
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling the significance of adduct formation between thiocarbonyl Lewis donors and diiodine for the structural organization of rhodanine-based small molecule semiconductors†
Anna Laura
Sanna
 a,
Simone
Acca
a,
Enrico
Podda
b,
Antonello
Mascia
c,
Anna
Pintus
a,
M. Carla
Aragoni
a,
Vito
Lippolis
a,
Carlo
Ricci
d,
Piero
Cosseddu
c,
Massimiliano
Arca
*a and
Giuseppe
Sforazzini
*a
a,
Simone
Acca
a,
Enrico
Podda
b,
Antonello
Mascia
c,
Anna
Pintus
a,
M. Carla
Aragoni
a,
Vito
Lippolis
a,
Carlo
Ricci
d,
Piero
Cosseddu
c,
Massimiliano
Arca
*a and
Giuseppe
Sforazzini
*a
aDepartment of Chemical and Geological Sciences, University of Cagliari, S.P. no. 8 km 0.700, Monserrato 09042, Cagliari, Italy. E-mail: giuseppe.sforazzini@unica.it; marca@unica.it
bCentro Servizi di Ateneo per la Ricerca - CeSAR, University of Cagliari, S.P. no. 8 Km 0.700, Monserrato 09042, Cagliari, Italy
cDepartment of Electrical and Electronic Engineering, University of Cagliari, Piazza d’Armi, Cagliari 09123, Italy
dDepartment of Physics, University of Cagliari, S.P. no. 8 Km 0.700, 09042, Monserrato, CA, Italy
First published on 8th July 2024
Abstract
Rhodanine vinyl bithiophene (BTR) was synthesized and characterized both spectroscopically and structurally. The reaction of BTR with molecular iodine led to the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 “spoke” adduct BTR·I2, formed by interaction of the rhodanine thiocarbonyl group with a diiodine (I2) molecule. The elongation of the I–I bond in the adduct with respect to solid-state I2 and the Raman response in the low-energy region (ν = 150 cm−1) clearly indicate BTR·I2 to be a weak CT adduct. Hybrid-DFT calculations showed that the adduct formation narrowed the HOMO–LUMO gap in BTR·I2 as compared to BTR, while the extended network of secondary interactions, including type-I halogen bonds (XB), results in the formation of an extended 3D network. As a consequence, the room temperature conductivity of BTR·I2 increased with respect to BTR, testifying for a more efficient molecular packing for charge percolation, with the formation of charge carriers in the crystal being facilitated by the presence of I2. It is worth noting that the single-crystal junction device operates at room temperature, in air, and no variation of conductivity over time was observed, indicating that no loss of diiodine occurred during measurements. These results clearly indicate the formation of thiocarbonyl–diiodine CT adducts and their potential as a solid additive for modulating the organization of small molecule semiconductors.
:1 “spoke” adduct BTR·I2, formed by interaction of the rhodanine thiocarbonyl group with a diiodine (I2) molecule. The elongation of the I–I bond in the adduct with respect to solid-state I2 and the Raman response in the low-energy region (ν = 150 cm−1) clearly indicate BTR·I2 to be a weak CT adduct. Hybrid-DFT calculations showed that the adduct formation narrowed the HOMO–LUMO gap in BTR·I2 as compared to BTR, while the extended network of secondary interactions, including type-I halogen bonds (XB), results in the formation of an extended 3D network. As a consequence, the room temperature conductivity of BTR·I2 increased with respect to BTR, testifying for a more efficient molecular packing for charge percolation, with the formation of charge carriers in the crystal being facilitated by the presence of I2. It is worth noting that the single-crystal junction device operates at room temperature, in air, and no variation of conductivity over time was observed, indicating that no loss of diiodine occurred during measurements. These results clearly indicate the formation of thiocarbonyl–diiodine CT adducts and their potential as a solid additive for modulating the organization of small molecule semiconductors.
Introduction
The molecular organization of π-conjugated compounds in the solid-state plays a crucial role in organic electronics.1–3 Intermolecular interactions, including π–π stacking, dipole–dipole, and charge transfer interactions, significantly influence the arrangement of molecules, thereby defining pathways for charge percolation.4–7 Understanding and controlling these interactions is essential for improving the energy efficiency, stability, and overall performance of organic electronic devices. Over the past decade, there has been an increasing interest in the development of a class of π-conjugated compounds commonly referred to as “small molecules” (SMs).8–10 These compounds are currently widely investigated for their potential in creating n-type semiconductor materials for highly efficient and sustainable organic solar cells (OSCs), among other applications.9–13 The typical design of a SM consists of a polycyclic aromatic π-system arranged in a linear or fused fashion, along with solubilizing side chain and electron directing functionality (Fig. 1). Widespread research efforts have been devoted to enhancing the efficiency of OSCs by thoughtfully tuning the energy level of the π-conjugated compounds and carefully selecting solubilizing chains to control their arrangement in the solid state once they are processed from solution.14–16 However, it is essential to note that an inappropriate choice of solubilizing groups can have a detrimental impact on the performance of an optoelectronic device. This occurs because the lateral side chains might promote molecular assemblies with good local charge mobility but an overall modest charge carrier percolation.17–21 Exploring post-synthetic approaches for controlling molecular organization is of particular interest. Common strategies involve the use of processing additives in the solution of organic semiconductors during the drying process, and thermal post-treatment of the formed thin films. Based on their distinct properties and effects, additives can be categorized into two categories: solvent additives and solid additives. The first category typically consists of solvents that primarily impact the solution state and evaporation dynamics in the film formation process. These additives have a higher boiling point as compared to the main solvent, leading to a slower evaporation rate during the film preparation, thus allowing for fine-tuning of the film crystallinity, as well as the phase separation in the case of blends.15,22–25 | ||
| Fig. 1 Schematic representation of the molecular structure of small molecule semiconductors (top). Selected representative examples of the bridge–acceptor molecular structure segment (bottom). BTR = bithiophene vinyl rhodanine; BTMN = bithiophene vinyl rhodanine malonitrile. | ||
The second category, solid additives, operates distinctively from their solvent counterparts, first because they alter the intermolecular interaction among π-conjugated compounds, and second because they are incorporated into the films, so in principle they do not require high temperature for their removal.26–28 Such a scenario is particularly interesting as it avoids the risk of film degradation which inevitably leads to a reduction in device performance. Examples of solid additives include 7-dibromo-9,9-dimethyl fluorene (DBDMF), hydroxy-methylpyrimidine derivatives, 9-fluorenone-1-carboxylic acid (FCA), 2-benzylidene-1-indene-1,3-dione and its derivatives, and diiodine.26–30 The latter additive is particularly intriguing because it is commonly employed also as a p-type chemical dopant for both small molecules and π-conjugated polymers, contributing to the enhancement of charge carriers in the semiconductor layer.29,30 However, the optimal generation of charge carriers relies heavily on the homogeneous distribution of the dopant throughout the material. To achieve this objective, various parameters must be carefully tuned during the film deposition process. Studies have demonstrated that employing higher temperatures is advantageous for this purpose.29 Consequently, the use of a solid dopant may prove to be less energy-effective if heat must be applied during the fabrication of optoelectronic devices. As a possible solution to this problem, our vision is to leverage the terminal functionality of semiconductors as coordination points for solid additives/dopants, so to reach a homogeneous distribution at the molecular level in the materials also at room temperature. In this context, intermolecular charge-transfer (CT)31 and σ-hole interactions,32 such as hydrogen, halogen, and chalcogen bonding interactions,33 are very promising to establish directional secondary bonds between semiconducting compounds and solid additives. In particular, contrary to carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), chalcocarbonyl functionality CCh (Ch = S, Se) is known to promote intermolecular CT with I2.31 At the same time, the CCh unit is of common occurrence in the terminal unit of SMs as it is intrinsically included in the molecular architecture of rhodanine electron-directing groups.9,11–13 To the best of our knowledge, no examples of CCh⋯I2 interactions in the rhodanine functionality and in the presence of π-conjugated compounds have been reported in the past. Therefore, we have focused our attention on investigating CCh⋯I2 bond formation in a model compound consisting of a π-conjugated segment directly connected to a rhodanine functionality. In this work, we report on the synthesis and structural and spectroscopic characterization of a bithiophene derivative featuring a N-ethylrhodanine (BTR, Fig. 1), and its CT adduct with molecular iodine. By utilizing a single crystal junction set-up, the effect of solid-state molecular organization on charge percolation through the crystals of the prepared compounds and adducts is investigated.
