
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of hydrogen bond interactions on the electronic nature of DPP-based organic semiconductors: implications on charge transport†
Raúl
González-Núñez‡
 a,
Gabriel
Martinez‡
b,
Nelson Ricardo
Avila-Rovelo§
b,
Kyeog-Im
Hong¶
bc,
Amparo
Ruiz-Carretero
*bc and
Rocío
Ponce Ortiz
*a
a,
Gabriel
Martinez‡
b,
Nelson Ricardo
Avila-Rovelo§
b,
Kyeog-Im
Hong¶
bc,
Amparo
Ruiz-Carretero
*bc and
Rocío
Ponce Ortiz
*a
aDepartment of Physical Chemistry, University of Málaga, Campus de Teatinos s/n, Málaga 29071, Spain. E-mail: rocioponce@uma.es
bUniversity of Strasbourg, Institute Charles Sadron, CNRS, UPR22, 23 Rue du Loess, 67034, Strasbourg Cedex 2, France. E-mail: amparo.ruiz@ics-cnrs.unistra.fr
cInstituto de Ciencia de Materiales de Madrid, Consejo Superior de Investigaciones Científicas CSIC, Sor Juana Inés de la Cruz 3, 28049, Madrid, Spain
First published on 7th October 2024
Abstract
In this work, we show that hydrogen bond interactions, in addition to directing supramolecular order, can have an intriguing effect on the molecular and electronic properties of DPP derivatives, facilitating redox processes in the solid state. As a result, charge carrier formation can be enhanced in devices, contributing to the improvement of electrical performances of two exceptionally small molecular semiconductors based on a thiophene-capped diketopyrrolopyrrole (DPP) as the electroactive component and amide groups as hydrogen-bonding units. Two different topologies of the amide groups were explored, C-centered and N-centered (HDPPBA-C and HDPPBA-N, respectively), and the results were compared to the HDPPH molecule with no amide groups. Spectroelectrochemical experiments, in conjunction with vibrational spectroscopies and DFT calculations, demonstrate that hydrogen bond formation promotes modifications on the effective conjugation length of the studied semiconductors, which may facilitate the generation of free charge carriers in organic field effect transistors (OFETs). These results open a new strategy towards the simple design of organic semiconductors and control over their morphological and electrical properties by simple hydrogen bond formation.
Introduction
Organic materials have appeared in the last few decades as great alternatives for the fabrication of electronic devices, such as organic field effect transistors (OFETs)1 or organic solar cells.2 The versatility of organic synthesis allows us to provide small molecules or polymers with the desired optoelectronic properties.3 Interestingly, the final efficiency of a given device does not only depend on the molecular design but also, very importantly, on the way the materials assemble.4 It is here that noncovalent interactions play a crucial role, since they guide the assembly process in solution and when depositing molecules on thin films. In the case of organic semiconductors, since they are based on conjugated segments and aromatic moieties, π stacking is the main non-covalent interaction guiding their molecular packing. Nevertheless, the interplay of π stacking with other non-covalent interactions, such as hydrogen-bonding, has been proven a promising strategy to improve the morphology of organic electronic devices by creating well-connected domains of semiconductors able to transport charges efficiently.5–12Despite the exciting results, where the beneficial role of hydrogen bonding is highlighted, the incorporation of such moieties in organic semiconductors for the fabrication of different organic electronic devices is still underexplored. In addition, in the search of efficient organic semiconductors reaching the performance required for device commercialization, a great effort has been mostly devoted over the years to the optimization of structure–property relationships of pristine organic semiconducting materials, looking for a proper balance between molecular and intermolecular characteristics for efficient charge transport. However, in the last few years, enormous interest has emerged in the use of molecular dopants, which allows tuning the energy gap and the optical and electrical properties of semiconducting materials.13 Thus, this strategy has been proved to tremendously boost the materials’ electrical performances, through significantly improving charge injection and transport in devices.14–21 However, the rational selection and design of efficient dopants in organic materials is still challenging. In this sense, self-doping in molecular materials is a more straightforward approach since it avoids the need for dopant synthesis. Self-doping in luminescence organic semiconductors has been achieved by incorporating longer conjugation impurities (normally a by-product of the reaction) that acts as the molecular dopant.22,23 Other strategies entail the doping of the semiconductor backbone by introducing functional groups into the side chains24 or the use of quinoidal semiconductors, where the coexistence of closed-shell or open-shell structures could give rise to self-doping characteristics.25–27 In any way, these last strategies give rise to the generation of free charge carriers, which improves electrical performances. In addition, few examples of hydrogen-bonding assisted self-doped conductors can be found in the literature, where the supramolecular interaction was proved to be responsible for the self-doping mechanism.28–30 However, in organic transistors, self-doping, understood as the formation of unintentional doped species in the pristine material, gives usually results that are not ideal, since improving the charge field-effect mobility without decreasing the ON/OFF ratio is challenging.31 Thus, it is necessary to search for alternatives able to modulate the electronic characteristics of the materials and facilitate charge formation without the need of adding dopants.
In this work, we demonstrate that good values of charge carrier mobility can be achieved with discrete small basic π-conjugated cores containing hydrogen-bonding units. We have chosen a simple thiophene-capped diketopyrrolopyrrole (DPP) as the electroactive segment and amide groups as the hydrogen-bonding units (Scheme 1) and fabricated OFETs processed from solution. DPP is a very well-known π-conjugated system found in a plethora of organic electronic devices due to its excellent electronic properties and stability.32,33 Usually, such a conjugated system appears as a component of semiconducting polymers of high molecular weight or as a part of more sophisticated discrete molecules used in organic electronic devices.34–36 In other cases, devices fabricated from single core DPPs have been reported in the literature,37–39 using their ability to form hydrogen bonds through their non-alkylated or mono-alkylated lactams to grow molecular crystals. Even though the hydrogen bonded structures achieved with this technique are well-organized, the high insolubility of non-alkylated systems requires processing by sublimation or the application of the latent pigment technique (protection and thermal/acidic post-treatment) to process them from solution. In addition, this strategy is not versatile since the assembly process always follows the same trend, thus limiting the possibility to tune the optoelectronic properties of the materials based on their aggregation.
