
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding the horizons for viable precursors and liquid fluxes for the synthesis of BaZrS3 and related compounds†
Kiruba Catherine
Vincent‡
 a,
Shubhanshu
Agarwal‡
a,
Zirui
Fan
a,
Alison Sofia Mesa
Canizales
ab and
Rakesh
Agrawal
*a
a,
Shubhanshu
Agarwal‡
a,
Zirui
Fan
a,
Alison Sofia Mesa
Canizales
ab and
Rakesh
Agrawal
*a
aDavidson School of Chemical Engineering, Purdue University, West Lafayette, IN 47907, USA. E-mail: agrawalr@purdue.edu
bChemical and Environmental Engineering, Universidad Nacional de Colombia, Bogota DC, Colombia
First published on 11th July 2024
Abstract
Chalcogenide perovskites represent a prominent class of emerging semiconductor materials for photovoltaic applications, boasting excellent optoelectronic properties, appropriate bandgaps, and remarkable stability. Among these, BaZrS3 is one of the most extensively studied chalcogenide perovskites. However, its synthesis typically demands high temperatures exceeding 900 °C. While recent advancements in solution-processing techniques have mitigated this challenge, they often rely on costly and difficult-to-find organometallic precursors. Furthermore, there is a notable gap in research regarding the influence of the Ba/Zr ratio on phase purity. Thus, our study explores solid-state reactions to investigate the impact of metal ratios and sulfur pressure on the phase purity of BaZrS3. Expanding upon this investigation, we aim to leverage cost-effective metal halide and metal sulfide precursors for the solution-based synthesis of BaMS3 (M = Ti, Zr, Hf) compounds. Additionally, we have devised a bilayer stacking approach to address the halide affinity of alkaline earth metals. Moreover, we introduce a novel solution-chemistry capable of dissolving alkaline earth metal sulfides, enabling the synthesis of BaMS3 compounds from metal sulfide precursors. While the BaSx liquid flux has shown promise, we identify the selenium liquid flux as an alternative method for synthesizing BaMS3 compounds.
1. Introduction
Over the past two decades, there has been a notable surge in the development of new solar absorbers for photovoltaic applications. Lead halide perovskites have been particularly exciting due to their optoelectronic properties and the efficiencies they have achieved in photovoltaic devices.1–3 However, they often suffer from increased instability to air, moisture, and heat, rendering them unsuitable for large-scale deployment in their current form.4 Chalcogenide semiconductor materials, such as Cu(In,Ga)Se2 and Cu2ZnSnSe2, have also shown promise as photovoltaic materials with suitable bandgaps for single-junction applications, but they lag behind lead halide perovskites in terms of efficiencies.5,6 Nevertheless, these materials offer increased stability to air, moisture, and thermal degradation compared to lead halide perovskites.7 They could also be readily mass-produced and deployed in photovoltaic farms, provided their device efficiencies are improved.The possible research avenues in solar absorber research include improving the stability of halide perovskites, enhancing the device efficiencies of existing chalcogenide materials, or exploring stable, high-performing alternatives to lead halide perovskites. The latter has drawn the attention of the photovoltaic research community toward chalcogenide perovskites. These materials crystallize in a similar ABX3 structure as lead halide perovskites, where A represents an alkaline earth metal (Ca, Sr, Ba), B denotes an early transition metal (Ti, Zr, Hf), and X is predominantly sulfur. While the Zr and Hf versions crystallize in a distorted perovskite structure, with the [BX6]2− octahedra slightly tilted, the Ti versions crystallize in a distinct hexagonal structure.8–10 Chalcogenide perovskites exhibit extremely high light absorption coefficients (>105 cm−1), surpassing even lead halide perovskites, high dielectric constants, reasonable carrier mobilities, and strong photoluminescence.8,11–13 Some of them feature suitable bandgaps for the top layer in tandem solar cell applications, while others show promise for solar water splitting and light-emitting diode applications.8,12–15 ATiS3 compounds possess a quasi-1-D structure with low thermal conductivity, making them promising candidates for thermoelectric applications.16,17
Despite their considerable promise, progress in researching these materials has been slow, primarily due to the high synthesis temperatures, often exceeding 900 °C.12,18–21 Among these materials, BaZrS3 has been the focus of much attention owing to its particularly suitable bandgap for photovoltaic applications and its synthesis at relatively lower temperatures than other chalcogenide perovskites.22,23 However, research on BaZrS3 has predominantly centered on solid-state forms, with few reports on film synthesis, most of which rely on high-temperature vacuum deposition and annealing processes, which are less appealing.23–26
Recent studies from our group and others have made significant strides in lowering the synthesis temperature of BaMS3 (M = Ti, Zr, Hf) compounds through moderate-temperature solution processing. These methods use a BaSx liquid flux in a sulfur-rich environment but still rely on complex and expensive organometallic precursors for film casting.27–31 Historically, solution-processed synthesis of chalcogenide materials has utilized cost-effective metal salts and metal sulfide precursors.32–35 However, challenges such as the high oxophilicity of transition metals, the strong affinity of alkaline earth metals for halides, and the low solubility of alkaline-earth and transition metal sulfides in traditional solution chemistries have complicated these efforts.30,36,37 A recent study from our group addressed the issue of oxophilicity by utilizing an HfH2-based oxygen trap to remove oxide impurities from the film and the ampule environment during synthesis.38 However, other challenges persist, requiring further investigation.
In this study, we have developed innovative approaches to address the halide affinity of alkaline-earth metals and synthesize BaMS3 compounds from cost-effective metal halide precursors. We also demonstrate chemistry to dissolve alkaline-earth metal sulfides and propose a strategy to utilize metal sulfides for casting BaMS3 films. Additionally, this study provides an example of using a selenium liquid flux, which could facilitate the synthesis of BaMS3 and related compounds. Our study explores cost-effective solution-deposition routes for BaMS3 synthesis by leveraging critical insights from previous reports.
2. Results and discussion
2.1. Does BaZrS3 allow off-stoichiometric compositions?
Chalcogenide semiconductors like Cu(In,Ga)Se2 and Cu2ZnSnSe4 exhibit off-stoichiometry in their structure, where Cu-poor compositions up to a certain I/III or I/(II + IV) ratio do not induce phase segregation.39,40 This is significant because Cu vacancies are shallow acceptor defects, imparting p-type conductivity to these materials.41 Notably, the record efficiency Cu(In,Ga)Se2 device possesses a Cu/(In + Ga) ratio of 0.93.42 Conversely, Cu-rich compositions often result in secondary Cu2Se impurities, which may pose challenges if not easily eliminated.41 This spurred our investigation into off-stoichiometric compositions in BaZrS3, which remains largely unexplored. Similar to Cu-poor compositions in Cu(In,Ga)Se2, Ba-poor compositions could potentially alter the carrier concentrations in BaZrS3. The defects arising from such conditions, including VBa, ZrBa, SBa, and other defect complexes, warrant thorough examination. Preliminary indications from defect calculations suggest that these defects are either shallow or possess high defect formation energies.43,44
Table 1 enumerates the powder reactions where we mixed BaS and ZrS2 in different molar ratios and sulfurized at 575 °C for 12 h in the presence of sulfur vapor. It is evident from Table 1 and Fig. S1 (ESI†) that increasing the Ba:Zr ratio above 1 in the presence of excess sulfur led to the formation of the BaS3 secondary phase alongside crystalline BaZrS3. This indicates that BaZrS3 does not accommodate Zr-poor off-stoichiometries, perhaps because VZr would disrupt the [ZrS6]2− octahedra and destabilize the BaZrS3 crystal lattice. It is plausible that in Ba-rich compositions, barium polysulfide was initially formed, but it decomposed into BaS3 and sulfur upon cooling.27 Consequently, crystalline BaS3 and BaZrS3 were observed under barium-rich conditions. Previous studies have demonstrated the solubility of BaS3 in water.28 Thus, a gentle water wash could eliminate all BaS3 impurities from the sample. In our investigation, we followed a water-washing procedure outlined in the experimental section, resulting in the phase-pure synthesis of BaZrS3, as validated by X-ray diffraction in Fig. 1(a) and Raman spectroscopy in Fig. S2 (ESI†). Regarding the Ba-poor compositions, no secondary phases were observed for Ba:Zr ∼ 0.9, 0.8, and 0.7, yielding phase-pure BaZrS3 as confirmed by X-ray diffraction and Raman spectroscopy. However, a further decrease to 0.6 metal ratio resulted in slight ZrS3 impurities, evident in the X-ray diffraction spectra and Raman analysis (see Fig. 1(b) and Fig. S3, ESI†). As the Ba:Zr ratio decreased, the prevalence of secondary phases increased as anticipated. Interestingly, our study indicates that BaZrS3 can accommodate Ba-poor compositions up to Ba:Zr ∼ 0.7. It would be intriguing to investigate the impact of these compositions on the optoelectronic properties, although this falls beyond the scope of our current work. The off-stoichiometry of BaZrS3 could potentially impact the carrier concentration, which may vary depending on the Ba![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zr ratio in the material. Consequently, a wide range of carrier concentrations and both p-type and n-type conductivities have been reported for BaZrS3.12,45 Nevertheless, these findings will be helpful in the subsequent section, where we will elaborate on our approaches to fabricating BaZrS3 films.
