksandr.kalinko/xaesa.
- A. Kuzmin and J. Chaboy, IUCrJ, 2014, 1, 571–589 CrossRef CAS PubMed.
- J. J. Rehr and R. C. Albers, Rev. Mod. Phys., 2000, 72, 621–654 CrossRef CAS.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7565–7576 CrossRef CAS.
- D. Kim, D. Lim, H. Ryu, J. Lee, S. Il Ahn, B. S. Son, S.-J. Kim, C. H. Kim and J.-C. Park, Inorg. Chem., 2017, 56, 12116–12128 CrossRef CAS PubMed.
- L. Hedin and B. I. Lundqvist, J. Phys. C: Solid State Phys., 1971, 4, 2064–2083 CrossRef.
- C.-H. Park, S.-T. Hong and D. A. Keszler, J. Solid State Chem., 2009, 182, 496–501 CrossRef CAS.
- M. Fang, S. Mahlik, A. Lazarowska, M. Grinberg, M. S. Molokeev, H. Sheu, J. Lee and R. Liu, Angew. Chem., Int. Ed., 2019, 58, 7767–7772 CrossRef CAS PubMed.
- Y. Wei, H. Jia, H. Xiao, M. M. Shang, C. C. Lin, C. Su, T.-S. Chan, G. G. Li and J. Lin, RSC Adv., 2017, 7, 1899–1904 RSC.
- J. Rakovan, M. Newville and S. Sutton, Am. Mineral., 2001, 86, 697–700 CrossRef CAS.
- F. W. Lytle, Ber. Bunsen-Ges. Phys. Chem., 1987, 91, 1251–1257 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- L. Havlák, J. Bárta, M. Buryi, V. Jarý, E. Mihóková, V. Laguta, P. Boháček and M. Nikl, J. Phys. Chem. C, 2016, 120, 21751–21761 CrossRef.
- D. K. Patel, B. Rajeswari, V. Sudarsan, R. K. Vatsa, R. M. Kadam and S. K. Kulshreshtha, Dalton Trans., 2012, 41, 12023–12030 RSC.
- M. Buryi, T. Salamakha, V. Babin, J. Paterek, F. Hájek, Z. Remeš, L. Landová, E. Trusova and Y. Tratsiak, Ceram. Int., 2021, 47, 29232–29252 CrossRef CAS.
- A. Antuzevics, G. Krieke, E. Pavlovska and U. Rogulis, J. Non-Cryst. Solids, 2019, 522, 119548 CrossRef CAS.
- H. W. den Hartog, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 27, 20–26 CrossRef CAS.
- A. Laurikenas, D. Sakalauskas, A. Marsalka, R. Raudonis, A. Antuzevics, V. Balevicius, A. Zarkov and A. Kareiva, J. Sol-Gel Sci. Technol., 2021, 97, 479–487 CrossRef CAS.
- A. S. Marfunin, Spectroscopy, luminescence and radiation centers in minerals, Springer Science & Business Media, 2012 Search PubMed.
- A. E. Hughes and B. Henderson, Point Defects in Solids, 1972, pp. 381–490 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc03091e |
| This journal is © The Royal Society of Chemistry 2024 |