O), chalcocarbonyl functionality CCh (Ch = S, Se) is known to promote intermolecular CT with I2.31 At the same time, the CCh unit is of common occurrence in the terminal unit of SMs as it is intrinsically included in the molecular architecture of rhodanine electron-directing groups.9,11–13 To the best of our knowledge, no examples of CCh⋯I2 interactions in the rhodanine functionality and in the presence of π-conjugated compounds have been reported in the past. Therefore, we have focused our attention on investigating CCh⋯I2 bond formation in a model compound consisting of a π-conjugated segment directly connected to a rhodanine functionality. In this work, we report on the synthesis and structural and spectroscopic characterization of a bithiophene derivative featuring a N-ethylrhodanine (BTR, Fig. 1), and its CT adduct with molecular iodine. By utilizing a single crystal junction set-up, the effect of solid-state molecular organization on charge percolation through the crystals of the prepared compounds and adducts is investigated.
Results and discussion
Design and synthesis
We designed a bithiophene π-conjugated backbone decorated with a rhodanine electron-withdrawing group as a model of the bridge–acceptor segment of a SM semiconductor (Fig. 1). The bithiophenic segment was deliberately incorporated in the design to mimic the behaviour of the bridging linker between a hypothetical central core of a small molecule and the acceptor terminal group. Thus, the bithiophene moiety serves a dual purpose: first, it helps establish a reasonable electronic density in proximity to the electron-withdrawing group (rhodanine), and second, it functions as a means to verify the absence of side reactions, such as halogenation in positions 2 and 3, when the compounds undergo treatment with halogens. The synthetic pathway of the model compound bithiophene vinyl rhodanine (BTR) is depicted in Scheme 1. BTR was synthesized by Vilsmeier reaction34 followed by Knoevenagel condensation.35 Commercially available 2,2′-bithiophene (BT) underwent an initial reaction with phosphorus oxychloride (V), POCl3, and dimethylformamide (DMF) in 1,2-dichloroethane (1,2-DCE), resulting in quantitative yield of 2,2′-bithiophene-5-carbaldehyde (BTA). | ||
| Scheme 1 (a) POCl3 (i), DMF (ii), 1,2-DCE, reflux (83 °C), 24 h; (b) 3-ethyl-2-thioxo-1,3-thiazolidin-4-one, piperidine, ethanol, reflux (78 °C), 24 h. | ||
Subsequently, the obtained acylated product was reacted with N-ethylrhodanine, in the presence of piperidine and ethanol, to give the desired product in quantitative yields. BTR was then treated with I2 to form CT adducts through the interaction of the rhodanine CS functionality and I2.
X-ray diffraction analysis
The reactions between thiocarbonyl donors and I2 allow for the formation of a variety of products, including simple CT “spoke” adducts CS⋯I–I, “extended” CT adducts CS⋯I–I–(I–I)n, iodonium compounds CS⋯I+⋯SC, in addition to donor oxidation products such as [C–S–S–C]2+ dications.31 Cationic products can be counterbalanced by discrete or extended (poly)iodides with the general formula [I2m+n]n− (n, m > 0), such as [I4]2−, [I5]−, and [I7]−, with the largest reported so far being the ferrocenium salt of [I29]3−.36,37 To investigate the potential scenarios involving rhodanine-containing thiophene derivatives, BTR and I2 were made to react under various reaction conditions, systematically changing both the solvents and the molar ratios of reactants. Single crystals of BTR were grown by slow evaporation of an ethanol solution (Table S1, ESI†). The molecular unit is nearly planar except for the ethyl substituent at the rhodanine ring (Fig. 2a). Notably, the rhodanine ring shows the endocyclic sulfur atom periplanar with respect to the central thiophene ring, while the terminal thiophene is disordered over two positions, periplanar (64%) and antiperiplanar (36%) with respect to the central ring. Parallel BTR units (angle between the molecular planes 1.7°) are packed in a head-to-tail fashion based on π–π slipped stacking interactions between rhodanine and the terminal thiophene rings (intercentroid distance = 3.68 Å considering the major component) to form a pile developing along the [001] direction (Fig. 2b). Parallel piles pack along the [010] direction through interactions involving the ethyl substituents and the thiophene and the rhodanine rings. By either diffusion of diiodine vapors into a BTR hexane solution or direct mixing of hexane solutions of BTR and I2, a single type of product was isolated, identified as BTR·I2 by single-crystal X-ray diffraction analysis, and consisting of a CT “spoke” adduct showing a (C)S⋯I–I linear moiety [S1⋯I1, 2.7576(5); I1–I2, 2.8067(4) Å; S1–I1–I2, 164.70(1)°; Fig. 2c]. Analogous to the crystal structure of BTR, the disorder of the terminal thiophene was modelled over two sites with atomic occupancies of 85:15 with the predominance of the antiperiplanar conformation. Interestingly, the planarity observed for the BRT units in its crystal structure is retained in the CT adduct with I2. As typical of CT interactions, the transfer of electron density from the thiocarbonyl donor to I2 results in a slight elongation of the CS bond [C1–S1 = 1.632(2) and 1.666(1) Å in BTR and BTR·I2, respectively]. Accordingly, the I–I bond in BTR·I2 is elongated as compared to free I2 [I1–I2 = 2.806(1) and 2.715(6) Å in BTR·I2 and in the free I2,38 respectively], as a consequence of the CT to the I2 antibonding σ* MO. Accordingly, the I–I bond length in BTR·I2 corresponds to a Pauling bond order of 0.691.39 The strength of the CT interaction in BTR·I2 is weaker than the average of the related CT adducts deposited in the Cambridge Structural Database (CSD) between thiocarbonyl donors embedded in pentatomic rings and I2 featuring a S⋯I–I moiety [angle larger than 170°, 44 hits; S⋯I and I–I average distances = 2.695(7) and 2.879(5) Å, respectively]. The weakness of the interaction can be attributed to the presence of the electron withdrawing carbonyl group in the rhodanine ring. The bond distances and angles in the >CS⋯I–I fragment of BTR·I2 strictly recall those observed in the adduct (mdth)·I2 (mdth = 1,5,5-trimethyl-2-thiohydantoin) [S⋯I 2.818(2), I–I 2.795(1) Å, S⋯I–I 178.03(3)°].40 It is worth noting that the S⋯I and I–I relative bond distances in the S⋯I–I moiety clearly fall within the correlation proposed by Devillanova for