:Zr ratio in the material. Consequently, a wide range of carrier concentrations and both p-type and n-type conductivities have been reported for BaZrS3.12,45 Nevertheless, these findings will be helpful in the subsequent section, where we will elaborate on our approaches to fabricating BaZrS3 films.
| S. no. | Ba:Zr (molar ratio) |
X-ray diffraction phases (without water wash) | Raman phases (without water wash) | Estimated sulfur pressure (atm) |
|---|---|---|---|---|
| 1 | 2:1 |
BaZrS3 + BaS3 | BaZrS3 + BaS3 | 0.79 |
| 2 | 1.5:1 |
BaZrS3 + BaS3 | BaZrS3 + BaS3 | 0.79 |
| 3 | 1:1 |
BaZrS3 | BaZrS3 | 0.79 |
| 4 | 0.9:1 |
BaZrS3 | BaZrS3 | 0.79 |
| 5 | 0.8:1 |
BaZrS3 | BaZrS3 | 0.79 |
| 6 | 0.7:1 |
BaZrS3 | BaZrS3 | 0.79 |
| 7 | 0.6:1 |
BaZrS3 + ZrS3 | BaZrS3 + ZrS3 | 0.79 |
| 8 | 0.5:1 |
BaZrS3 + ZrS3 | BaZrS3 + ZrS3 | 0.79 |
| 9 | 0.4:1 |
ZrS3 + BaZrS3 | ZrS3 | 0.79 |
| 10 | 1.5:1 |
BaZrS3 + BaS3 | BaZrS3 + BaS3 | 0.79 |
| 11 | 1.5:1 |
BaZrS3 + BaS3 | BaZrS3 + BaS3 | 0.59 |
| 12 | 1.5:1 |
BaZrS3 + BaS3 + Ba3Zr2S7 | BaZrS3 + BaS3 + Ba3Zr2S7 | 0.46 |
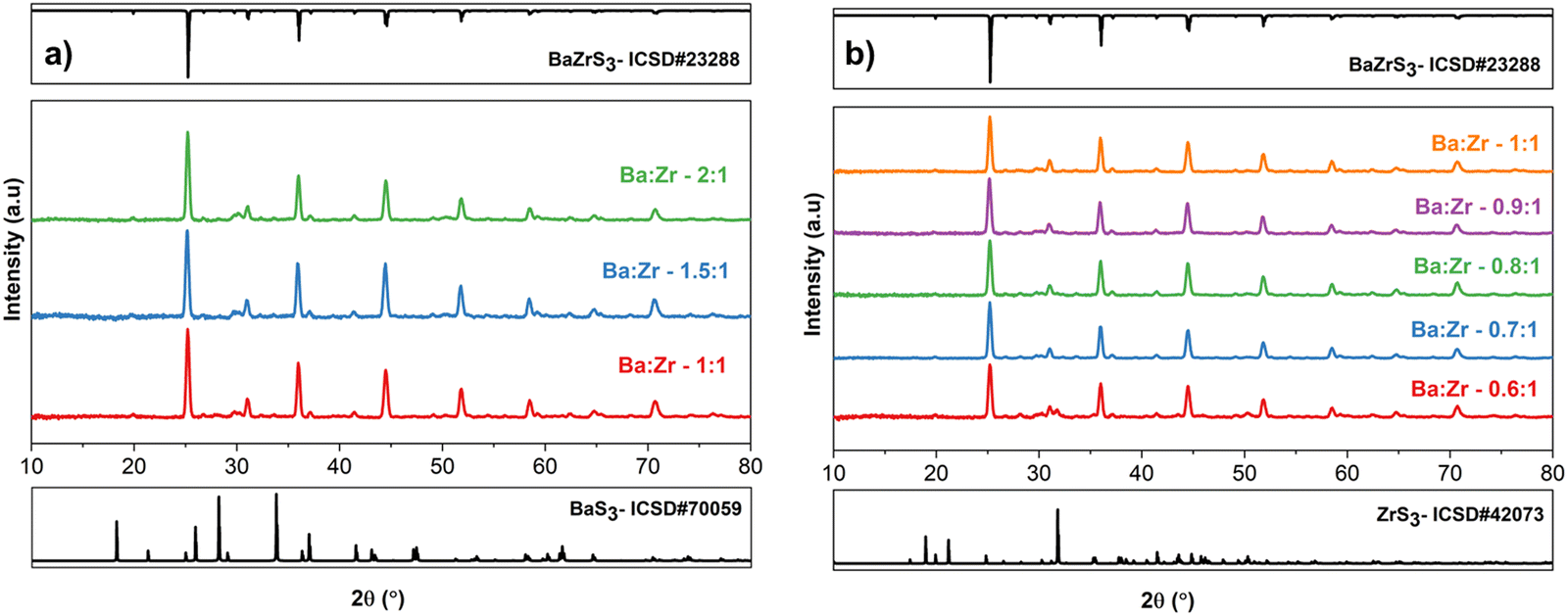 | ||
| Fig. 1 (a) and (b) X-ray diffraction pattern of the Ba–Zr–S powder synthesized with different Ba:Zr ratios at 575 °C for 12 h with a sulfur pressure of 0.79 atm. BaS and ZrS2 were used as barium and zirconium precursors. The synthesized powders in (a) were water-washed to remove any water-soluble impurities (BaS3). The sample with Ba:Zr 0.6:1 in (b) has ZrS3 impurities. | ||
It is essential to highlight that compositions with excess Ba under sulfur-poor conditions were also investigated. Despite the common expectation that Ruddlesden–Popper (RP) phases of BaZrS3 would form in Ba-excess compositions due to their formation energies being similar to those of the perovskite phase,46 our previous discussion elucidated that BaZrS3, along with BaS3, was formed in Ba-excess compositions under sulfur-rich conditions. However, as the sulfur pressure decreased during synthesis, RP phases, notably Ba3Zr2S7, reported to be the most stable among all Ba–Zr–S RP phases,45,47 emerged alongside BaZrS3, as confirmed by X-ray diffraction and Raman spectroscopy (see Fig. S4 and S5, ESI†). At even lower sulfur pressures, starting binaries remained partially unreacted. Hence, it is imperative to operate under sulfur-excess conditions to achieve phase-pure BaZrS3.
These observations indicate that, in a saturated sulfur environment with barium above stochiometric amount, BaZrS3 does not accommodate Zr-deficient off-stoichiometric compounds. Instead, it leads to the formation of BaZrS3 and BaS3 secondary phases. Conversely, Ba-deficient off-stoichiometric compounds appear stable up to a Ba:Zr ratio of 0.7. Building on these findings, we have developed several solution processing routes for the synthesis of chalcogenide perovskites.
2.2. Stacked binary metal sulfides enabling the use of transition metal halides
Metal halides are widely available and relatively inexpensive, making them preferred precursors for many material systems. For instance, metal chlorides of certain late and post-transition elements, dissolved in dimethyl formamide (DMF) with thiourea as a sulfur source, can be utilized to synthesize metal sulfide semiconductors.33,48 However, the affinity of barium and other alkaline earth metals for chlorine hindered our group's initial attempts to use these compounds in synthesizing BaZrS3. As further confirmation, we investigated using a dimethylformamide ink containing BaCl2, ZrCl4, and thiourea. As anticipated, the resulting film contained BaCl2 and ZrS3 after sulfurization, as depicted in Fig. S6 (ESI†), underscoring barium's affinity for chlorine. We encountered similar challenges with bromide and iodide precursors, discouraging using halides for mixed precursor inks. A potential alternative to bypass alkaline earth element's chlorine affinity involves employing a bilayer stacking approach with BaSx/ZrSx films, where ZrSx could be synthesized from zirconium halide precursors and then sulfurized to achieve BaZrS3.While some researchers previously regarded ZrS3 as a dissociation product of BaZrS3, our earlier suggestion posited ZrS3 as a feasible zirconium precursor for synthesizing BaZrS3.27 In this scenario, ZrS3 could undergo a reaction with BaS in the presence of excess sulfur, leading to the formation of BaZrS3.27 Based on this, our proposed method involves creating a binary sulfide stack with a bottom layer of ZrS3 and a top layer of BaS, illustrated in Fig. 2(a). Subsequently, this stack is heated in a sulfur-rich environment to yield BaZrS3.
 | ||
| Fig. 2 (a) Reaction schematic depicting the synthesis of BaMS3 by layering binary sulfides. (b) Various molecular precursor chemistries employed for depositing ZrS3 film. (c) Plot comparing the X-ray diffraction patterns of ZrS3 film prepared using different solution chemistries, (i) ZrCl4 + thiourea (TU) + dimethyl formamide (DMF), (ii) ZrCl4 + TU + butylamine (BA), (iii) ZrCl4 + 2-methyl-2-propanethiol (MePT) + BA and (iv) ZrCl4 + BA + CS2 + pyridine (Pyd). | ||
We dissolved Zr, Hf, and Ti halides using four distinct solution chemistries, resulting in metal–sulfur coordinated complexes, as depicted in Fig. 2(b). The confirmation of metal–sulfur interactions in these chemistries was studied through liquid Raman and 1H-NMR (nuclear magnetic resonance) analyses. While amine–carbon disulfide chemistry has been extensively studied for dissolving various metal oxides, metal acetylacetonates, and metal hydroxides, limited research has focused on the dissolution of metal halides, particularly those of early transition metals. Liquid Raman analysis of a 0.3 M solution of ZrCl4 in butylamine–CS2–pyridine (Fig. S7, ESI†) revealed broadening of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S peak at 656 cm−1, indicating potential metal–sulfur interaction, alongside a new peak at 2648 cm−1, possibly attributed to the S–H bond from CS2 insertion in butylamine. Similarly, for ZrCl4 in 2-methyl-2-propanethiol-butylamine solution, Fig. S8 (ESI†) shows a slight shift and broadening in the C–S peak at 590 cm−1 and the S–H peak at 2577 cm−1, indicating sulfur coordination to the zirconium through a dative bond.49 However, the observed peak shift was minimal, and the ratio of the S–H peak to the C–S peak did not decrease significantly, suggesting incomplete substitution of chlorine with thiolate due to thiolate formation. This finding aligns with observations by Murria et al., who reported similar incomplete substitution when dissolving copper chlorides in amine–thiol systems.32
S peak at 656 cm−1, indicating potential metal–sulfur interaction, alongside a new peak at 2648 cm−1, possibly attributed to the S–H bond from CS2 insertion in butylamine. Similarly, for ZrCl4 in 2-methyl-2-propanethiol-butylamine solution, Fig. S8 (ESI†) shows a slight shift and broadening in the C–S peak at 590 cm−1 and the S–H peak at 2577 cm−1, indicating sulfur coordination to the zirconium through a dative bond.49 However, the observed peak shift was minimal, and the ratio of the S–H peak to the C–S peak did not decrease significantly, suggesting incomplete substitution of chlorine with thiolate due to thiolate formation. This finding aligns with observations by Murria et al., who reported similar incomplete substitution when dissolving copper chlorides in amine–thiol systems.32
1H-NMR studies also show a likely metal–sulfur interaction between ZrCl4 and thiol (Fig. S9, ESI†). The Zr–Cl–thiolate, however, did not dissolve significantly in pyridine-d5 initially. But, adding a stoichiometric amount of butylamine resulted in complete dissolution with a broadening in the 1H-NMR peak of H bounded to sulfur, hinting towards some interaction between butylamine and 2-methyl-2-propanethiol likely forming [CH3(CH2)3NH4]+[SC(CH3)3]−. The 1H-NMR results in Fig. S10 (ESI†) show the broadening of the thiol proton peak after adding ZrCl4, but the peak did not disappear. Notably, exchangeable protons, such as those in the thiol group, are not always accurately quantified through peak integration. The peak broadening indicates a dynamic thiol proton. We hypothesize that this suggests the thiolate is in equilibrium between its protonated and deprotonated forms. When it is deprotonated, we expect it to be bound to the Zr4+. This observation could also indicate that not all Zr–Cl bonds underwent replacement with thiol; alternatively, it might have resulted in the formation of an adduct.