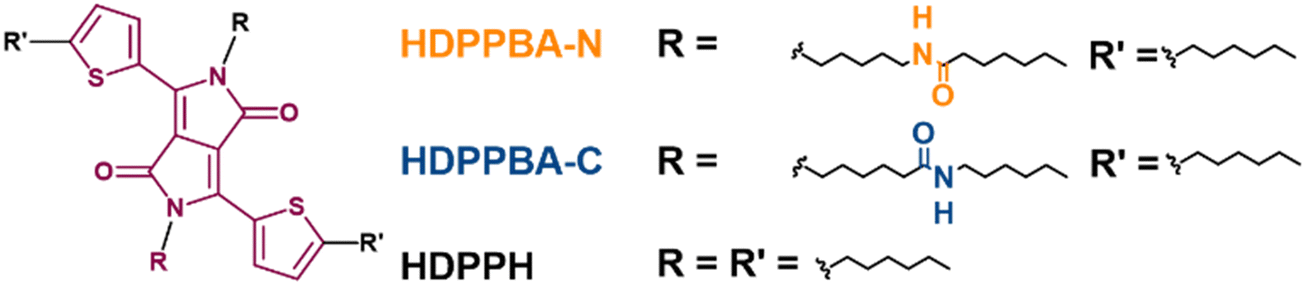 | ||
| Scheme 1 Chemical structures of HDPPBA-N, HDPPBA-C and HDPPH semiconductors. | ||
Results and discussion
Molecular and supramolecular characteristics
The synthesis and self-assembly behavior of the derivatives explored in this work, HDPPBA-N, HDPPBA-C and HDPPH, have been previously reported by us.40 Ground-state density functional theory (DFT) calculations in a vacuum were performed to analyze the molecular structures and properties of the amide derivatives and the control molecule, using the B3LYP functional and the 6-31G** basis set. The results display the same molecular skeleton for HDPPH, HDPPBA-C and HDPPBA-N, having similar dihedral angles as seen in Fig. S1 (ESI†). Thus, DFT calculations show a quite planar structure with dihedral angles between the DPP core and the thiophene groups of approx. 15°. As expected, the frontier molecular orbitals in the three studied molecules are delocalized over the whole conjugated skeleton with a higher contribution on the DPP core. All the derivatives show comparable HOMO–LUMO gaps of around 2.49 eV (see Fig. S2, ESI†), although slightly destabilized HOMO and LUMO orbitals are predicted for the C-centered molecule, while the opposite trend is found for the N-centered derivative compared to the control molecule. Nonetheless, theoretical calculations show comparable structures and electronic gaps, so differences in electronic properties are not been related to molecular structure modifications.The optical properties in solution and as thin films of HDPPH, HDPPBA-C and HDPPBA-N were studied by UV-Vis absorption (Fig. 1) and rationalised with the help of the time-dependent density functional theory (TD-DFT). All three compounds show a similar spectrum in solution, due to the minimisation of intermolecular interactions between the molecules, showing a main band at around 560 nm, with vibronic features at 520 nm and 482 nm. DFT calculations correctly predict these experimental results and assign the lowest energy band to one electron excitation from the HOMO to the LUMO (Fig. S3, ESI†). In addition, the thin film spectra of the studied materials show a broader and red-shifted spectral profile with two main bands at around 490 nm and 610 nm.
 | ||
| Fig. 1 UV-Vis absorption spectra in chloroform solutions (black) and thin films (red) of (a) HDPPH, (b) HDPPBA-C and (c) HDPPBA-N. | ||
The effect of side alkyl chain engineering was analyzed by recording emission spectra and measuring the corresponding Stokes shifts, both in solution and in the solid state (see Fig. S5, ESI†). While small Stokes shifts, around 11–15 nm, are recorded in solution, large Stokes shifts are found in the solid state, particularly for the molecules having hydrogen bond interactions (141 nm and 134 nm for HDPPBA-C and HDPPBA-N, respectively versus 110 nm for HDPPH). These large Stokes shifts upon hydrogen bond interactions have been previously reported.41
The redox properties of HDPPH, HDPPBA-C and HDPPBA-N in solution were experimentally evaluated by cyclic voltammetry (CV) using 1 mM dichloromethane (CH2Cl2) for the three derivatives, which undergo similar reduction and oxidation processes. The compounds exhibit two one-electron oxidation peaks reversible in nature at E1/2 0.36, 0.38 V and 0.41 V and E1/2 0.72, 0.71 and 0.77 V for HDPPH, HDPPBA-C and HDPPBA-N, respectively (Fig. S6, ESI†). This observation suggests that the three DPP-based compounds are electron-rich, as they demonstrate the ability to donate two electrons. The oxidation processes are accompanied by a one-electron reduction at E1/2 −1.73 V (C-centered) or Epa −1.82 and −1.74 V (control and N-centered), which presents reversibility shown by the returning wave in the case of HDPPBA-C, but irreversible for HDPPH and HDPPBA-N. The HOMO and LUMO energy levels were experimentally calculated from the potential at the onset of oxidation and reduction, respectively, using the frontier orbitals of ferrocene (−4.8 eV below the vacuum level) as the reference. The HOMO levels of HDPPH, HDPPBA-C and HDPPBA-N were calculated to be −5.08, −5.08 and −5.16 eV, respectively. On the other hand, the obtained LUMO values were −3.50, −3.49 and −3.50 eV, respectively. Accordingly, all three derivatives have similar band gaps ranging from 1.59 to 1.64 eV. The minor distinction in the electronic nature of the materials is reasonable, considering that the electron richness of the alkyl (in thiophenes) and amide chains (in lactams) is similar.