linear three-body systems A–B–C involving either halogens, X–X–X (X = Br, I), or halogen(s) and chalcogen(s) atoms, Ch–X–Y, X–Ch–Y, and Ch–X–Ch (Ch = S, Se; X = Y = Cl, Br, I; X = I, Y = Cl, Br, I), which correlates the relative elongations δ1 and δ2 of the two interatomic distances with respect to the sum of the relevant covalent radii (Fig. S1 in the ESI†). Pairs of planar adduct units show π–π stacking interactions between the rhodanine ring of one unit and the terminal thiophene ring of a symmetry related one (intercentroid distance 3.67 Å; Fig. 2d). Dimers formed by adjacent pairs interact through C–S⋯I and C–H⋯O weak contacts, the latter defining a R22(10) hydrogen bonding motif as the result of two interacting symmetry-related rhodanine groups [C11i–S4 = 1.721(2); S4Ai⋯I1 = 3.660(1) Å; C11i–S4Ai⋯I1 = 172.66(8); C1–S1 = 1.666(2); C6–H6 = 0.95 Å; H6ii⋯O1 = 2.34; C6ii⋯O1 = 3.210(3) Å; C6ii–H6ii ⋯O1 = 151°; i = 1 + x, 1 + y, +z; ii = 1 − x, 2 − y, 2 − z; Fig. 2e and S2 in the ESI†]. In addition, the terminal I2 units show very weak I⋯I contacts, close to the sum of the relevant van der Waals radii, with a topology typical of bent type-I halogen bonds (XB) [I2⋯I2iii = 4.1267(5) Å; I1–I2⋯I2iii = 124.57(1)°; iii = 1–x, 1–y, –z; Fig. 2e].
 | ||
| Fig. 2 (a) Ellipsoid plot and atom labelling scheme of compound BTR. The terminal thiophene ring is disordered over two positions with fractional occupancies 64:36. (b) Intermolecular interactions in the crystal packing of compound BTR (intercentroid distance of 3.68 Å). The minor component of the disordered moiety was omitted for clarity. (c) Ellipsoid plot and atom labelling scheme of compound BTR·I2. The terminal thiophene ring is disordered over two positions with fractional occupancies 85:15. (d) π–π stacking interactions between pairs of BTR·I2 units featuring an intercentroid distance of 3.67 Å when considering the most abundant position of the disordered terminal thiophenyl moiety. (e) View along the [101] direction of a portion of a ribbon motif found in the crystal structure of BTR·I2 (see the text for a description of weak interactions). Ellipsoids were drawn at the 50% probability level. Hydrogen atoms were omitted for clarity in (b) and (d). Symmetry operations: i = 1 + x, 1 + y, +z; ii = 1 − x, 2 − y, 2 − z; iii = 1 − x, 1 − y, −z. See Tables S2–S5 (ESI†) for selected bond angles (°) and distances (Å). | ||
Raman spectroscopy
Raman spectroscopy is a sensitive technique providing structural information on dihalogens, interhalogens, and (inter)polyhalides.41 In particular, in “spoke” I2-adducts the extent of the elongation of perturbed diiodine is reflected in the shifting of the Raman peak due to the I–I σg stretching vibration. Molecular iodine (bond order nI–I = 1) shows a peculiar peak at 214.5 cm−1 in the gas phase, due to the I–I stretching vibration.42–45 The same vibration can be envisaged in the range 203–211 cm−1 in low-polar organic solvents.45 Accordingly, nearly monomeric I2 trapped in solid Kr displayed a distinct Raman band at 210.8 cm−1 at 32 K.44 In crystalline I2, two molecular units are present in the unit cell, and two vibrations can be envisaged, attributed to the in-phase (Ag, 180 cm−1) and out-of-phase (B3g, 189 cm−1) stretching modes.46,47 In solid state I2 at room temperature, only the former band is often observed, so that I2 is reported to feature a single Raman band.45,48 In molecular systems containing perturbed diiodine (nI–I < 1) the weakening of the I–I bond is reflected in a linearly dependent shift of the Raman Ag band toward lower wavenumbers. The Raman spectrum of BTR·I2 shows a single peak at 150 cm−1 (Fig. S3 in the ESI†), clearly absent in the Raman spectrum of BTR. The shift of this peak (about 30 cm−1) with respect to the peak observed for solid I2, together with the moderate elongation of the I–I bond, classifies BTR·I2 as a weak CT adduct (nI–I > 0.6).49 In fact, the Raman shift recorded for BTR·I2 falls within the linear correlation between the measured Raman frequency and the I–I bond distances reported for weak I2 CT adducts.49Computational study
To better understand the reactivity of BTR towards diiiodine, a set of theoretical calculations were performed on both BTR and its 1:1 diiodine CT adduct. Based on comparative investigations reported previously,50,51 the hybrid mPW1PW52 functional was paralleled by Schäfer, Horn and Ahlrichs split-valence basis sets including polarization functions (SVP),53 in the formulation of Weigend (def2-SVP)54 for all atoms except iodine, for which the LANL08(d)55 basis set including relativistic effective core potentials (RECPs) was adopted. Over the past few decades, the interaction of chalcogen donors with diiodine has been described by using two apparently opposite descriptions. Traditionally, this interaction was interpreted on the basis of an orbital-mixing model as a charge-transfer (CT) interaction between the Lewis base L, featuring a lone pair of electrons (LP) on the donor atom, and the I2 Lewis acid, featuring a σ* antibonding molecular orbital located along the I–I bond, so that the CT results in a lowering of the I–I bond order and an elongation of the bond as compared to that of unperturbed I2.31,43,48
More recently, the same type of interactions has been considered to be governed by the anisotropy of electrostatic potential on the interacting halogen atom, whose charge depletion sites (σ-holes),32 located opposite to the I–I bond in I2 (Fig. 3b), would orient the halogen towards more nucleophilic sites in the organic molecule, i.e. those displaying a more negative electrostatic potential. In this context, the spoke adduct L·I2 has been considered as the result of a halogen bond (XB)56,57 interaction between the I2 XB donor and the L acceptor.58 Recent theoretical studies carried out often at the DFT level have agreed that σ-hole interactions result from different contributions. Energy decomposition analyses (EDAs) have shown that the energy of σ-hole interactions can be decomposed into an electrostatic term and an MO-mixing term,59,60 in addition to dispersion.61 The relative weight of each contribution depends largely on the nature of the involved groups, so that the nature of the σ-hole interactions spans from almost purely ionic, especially at longer distances, to largely covalent, in which case a three-body system, featuring complementary bond orders, can be envisaged.