Interactions were also observed in the ZrCl4–thiourea–dimethyl formamide and ZrCl4–thiourea–butylamine systems, leading to shifts and broadening in the CS Raman peak, as illustrated in Fig. 3 and 4. Similar dissolutions can also be achieved with ZrBr4 and ZrI4, yielding soluble complexes. All four Zr–S coordinated solutions were then doctor blade-coated onto alumina-coated Eagle XG glass substrates and annealed at 350 °C on a hot plate to remove the excess solvent, resulting in an amorphous ZrSx compound. Subsequently, the resulting film underwent sulfurization in an ampule containing HfH2 and sulfur for 6 h at 575 °C to form a ZrS3 film, as depicted in the X-ray diffraction patterns and Raman spectra in Fig. 2(c) and Fig. S11 (ESI†). The role of HfH2 in removing any residual oxygen in the film is elaborated in our previous work.38 It is important to note that although ZrS3 could be synthesized at much lower temperatures of 475 °C, we chose 6 h at 575 °C to ensure complete removal of residual oxygen (see Fig. S12, ESI†). Further optimization strategies such as rigorous solvent drying, use of anhydrous precursors, processing in ultra-oxygen-free glove boxes, and avoiding exposure to air during the entire processing could significantly reduce the synthesis time and temperature. The X-ray diffraction patterns and Raman spectroscopy confirmed the production of phase-pure ZrS3 in all cases. The secondary phase of ZrO2 was eliminated with prolonged sulfurization time and careful handling of precursors and solutions. Similar to previous studies, the ZrS3 films exhibited non-continuous morphology due to the ribbon structure of ZrS3 (refer to Fig. 5(b) and Fig. S13, S14, ESI†).38 Energy dispersive X-ray spectroscopy (EDX) maps in Fig. S15 and S16 (ESI†) confirm the uniform distribution of Zr and S in the grains. Furthermore, ZrS3 synthesis from soluble inks of ZrBr4 and ZrI4 was also demonstrated (see Fig. S17, ESI†). It is worth noting that after annealing the film on the hot plate, trace amounts of residual chlorine, bromine, or iodine were detected in the film via X-ray Fluorescence measurements, but no halide impurities remained after sulfurization.
 | ||
| Fig. 3 Liquid Raman on only (a) dimethyl formamide (DMF), (b) thiourea (TU) + DMF, and (c) TU + DMF + ZrCl4 (0.3 M). The peak shift in the CS bond stretch of TU suggests a metal–sulfur interaction. (ii) presents the zoomed-in representation of (i). | ||
 | ||
| Fig. 4 Liquid Raman on only (a) butylamine (BA), (b) thiourea (TU) + BA, (c) TU (2 M) + BA + ZrCl4 (0.5 M), and (d) TU (3 M) + BA + ZrCl4 (0.5 M). The peak shift in the CS bond stretch of TU suggests a metal–sulfur interaction. (ii) presents the zoomed-in representation of (i). | ||
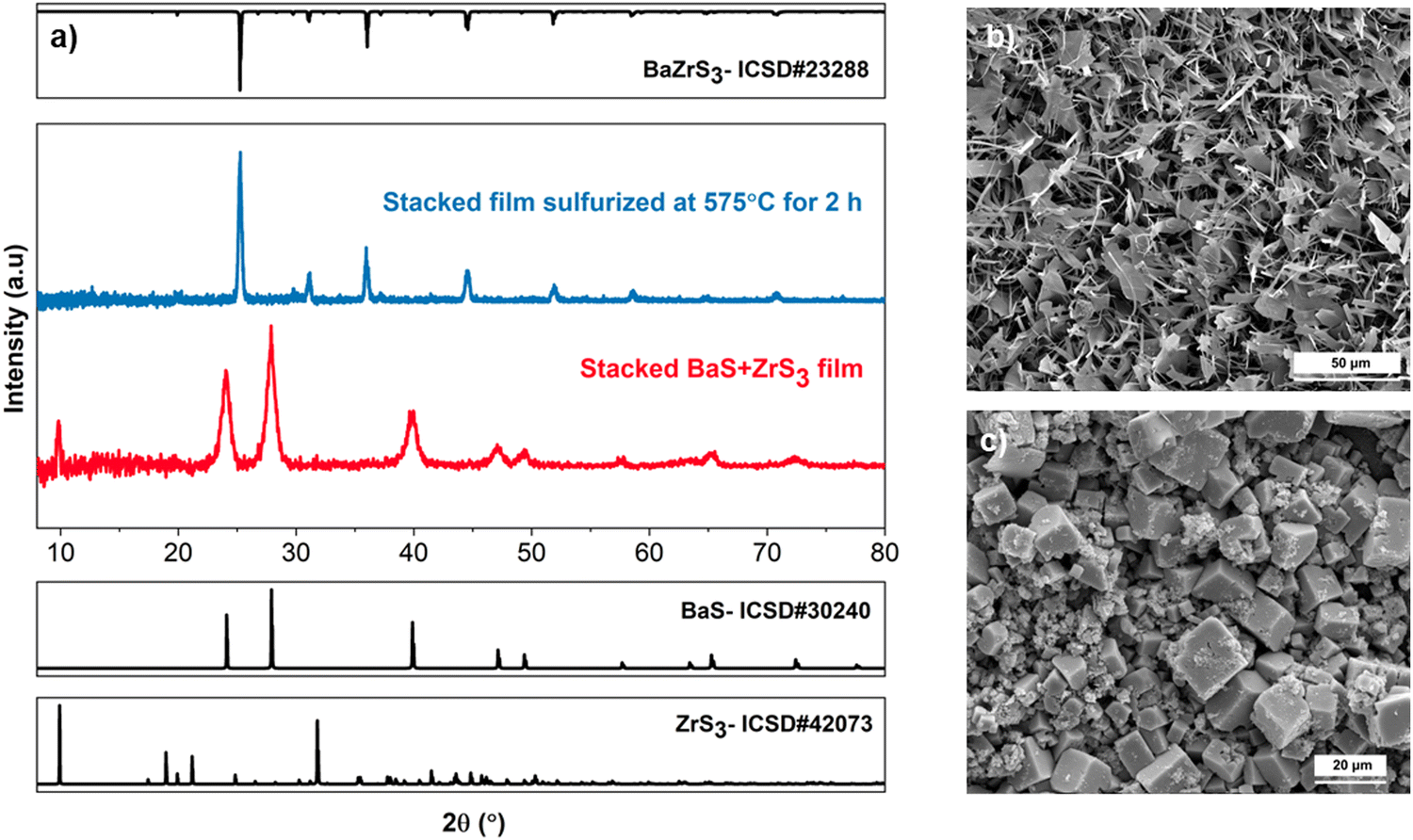 | ||
| Fig. 5 (a) X-ray diffraction pattern showcasing the as-annealed stacked bilayer film of ZrS3 and BaS before ampule sulfurization and the BaZrS3 film after sulfurization. (b) SEM top-view image depicting the ZrS3 film prepared from the ZrCl4–butylamine–CS2–pyridine solution chemistry. (c) SEM top-view image illustrating the BaZrS3 film derived from the ZrS3–BaS bilayer stack. | ||
Subsequently, a thin layer of BaS was coated onto these ZrS3 films using the soluble barium thiolate ink obtained by dissolving Bis(pentamethylcyclopentadienyl)barium (Cp*2Ba) in 2-methyl-2-propanethiol and butylamine (see experimental section for details), and the resulting stacked film of BaS/ZrS3 was sulfurized with sulfur and HfH2. The as-annealed stacked films exhibit crystalline phases of both BaS and ZrS3, as depicted in Fig. 5(a) and Fig. S18 (ESI†). While care was taken to ensure that approximately equal amounts of Ba and Zr molar ratios were present in the film by maintaining similar concentrations of Ba and Zr inks, previous discussions in the study indicate that a barium-excess film could also be utilized. This is because it would form BaZrS3 and BaS3 upon sulfurization, and the BaS3 component could be easily washed away. During sulfurization at 575 °C, this process utilized the BaSx liquid flux to accelerate mass transfer, and although shorter times should be feasible, sulfurization was performed for 2 h to minimize undesirable oxide phases. As illustrated in Fig. 5(a) and Fig. S18, S19 (ESI†), we obtained a pure BaZrS3 phase, as confirmed by X-ray diffraction and Raman spectroscopy. The diffuse reflectance spectroscopy also confirmed a bandgap of around 1.85 eV (see Fig. S20, ESI†). Nevertheless, the film exhibited cracking and isolated grains (see Fig. 5(c) and Fig. S21, ESI†), suggesting a need for further research to optimally control the BaSx liquid flux. However, this could be an enticing way to make micron-scale single crystals without residual impurities. EDX analysis confirmed the uniform distribution of Ba, Zr, and S, as shown in Fig. S22 and S23 (ESI†). Additionally, the BaZrS3 grains show no residual chlorine (see Fig. S24, ESI†). This versatile method can be readily applied to Ba–Hf–S and Ba–Ti–S systems, yielding Ba6Hf5S16 and BaTiS3, as demonstrated in Fig. S25–S27 (ESI†). However, achieving stoichiometric BaHfS3 using this route proved challenging due to the competing Ruddlesden–Popper phases of the Ba–Hf–S system. In all the cases, an equal number of layers with equally concentrated barium and zirconium/hafnium/titanium solutions were blade-coated to ensure a nearly stoichiometric film.
The success of this approach is noteworthy as it enables the utilization of easily manageable and cost-effective halide precursors. While our primary focus was on one stacking method utilizing the halide precursors, alternative approaches could also be explored. In another instance, we applied a ZrCl4-containing ink at the bottom to form an amorphous ZrSx layer without sulfurizing it to ZrS3. The coated film underwent annealing at 450 °C for 30 minutes to eliminate residual chlorine. Subsequently, BaS was applied to the top using the barium thiolate solution, and the stacked film was sulfurized, resulting in BaZrS3, as shown in Fig. S28 (ESI†). In an alternate scenario, BaS was coated at the bottom, and ZrCl4-containing ink was applied at the top. After annealing and sulfurization of this stack, Ba3Zr2S7 was obtained, albeit with significant secondary phases, including BaCl2 and ZrS3, as illustrated in Fig. S29 (ESI†). This outcome was anticipated, as barium likely reacted with chlorine during the annealing process of the stacked film on the hot plate. These findings underscore the significance of selecting suitable precursors and optimizing the sequence of processing steps to establish a thermodynamic driving force for synthesizing chalcogenide perovskites.