CV experiments on thin films were carried out in acetonitrile containing 0.2 M TBAPF6 as a supporting electrolyte (Fig. 2). Similar oxidation and reduction processes are recorded for the three molecules. However, it should be noted that, for both HDPPBA-C and HDPPBA-N compounds, a shoulder is detected from which the HOMO energy levels are calculated (explanation of the origin of the shoulder will be shown below). Slightly destabilized energy values are found for HDPPBA-C (−5.13 eV) and HDPPBA-N (−5.16 eV) compared to HDPPH (−5.21 eV). On the other hand, for LUMO energy levels, we found the same tendency, slightly destabilized energy values for HDPPBA-C (−3.63 eV) and HDPPBA-N (−3.59) compared to HDPPH (−3.68 eV).
 | ||
| Fig. 2 Thin film cyclic voltammetry spectra recorded in acetonitrile/TBAPF6 (0.2 M) for HDPPH, HDPPBA-C and HDPPBA-N. | ||
Thin films of HDPPH, HDPPBA-C and HDPPBA-N were deposited by drop casting solutions of the semiconducting materials in chloroform on Si/SiO2 substrates. After thermal annealing at 120° for 3 hours, their morphologies were studied by atomic force microscopy (AFM). Clear differences were found comparing HDPPH with the amide derivatives. While the control molecule forms large crystalline structures (Fig. 3a), HDPPBA-C and HDPPBA-N display a morphology composed of smaller fibers (Fig. 3b and c). This is ascribed to the formation, in HDPPBA-N and HDPPBA-C, of J-type aggregates in solution and in thin films formed by hydrogen bonding while the control molecule only assembles thanks to π–π stacking.40
 | ||
| Fig. 3 AFM images of semiconducting thin films deposited on substrates at room temperature with a posterior thermal treatment at 120° for 3 hours: (a) HDPPH, (b) HDPPBA-C and (c) HDPPBA-N. | ||
Analysing the AFM data, we found a decrease of the average roughness for HDPPBA-N (104 nm) and HDPPBA-C (45 nm), in accordance with the formation of small fibres in these compounds, with respect to HDPPH (169 nm). It should be noted that, in the area analysed, the maximum peak height is found in HDPPH (931 nm), which forms a large crystalline structure, while the values of maximum peak heights, for HDPPBA-C and HDPPBA-N, are 169 nm and 532 nm, respectively.
XRD measurements were also carried out on the thin films (Fig. S7, ESI†), indicating a more crystalline structure for HDPPH compared to HDPPBA-C and HDPPBA-N. Even though crystallinity is desirable to achieve efficient charge transport, HDPPBA-C and HDPPBA-N provide interconnected amorphous fibrillar networks, while HDPPH shows large crystalline structures with sharp boundaries among crystallites. We believe that these two morphological scenarios are crucial to compare their charge transport properties since the interconnection among amorphous aggregates has been proven beneficial in polymeric semiconductors42 and other hydrogen-bonded oligomers,5 but in this case, we are pushing the limits by miniaturizing the conjugated skeleton of the derivatives explored.
Electrical properties of thin film transistors
The photoconductivity properties of HDPPBA-N, HDPPBA-C and HDPPH measured at the nanoscale by electrodeless techniques based on microwave conductivity have been already reported by us. However, their charge mobility in full devices was not measured, having only a previous screening indicating that the hydrogen-bonded derivatives had superior values of photoconductivity, and charge carrier lifetimes are one order of magnitude larger than the control derivative.40In this work, in order to measure the electrical characteristics of the studied compounds, solution processed OFETs were fabricated in a top-contact/bottom-gate structure (see the detailed procedure in the ESI†). The charge carrier mobilities (μ), threshold voltage (VT) and current on/off ratio (ION/IOFF) were extracted from the saturated region in the device transfer curves and the electrical parameters of the best performing devices are summarized in Table 1. Complete device characterization can be found in the ESI.† Compounds HDPPBA-C and HDPPBA-N show similar maximum p-type field effect mobilities, with values of 2 × 10−2 cm2 V−1 s−1 and 1 × 10−2 cm2 V−1 s−1, respectively (see output and transfer plots in Fig. 4). In contrast, the HDDPH compound is not active under any tested conditions (see details in the ESI†), which indicates that the introduction of amide groups in these molecules promotes and favors charge transport.
 | ||
| Fig. 4 Transfer (a) and (b) and output (c) and (d) of HDPPBA-C (a) and (c) and HDPPBA-N (b) and (d). The transfer characteristics were measured at a constant source–drain voltage of −80 V. The gate voltage in the output plots varies from 0 to −80 V in steps of 10 V. | ||
In order to understand the extent and reasons behind this improvement, which may not be solely related to morphological and crystallinity changes due to hydrogen bond formation, a physical chemistry investigation including vibrational spectroscopy and spectroelectrochemistry was performed, as shown in the following sections.
Analysis of electronic properties by spectroscopy.
Attenuated total reflectance (ATR) IR spectra of HDPPBA-N, HDPPBA-C and HDPPH semiconductors as powder samples were recorded to account for the hydrogen bond formation and its implications on the electronic structure of the conjugated skeletons (Fig. 5, left). It should be noted that while a single ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) vibration is found in HDPPH at 1656 cm−1 arising from the cross-conjugated carbonyl group of the DPP lactam group, in the case of HDPPBA-N and HDPPBA-C, as expected, two distinct bands appear at around 1657 and 1639–1642 cm−1. The IR band recorded at 1657 cm−1 is associated with the cross-conjugated lactam ring while the one recorded at 1639–1642 cm−1 is due to the alkyl chain amide groups forming hydrogen bonds. As expected, the value of the latter is significantly lower than the one predicted by theoretical calculations on isolated molecules of both HDPPBA-N and HDPPBA-C (1706 and 1708 cm−1), which indicates a lengthening of the CO double bond due to intramolecular interactions (see Fig. S10–S21, ESI†).