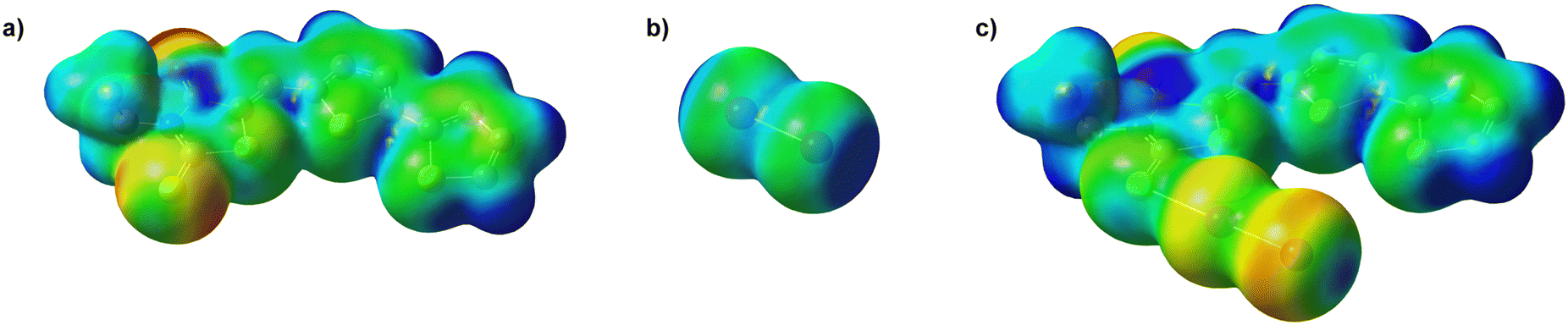 | ||
| Fig. 3 Molecular electrostatic potential (MEP) mapped on the electron density surface (5 × 10−3 |e| bohr−3) in BTR (a; periplanar conformation), I2 (b), and BTR·I2 (c; periplanar conformer). MEP range −0.05 (red) – +0.05 (blue) a.u. Hydrogen atoms were omitted for clarity. | ||
With the aim to get an insight into the interaction between BTR and I2, DFT calculations were carried out on the isolated BTR and I2 molecules, and the BTR·I2 “spoke” adduct (see Tables S6–S8 in the ESI† for the orthogonal Cartesian coordinates in the optimized geometries). As described above, the crystal structure of BTR shows the terminal thiophene ring disordered over the two possible planar and antiperiplanar conformations. Both planar conformers were optimized in the gas phase showing a negligible total electronic energy difference (0.8 kcal mol−1), thus justifying the presence of both conformers in the crystal structure. In Table 1, a selection of the natural charges62–64Q calculated for BTR at the optimized geometry is reported (see Tables S9–S11 for a full list in the ESI†). While thiophene and the rhodanine endocyclic S-atoms are positively charged, the rhodanine thiocarboxylic sulfur atom features a small negative charge. The Kohn–Sham (KS) HOMO and HOMO-1 calculated for BTR (Fig. S4 in the ESI†), very close in energy to each other (ε = –6.344 and –6.542 eV, respectively), show a significant contribution from the exocyclic S-atom. In particular, HOMO-1 is a 3p lone pair of electrons lying on the molecular plane. Therefore, the frontier MO composition and the natural population analysis (NPA)62–64 indicate the exocyclic sulfur atom of the rhodanine ring as the preferred donor site towards I2 in a CT interaction. On the other hand, Fig. 3a shows the electrostatic potential calculated for BTR, mapped on the electron density, clearly showing that the most negative electrostatic potential regions on the sulfur electron density are located on the molecular plane, in accordance with the structural data collected for BTR·I2. Thus, the two limit views of MO interpretation and electrostatic potential map converge to the same result. Indeed, the regions on the sulfur atom showing the largest negative electrostatic potential coincide with those where the lobes of KS-HOMO-1 isosurfaces are located (see Fig. S5 in the ESI†).
| Q | BTR | BTR·I2 |
|---|---|---|
| O1 | −0.591 | −0.581 |
| N1 | −0.502 | −0.491 |
| S1 | −0.064 | −0.020 |
| S2 | 0.374 | 0.417 |
| S3 | 0.430 | 0.445 |
| S4 | 0.430 | 0.439 |
| I1 | — | −0.028 |
| I2 | — | −0.183 |
The geometry of the 1:1 “spoke” adduct BTR·I2 was successfully optimized (Fig. S6 in the ESI†), with the diiodine unit lying on the molecular plane, in agreement with the structural data discussed above. A very good agreement between calculated and experimental bond lengths and angles was found. In particular, the interaction between the thiocarbonyl rhodanine group and diiodine shows metric parameters (CS, 1.661; S⋯I, 2.951; I–I, 2.780 Å; S⋯I–I, 173.2°), close to those determined by single crystal X-ray diffraction analysis (see above). This notwithstanding, the optimized I–I bond distance is slightly longer than the experimental value (by 0.027 Å), suggesting a modest underestimation of the CT interaction. The moderate elongation of the I–I bond distance dI–I in the adduct as compared with that optimized at the same level of theory for I2 (ΔdI–I = 0.114 Å) is in agreement with the weak nature of the interaction. The interaction energy in the “spoke” adduct associated with the orbital mixing and CT can be evaluated by means of a second order perturbation theory analysis of the Fock matrix in the Natural Bond Orbital basis at the optimized geometry. This is achieved by examining all possible interactions between occupied (donor) NBOs and virtual (acceptor) non-Lewis NBOs and evaluating their interaction energy by means of the second order perturbation theory.64 For each donor NBO (i) and acceptor NBO (j), the associated stabilization energy ECT can be estimated as
| ECT = qi·F(i,j)/(εj − εi) |
As a consequence of the CT, the I–I Wiberg bond65 index in the I2 unit is lowered to 0.793, which is slightly larger than the value calculated from the structural data using Pauling's equation.