2.3. Hybrid colloidal molecular precursor inks
In addition to pure metals, metal chalcogenides are preferred as low-cost, anionic, impurity-free metal precursors for solution-processed chalcogenide thin films.50,51 Employing metal sulfides as precursors for synthesis would restrict the incorporation of unwanted anionic impurities and offer a cost-effective, streamlined route to synthesize BaMS3 compounds. However, the dissolution of alkaline earth metal sulfides and transition metal sulfides has historically been challenging, with no reports of ZrS2 or ZrS3 dissolution in any solution chemistry and only one report on co-dissolution of BaS alongside Cu2S and SnO in ethylenediamine–ethanedithiol (EDA–EDT) at 60 °C for 11 days.52 This dissolution was arduous, and attempts to dissolve only BaS in EDA–EDT were unsuccessful in our experiments, even after multiple attempts. Consequently, further investigation was necessary to identify solution chemistry capable of dissolving alkaline earth metal sulfides. As a result, various combinations of amine–thiol with different concentrations were explored, yet no success was achieved in room-temperature dissolutions. Notable solution chemistries such as hydrazine–sulfur also failed to dissolve BaS and ZrS2.However, combining amine and CS2 to produce alkyldithiocarbamic acid opened new possibilities for reactive solvent systems as we investigated this less-studied chemistry. Previously, butyldithiocarbamic acid had been shown to dissolve metal salts such as metal oxides,34 but we notably extended this chemistry by dissolving alkaline earth metal sulfides in propyldithiocarbamic acid in pyridine, resulting in a clear solution. This marks the first instance of standalone dissolution of BaS and SrS in solution chemistry and could play a pivotal role in the solution-processed synthesis of several alkaline earth metal-based chalcogenide compounds, including BaMS3 chalcogenide perovskites, Cu2BaSnS4, and Cu2SrSnS4. As a side note, due to the current interest in forming thin films of Cu2BaSnS4 for solar cells,53,54 we demonstrate solution-deposited synthesis of Cu2BaSnS4 using this chemistry in Fig. S30 (ESI†), where we co-dissolved Cu2S, BaS, and Sn in propylamine–CS2–pyridine to create a mixed precursor ink.
While our newly enhanced solution chemistry for BaS could be used with the previously described routes for homogeneous precursor inks and stacked films as Ba source, here we describe a new hybrid molecular precursor route employing dissolved BaS and colloidal suspension of ZrS3. ZrS2 and ZrS3 are van der Waals bonded 2D materials, suggesting the potential for colloidal suspensions in a suitable solvent. However, ZrS2 is prone to oxidation upon exposure to air, prompting us to utilize ZrS3 as the Zr precursor. Compared to a complete molecular precursor approach, colloidal particles in ink could facilitate nucleation in the coating process, leading to favorable film morphology. The hybrid molecular precursor route featuring dissolved BaS in an amine–CS2 reactive solvent system with pyridine as the bulk solvent and colloidal ZrS3 flakes dispersed also builds upon the concept of ZrS3 as a plausible precursor outlined in our previous publication.27 The resulting ink underwent sonication for several days to achieve a homogeneous dispersion solution, with the Ba:Zr ratio adjusted to be stoichiometric. Subsequently, the ink was coated and sulfurized (refer to Fig. 6(a) for a schematic of the synthesis process). Despite the reasonable air stability of BaS and ZrS3, the solution was coated in an oxygen-free glovebox in this study. The as-coated film contained both BaS and ZrS3, as confirmed in Fig. 6(b), and upon sulfurization, this method yielded phase-pure BaZrS3 at 575 °C (refer to Fig. 6(c) and Fig. S31, ESI†). EDX measurements of this film (depicted in Fig. S32, ESI†) verified the presence of Ba, Zr, and S in the grains. However, we observed that the size of the ZrS3 colloidal particles influenced the reaction time. In one instance, ZrS3 powder was synthesized by sulfurizing Zr nanopowder, while in another case, bulk ZrS2 powder was sulfurized to form ZrS3 powder, both at 575 °C for 18 h. The average grain size of ZrS3 produced from Zr nanopowder was significantly smaller than that from ZrS2 powder, resulting in a faster reaction to form BaZrS3. With the larger ZrS3 particles, we observed some unreacted BaS3 and ZrS3, along with BaZrS3, in the film (refer to Fig. S33, ESI†). However, in both scenarios, the targeted continuous thin film was not achieved, leading to the formation of isolated large grains of BaZrS3 (as indicated in the inset of Fig. 6(c) and Fig. S34, ESI†) rather than the intended continuous thin film. As previously noted, the overgrowth to form material islands is a common challenge for methods relying on a BaSx liquid flux. Therefore, controlling the quantity of liquid flux is crucial for this approach. Additionally, an alternative synthesis of ZrS3 to produce smaller particles in the suspension could further decrease reaction time and improve film morphology. Nevertheless, the diffuse reflectance measurements confirmed a bandgap of 1.86 eV for the BaZrS3 film, promising its potential utilization in tandem solar cell applications (depicted in Fig. 6(d)). Notably, this method was successfully extended for synthesizing related chalcogenide perovskite BaHfS3 and hexagonal BaTiS3, as confirmed by the X-ray diffraction and Raman spectra (see Fig. 6(a), 7(a) and S35, S36, ESI†). Moreover, the diffuse reflectance indicated a bandgap of approximately 2.16 eV for BaHfS3, promising its potential utility in optoelectronic applications (shown in Fig. 7(b)).
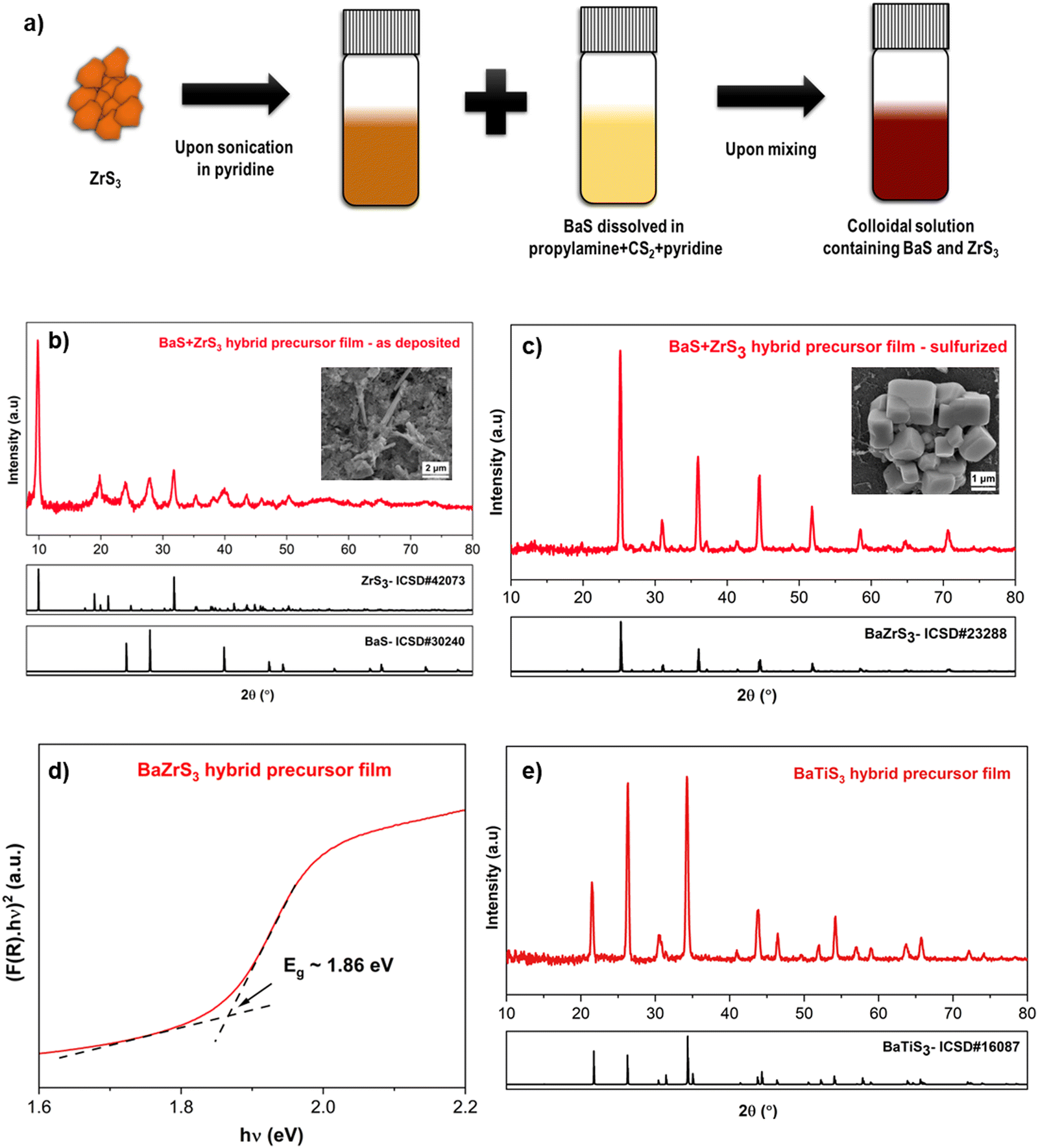 | ||
| Fig. 6 (a) Reaction schematic illustrating the preparation process of a hybrid colloidal molecular precursor ink using binary metal sulfides. (b) X-ray diffraction pattern of the as-coated hybrid precursor film, with an inset displaying the SEM top view of the as-annealed film. (c) X-ray diffraction pattern of the BaZrS3 film obtained after the sulfurization of the as-coated film, with an inset showing the SEM top view of the sulfurized film. (d) Kubelka–Munk transformation applied to the diffuse reflectance spectra of the BaZrS3 film derived from hybrid precursor film (considering direct bandgap)8 (e) X-ray diffraction pattern of the BaTiS3 film obtained after sulfurizing the as-coated BaS–TiS2 film from the hybrid precursor route. | ||
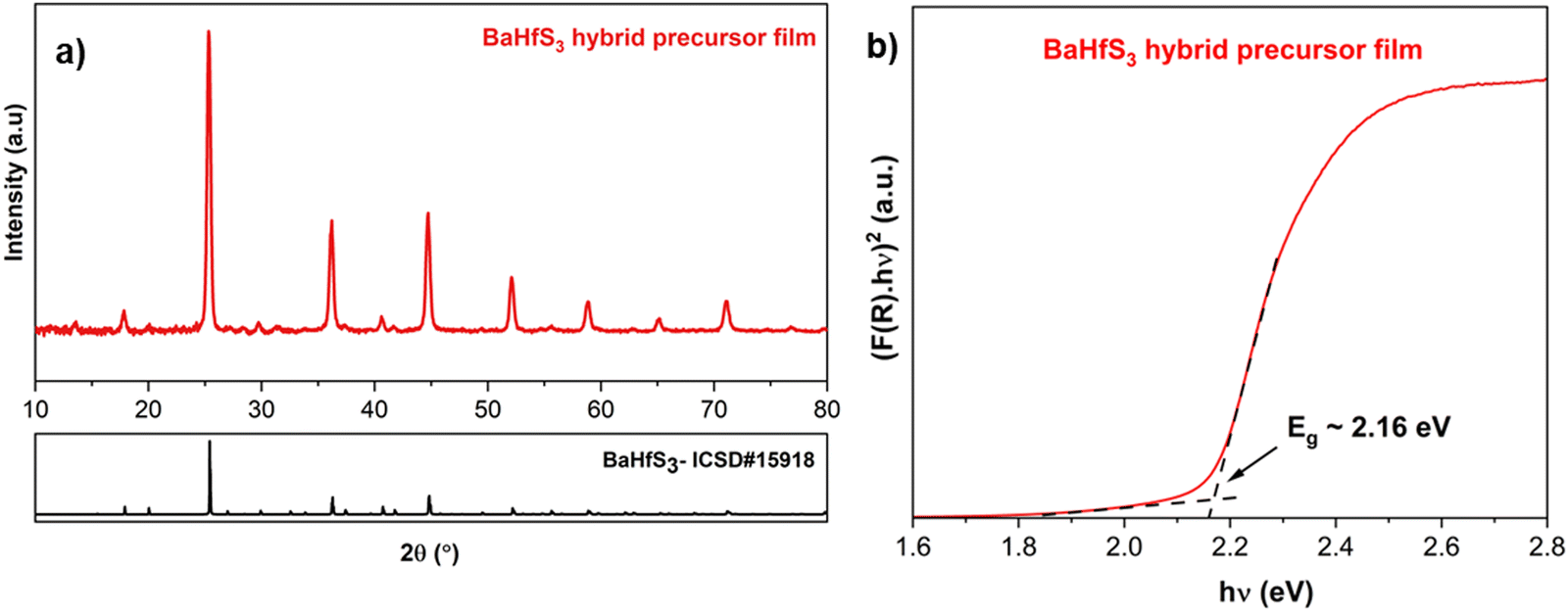 | ||
| Fig. 7 (a) X-ray diffraction pattern of the BaHfS3 film obtained after sulfurizing the as-coated BaS–HfS3 film from the hybrid precursor route. (b) Kubelka–Munk transformation applied to the diffuse reflectance spectra of the BaHfS3 film derived from hybrid precursor film (considering direct bandgap).8 | ||
This study represents the first account of a pathway solely employing metal sulfide precursors for the solution-processed synthesis of BaZrS3 and other related compounds. Furthermore, the dissolution of BaS in propylamine–CS2 exhibits novelty. Subsequent studies could explore solution chemistry aimed at co-dissolving BaS and ZrS2/ZrS3 to establish a fully dissolved molecular precursor route, which would also aid in limiting excess BaSx flux and controlling grain growth.