O) vibration is found in HDPPH at 1656 cm−1 arising from the cross-conjugated carbonyl group of the DPP lactam group, in the case of HDPPBA-N and HDPPBA-C, as expected, two distinct bands appear at around 1657 and 1639–1642 cm−1. The IR band recorded at 1657 cm−1 is associated with the cross-conjugated lactam ring while the one recorded at 1639–1642 cm−1 is due to the alkyl chain amide groups forming hydrogen bonds. As expected, the value of the latter is significantly lower than the one predicted by theoretical calculations on isolated molecules of both HDPPBA-N and HDPPBA-C (1706 and 1708 cm−1), which indicates a lengthening of the CO double bond due to intramolecular interactions (see Fig. S10–S21, ESI†).
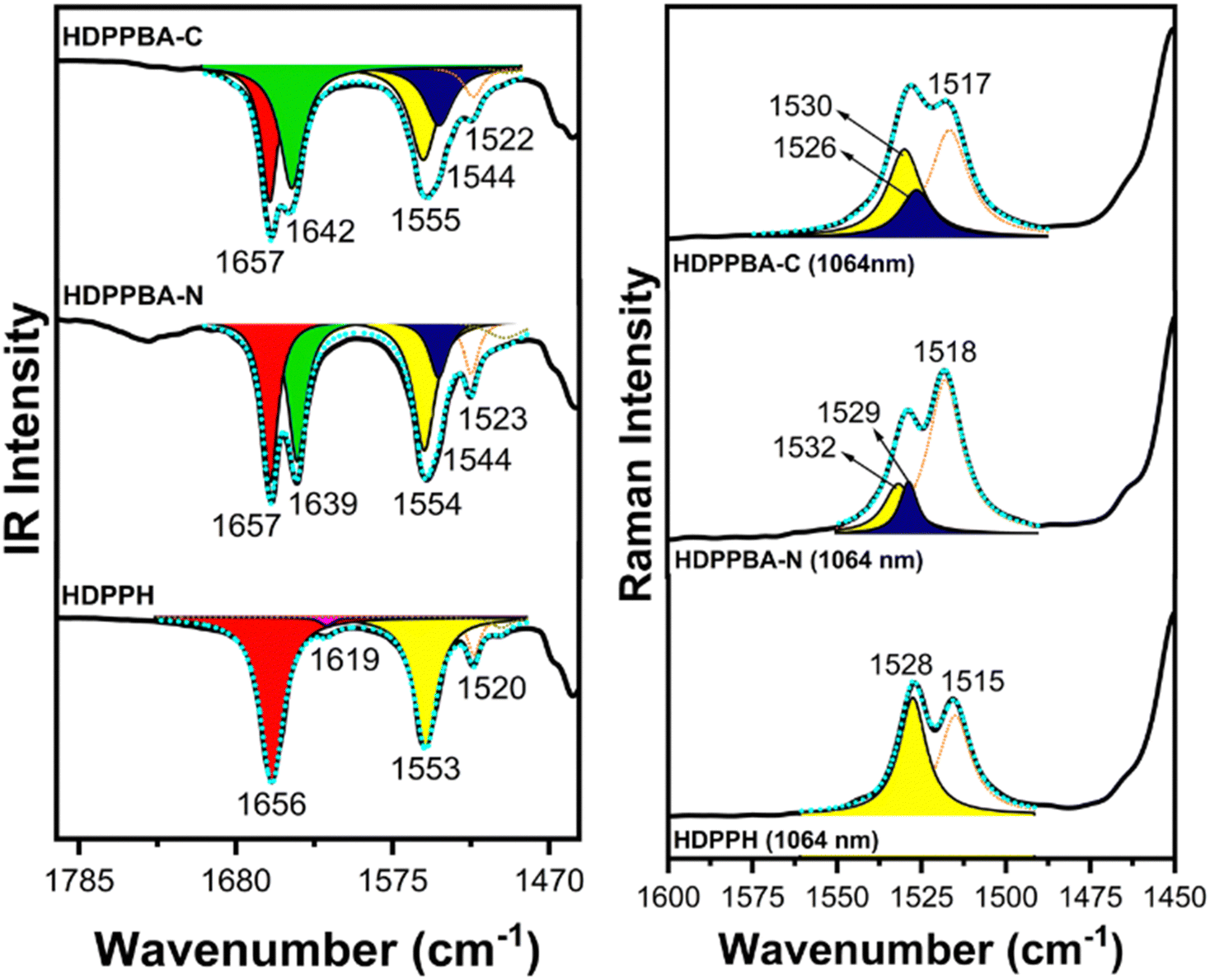 | ||
| Fig. 5 IR spectra of HDPPBA-C, HDPPBA-N, and HDPPH (left) and Raman spectra of HDPPBA-C, HDPPBA-N, and HDPPH (right) as solid powders. | ||
In addition, and unexpectedly, IR spectra also point out to changes on the electronic properties of the conjugated skeletons due to hydrogen bond formation. To analyze this, we now focus our attention on the collective vibration over the thiophene-DPP-thiophene chain. This collective vibration appears at 1553 cm−1, being a quite sharp band in HDPPH. However, when hydrogen bonds take part in HDPPBA-N and HDPPBA-C, the aforementioned IR band widens showing two clear contributions at 1555–1554 cm−1 and 1544 cm−1. The latter indicating some participation of the carbonyl group of the lactam ring in the supramolecular hydrogen bond interaction, which promotes enhanced π-conjugation in molecules having such interactions.