This is consistent with the weak adduct nature of the BTR·I2 adduct, evidenced by the optimized metric parameters (see above) and experimentally confirmed by Raman spectroscopy (see above). The molecular electron density mapped on the density surface in BTR·I2 clearly shows the most negative potential on the most electronegative oxygen atoms, in accordance with the NPA (Fig. 3c). It is worth noting that the terminal iodine atom in the adduct displays a distribution of the electrostatic potential pointing to a σ-hole topology, in perfect agreement with the long XB interactions connecting the stacks of BTR·I2 in the solid state, responsible for the 3D lattice dimensionality.
Although Koopmans’ theorem cannot be applied to density functional theory, the Kohn–Sham eigenvalues can be considered as useful parameters66 in evaluating the variations in the HOMO–LUMO energy gap induced by the interaction of BTR with I2. In fact, the gas-phase ΔεHL HOMO–LUMO energy gap is in good agreement with the band gap of small π-conjugated compounds.57,67 In the case of BTR and BTR·I2 ΔεHL values of 3.26 and 3.16 eV are calculated (Fig. 4), falling within the range –2.5 ∼ –4.0 eV, typical of a small organic semiconductor.67 It is noteworthy that the case of BTR clearly demonstrates how I2-adduct formation can efficiently reduce the HOMO–LUMO energy gap in the resulting adducts compared to the pristine substrates. The case of study shows that this strategy holds not only when Lewis donors with a large Mulliken hardness are considered, as in the case of N-donors,51 but also when softer S-donors are considered, as in the case of BTR. Finally, a vibrational analysis carried out on BTR·I2 confirms the attribution of the Raman peak at 150 cm−1 as due to the stretching of the perturbed I2 unit.
 | ||
| Fig. 4 Kohn–Sham frontier orbital energy levels for BTR (left) and BTR·I2 (right). Color: carbon, gray; nitrogen, blue; oxygen, red; sulfur, yellow; hydrogen, white. Cutoff value = 0.05 |e|. | ||
Single crystal electrical characterization
To investigate the impact of the molecular organization, induced by the thiocarbonyl–diiodine interactions, on the charge transport properties of the bulk material, we fabricated single crystal junction devices (see the ESI† for details). Single crystals of BTR and BTR·I2 were carefully transferred to the top of a glass substrate grafted with two gold pads. Each crystal was disposed to bridge the two electrodes, and a droplet of silver paste was applied in each crystal–metal junction to obtain a more stable electrical contact (Fig. 5a). In order to evaluate the stability and the reproducibility of the measurements, the two devices were measured on two different days and each measuring run was cycled five times. The average results of the electrical characterization are reported in Fig. 5b. From the reported plots we clearly observe that the different runs of measurements are very reproducible, as demonstrated by the limited standard deviation (see error bars) and, most importantly, that the BTR·I2 adduct is characterized by a much higher conductivity with respect to the crystals of the pristine BTR. It is worth noting that the distance between the two electrodes was kept constant in the two sets of devices, and that the crystals have been properly selected to have comparable geometrical dimensions, i.e. width and height. The enhanced electrical current of the adducts might suggest that molecules have adopted a more efficient packing for charge percolation, and at the same time diiodine may have facilitated the formation of charge carriers in the crystal. It is noteworthy to observe that diiodine was not displaced upon electric current application, as the value of conductivity is steady over time.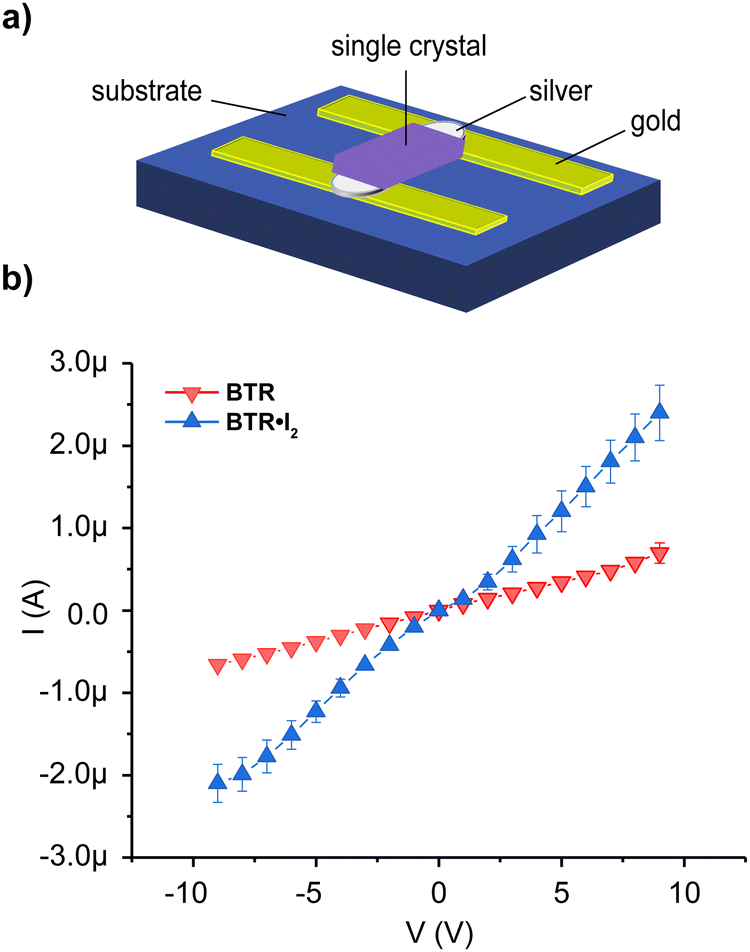 | ||
| Fig. 5 (a) Device architecture for single-crystal electrical characterization. (b) Average current–voltage plots of BTR (red) and BTR·I2 (blue). | ||
Conclusions
In summary, we have presented a novel approach to regulate the molecular organization of rhodanine-containing π-conjugated compounds. Our study demonstrates that I2 can interact at the molecular level, forming stable adducts through interaction with the rhodanine thiocarbonyl terminal groups. Furthermore, preliminary study on the electrical conductivity of single crystals suggests that the molecular arrangement in BTR·I2 leads to an enhancement of the flow of electric current. Therefore, the following conclusions can be drawn:(1) I2 can be used as a solid additive to tailor the molecular organization of rhodanine-containing π-conjugated thiophene derivatives.