2.4. Utilizing selenium liquid flux: solid-state synthesis of BaMS3
From the above discussions, it is evident that a liquid flux is paramount in synthesizing chalcogenide perovskites at lower temperatures. BaSx liquid flux has been pivotal in the previously discussed solution-processing routes for synthesizing BaMS3 compounds. This section explores other potential liquid fluxes for synthesizing BaMS3 and related compounds. Prior studies on chalcogenide materials have employed various liquid fluxes to lower synthesis temperatures. Examples include Cu2−xSe flux in Cu-based chalcogenide semiconductors, Sb2S3 liquid flux in antimony-based semiconductors, and BaSx in barium-based chalcogenide semiconductors.27,29,55–57 As mentioned earlier, an ideal liquid flux should meet strict requirements: it should be in a liquid state at the reaction temperature, partially dissolve the metal precursors to reduce diffusion barriers, and avoid reacting with precursors to form undesired secondary phases. A selenium liquid flux can likely fulfill these requirements—it remains liquid above 220 °C, partially dissolves metal sulfides, and the instability of BaMSe3 compounds could potentially prevent the formation of ternary selenide secondary phases. As a proof of concept, solid-state samples were synthesized by combining BaS, ZrS2, and elemental Se in a sealed ampule and heating to a target temperature (refer to Fig. 8(a)). X-ray diffraction measurements from samples quenched at different temperatures showed that BaS and ZrS2 dissolved in the selenium liquid, as evidenced by the absence of BaS diffraction peaks at 475 °C and diminished ZrS2 peaks. However, the emergence of BaSe3 peaks indicated the partial breakdown of BaS to form BaSe3. As the temperature increased to 530 °C, intense peaks of BaZrS3 were observed, accompanied by a diminished secondary phase of BaSe3. This increase in BaZrS3 intensity continued to 575 °C, see Fig. 8(b). One hypothesis for this success is that BaS and ZrS2 do not significantly break down into BaSe/BaSe3 and ZrSe2/ZrSe3 at the temperatures used. If they did, at least some byproduct sulfur would have escaped to the vapor phase, resulting in insufficient sulfur for BaZrS3 formation. The absence of significant binary selenides supports this hypothesis—binary sulfides simply dissolve as binaries and reconstitute as BaZrS3 without the need for additional sulfur in the mixture.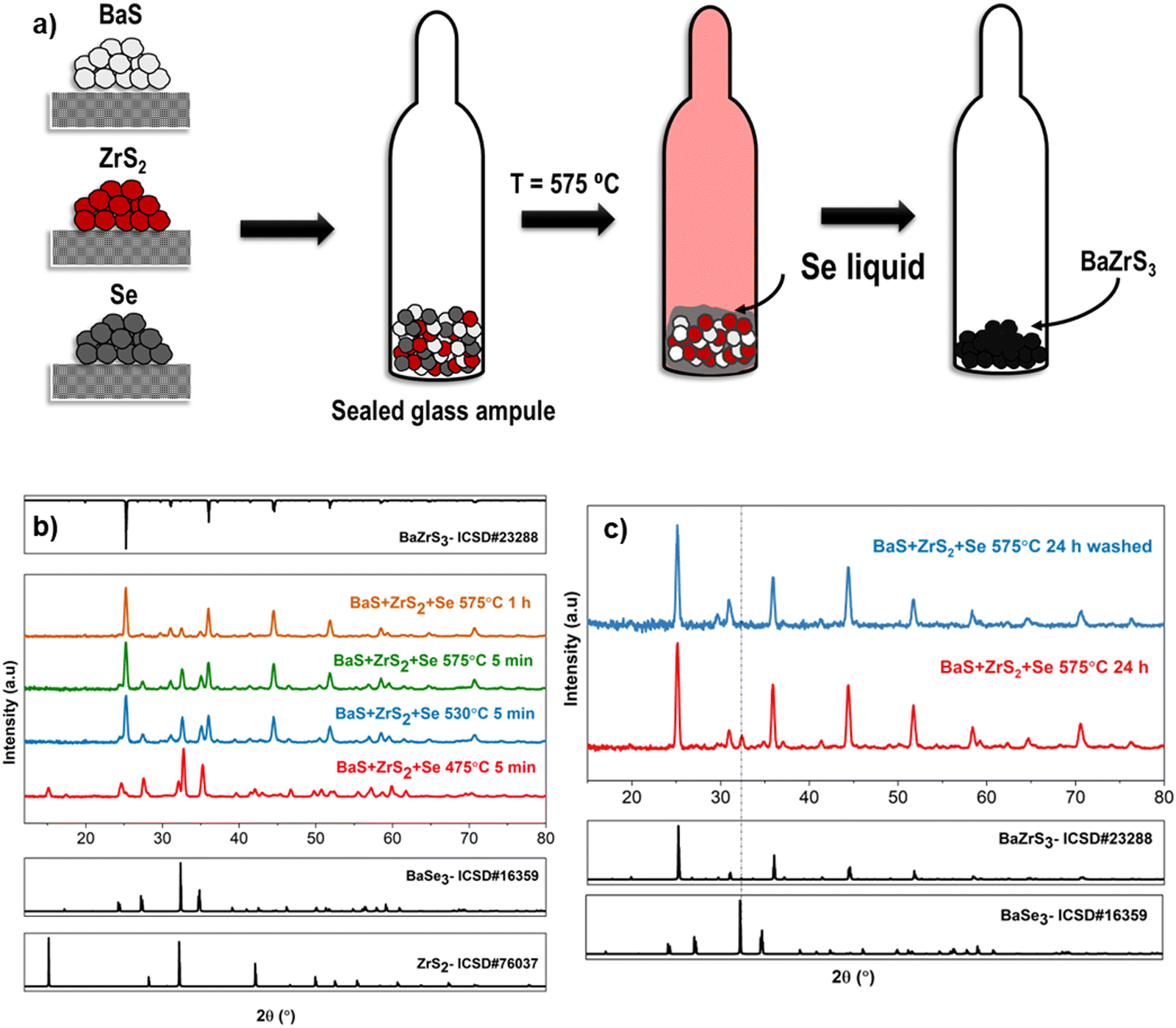 | ||
| Fig. 8 (a) Reaction schematic depicting the synthesis of BaMS3 utilizing a selenium liquid flux. (b) X-ray diffraction patterns illustrating the evolution of intermediate phases with temperature for binary sulfides when subjected to the selenium liquid flux. (c) Comparison plot of the X-ray diffraction pattern for the water-washed and unwashed BaZrS3 powder. | ||
With increased reaction time, the relative peak intensity of BaZrS3 to BaSe3 continued to rise, albeit with a small amount of residual BaSe3 secondary phase remaining after 24 h of reaction at 575 °C. This residual BaSe3 was found to be readily soluble in water. Consequently, the BaZrS3 powder, containing the BaSe3 secondary phase, underwent two rinses with deionized water to achieve a pure BaZrS3 phase, as confirmed by X-ray diffraction analysis (see Fig. 8(c)). Interestingly, in the absence of excess sulfur and the presence of a liquid flux, BaZrS3 formed instead of the Ruddlesden–Popper phase. This finding and previous results in this study suggest that Ruddlesden–Popper phases form in a sulfur-poor environment only when there are diffusional limitations. However, Raman analysis in Fig. S37 (ESI†) suggested the presence of residual melt from the selenium liquid flux, encapsulating the BaZrS3 grains. EDX analysis of the water-washed BaZrS3 powder further supported this observation. The BaZrS3 powder exhibited lumping and possibly contained agglomerates of finer BaZrS3 grains in the nanometer range, as depicted in Fig. S38 (ESI†), with small selenium-rich regions illustrated in Fig. S39 (ESI†). However, this excess selenium could be readily dissolved in various solution chemistries, with amine–thiol mixtures and trioctylphosphine being some solvents.58,59 Fig. S40 (ESI†) highlights our attempt to dissolve this residual selenium. Notably, compositional analysis of the BaZrS3 powder revealed negligible selenium content (see Fig. S41, ESI†).