This can be understood considering that, when the lactam carbonyl group is involved in the hydrogen bond formation, the cross-conjugated path due to the carbonyl lactam is blocked and thus the molecular π-conjugation improves through the linear conjugated path (see Scheme 2). Nonetheless, it seems reasonable that only a fraction of the molecules may interact through those carbonyl groups, being the hydrogen bonding through lateral alkyl chains with amide groups the most probable one. The same conclusion is gathered from the FT-Raman spectra of the semiconductors as bulk samples, where two distinct contributions are found in the Raman band due to the collective (CC/C–C) of the linear conjugated path (Fig. 5, right and Fig. S22–S33, ESI†).
 | ||
| Scheme 2 Schematic representation of molecular π-conjugation showing: (left) the presence of different competing paths (linear and cross-conjugated) in molecules with no hydrogen bond formation, and (right) the enhanced π-conjugated linear path after hydrogen bond formation through the carbonyl group of the lactam ring. | ||
It is expected that the hydrogen bonding through the imide carbonyl group may influence the electronic properties of the molecules, affecting the energies of the frontier molecular orbitals as previously shown in thin film CV spectra. It should be noted that, for both HDPPBA-C and HDPPBA-N compounds, shoulders were detected in the oxidation waves, which can be ascribed to the oxidation potential of those molecules forming hydrogen bonds through the carbonyl group of the imide DPP unit. Calculating the HOMO potential from the onset of the aforementioned shoulders, slightly destabilized energy values are found for HDPPBA-C (−5.13 eV) and HDPPBA-N (−5.16 eV) compared to HDPPH (−5.21 eV). This result may facilitate charge formation in field effect transistor devices. In fact, while the control molecule HDPPH was inactive in OFETs, field-effect mobilities with values of 2 × 10−2 cm2 V−1 s−1 and 1 × 10−2 cm2 V−1 s−1 were recorded for HDPPBA-C and HDPPBA-N, respectively. Although, in thin film devices, a correct supramolecular organization is commonly crucial to boost the final electrical performance, this molecular electronic property modulation by intermolecular interactions may have a positive impact on the device performance.
Analysis of electronic properties by spectroelectrochemistry. Charge carriers’ stabilization
In order to evaluate the ability of the studied semiconductors to stabilize charge carriers and to probe if non-covalent interactions play a role in the formation of these charged species, as indicated by thin film CV, in situ spectroelectrochemical studies were first conducted in dilute dichloromethane solution (10−4 M) at room temperature using 0.1 M Bu4NPF6 as the supporting electrolyte and an optically transparent thin-layer electrochemical (OTTLE) cell. As shown in Fig. 6 and Fig. S34–S36 (ESI†), the three compounds behave similarly in solution, as expected, due to the absence or minimization of intermolecular interactions in diluted solutions, and all stabilize one positive charge at identical redox potentials. TD-DFT calculations support the experimental results. Therefore, no discernible changes on the molecular and electronic structures of the isolated molecules are found by the inclusion of amide groups on the lateral groups. | ||
| Fig. 6 (a) A scheme of optically transparent thin layer electrochemical cell (OTTLE). UV-Vis-NIR absorption spectra of (b) HDPPH (c) HDPPBA-C, and (d) HDPPBA-N electrochemically oxidized by the progressive increase of the oxidation potential. | ||
In contrast, the enhanced effective π-conjugation imparted by hydrogen bond formation, evidenced in the previous section and shown in Scheme 2, is expected to influence charge injection, stabilization and transport in the solid state, where intermolecular interactions are crucial. Thus, thin film oxidation was also attempted by exposing the films deposited onto the quartz substrate to iodine vapour (a very weak oxidant) for 24 hours (Fig. 7). Remarkably, different results are found for semiconductors having hydrogen-bonding units, HDPPBA-N and HDPPBA-C, compared to the control material, HDPPH. It should be noted that, while the formation of a positive charged species is found for derivatives with hydrogen bonding abilities, no oxidation is found in the case of HDPPH, where no hydrogen bond interactions are possible.
 | ||
| Fig. 7 (a) Schematic representation of the oxidation process with iodine vapour in a gas cell. Solid state oxidation of (b) HDPPH, (c) HDPPBA-C and (d) HDPPBA-N, upon iodine vapour exposition. | ||
To analyze this, the IR spectra of the semiconductor thin films, prior and after exposition to iodine vapour, were recorded. As shown in Fig. 8, no remarkable differences are found on the IR spectra of HDPPH before and after iodine exposition. However, in the case of HDPPBA-N and HDPPBA-C, great changes are recorded according to the radical cation formation. Therefore, the enhanced π-conjugated path, due to hydrogen bond formation through the imide carbonyl group, has indeed an effect on the oxidative potential of the semiconducting thin films, and thus it should facilitate the generation of free charge carriers in OFETs, contributing to the charge carrier mobility improvement shown above.
 | ||
| Fig. 8 Film IR spectra of HDPPBA-C (left), HDPPBA-N (center) and HDPPH (right). Recorded spectra before iodine vapour exposition (in black) and after 24 h exposition to iodine vapour (in red). | ||
It should be noted that, while HDPPH was not active in OFETs, field effect mobilities of 2 × 10−2 cm2 V−1 s−1 and 1 × 10−2 cm2 V−1 s−1 were recorded for HDPPBA-C and HDPPBA-N, respectively. As far as we know, this is the first time that an enhancement of the effective π-conjugation length of organic semiconductors, by a simple chemical design promoting non-covalent interactions, has been shown to boost the ability of organic semiconductors to stabilize charge carriers and ultimately enhance their electrical performance.