(2) The diiodine adduct undergoes a reduction in the HOMO–LUMO energy gap compared to the pristine compound.
(3) FT-Raman spectroscopy represents an effective and fast non-destructive approach to reveal the coordination of diiodine and can therefore be used in semiconductors to verify the adduct formation and its strength in the absence of a single crystal X-ray diffraction analysis, in particular when non-crystalline materials are considered.
(4) Finally, DFT calculations can model both the CT/σ-hole interaction between the Lewis donor and diiodine and the XB intermolecular interactions. In this context, neglecting dispersion effects, two concurrent interpretation models for σ-hole interactions have been proposed, i.e. a MO-mixing approach, consisting of a partial charge-transfer from a nucleophile (Lewis base) to an electrophile (Lewis acid), or a noncovalent σ-hole interpretation based on the topology of electrostatic potential. Energy decomposition schemes have shown that both aspects contribute to XB and ChB interactions. As previously shown for different systems showing XB and ChB interactions involving chalcogen donors interacting with dihalogens/interhalogens the two models converge to the same conclusions, with the depletion areas of electrostatic potential (σ-holes) being topologically related to the direction of antibonding natural orbitals.
The case study of BTR/BTR·I2 shows for the first time the role played by the introduction of chalcocarbonyl groups in π-conjugated derivatives in the formation of supramolecular architectures based on XB/ChB interactions capable of extending the dimensionality of their lattice, reducing the HOMO–LUMO energy gap, and ultimately increasing their semiconducting behaviour.
These results pave the way for the development of novel processing methods that can be directly applied to SM semiconductors, facilitating the fabrication of optoelectronic devices, such as organic solar cells based on non-fullerene small-molecule acceptors, organic field-effect transistors (OFETs), and organic memory devices (OMDs).
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and/or its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. S. would like to thank PON Ricerca e Innovazione (annualità 2014-2020, grant no. DOT1304455-2), Fondazione di Sardegna (annualità 2020, FdS grant no. F75F21001250007) and the Italian Ministry of University and Research (MUR) (National Recovery and Resilience Plan (NRRP) funded by the European Union – Next Generation EU; annualità 2022, grant no. P2022ZHBZE) for the financial support. MA, MCA, VL, and AP acknowledge the Ministero per l’Ambiente e la Sicurezza Energetica (MASE; formerly Ministero della Transizione Ecologica, MITE) – Direzione generale Economia Circolare for funding (RAEE – Edizione 2021). M. A. acknowledges the CINECA award under the ISCRA initiative, for the availability of high-performance computing resources and support (project ChWEEE, HP10CDTEF0). We acknowledge the CeSAR (Centro Servizi di Ateneo per la Ricerca) of the University of Cagliari, Italy for providing access to the SC-XRD facility.Notes and references
- M. Sawatzki-Park, S. Wang, H. Kleemann and K. Leo, Highly Ordered Small Molecule Organic Semiconductor Thin-Films Enabling Complex, High-Performance Multi-Junction Devices, Chem. Rev., 2023, 123, 8232–8250 CrossRef CAS PubMed.
- S. Fratini, M. Nikolka, A. Salleo, G. Schweicher and H. Sirringhaus, Charge transport in high-mobility conjugated polymers and molecular semiconductors, Nat. Mater., 2020, 19, 491–502 CrossRef CAS PubMed.
- K. Gu and Y. Loo, The Polymer Physics of Multiscale Charge Transport in Conjugated System, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 1559–1571 CrossRef CAS.
- K. Kim, H. Yu, H. Kang, D. J. Kang, C. Cho, H. Cho, J. H. Oh and B. J. Kim, Influence of intermolecular interactions of electron donating small molecules on their molecular packing and performance in organic electronic devices, J. Mater. Chem. A, 2013, 1, 14538–14547 RSC.
- F. Chen, Y. Wang, W. Zhang, T. Tian, B. Bai, H. Wang, F. Bai and M. Li, Role of Intermolecular Interactions in Molecular Packing of Alkoxy-Substituted Bis-1,3,4-oxadiazole Derivatives, Cryst. Growth Des., 2019, 19, 6100–6113 CrossRef CAS.
- M. Zhu, P. Li, J. Li and T. Lei, Molecular packing and film morphology control in organic electrochemical transistors, Mol. Syst. Des. Eng., 2022, 7, 6–20 RSC.
- P. A. Banks, A. M. Dyer, A. C. Whalley and M. T. Ruggiero, Side-chain torsional dynamics strongly influence charge transport in organic semiconductor, Chem. Commun., 2022, 58, 12803–12806 RSC.
- J. Chen, W. Zhang, L. Wang and G. Yu, Recent Research Progress of Organic Small-Molecule Semiconductors with High Electron Mobilities, Adv. Mater., 2023, 35, 2210772 CrossRef CAS PubMed.
- J. Yi, G. Zhang, H. Yu and H. Yan, Advantages, challenges and molecular design of different material types used in organic solar cell, Nat. Rev. Mater., 2024, 9, 46–62 CrossRef CAS.
- X. Li, X. Kong, G. Sun and Y. Li, Organic small molecule acceptor materials for organic solar cells, eScience, 2023, 3, 100171 CrossRef.
- B. Kan, Y. Kan, L. Zuo, X. Shi and K. Gao, Recent progress on all-small molecule organic solar cells using small-molecule nonfullerene acceptors, InfoMat, 2021, 3, 175–200 CrossRef CAS.
- R. Zhoul, Z. Jiang, C. Yang, J. Yu, J. Feng, M. A. Adil, D. Deng, W. Zou, J. Zhang, K. Lu, W. Ma, F. Gao and Z. Wei, All-small-molecule organic solar cells with over 14% efficiency by optimizing hierarchical morphologies, Nat. Commun., 2019, 10, 5393 CrossRef PubMed.
- J. Ahn, et al., Simple and Versatile Non-Fullerene Acceptor Based on Benzothiadiazole and Rhodanine for Organic Solar Cells, ACS Appl. Mater. Interfaces, 2019, 11, 30098–30107 CrossRef CAS PubMed.
- C. Yan, S. Barlow and Z. Wang, et al., Non-fullerene acceptors for organic solar cells, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- S. Shoaee, A. L. Sanna and G. Sforazzini, Elucidating Charge Generation in Green-Solvent Processed Organic Solar Cells, Molecules, 2021, 26, 7439 CrossRef CAS PubMed.