A similar procedure led to the synthesizing of ternary perovskite BaHfS3 and hexagonal BaTiS3, as evidenced in Fig. S42 and S45 (ESI†). This approach's success lies in synthesizing sulfide perovskites and in the potential for selenium alloying at moderate temperatures by heating a mixture of BaS, ZrS2, Zr, and selenium powder together. However, this process would require optimization and, therefore, would be investigated as part of a separate study. Although BaMS3 compounds have been successfully synthesized at temperatures below 600 °C, attempts to synthesize SrMS3 compounds via sulfurization of respective binary sulfides with excess sulfur have not yielded successful results (see Fig. S46, ESI†). One reason could be that, unlike BaSx liquid flux at temperatures above 525 °C, Sr has not been reported to form any liquid strontium polysulfides at temperatures below 600 °C.
When the binary sulfides of strontium and zirconium were heated with selenium at 575 °C, we observed the emergence of α-SrZrS3 peaks (needle-like crystal structure), as depicted in Fig. S47 (ESI†). However, achieving phase pure α-SrZrS3 proved to be difficult in this case. It should be noted that single crystal analysis on a similar sample subjected to 650 °C revealed a striking similarity in the diffraction pattern between α-SrZrS3 and the somewhat unlikely compound SrSeS3. Rigorous elemental characterizations are needed in the future to differentiate between these phases accurately. We noted a gradual increase in peak intensity from 48 h to 7 days and 21 days, as illustrated in Fig. S47 (ESI†). These results are promising and encourage further exploration of other liquid fluxes. Moreover, this outcome underscores the utility of selenium liquid flux and highlights that the synthesis of Sr ternaries might be thermodynamically favorable at these temperatures but suffers from diffusional limitations.
3. Conclusions
Chalcogenide perovskites, while promising, have posed challenges in the synthesis, hindering progress in this material system. BaZrS3, particularly suitable for photovoltaic applications, has been synthesized at temperatures unsuitable for solar cell integration. Our study aimed to ascertain the optimal metal ratios and sulfur pressure during the synthesis of BaZrS3 to prevent phase disintegration. Subsequently, we proposed methods to address limitations arising from the halide affinity of alkaline earth metals and the unavailability of solution chemistries for alkaline earth metal sulfides to fabricate solution-deposited BaZrS3 thin films.We successfully dissolved alkaline earth sulfides such as BaS and SrS, demonstrating their utility in synthesis. Additionally, we showcased the successful synthesis of BaMS3 by employing stacked films of appropriate precursors. Notably, the sequencing of stacked layers allowed the use of ZrCl4 precursor without forming BaCl2 impurity in the final BaZrS3 film. Furthermore, we identified an alternative liquid in Se that facilitates the formation of BaMS3 at temperatures below 600 °C. The versatility of our methods extends to the synthesis of other BaMS3 (M = Hf, Ti) compounds, enabling low-cost and facile production of this class of chalcogenide perovskites.
4. Experimental methods
4.1. Materials and characterization
Barium sulfide (BaS, 99.9%), titanium(IV) sulfide (TiS2, 99.9%), selenium powder (99.99%), zirconium(IV) chloride (ZrCl4, anhydrous powder, >99.99%), hafnium(IV) chloride (HfCl4, purified by sublimation, 99.9), 2-methyl-2-propanethiol (MePT, 99%), butylamine (BA, 99.5%), carbon disulfide (CS2, anhydrous, >99%), pyridine (Pyd, anhydrous, 99.8%), N,N dimethyl formamide (DMF, anhydrous, 99.8%), thiourea (TU, ACS Reagent, >99%), sulfur flakes (>99.99%), molecular sieves (Type 3A, bead size 8–12 mesh), propylamine (PA, >99%) and alumina dispersed in isopropanol (20 wt%) were purchased from Sigma Aldrich. Bis(pentamethylcyclopentadienyl)barium (Cp*2Ba), zirconium(IV) sulfide (ZrS2, 99%), zirconium(IV) bromide (ZrBr4, 98%) and zirconium(IV) iodide (ZrI4, 99.5%) were purchased from STREM chemicals. Titanium(IV) chloride (TiCl4, 99.99%) and pyridine-d{5} (99.5% Isotopic) were purchased from Fisher Scientific. Titanium hydride (99.9%) and hafnium hydride (99.9%) were purchased from Nano Research Elements. Borosilicate glass ampules were procured from Chemglass. Hafnium disulfide (>99.995%) was purchased from Ossila.Solvents without sure seal were stored over molecular sieves for several days before use. Thiourea was recrystallized through a two-step process, using 18.2 MΩ deionized water and was dried under vacuum overnight. All other chemicals were used as received.
Alkali-free Eagle XG glass substrates were purchased from Stemmerich. It should be noted that the glass substrate itself contains traces of zirconium. Selenium powder was vacuum dried at 100 °C overnight before use. Sulfur flakes were finely ground inside a nitrogen-filled glovebox and vacuum-dried overnight at room temperature. All chemicals were stored in nitrogen-filled glove boxes. Sulfurization at 650 °C was carried out in a carbon coated quartz ampule sealed using a propane-oxygen torch.
Raman spectroscopy was performed using a Horiba/Jobin-Yvon HR800 Raman spectrometer with a 632.8 nm excitation laser wavelength. Spectra on liquid solutions were collected using a quartz cuvette enclosed in a nitrogen atmosphere.
X-ray diffraction analysis was conducted using a Rigaku SmartLab Diffractometer under ambient conditions, employing parallel beam geometry with an incident beam angle of 0.5 degrees. Data collection utilized a Cu Kα source (λ = 1.5406 Å) operated at 40 kV and 44 mA.
Scanning electron microscopy (SEM) and energy dispersive X-ray measurements were performed using the FEI Nova three-dimensional system equipped with an Everhart Thornley detector. The measurements were carried out at an accelerating voltage of 10 kV with a working distance of 5 mm. The samples were coated with ∼10 nm of platinum for better image resolution.
Reflectance data were obtained employing a PerkinElmer Lambda 950 spectrometer equipped with an integrating sphere.
NMR spectra were recorded on a Bruker AVANCE III 400 MHz spectrometer, and chemical shifts were referenced to residual solvent signals for both 1H. For 1H NMR, the zg30 pulse sequence was utilized with 9 scans, a 1-second relaxation delay, and an acquisition time of 2.1955 seconds.
4.2. Alumina deposition
A thin layer of alumina was coated onto the glass substrate to improve the surface wettability during the coating process. The alumina also acts as a barrier and prevents cationic diffusion from substrate. Eagle XG glass was thoroughly rinsed with DI water, 2-propanol, and methanol, followed by sonication in an alconox bath and DI water. The substrate was blow dried using a nitrogen gun. Subsequently, the glass was cleaned in a UV-ozone cleaner for 30 minutes. Commercial alumina nanoparticle solution was diluted by combining 0.2 ml of the commercial solution with 1.8 ml of Isopropanol. The resulting solution was spin-coated onto the cleaned EXG glass at 1000 RPM for 1 minute, followed by annealing at 100 °C for 1 minute and then at 500 °C for 30 minutes.4.3. Sample preparation
All chemical storage, solution preparation, thin film coating, and sample handling were conducted inside nitrogen- and ultra-high purity (UHP) argon-filled gloveboxes. The ampules were sealed using a Schlenk line with UHP argon as the purging gas. Table 2 summarizes the four different synthesis methods discussed in this work.| Method | Precursors used | Powder/thin film | Product |
|---|---|---|---|
| Solid-state synthesis | BaS + ZrS2 powders | Powders | BaZrS3 and RP phases |
| Stacked metal sulfides | MS3 using halide precursors + BaS stack | Thin film | BaMS3 M = Ti, Zr, Hf |
| Hybrid colloidal inks | Colloidal MS3 + BaS using sulfide precursors | Thin film | BaMS3 M = Ti, Zr, Hf |
| Selenium flux | BaS + MS2 powders with Se | Powders | BaMS3 M = Ti, Zr, Hf |
The sulfur pressure was calculated at the synthesis temperature using the ideal gas law, with S6 assumed to be the predominant sulfur species. Borosilicate ampules with a nominal volume of 10 ml were used; however, for the calculations, the volume was considered slightly larger to account for the additional volume contributed by the ampule stem.
The synthesized powder was subjected to two rinses with DI water to eliminate water-soluble secondary phases such as BaS3. The powder was then dried with flowing argon gas.
Amine–thiol. A 0.3 M solution of ZrCl4 was prepared with the butylamine: 2-methyl-2-propanethiol molar ratio of 1
:2.2. 2-methyl-2-propanethiol was added first to the metal precursor, followed by butylamine. The solution was stirred overnight at 35 °C and used for coating.
Amine–CS2. A 0.3 M solution of ZrCl4 was prepared with a butylamine
:CS2:pyridine molar ratio of 1:1:1. Butylamine was added first to the metal precursor, followed by CS2 and pyridine. The solution was stirred overnight at 35 °C and used for coating. Similar dissolution procedures were followed for ZrBr4, ZrI4, TiCl4, and HfCl4.
Amine–thiourea. A 0.3 M solution of ZrCl4 and 2 M thiourea in butylamine was stirred overnight at 35 °C and used. ZrCl4 and thiourea were mixed together, and butylamine was added.
DMF–thiourea. A 0.3 M solution of ZrCl4 and 3 M thiourea in dimethylformamide (DMF) was stirred overnight at 35 °C and used. ZrCl4 and thiourea were mixed together, and DMF was added.
Cp*2Ba dissolution. A 0.3 M solution of Cp*2Ba was prepared with the butylamine
:2-methyl-2-propanethiol molar ratio of 1:1. 2-methyl-2-propanethiol was added first to the metal precursor, followed by butylamine.
Bilayer fabrication. In one route, the four ZrCl4 solutions routes discussed above were coated, annealed, and sulfurized to fabricate the bottom layer of ZrS3. The samples were sealed in an ampule with HfH2 and sulfur and heated to 575 °C for 6 h to synthesize the bottom ZrS3 layer. Cp*2Ba was blade coated on top of ZrS3, sealed in an ampule, and heated at 575 °C for 2 h to synthesize BaZrS3. Similar procedure was adapted for the iodide and bromide salts as well as BaHfS3 and BaTiS3.