Conclusions
In conclusion, we present here a molecular engineering strategy that allows simultaneously control over the semiconductors morphological and intrinsic electronic properties by simply promoting hydrogen-bond formation in remarkably small DPP derivatives. We show that the electrical improvement found in semiconductors having C- or N-centered amide groups can not be solely related to supramolecular characteristics in the thin film, but it could be also ascribed to the modulation of the molecular electronic properties by the hydrogen-bond interactions. In fact, a comprehensive spectroelectrochemical and spectroscopic study points out to a complex scenario in which hydrogen-bonds formation promote the linear conjugated path in DPP semiconductors respect to the cross-conjugated path, when the carbonyl of the DPP lactam group is involved in the hydrogen-bonds formation. We have demonstrated that this enhancement of the semiconductor effective-conjugation facilitates charge carriers formation in the thin film, as shown by cyclic voltammetry and upon iodine oxidation, which may also contribute to the boost of the electrical performance in OFETs. We feel that this synthetic approach could be extrapolated to many already existing efficient DPP-based conjugated cores, boosting their electronic performance by simply modifying their pending alkyl solubilizing units.Author contributions
G. M. synthesized and provided the molecules for the study and interpreted the results. R. G.-N. fabricated all the devices, performed the electrochemical oxidation experiments, interpreted the results. N. R. A. R. synthesized and provided the C-centered derivative. K. H. performed the cyclic voltammetry measurements on thin film. A. R. C. and R. P. O. supervised the work, interpreted the results, and wrote the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work at the University of Málaga was supported by the MICINN (project PID2022-139548GB-I00). R. G.-N. thanks the MICINN for a FPI predoctoral fellowship (PRE2020-092327). A. R. C. and N. R. A. R. thank the Foundation for Frontier Research in Chemistry (FRC) LabEx Emerging Investigators Grant 2018 (CSC-ARC-18). A. R. C. and G. M. thank the Graduate School of Complex Systems Chemistry of Strasbourg for his doctoral fellowship funded by the French National Research Agency (CSC-IGSANR-17-EURE-0016). The authors thank the Agence Nationale de la Recherche (ANR JCJC TOTALBOND 2020). R. P. O and A. R. C. acknowledge the MICIIN for the REDES project “RED2022-134503-T”. The authors would also like to thank the computer resources, technical expertise and assistance provided by the SCBI (Supercomputing and Bioinformatics) centre of the University of Málaga. The Vibrational spectroscopy (EVI) lab of the Research Central Services (SCAI) of the University of Málaga is also gratefully acknowledged.References
- M. Muccini, A Bright Future for Organic Field-Effect Transistors, Nat. Mater., 2006, 5(8), 605–613, DOI:10.1038/nmat1699.
- C. J. Brabec, N. S. Sariciftci and J. C. Hummelen, Plastic Solar Cells, Adv. Funct. Mater., 2001, 11(1), 15–26 CrossRef CAS.
- Z. B. Henson, K. Müllen and G. C. Bazan, Design Strategies for Organic Semiconductors beyond the Molecular Formula, Nat. Chem., 2012, 4(9), 699–704, DOI:10.1038/nchem.1422.
- F. Liu, C. Wang, J. K. Baral, L. Zhang, J. J. Watkins, A. L. Briseno and T. P. Russell, Relating Chemical Structure to Device Performance via Morphology Control in Diketopyrrolopyrrole-Based Low Band Gap Polymers, J. Am. Chem. Soc., 2013, 135(51), 19248–19259, DOI:10.1021/ja408923y.
- T. Aytun, L. Barreda, A. Ruiz-Carretero, J. A. Lehrman and S. I. Stupp, Improving Solar Cell Efficiency through Hydrogen Bonding: A Method for Tuning Active Layer Morphology, Chem. Mater., 2015, 27(4), 1201–1209, DOI:10.1021/cm503915t.
- A. Ruiz-Carretero, T. Aytun, C. J. Bruns, C. J. Newcomb, W.-W. Tsai and S. I. Stupp, Stepwise Self-Assembly to Improve Solar Cell Morphology, J. Mater. Chem. A, 2013, 1(38), 11674, 10.1039/c3ta12411h.
- X. Shi and W. Bao, Hydrogen-Bonded Conjugated Materials and Their Application in Organic Field-Effect Transistors, Front. Chem., 2021, 9 Search PubMed.
- J. H. Oh, W.-Y. Lee, T. Noe, W.-C. Chen, M. Könemann and Z. Bao, Solution-Shear-Processed Quaterrylene Diimide Thin-Film Transistors Prepared by Pressure-Assisted Thermal Cleavage of Swallow Tails, J. Am. Chem. Soc., 2011, 133(12), 4204–4207, DOI:10.1021/ja110486s.
- J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, Significant Improvement of Semiconducting Performance of the Diketopyrrolopyrrole–Quaterthiophene Conjugated Polymer through Side-Chain Engineering via Hydrogen-Bonding, J. Am. Chem. Soc., 2016, 138(1), 173–185, DOI:10.1021/jacs.5b09737.
- J. Ma, Z. Liu, J. Yao, Z. Wang, G. Zhang, X. Zhang and D. Zhang, Improving Ambipolar Semiconducting Properties of Thiazole-Flanked Diketopyrrolopyrrole-Based Terpolymers by Incorporating Urea Groups in the Side-Chains, Macromolecules, 2018, 51(15), 6003–6010, DOI:10.1021/acs.macromol.8b01020.
- C. Wang, M. Liu, S. Rahman, H. P. Pasanen, J. Tian, J. Li, Z. Deng, H. Zhang and P. Vivo, Hydrogen Bonding Drives the Self-Assembling of Carbazole-Based Hole-Transport Material for Enhanced Efficiency and Stability of Perovskite Solar Cells, Nano Energy, 2022, 101, 107604, DOI:10.1016/j.nanoen.2022.107604.
- Y.-C. Lin, C.-C. Shih, Y.-C. Chiang, C.-K. Chen and W.-C. Chen, Intrinsically Stretchable Isoindigo–Bithiophene Conjugated Copolymers Using Poly(Acrylate Amide) Side Chains for Organic Field-Effect Transistors, Polym. Chem., 2019, 10(38), 5172–5183, 10.1039/C9PY00845D.
- Z. Qin, C. Gao, W. W. H. Wong, M. K. Riede, T. Wang, H. Dong, Y. Zhen and W. Hu, Molecular Doped Organic Semiconductor Crystals for Optoelectronic Device Applications, J. Mater. Chem. C, 2020, 8(43), 14996–15008, 10.1039/D0TC02746D.
- A. D. Scaccabarozzi, A. Basu, F. Aniés, J. Liu, O. Zapata-Arteaga, R. Warren, Y. Firdaus, M. I. Nugraha, Y. Lin, M. Campoy-Quiles, N. Koch, C. Müller, L. Tsetseris, M. Heeney and T. D. Anthopoulos, Doping Approaches for Organic Semiconductors, Chem. Rev., 2022, 122(4), 4420–4492, DOI:10.1021/acs.chemrev.1c00581.
- A. F. Paterson, N. D. Treat, W. Zhang, Z. Fei, G. Wyatt-Moon, H. Faber, G. Vourlias, P. A. Patsalas, O. Solomeshch, N. Tessler, M. Heeney and T. D. Anthopoulos, Small Molecule/Polymer Blend Organic Transistors with Hole Mobility Exceeding 13 Cm2 V−1 S−1, Adv. Mater., 2016, 28(35), 7791–7798, DOI:10.1002/adma.201601075.
- S. Hunter, A. D. Mottram and T. D. Anthopoulos, Temperature and Composition-Dependent Density of States in Organic Small-Molecule/Polymer Blend Transistors, J. Appl. Phys., 2016, 120(2), 025502, DOI:10.1063/1.4955282.
- A. F. Paterson, L. Tsetseris, R. Li, A. Basu, H. Faber, A.-H. Emwas, J. Panidi, Z. Fei, M. R. Niazi, D. H. Anjum, M. Heeney and T. D. Anthopoulos, Addition of the Lewis Acid Zn(C6F5)2 Enables Organic Transistors with a Maximum Hole Mobility in Excess of 20 Cm2 V−1 S−1, Adv. Mater., 2019, 31(27), 1900871, DOI:10.1002/adma.201900871.
- J. Smith, W. Zhang, R. Sougrat, K. Zhao, R. Li, D. Cha, A. Amassian, M. Heeney, I. McCulloch and T. D. Anthopoulos, Solution-Processed Small Molecule-Polymer Blend Organic Thin-Film Transistors with Hole Mobility Greater than 5 Cm2/Vs, Adv. Mater., 2012, 24(18), 2441–2446, DOI:10.1002/adma.201200088.
- J. Panidi, A. F. Paterson, D. Khim, Z. Fei, Y. Han, L. Tsetseris, G. Vourlias, P. A. Patsalas, M. Heeney and T. D. Anthopoulos, Remarkable Enhancement of the Hole Mobility in Several Organic Small-Molecules, Polymers, and Small-Molecule:Polymer Blend Transistors by Simple Admixing of the Lewis Acid p-Dopant B(C6F5)3, Adv. Sci., 2018, 5(1), 1700290, DOI:10.1002/advs.201700290.
- S. Lan, Y. Yan, H. Yang, G. Zhang, Y. Ye, F. Li, H. Chen and T. Guo, Improving Device Performance of N-Type Organic Field-Effect Transistors via Doping with a p-Type Organic Semiconductor, J. Mater. Chem. C, 2019, 7(15), 4543–4550, 10.1039/C8TC05740K.
- L. Cao, C. Ren and T. Wu, Recent Advances in Doped Organic Field-Effect Transistors: Mechanism, Influencing Factors, Materials, and Development Directions, J. Mater. Chem. C, 2023, 11(10), 3428–3447, 10.1039/D2TC05035H.
- A. D. Kuimov, C. S. Becker, N. A. Shumilov, I. P. Koskin, A. A. Sonina, V. Y. Komarov, I. K. Shundrina and M. S. Kazantsev, Synthetic Approach for the Control of Self-Doping in Luminescent Organic Semiconductors, Mater. Chem. Front., 2022, 6(16), 2244–2255, 10.1039/D2QM00345G.
- O. D. Parashchuk, A. A. Mannanov, V. G. Konstantinov, D. I. Dominskiy, N. M. Surin, O. V. Borshchev, S. A. Ponomarenko, M. S. Pshenichnikov and D. Y. Paraschuk, Molecular Self-Doping Controls Luminescence of Pure Organic Single Crystals, Adv. Funct. Mater., 2018, 28(21), 1800116, DOI:10.1002/adfm.201800116.
- P. Chen, D. Wang, L. Luo, J. Meng, Z. Zhou, X. Dai, Y. Zou, L. Tan, X. Shao, C. Di, C. Jia, H.-L. Zhang and Z. Liu, Self-Doping Naphthalene Diimide Conjugated Polymers for Flexible Unipolar n-Type OTFTs, Adv. Mater., 2023, 35(20), 2300240, DOI:10.1002/adma.202300240.
- K. Yang, X. Zhang, A. Harbuzaru, L. Wang, Y. Wang, C. Koh, H. Guo, Y. Shi, J. Chen, H. Sun, K. Feng, M. C. Ruiz Delgado, H. Y. Woo, R. P. Ortiz and X. Guo, Stable Organic Diradicals Based on Fused Quinoidal Oligothiophene Imides with High Electrical Conductivity, J. Am. Chem. Soc., 2020, 142(9), 4329–4340, DOI:10.1021/jacs.9b12683.
- Y. Zhang, Y. Zheng, H. Zhou, M.-S. Miao, F. Wudl and T.-Q. Nguyen, Temperature Tunable Self-Doping in Stable Diradicaloid Thin-Film Devices, Adv. Mater., 2015, 27(45), 7412–7419, DOI:10.1002/adma.201502404.
- D. Yuan, Y. Guo, Y. Zeng, Q. Fan, J. Wang, Y. Yi and X. Zhu, Air-Stable n-Type Thermoelectric Materials Enabled by Organic Diradicaloids, Angew. Chem., Int. Ed., 2019, 58(15), 4958–4962, DOI:10.1002/anie.201814544.
- Y. Kobayashi, M. Yoshioka, K. Saigo, D. Hashizume and T. Ogura, Hydrogen-Bonding-Assisted Self-Doping in Tetrathiafulvalene (TTF) Conductor, J. Am. Chem. Soc., 2009, 131(29), 9995–10002, DOI:10.1021/ja809425b.