- Z. Zhang, J. Yuan, Q. Wei and Y. Zou, Small-Molecule Electron Acceptors for Efficient Non-fullerene Organic Solar Cells, Front. Chem., 2018, 6, 414 CrossRef PubMed.
- X. Yao, W. Shao, X. Xiang, W. Xiao, L. Liang, F. Zhao, J. Lingd, Z. Lu, J. Li and W. Li, Side chain engineering on a small molecular semiconductor: Balance between solubility and performance by choosing proper positions for alkyl side chains, Org. Electron., 2018, 61, 56–64 CrossRef CAS.
- A. Khot and B. M. Savoie, How side-chain hydrophilicity modulates morphology and charge transport in mixed conducting polymers, J. Polym. Sci., 2022, 60, 610–620 CrossRef CAS.
- I. E. Kuznetsov, et al., Tailoring the charge transport characteristics in ordered small-molecule organic semiconductors by side-chain engineering and fluorine substitution, Phys. Chem. Chem. Phys., 2022, 24, 16041–16049 RSC.
- P. J. W. Sommerville, A. H. Balzer, G. Lecroy, L. Guio, Y. Wang, J. W. Onorato, N. A. Kukhta, X. Gu, A. Salleo, N. Stingelin and C. K. Luscombe, ACS Polym. Au, 2023, 3, 59–69 CrossRef CAS PubMed.
- Y. He, N. A. Kukhta, A. Marks and C. K. Luscombe, The effect of side chain engineering on conjugated polymers in organic electrochemical transistors for bioelectronic applications, J. Mater. Chem. C, 2022, 10, 2314–2332 RSC.
- R. Datt, Suman, A. Bagui, A. Siddiqui, R. Sharma, V. Gupta, S. Yoo, S. Kumar and S. P. Singh, Effectiveness of solvent vapor annealing over thermal annealing on the photovoltaic performance of non-fullerene acceptor based BHJ solar cells, Sci. Rep., 2019, 9, 8529 CrossRef PubMed.
- T. Shan, Y. Zhang, Y. Wang, Z. Xie, Q. Wei, J. Xu, M. Zhang, C. Wang, Q. Bao, X. Wang, C.-C. Chen, J. Huang, Q. Chen, F. Liu, L. Chen and H. Zhong, Universal and versatile morphology engineering via hot fluorous solvent soaking for organic bulk heterojunction, Nat. Commun., 2020, 11, 5585 CrossRef CAS PubMed.
- H. Liao, C. Ho, C. Chang, M. Jao, S. B. Darling and W. Su, Additives for morphology control in high-efficiency organic solar cells, Mater. Today, 2013, 16, 326–336 CrossRef CAS.
- H. Lin, X. Yao, M. Li, X. Yu, X. Du, G. Yang, C. Zheng and S. Tao, Volatile Solvent Additives Enabling High-Efficiency Organic Solar Cells without Thermal Annealing, ACS Appl. Energy Mater., 2022, 5, 15529–15537 CrossRef CAS.
- Y. Jiang and Z. Li, The effect of additives on the morphology of organic solar cells: A brief review, Synth. Met., 2023, 299, 117480 CrossRef CAS.
- M. Choi, H. Jeong, J. Lee, Y. Choi, I. Kim, D. Kim, H. Kang and S. Jang, Non-halogenated and non-volatile solid additive for improving the efficiency and stability of organic solar cells, J. Mater. Chem. A, 2024, 12, 8963–8971 RSC.
- Y. Ma, Y. Zhang and H. Zhang, Solid additives in organic solar cells: progress and perspective, J. Mater. Chem. C, 2022, 10, 2364–2374 RSC.
- S. Yoon, J. Cho, S. H. Yu, Hae J. Son and D. S. Chung, Effects of iodine doping on small molecule organic semiconductors for high charge carrier mobility and photoconductivity, Org. Electron., 2016, 34, 28e32 CrossRef.
- C. J. Boyle, et al., Tuning charge transport dynamics via clustering of doping in organic semiconductor thin films, Nat. Commun., 2019, 10, 2827 CrossRef PubMed.
- M. C. Aragoni, M. Arca, F. A. Devillanova, A. Garau, F. Isaia, V. Lippolis and G. Verani, Charge-transfer adducts between donors containing chalcogens (S and Se) and di-iodine: solution studies, Coord. Chem. Rev., 1999, 184, 271–290 CrossRef CAS.
- P. Politzer, J. S. Murray and T. Clark, Halogen bonding and other sigma-hole interactions: a perspective, Phys. Chem. Chem. Phys., 2013, 15, 11178–11189 RSC.
- M. C. Aragoni, M. Arca, V. Lippolis, A. Pintus, Y. Torubaev and E. Podda, A structural approach to the strength evaluation of linear chalcogen bonds, Molecules, 2023, 28, 3133 CrossRef CAS PubMed.
- A. Vilsmeier and A. Haack, Über die Einwirkung von Halogenphosphor auf Alkyl-formanilide. Eine neue Methode zur Darstellung sekundärer und tertiärer p-Alkylamino-benzaldehyde, Ber. Dtsch. Chem. Ges., 1927, 60, 119–122 CrossRef.
- K. van Beurden, S. de Koning, D. Molenndijk and J. van Schijndel, The Knoevenagel reaction: review of the unfinished treasure map to forming carbon–carbon bonds, Green Chem. Lett. Rev., 2020, 13, 349–364 CrossRef CAS.
- M. C. Aragoni, M. F. Cherchi, V. Lippolis, A. Pintus, E. Podda, A. M. Z. Slawin, J. D. Woollins and M. Arca, Self-Assembly of Supramolecular Architectures Driven by σ-Hole Interactions: A Halogen-Bonded 2D Network Based on a Diiminedibromido Gold(III) Complex and Tribromide Building Blocks, Molecules, 2022, 27, 6289 CrossRef CAS PubMed.
- H. Svensson and L. L. Kloo, Synthesis, Structure, and Bonding in Polyiodide and Metal Iodide−Iodine Systems, Chem. Rev., 2003, 103, 1649–1684 CrossRef PubMed.
- F. van Bolhuis, P. B. Koster and T. Migchelsen, Refinement of the crystl structure of iodine at 110°K, Acta Crystallogr., 1967, 23, 90–91 CrossRef CAS.
- L. Pauling, The Nature of the Chemical Bond, 3rd ed., Cornell University Press, Ithaca, New York, 1960 Search PubMed.