In another route, the ZrCl4–amine–CS2 ink was coated and annealed on the hotplate at 450 °C for 30 minutes. Cp*2Ba was blade coated on top, sealed in an ampule, and heated at 575 °C for 2 h to synthesize BaZrS3.
:CS2:pyridine molar ratio of 1:1:1.5. Propylamine was added first to the metal precursor, followed by CS2 and pyridine. The solution was stirred for 3–4 days at 35 °C and combined with the ZrS3 colloidal solution. The solution was then blade-coated, sealed, and sulfurized.
Similar procedures were repeated for Ba–Hf–S and Ba–Ti–S synthesis, with the only difference being the starting precursors. HfH2 and TiH2 were used as precursors to synthesize binary sulfides.
Solutions were blade-coated using an automated blade coater at a speed of 15 mm per second with a single pass, followed by annealing at 350 °C for 2 minutes on a hot plate and cooling for 1 minute. The coating was repeated 6–8 times on alumina-coated Eagle XG substrates. The resulting films were transferred to a 5 ml ampule containing 5 mg of HfH2 and 15 mg of sulfur. The ampules underwent three purging cycles with vacuum-UHP argon and were sealed using a butane-fueled blowtorch. Subsequently, the ampules were heated in a refractory furnace to the desired temperature and cooled naturally under a slow argon flow.
The synthesized BaMS3 (M = Ti, Zr, Hf) powder was subjected to two rinses with DI water to eliminate any water-soluble secondary phases. The water-washed powder was further washed in a 1:5 vol:vol mixture of ethanedithiol–propylamine to wash away any residual selenium. The powder was stirred in amine–thiol solvent overnight at room temperature. The powder was afterward dried with flowing argon gas.
Data availability statement
The data supporting this article have been included in the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the National Science Foundation's financial support through grants 1735282-NRT (SFEWS) and 10001536 (INFEWS). Special thanks are extended to Prof. Suzanne C. Bart, Dr Apurva A. Pradhan, Dr Jonathan W. Turnley, and Dr Madeleine C. Uible for their valuable discussions regarding the project.References
- A. Wang, C. Zuo, X. Niu, L. Ding, J. Ding and F. Hao, Recent Promise of Lead-Free Halide Perovskites in Optoelectronic Applications, Chem. Eng. J., 2023, 451, 138926, DOI:10.1016/j.cej.2022.138926.
- H. Hu, B. Dong and W. Zhang, Low-Toxic Metal Halide Perovskites: Opportunities and Future Challenges, J. Mater. Chem. A, 2017, 5(23), 11436–11449, 10.1039/C7TA00269F.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Properties and Potential Optoelectronic Applications of Lead Halide Perovskite Nanocrystals, Science, 2017, 358(6364), 745–750, DOI:10.1126/science.aam7093.
- B. Park and S. Il Seok, Intrinsic Instability of Inorganic–Organic Hybrid Halide Perovskite Materials, Adv. Mater., 2019, 31(20), 1805337, DOI:10.1002/adma.201805337.
- T. Feurer, P. Reinhard, E. Avancini, B. Bissig, J. Löckinger, P. Fuchs, R. Carron, T. P. Weiss, J. Perrenoud, S. Stutterheim, S. Buecheler and A. N. Tiwari, Progress in Thin Film CIGS Photovoltaics – Research and Development, Manufacturing, and Applications, Prog. Photovoltaics Res. Appl., 2017, 25(7), 645–667, DOI:10.1002/pip.2811.
- S. Giraldo, Z. Jehl, M. Placidi, V. Izquierdo-Roca, A. Pérez-Rodríguez and E. Saucedo, Progress and Perspectives of Thin Film Kesterite Photovoltaic Technology: A Critical Review, Adv. Mater., 2019, 31(16), 1806692, DOI:10.1002/adma.201806692.
- M. Theelen and F. Daume, Stability of Cu(In,Ga)Se2 Solar Cells: A Literature Review, Sol. Energy, 2016, 133, 586–627, DOI:10.1016/j.solener.2016.04.010.
- K. V. Sopiha, C. Comparotto, J. A. Márquez and J. J. S. Scragg, Chalcogenide Perovskites: Tantalizing Prospects, Challenging Materials, Adv. Opt. Mater., 2022, 2101704, DOI:10.1002/adom.202101704.
- M. Ju, J. Dai, L. Ma and X. C. Zeng, Perovskite Chalcogenides with Optimal Bandgap and Desired Optical Absorption for Photovoltaic Devices, Adv. Energy Mater., 2017, 7(18), 1700216, DOI:10.1002/aenm.201700216.
- Y.-Y. Sun, M. L. Agiorgousis, P. Zhang and S. Zhang, Chalcogenide Perovskites for Photovoltaics, Nano Lett., 2015, 15(1), 581–585, DOI:10.1021/nl504046x.
- S. Filippone, B. Zhao, S. Niu, N. Z. Koocher, D. Silevitch, I. Fina, J. M. Rondinelli, J. Ravichandran and R. Jaramillo, Discovery of Highly Polarizable Semiconductors BaZrS3 and Ba3Zr2S7, Phys. Rev. Mater., 2020, 4(9), 091601, DOI:10.1103/PhysRevMaterials.4.091601.
- X. Wei, H. Hui, C. Zhao, C. Deng, M. Han, Z. Yu, A. Sheng, P. Roy, A. Chen, J. Lin, D. F. Watson, Y. Y. Sun, T. Thomay, S. Yang, Q. Jia, S. Zhang and H. Zeng, Realization of BaZrS3 Chalcogenide Perovskite Thin Films for Optoelectronics, Nano Energy, 2020, 68, 104317, DOI:10.1016/j.nanoen.2019.104317.
- K. Hanzawa, S. Iimura, H. Hiramatsu and H. Hosono, Material Design of Green-Light-Emitting Semiconductors: Perovskite-Type Sulfide SrHfS3, J. Am. Chem. Soc., 2019, 141(13), 5343–5349, DOI:10.1021/jacs.8b13622.
- Y. Liang, J. Li, Z. Chen, G. Li, M. Li, M. Jia, X. Chen, X. Li, Y. Han and Z. Shi, Tapping the Light Emitting Potential of Chalcogenide Perovskite SrHfS3via Eu2+ Doping, Adv. Opt. Mater., 2024, 12(6), 2301977, DOI:10.1002/adom.202301977.
- Y. Han, J. Fang, Y. Liang, H. Gao, J. Yang, X. Chen, Y. Yuan and Z. Shi, Preparation of Chalcogenide Perovskite SrHfS3 and Luminescent SrHfS3:Eu2+ Thin Films, Appl. Phys. Lett., 2024, 124(13), 131902, DOI:10.1063/5.0200555.
- M. Ishii and M. Saeki, Raman and Infrared Spectra of BaTiS3 and BaNbS3, Phys. Status Solidi B, 1992, 170(1), K49–K54, DOI:10.1002/pssb.2221700149.
- T. R. Paudel and E. Y. Tsymbal, Evaluating the Thermoelectric Properties of BaTiS3 by Density Functional Theory, ACS Omega, 2020, 5(21), 12385–12390, DOI:10.1021/acsomega.0c01139.
- C. Comparotto, A. Davydova, T. Ericson, L. Riekehr, M. V. Moro, T. Kubart and J. Scragg, Chalcogenide Perovskite BaZrS3: Thin Film Growth by Sputtering and Rapid Thermal Processing, ACS Appl. Energy Mater., 2020, 3(3), 2762–2770, DOI:10.1021/acsaem.9b02428.
- S. Sharma, Z. Ward, K. Bhimani, K. Li, A. Lakhnot, R. Jain, S.-F. Shi, H. Terrones and N. Koratkar, Bandgap Tuning in BaZrS3 Perovskite Thin Films, ACS Appl. Electron Mater., 2021, 3(8), 3306–3312, DOI:10.1021/acsaelm.1c00575.
- I. Sadeghi, K. Ye, M. Xu, Y. Li, J. M. LeBeau and R. Jaramillo, Making BaZrS3 Chalcogenide Perovskite Thin Films by Molecular Beam Epitaxy, Adv. Funct. Mater., 2021, 31(45), 2105563, DOI:10.1002/adfm.202105563.
- J. A. Márquez, M. Rusu, H. Hempel, I. Y. Ahmet, M. Kölbach, I. Simsek, L. Choubrac, G. Gurieva, R. Gunder, S. Schorr and T. Unold, BaZrS3 Chalcogenide Perovskite Thin Films by H2S Sulfurization of Oxide Precursors, J. Phys. Chem. Lett., 2021, 12(8), 2148–2153, DOI:10.1021/acs.jpclett.1c00177.
- Y. Wang, N. Sato and T. Fujino, Synthesis of BaZrS3 by Short Time Reaction at Lower Temperatures, J. Alloys Compd., 2001, 327(1–2), 104–112, DOI:10.1016/S0925-8388(01)01553-5.
- R. Bystrický, S. K. Tiwari, P. Hutár, L. Vančo and M. Sýkora, Synthesis of Sulfide Perovskites by Sulfurization with Boron Sulfides, Inorg. Chem., 2022, 61, 18823–18827, DOI:10.1021/acs.inorgchem.2c03200.
- S. Niu, J. Milam-Guerrero, Y. Zhou, K. Ye, B. Zhao, B. C. Melot and J. Ravichandran, Thermal Stability Study of Transition Metal Perovskite Sulfides, J. Mater. Res., 2018, 33(24), 4135–4143, DOI:10.1557/jmr.2018.419.
- S. Niu, B. Zhao, K. Ye, E. Bianco, J. Zhou, M. E. McConney, C. Settens, R. Haiges, R. Jaramillo and J. Ravichandran, Crystal Growth and Structural Analysis of Perovskite Chalcogenide BaZrS3 and Ruddlesden–Popper Phase Ba3Zr2S7, J. Mater. Res., 2019, 34(22), 3819–3826, DOI:10.1557/jmr.2019.348.
- C. Comparotto, P. Ström, O. Donzel-Gargand, T. Kubart and J. J. S. Scragg, Synthesis of BaZrS3 Perovskite Thin Films at a Moderate Temperature on Conductive Substrates, ACS Appl. Energy Mater., 2022, 5(5), 6335–6343, DOI:10.1021/acsaem.2c00704.
- K. C. Vincent, S. Agarwal, J. W. Turnley and R. Agrawal, Liquid Flux–Assisted Mechanism for Modest Temperature Synthesis of Large-Grain BaZrS3 and BaHfS3 Chalcogenide Perovskites, Adv. Energy Sustainability Res., 2023, 2300010, 2300010, DOI:10.1002/aesr.202300010.
- R. Yang, A. D. Jess, C. Fai and C. J. Hages, Low-Temperature, Solution-Based Synthesis of Luminescent Chalcogenide Perovskite BaZrS3 Nanoparticles, J. Am. Chem. Soc., 2022, 144(35), 15928–15931, DOI:10.1021/jacs.2c06168.