- Y. Kobayashi, M. Yoshioka, K. Saigo, D. Hashizume and T. Ogura, Hydrogen-Bonding Tetrathiafulvalene (TTF) Conductors: Carrier Generation by Self-Doping, Phys. B, 2010, 405(11, Supplement), S23–S26, DOI:10.1016/j.physb.2009.10.049.
- S.-C. Kim, J. Whitten, J. Kumar, F. F. Bruno and L. A. Samuelson, Self-Doped Carboxylated Polyaniline: Effect of Hydrogen Bonding on the Doping of Polymers, Macromol. Res., 2009, 17(9), 631–637, DOI:10.1007/BF03218921.
- Z. B. Henson, Y. Zhang, T.-Q. Nguyen, J. H. Seo and G. C. Bazan, Synthesis and Properties of Two Cationic Narrow Band Gap Conjugated Polyelectrolytes, J. Am. Chem. Soc., 2013, 135(11), 4163–4166, DOI:10.1021/ja400140d.
- D. Chandran and K.-S. Lee, Diketopyrrolopyrrole: A Versatile Building Block for Organic Photovoltaic Materials, Macromol. Res., 2013, 21(3), 272–283, DOI:10.1007/s13233-013-1141-3.
- M. Grzybowski and D. T. Gryko, Diketopyrrolopyrroles: Synthesis, Reactivity, and Optical Properties, Adv. Opt. Mater., 2015, 3(3), 280–320, DOI:10.1002/adom.201400559.
- Q. Liu, H. Sun, C. Blaikie, C. Caporale, S. Manzhos, K. Feron, J. M. MacLeod, M. Massi, S. E. Bottle, J. Bell, Y.-Y. Noh and P. Sonar, Naphthalene Flanked Diketopyrrolopyrrole Based Organic Semiconductors for High Performance Organic Field Effect Transistors, New J. Chem., 2018, 42(15), 12374–12385, 10.1039/C8NJ01453A.
- Q. Liu, S. Chavhan, H. Zhang, H. Sun, A. J. Brock, S. Manzhos, Y. Chen, K. Feron, S. E. Bottle, J. C. McMurtrie, J.-H. Jou, H.-S. Chen, M. R. Nagar, W. Hu, Y.-Y. Noh, Y. Zhen and P. Sonar, Short Alkyl Chain Engineering Modulation on Naphthalene Flanked Diketopyrrolopyrrole toward High-Performance Single Crystal Transistors and Organic Thin Film Displays, Adv. Electron. Mater., 2021, 7(1), 2000804, DOI:10.1002/aelm.202000804.
- Q. Liu, H. Sun, S. P. Ponnappa, K. Feron, S. Manzhos, M. W. M. Jones, S. E. Bottle, J. Bell, Y.-Y. Noh and P. Sonar, Naphthalene Flanked Diketopyrrolopyrrole: A New DPP Family Member and Its Comparative Optoelectronic Properties with Thiophene- and Furan- Flanked DPP Counterparts, Org. Electron., 2019, 74, 290–298, DOI:10.1016/j.orgel.2019.07.017.
- E. D. Głowacki, H. Coskun, M. A. Blood-Forsythe, U. Monkowius, L. Leonat, M. Grzybowski, D. Gryko, M. S. White, A. Aspuru-Guzik and N. S. Sariciftci, Hydrogen-Bonded Diketopyrrolopyrrole (DPP) Pigments as Organic Semiconductors, Org. Electron., 2014, 15(12), 3521–3528 CrossRef PubMed.
- A. Ruiz-Carretero, N. R. Á. Rovelo, S. Militzer and P. J. Mésini, Hydrogen-Bonded Diketopyrrolopyrrole Derivatives for Energy-Related Applications, J. Mater. Chem. A, 2019, 7(41), 23451–23475, 10.1039/C9TA05236D.
- J. Dhar, D. Prasad Karothu and S. Patil, Herringbone to Cofacial Solid State Packing via H-Bonding in Diketopyrrolopyrrole (DPP) Based Molecular Crystals: Influence on Charge Transport, Chem. Commun., 2015, 51(1), 97–100, 10.1039/C4CC06063F.
- N. R. Ávila-Rovelo, G. Martinez, W. Matsuda, S. Sinn, P. Lévêque, D. Schwaller, P. Mésini, S. Seki and A. Ruiz-Carretero, Hydrogen-Bonded Organic Semiconductors with Long Charge Carrier Lifetimes, J. Phys. Chem. C, 2022, 126(26), 10932–10939, DOI:10.1021/acs.jpcc.2c03105.
- Y. Liu, X. Tao, F. Wang, J. Shi, J. Sun, W. Yu, Y. Ren, D. Zou and M. Jiang, Intermolecular Hydrogen Bonds Induce Highly Emissive Excimers:
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Enhancement of Solid-State Luminescence, J. Phys. Chem. C, 2007, 111(17), 6544–6549, DOI:10.1021/jp070288f.
Enhancement of Solid-State Luminescence, J. Phys. Chem. C, 2007, 111(17), 6544–6549, DOI:10.1021/jp070288f. - R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, A General Relationship between Disorder, Aggregation and Charge Transport in Conjugated Polymers, Nat. Mater., 2013, 12(11), 1038–1044, DOI:10.1038/nmat3722.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, and DFT calculation results. See DOI: https://doi.org/10.1039/d4tc02496f |
| ‡ R.G.-N and G.M. contributed equally to the manuscript. |
| § Current affiliation: Institute for Chemistry and Processes for Energy, Environment and Health, ICPEES, UMR 7515, 25 Rue Becquerel, 67087, Strasbourg Cedex 2, France |
| ¶ Current affiliation: Instituto Madrileño de Estudios Avanzados, IMDEA Nanoscience, Calle Faraday 9, 28049, Madrid, Spain |
| This journal is © The Royal Society of Chemistry 2024 |