- R. Montis, M. Arca, M. C. Aragoni, A. Bauzá, F. Demartin, A. Frontera, F. Isaia and V. Lippolis, Hydrogen- and halogen-bond cooperativity in determining the crystal packing of dihalogen charge-transfer adducts: a study case from heterocyclic pentatomic chalcogenone donors, CrystEngComm, 2017, 19, 4401–4412 RSC.
- M. C. Aragoni, M. Arca, F. A. Devillanova, M. B. Hursthouse, S. L. Huth, F. Isaia, V. Lippolis, A. Mancini, H. R. Ogilvie and G. Verani, Reactions of pyridyl donors with halogens and interhalogens: an X-ray diffraction and FT-Raman investigation, J. Organomet. Chem., 2005, 690, 1923–1934 CrossRef CAS.
- W. Holzer, W. F. Murphy and H. J. Bernstein, Resonance Raman Effect and Resonance Fluorescence in Halogen Gases, J. Chem. Phys., 1970, 52, 399–407 CrossRef CAS.
- O. Yildirim, A. Tsaturyan, A. Damin, S. Nejrotti, V. Crocellà, A. Gallo, M. R. Chierotti, M. Bonomo and C. Barolo, Quinoid-Thiophene-Based Covalent Organic Polymers for High Iodine Uptake: When Rational Chemical Design Counterbalances the Low Surface Area and Pore Volume, ACS Appl. Mater. Interfaces, 2023, 15, 15819–15831 CrossRef CAS PubMed.
- E. Hullko, T. Kiljunen, T. Kiviniemi and M. Pettersson, From monomer to bulk: Appearance of the Structural Motif of Solid Iodine in Small Clusters, J. Am. Chem. Soc., 2009, 131, 1050–1056 CrossRef PubMed.
- W. Kiefer and H. J. Bernstein, Resonance Raman spectroscopic study on iodine in various organic solvents: Spectroscopic constants and halfband widths of the I2 vibration, J. Raman Spectrosc., 1973, 1, 417–431 CrossRef CAS.
- A. Congeduti, M. Nardone and P. Postorino, Polarized Raman spectra of a single crystal of iodine, Chem. Phys., 2000, 256, 117–123 CrossRef CAS.
- M. Bulatova, D. M. Ivanov, J. M. Rautiainen, M. A. Kinzhalov, K.-N. Truong, M. Lahtinen and M. Haukka, Studies of Nature of Uncommon Bifurcated I–I⋯(I–M) Metal-Involving Noncovalent Interaction in Palladium(II) and Platinum(II) Isocyanide Cocrystals, J. Phys. Chem. A, 2019, 123, 4575–4580 CrossRef PubMed.
- P. D. Boyle, J. Christie, T. Dyer, S. M. Godfrey, I. R. Howson, C. McArthur, B. Omar, R. G. Pritchard and G. Rh Williams, Further structural motifs from the reactions of thioamides with diiodine and the interhalogens iodine monobromide and iodine monochloride: an FT-Raman and crystallographic study, J. Chem. Soc., Dalton Trans., 2000, 3106–3112 RSC.
- M. Arca, M. C. Aragoni, F. A. Devillanova, A. Garau, F. Isaia, V. Lippolis, A. Mancini and G. Verani, Reactions Between Chalcogen Donors and Dihalogens/Interalogens: Typology of Products and Their Characterization by FT-Raman Spectroscopy, Bioinorg. Chem. Appl., 2006, 58937, 1–12 Search PubMed.
- M. C. Aragoni, M. Arca, C. Caltagirone, C. Castellano, F. Demartin, P. G. Jones, T. Pivetta, E. Podda, V. Lippolis, S. Murgia and G. Picci, Role of the Solvent in the Reactivity of Bis-4-imidazoline-2-selone Derivatives toward I2: An Experimental and Theoretical Approach, J. Org. Chem., 2022, 87, 15448–15465 CrossRef CAS PubMed.
- M. C. Aragoni, M. Arca, F. Demartin, F. A. Devillanova, A. Garau, F. Isaia, V. Lippolis and G. Verani, DFT calculations, structural and spectroscopic studies on the products formed between IBr and N,N′-dimethylbenzoimidazole-2(3H)-thione and -2(3H)-selone, Dalton Trans., 2005, 2252–2258 RSC.
- C. Adamo and V. Barone, Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, Fully optimized contracted Gaussian basis sets for atoms Li to Kr, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- L. E. Roy, P. J. Hay and R. L. Martin, Revised Basis Sets for the LANL Effective Core Potentials, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, The halogen bond, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- Y. Zhang and W. Wang, Theoretical rationale for the role of strong halogen bond in the design of organic semiconductor materials, Comput. Theor. Chem., 2021, 1194, 113074 CrossRef CAS.
- L. J. McAllister, C. Präsang, J. P.-W. Wong, R. J. Thatcher, A. C. Whitwood, B. Donnio, P. O’Brien, P. B. Karadakov and D. W. Bruce, Halogen-bonded liquid crystals of 4-alkoxystilbazoles with molecular iodine: a very short halogen bond and unusual mesophase stability, Chem. Commun., 2013, 49, 3946–3948 RSC.
- L. de Azevedo Santos, S. C. C. van der Lubbe, T. A. Hamlin, T. C. Ramalho and F. M. Bickelhaupt, A quantitative molecular orbital perspective of the chalcogen bond, ChemistryOpen, 2021, 10, 391–401 CrossRef CAS PubMed.
- V. Oliveira, E. Kraka and D. Cremer, Phys. Chem. Chem. Phys., 2016, 18, 33031–33046 RSC.
- L. N. Anderson, F. W. Aquino, A. E. Raeber, X. Chen and B. M. Wong, Halogen Bonding Interactions: Revised Benchmarks and a New Assessment of Exchange vs Dispersion, J. Chem. Theory Comput., 2018, 14, 180–190 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural Population Analysis, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- A. E. Reed and F. Weinhold, Natural localised molecular orbital, J. Chem. Phys., 1985, 83, 1736–1740 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- K. B. Wiberg, Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to cyclobutane, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- J. Luo, Z. Q. Xue, W. M. Liu, J. L. Wu and Z. Q. Yang, Koopmans’ theorem for large molecular systems within density functional theory, J. Phys. Chem. A, 2006, 110(43), 12005–12009 CrossRef CAS PubMed.
- X. G. Yang, Y. Zhang, W. Wang and L. F. Ma, Enhanced Optoelectronic Performances of the Cocrystals between 2,2′-Bi (1,8-naphthyridine) and Iodine through Strong Halogen Bonds, Cryst. Growth Des., 2022, 22, 6863–6869 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, XRD analyses, computational details. CCDC 2341835 and 2341836. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4tc01791a |
| This journal is © The Royal Society of Chemistry 2024 |