- R. Yang, J. Nelson, C. Fai, H. A. Yetkin, C. Werner, M. Tervil, A. D. Jess, P. J. Dale and C. J. Hages, A Low-Temperature Growth Mechanism for Chalcogenide Perovskites, Chem. Mater., 2023, 35(12), 4743–4750, DOI:10.1021/acs.chemmater.3c00494.
- A. A. Pradhan, M. C. Uible, S. Agarwal, J. W. Turnley, S. Khandelwal, J. M. Peterson, D. D. Blach, R. N. Swope, L. Huang, S. C. Bart and R. Agrawal, Synthesis of BaZrS3 and BaHfS3 Chalcogenide Perovskite Films Using Single-Phase Molecular Precursors at Moderate Temperatures, Angew. Chem., Int. Ed., 2023, 62, e202301049, DOI:10.1002/anie.202301049.
- J. W. Turnley, K. C. Vincent, A. A. Pradhan, I. Panicker, R. Swope, M. C. Uible, S. C. Bart and R. Agrawal, Solution Deposition for Chalcogenide Perovskites: A Low-Temperature Route to BaMS3 Materials (M = Ti, Zr, Hf), J. Am. Chem. Soc., 2022, 144(40), 18234–18239, DOI:10.1021/jacs.2c06985.
- P. Murria, C. K. Miskin, R. Boyne, L. T. Cain, R. Yerabolu, R. Zhang, E. C. Wegener, J. T. Miller, H. I. Kenttämaa and R. Agrawal, Speciation of CuCl and CuCl2 Thiol-Amine Solutions and Characterization of Resulting Films: Implications for Semiconductor Device Fabrication, Inorg. Chem., 2017, 56(23), 14396–14407, DOI:10.1021/acs.inorgchem.7b01359.
- J. A. Clark, A. Murray, J. Lee, T. S. Autrey, A. D. Collord and H. W. Hillhouse, Complexation Chemistry in N,N-Dimethylformamide-Based Molecular Inks for Chalcogenide Semiconductors and Photovoltaic Devices, J. Am. Chem. Soc., 2019, 141(1), 298–308, DOI:10.1021/jacs.8b09966.
- S. Suresh and A. R. Uhl, Present Status of Solution-Processing Routes for Cu(In,Ga)(S,Se)2 Solar Cell Absorbers, Adv. Energy Mater., 2021, 11(14), 2003743, DOI:10.1002/aenm.202003743.
- J. W. Turnley and R. Agrawal, Solution Processed Metal Chalcogenide Semiconductors for Inorganic Thin Film Photovoltaics, Chem. Commun., 2024, 60(40), 5245–5269, 10.1039/D4CC01057D.
- K. P. Kepp, A Quantitative Scale of Oxophilicity and Thiophilicity, Inorg. Chem., 2016, 55(18), 9461–9470, DOI:10.1021/acs.inorgchem.6b01702.
- D. Zilevu and S. E. Creutz, Solution-Phase Synthesis of Group 3–5 Transition Metal Chalcogenide Inorganic Nanomaterials, Chem. Commun., 2023, 59(57), 8779–8798, 10.1039/D3CC01731A.
- S. Agarwal, J. W. Turnley, A. A. Pradhan and R. Agrawal, Moderate Temperature Sulfurization and Selenization of Highly Stable Metal Oxides: An Opportunity for Chalcogenide Perovskites, J. Mater. Chem. C, 2023, 11(45), 15817–15823, 10.1039/D3TC02716C.
- G. Gurieva, R. Ferreira, P. Knoll and S. Schorr, Cu2ZnSnSe4: How Far Does Off-Stoichiometry Go?, Phys. Status Solidi A, 2018, 215(17), 1700957, DOI:10.1002/pssa.201700957.
- K. V. Sopiha, J. K. Larsen, J. Keller, M. Edoff, C. Platzer-Björkman and J. J. S. Scragg, Off-Stoichiometry in I–III–VI2 Chalcopyrite Absorbers: A Comparative Analysis of Structures and Stabilities, Faraday Discuss., 2022, 239, 357–374, 10.1039/D2FD00105E.
- M. Monsefi and D.-H. Kuo, Defect State and Electric Transport of the Cu-Poor, Cu-Rich, and In-Rich Cu(In,Ga)Se2 Bulk Materials, Mater. Chem. Phys., 2014, 145(1–2), 255–259, DOI:10.1016/j.matchemphys.2014.02.019.
- M. Nakamura, K. Yamaguchi, Y. Kimoto, Y. Yasaki, T. Kato and H. Sugimoto, Cd-Free Cu(In,Ga)(Se,S)2 Thin-Film Solar Cell With Record Efficiency of 23.35%, IEEE J. Photovolt., 2019, 9(6), 1863–1867, DOI:10.1109/JPHOTOV.2019.2937218.
- X. Wu, W. Gao, J. Chai, C. Ming, M. Chen, H. Zeng, P. Zhang, S. Zhang and Y.-Y. Sun, Defect Tolerance in Chalcogenide Perovskite Photovoltaic Material BaZrS3, Sci. China Mater., 2021, 64(12), 2976–2986, DOI:10.1007/s40843-021-1683-0.
- W. Meng, B. Saparov, F. Hong, J. Wang, D. B. Mitzi and Y. Yan, Alloying and Defect Control within Chalcogenide Perovskites for Optimized Photovoltaic Application, Chem. Mater., 2016, 28(3), 821–829, DOI:10.1021/acs.chemmater.5b04213.
- Y. Han, J. Xu, Y. Liang, X. Chen, M. Jia, J. Zhang, L. Lian, Y. Liu, X. Li and Z. Shi, P-Type Conductive BaZrS3 Thin Film and Its Band Gap Tunning via Ruddlesden-Popper Ba3Zr2S7 and Titanium Alloying, Chem. Eng. J., 2023, 473, 145351, DOI:10.1016/j.cej.2023.145351.
- P. Kayastha, D. Tiwari, A. Holland, O. S. Hutter, K. Durose, L. D. Whalley and G. Longo, High-Temperature Equilibrium of 3D and 2D Chalcogenide Perovskites, Sol. RRL, 2023, 7(9), 2201078, DOI:10.1002/solr.202201078.
- W. Li, S. Niu, B. Zhao, R. Haiges, Z. Zhang, J. Ravichandran and A. Janotti, Band Gap Evolution in Ruddlesden-Popper Phases, Phys. Rev. Mater., 2019, 3(10), 101601, DOI:10.1103/PhysRevMaterials.3.101601.
- S. Agarwal, K. Weideman, D. Rokke, K. C. Vincent, D. Zemlyanov and R. Agrawal, Enhancing the Optoelectronic Properties of Solution-Processed AgInSe2 Thin Films for Application in Photovoltaics, J. Mater. Chem. C, 2024, 12(1), 325–336, 10.1039/D3TC03540A.
- E. M. Larsen, Zirconium and Hafnium Chemistry, Advances in Inorganic Chemistry and Radiochemistry, 1970, vol. 13, pp. 1–133 DOI:10.1016/S0065-2792(08)60335-0.
- D. B. Mitzi, M. Yuan, W. Liu, A. J. Kellock, S. J. Chey, L. Gignac and A. G. Schrott, Hydrazine-Based Deposition Route for Device-Quality CIGS Films, Thin Solid Films, 2009, 517(7), 2158–2162, DOI:10.1016/j.tsf.2008.10.079.
- X. Zhao, S. D. Deshmukh, D. J. Rokke, G. Zhang, Z. Wu, J. T. Miller and R. Agrawal, Investigating Chemistry of Metal Dissolution in Amine−Thiol Mixtures and Exploiting It toward Benign Ink Formulation for Metal Chalcogenide Thin Films, Chem. Mater., 2019, 31(15), 5674–5682, DOI:10.1021/acs.chemmater.9b01566.
- C. L. McCarthy and R. L. Brutchey, Solution Deposited Cu2BaSnS4–xSex from a Thiol–Amine Solvent Mixture, Chem. Mater., 2018, 30(2), 304–308, DOI:10.1021/acs.chemmater.7b03931.
- B. Teymur, Y. Zhou, E. Ngaboyamahina, J. T. Glass and D. B. Mitzi, Solution-Processed Earth-Abundant Cu2BaSn(S,Se)4 Solar Absorber Using a Low-Toxicity Solvent, Chem. Mater., 2018, 30(17), 6116–6123, DOI:10.1021/acs.chemmater.8b02556.
- D. Shin, T. Zhu, X. Huang, O. Gunawan, V. Blum and D. B. Mitzi, Earth-Abundant Chalcogenide Photovoltaic Devices with over 5% Efficiency Based on a Cu2BaSn(S,Se)4 Absorber, Adv. Mater., 2017, 29(24), 1606945, DOI:10.1002/adma.201606945.
- S. McLeod, E. Alruqobah and R. Agrawal, Liquid Assisted Grain Growth in Solution Processed Cu(In,Ga)(S,Se)2, Sol. Energy Mater. Sol. Cells, 2019, 195, 12–23, DOI:10.1016/j.solmat.2019.02.020.
- C. J. Hages, M. J. Koeper, C. K. Miskin, K. W. Brew and R. Agrawal, Controlled Grain Growth for High Performance Nanoparticle-Based Kesterite Solar Cells, Chem. Mater., 2016, 28(21), 7703–7714, DOI:10.1021/acs.chemmater.6b02733.
- S. R. Sashital and A. L. Gentile, Liquid Phase Epitaxial Growth of AgGaS2 Using Chalcogenide (Sulphide) Fluxes, J. Cryst. Growth, 1984, 69(2–3), 379–387, DOI:10.1016/0022-0248(84)90346-4.
- J. W. Turnley, S. D. Deshmukh, V. M. Boulos, R. Spilker, C. J. Breckner, K. Ng, J. K.-Y. Liu, J. T. Miller, H. I. Kenttämaa and R. Agrawal, A Selenium-Based “Alkahest”: Reactive Dissolutions of Metals and Metal Compounds with n-Alkylammonium Polyselenide Solutions, Inorg. Chem. Front., 2023, 10(20), 6032–6044, 10.1039/D3QI01632C.
- P. J. Newman and D. R. MacFarlane, Preparation of CdSe Quantum Dots in Ionic Liquids, Z. Phys. Chem., 2006, 220(10), 1473–1481, DOI:10.1524/zpch.2006.220.10.1473.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional X-ray diffraction, Raman, EDX, 1H NMR, and liquid Raman data. See DOI: https://doi.org/10.1039/d4tc02287d |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |