
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards photoelectrochromic modulation of NIR absorption in plasmonic ITO using pentacene films†
Anthony
Maho
 abc,
Dong Kuk
Kim
a,
Jessica
Wade
a,
Emma
Bryan
a,
Luc
Henrard
d,
Yoann
Olivier
d,
Stoichko D.
Dimitrov
e,
Rudi
Cloots
b and
Sandrine
Heutz
*a
abc,
Dong Kuk
Kim
a,
Jessica
Wade
a,
Emma
Bryan
a,
Luc
Henrard
d,
Yoann
Olivier
d,
Stoichko D.
Dimitrov
e,
Rudi
Cloots
b and
Sandrine
Heutz
*a
aDepartment of Materials and London Center for Nanotechnology, Imperial College London, London SW7 2AZ, UK. E-mail: s.heutz@imperial.ac.uk
bGroup of Research in Energy and Environment from Materials (GREENMAT), University of Liège, Quartier Agora – B6a, Sart-Tilman, 4000 Liège, Belgium
cUniv. Bordeaux, CNRS, Bordeaux INP, ICMCB, UMR 5026, 87 Avenue du Docteur Schweitzer, 33600 Pessac, France
dDepartment of Physics and Namur Institute of Structured Materials, University of Namur, Rue de Bruxelles 61, 5000 Namur, Belgium
eDepartment of Chemistry, Queen Mary University of London, London E1 4NS, UK
First published on 26th September 2024
Abstract
As a prototypical organic semiconductor material, pentacene is investigated in numerous electronic and optical systems for its high charge carrier mobility and attractive photophysical properties. However, charge generation, transfer and collection remain critical challenges, especially when considering hybrid structures with inorganic metal oxides as charge acceptors. Herein pentacene molecular layers are associated with plasmonic indium tin oxide (ITO) nanostructures, aiming to allow them to electrochromically modulate their near-infrared (NIR) absorption in the absence of external electric power. Multi-modal surface characterization shows that pentacene molecules transition from a perpendicular alignment when deposited on bare surfaces to more random organization, involving standing-up and face-on orientations, when deposited on ITO thin films. The optoelectronic properties of the interfaced materials are consequently impacted, with steady-state and ultrafast transient spectroscopies further highlighting how electrons are photogenerated in pentacene and consecutively transferred into plasmonic ITO, ultimately tuning its NIR optical response. Such ITO–pentacene bilayers therefore hold promise as novel heterojunction-like structures for the further innovative design of photoelectrochromic, self-powered smart windows.
1. Introduction
The remarkable structural, optical and electronic properties of pentacene make it an attractive material integrable to numerous device architectures that rely on highly ordered molecular thin films.1–3 Alongside efficient transport properties due to high charge carrier mobility, pentacene's unique photoactivity has notably attracted major interest in organic solar cells for its singlet fission activity, namely the spin-allowed photophysical process that enables the generation of up to two electrons from the absorption of a single photon via the specific conversion of one singlet excited state into two triplet states.4–8 Pentacene can therefore overcome the Shockley–Queisser theoretical limit of photoconversion efficiency in photovoltaic or photodetector devices, with singlet fission mechanisms leading to quantum yields over 100% with extremely fast kinetics (fs range) of charge generation. Various organic (fullerenes,9–13 phthalocyanines,14 tetracyanoquinodimethane (TCNQ), etc.15) and inorganic compounds (PbS,16 PbSe,17 MoS2,18,19etc.) have already shown their suitability as convenient charge acceptor materials as they are able to dissociate singlet and/or triplet excitons. The use of metal oxides as electron acceptors, being easily processable and bearing outstanding semiconducting properties thanks to their wide bandgaps, remains a significant opportunity in the field,20–24 especially as it involves the controlled design and in-depth characterization of potentially complex hybrid inorganic–organic structures. Studies focusing on pentacene interfaced with TiO2 and ZrO2 (nano)structures20–22 have recently highlighted the critical balance needed between efficient electron generation and suitable energetics for charge transfer between the organic donor and the inorganic acceptor. Charge generation, transfer and collection are ultimately shown to be highly dependent on the interface chemical, morphological and structural properties, considerably influencing both their dynamics and kinetics.Indium tin oxide (ITO), being widely used as a transparent electrode material of high conductivity and electrochemical tuneability,25–27 can also be considered as an inorganic electron acceptor in this context.28,29 Interestingly, ITO is subject to localized surface plasmon resonance (LSPR) when processed into highly crystalline nanoparticles through colloidal synthesis, which induces strong absorption of near-infrared (NIR) light in the 1.5–2.0 μm wavelength range. LSPR can be controlled by tuning the ITO nanostructure, i.e. size, shape, dopant concentration and distribution, depending on the synthetic protocols being used;30–33 it can also be modulated through electrical or electrochemical charging when exposed to an external bias, resulting in progressive and reversible changes in absorption frequency and intensity.34–38 When processed as a coating, plasmonic ITO therefore allows for NIR-selective electrochromic abilities of particular interest for the development of new-generation heat-filtering smart windows, being more optically functional and more durable than conventional electrochromic technologies. Indeed, whilst most electrochromic smart glazing systems traditionally rely on faradaic redox processes in metal oxides, the use of plasmonic nanocrystals allows for the occurrence of capacitive charging mechanisms which do not require cationic structural insertion/extraction – being potentially detrimental upon long-term cycling. The LSPR modulation in ITO is considered to be analogous to that found in metallic nanostructures, and is reported to result from the formation of accumulation and depletion regions near the surface of the nanocrystals. Such behavior implies larger ranges of modulation with smaller nanocrystals, i.e. 5 nm radius and below, as their entire volume can then lie within the accumulation/depletion region. Therefore, ITO coatings operate like supercapacitor electrodes whereas conventional electrochromic films are more analogous to battery electrodes, being subjected to cation migration through the active material; this capacitive mode specifically endows them with faster switching kinetics and enhanced stability upon reversible charge/discharge cycling.31,39
To strengthen the operational autonomy and bypass the need for an external power generator,40–42 we propose herein to study how plasmonic electrochromic materials can be combined with suitable photoactive components, namely, processing ITO–pentacene bilayers with the perspective of designing novel NIR-active photoelectrochromic systems. Photoelectrochromism refers to a change in color (or more generally, in optical properties) being produced within a considered material by charge carriers being photogenerated outside of this considered material. It constitutes a particular configuration of X-chromism to be distinguished from photochromism, where the change in color/optical properties is generated from light absorption within the considered material, and from electrochromism, referring to an electrically induced change in color/optical properties within the considered material. Here, relying on appropriate thin coating engineering, pentacene thin films are able to generate photoexcited electrons from visible (VIS) light absorption; these electrons can then be efficiently transferred and collected in ITO, which permits their NIR optical modulation.
Such a concept of structural architecture stands therefore as a disruptive alternative to previously designed photoelectrochromic systems, which typically require complex tandem configurations (i.e., a solar cell connected to an electrochromic device) or bulky multilayered assemblies associating electrochromic, electrolytic and photovoltaic components.43,44 In addition, the considered structure represents a heterojunction of pentacene as an organic molecular photodonor being interfaced with nanostructured ITO acting as an inorganic photoacceptor. This implies that an electrolyte, ion-conducting layer is presently not required as only electrons are involved in such capacitive form of electrochromism, and thus no insertion/extraction of cations takes place. All in all, the present approach of materials chemistry and structure design is therefore novel and can be transformative for the electrochromic field, with and additional impact expected in correlated fields of optoelectronics, semiconductors, and photovoltaics.
2. Experimental section
Unless specified otherwise, all chemicals are supplied by Sigma-Aldrich and used without further purification. Tin-doped (10 wt. %) indium oxide ITO nanocrystals are synthesized using previously established colloidal approaches.30,36 Briefly, indium and tin(II) acetates (99.99%) are mixed with oleylamine (70%), degassed under vacuum for 1 h at 120 °C, and then reacted at 230 °C under nitrogen for 1 h; after cooling to 60 °C, toluene (VWR, 99.9%) and oleic acid (90%) are added to the solution, and ITO nanocrystals are purified and recovered (five washing cycles) by adding absolute ethanol, centrifuging, and redispersing in toluene. The hydrophobic, insulating ligands bound to the ITO surface are then chemically removed using a nitrosonium tetrafluoroborate (NOBF4, 95%) solution in N,N-dimethylformamide (DMF, 99.8%); ligand stripping is induced by sonication, and bare ITO nanocrystals are recovered (seven washing cycles) by precipitating with toluene, centrifuging, and redispersing in DMF. Transmission electron microscopy (TEM) measurements on ITO nanocrystals are completed using a JEOL JEM-2100F instrument. ITO dispersions (45 mg mL−1) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMF:acetonitrile by volume are then spin-coated (40 μL) onto silicon, glass or quartz substrates at 5000 rpm for 30 s (three consecutive runs in total), resulting in uniform ITO layers. Pentacene thin films are grown by organic molecular beam deposition (OMBD) in a Kurt J. Lesker Spectros 100 system at a base pressure of 5 × 10−7 mbar, from pentacene powder (TCI, 99.999%) being sublimed at a rate of 0.5 Å s−1 on bare or ITO-coated silicon, glass or quartz substrates, kept at ambient temperature during deposition.
:1 DMF:acetonitrile by volume are then spin-coated (40 μL) onto silicon, glass or quartz substrates at 5000 rpm for 30 s (three consecutive runs in total), resulting in uniform ITO layers. Pentacene thin films are grown by organic molecular beam deposition (OMBD) in a Kurt J. Lesker Spectros 100 system at a base pressure of 5 × 10−7 mbar, from pentacene powder (TCI, 99.999%) being sublimed at a rate of 0.5 Å s−1 on bare or ITO-coated silicon, glass or quartz substrates, kept at ambient temperature during deposition.
Surface characterization is carried out on silicon substrates covered with ITO, pentacene and ITO–pentacene bilayers using tapping mode atomic force microscopy (AFM) on an Asylum Research MFP-3D instrument, and scanning electron microscopy (SEM) on a Zeiss LEO Gemini 1525 instrument. Root mean square roughness (Rq) values are determined from the total window of the AFM images using the open-source software Gwyddion, and calculated over three different images with the standard deviation quoted as an error. Top-view and cross-section SEM images are recorded on samples coated with a 10 nm conductive layer of chromium and grounded to the sample holder with silver paste. Optical microscopy images are recorded using an Olympus BX51 instrument. X-ray diffraction is conducted in the fixed θ–2θ geometry on a Philips X’Pert Pro Panalytical instrument using a Cu Kα source at a current of 40 A and a voltage of 40 V. The Scherrer equation D = Kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) cos(θ) is used to determine the relationship between the crystallite size D and the full width at half maximum (FWHM) β of a peak of the diffraction pattern, K is a dimensionless shape factor equaling 0.9, λ is the wavelength of the incident Cu Kα radiation, and θ is the Bragg angle. Raman spectra are recorded using a Renishaw inVia Raman microscope with a 50X objective lens, using a 633 nm excitation wavelength and a total acquisition time of 20 s. ATR-FTIR spectra are acquired using an Agilent Cary 630 instrument. VIS-NIR spectrophotometry is performed on an Agilent Cary 5000 instrument, using an integrating sphere configuration to measure total transmittance T(%) and total reflectance R(%) values. Transient absorption spectroscopy is conducted in an ambient environment with two instruments. For fs–ns experiments, a Ti:Sapphire amplifier (Spectra Physics’ Solstice) is used to generate the excitation (λ = 545 nm) and probe pulses (λ = 450–800 nm and 800–1500 nm), with 100 fs and 1 kHz repetition rate; the excitation is generated by a Light Conversion's TOPAS-NIRUVIS instrument, and the transient absorption signals are acquired with an Ultrafast Systems’ HELIOS using a 6 ns delay line. Microsecond transient absorption is carried out with a 355 ns pulse (390 μJ cm−2) generated by Litron laser's Aurora optical parametric oscillator passed through color filters and monochromator; detection is achieved ausing an InGaAs diode coupled to a Costronics amplifier, connected to Tektronics TDS 2012B for signal reading. Data analysis is conducted using Origin and Surface Xplorer.
cos(θ) is used to determine the relationship between the crystallite size D and the full width at half maximum (FWHM) β of a peak of the diffraction pattern, K is a dimensionless shape factor equaling 0.9, λ is the wavelength of the incident Cu Kα radiation, and θ is the Bragg angle. Raman spectra are recorded using a Renishaw inVia Raman microscope with a 50X objective lens, using a 633 nm excitation wavelength and a total acquisition time of 20 s. ATR-FTIR spectra are acquired using an Agilent Cary 630 instrument. VIS-NIR spectrophotometry is performed on an Agilent Cary 5000 instrument, using an integrating sphere configuration to measure total transmittance T(%) and total reflectance R(%) values. Transient absorption spectroscopy is conducted in an ambient environment with two instruments. For fs–ns experiments, a Ti:Sapphire amplifier (Spectra Physics’ Solstice) is used to generate the excitation (λ = 545 nm) and probe pulses (λ = 450–800 nm and 800–1500 nm), with 100 fs and 1 kHz repetition rate; the excitation is generated by a Light Conversion's TOPAS-NIRUVIS instrument, and the transient absorption signals are acquired with an Ultrafast Systems’ HELIOS using a 6 ns delay line. Microsecond transient absorption is carried out with a 355 ns pulse (390 μJ cm−2) generated by Litron laser's Aurora optical parametric oscillator passed through color filters and monochromator; detection is achieved ausing an InGaAs diode coupled to a Costronics amplifier, connected to Tektronics TDS 2012B for signal reading. Data analysis is conducted using Origin and Surface Xplorer.
3. Results
3.1. Fabrication of plasmonic ITO and pentacene layers
Colloidal synthesis of plasmonic ITO (10% wt. Sn) results in nanocrystals of 6.0 ± 1.0 nm diameter (average value on 40 measurements; TEM images in Fig. S1, ESI†), which are then ligand-stripped and spin-coated into thin films of 90 ± 10 nm thickness (average value on 10 measurements from SEM cross-section imaging). This coating methodology has been favored here for its simplicity and high degree of control. Such a thickness range represents an optimal trade-off at which the dynamic range of NIR absorbance is maximized with minimal impact on transparency in the VIS range.31,35,36 Pentacene thin films are grown by OMBD, selected and used here for delivering high coating material purity and strong adhesion with the substrate, while providing optimal control over layer thickness and structure. The used deposition growth rate of 0.5 Å s−1 is based on previous literature45–48 in order to obtain continuous layers predominantly in the thin film phase. A 120 ± 10 nm thickness (average value on 10 measurements) is implemented, again as an optimal trade-off between sufficient transparency/low opacity in VIS, and photon absorption (corresponding to a sufficient degree of charge generation/transfer). Bilayer architectures are then obtained through spin-coating of ∼90 nm thick layers of plasmonic ITO nanocrystals and subsequent deposition of ∼120 nm thick layers of pentacene by OMBD. It can be noted that the reverse order of deposition, i.e. spin-coating of ITO onto pentacene, causes the partial delamination of the pentacene layers due to the solvents used during spin-coating, and results in highly inhomogeneous bilayers with poor coverage.At this stage, it is key to analyze how the molecular packing and organization of pentacene molecules are affected by the presence of underlying ITO layers. The growth of pentacene thin films onto various substrates or sublayers is known to be strongly dependent on the structure of the initial monolayer, which in turn is governed by competing forces of intermolecular and/or interfacial type. When occurring, the nature and intensity of these molecule–molecule and/or molecule–substrate interactions can have a strong impact on the formation, stability, and optical/electrical properties of the bilayer structure. These could notably result in so-called templating effects, known to induce different molecular orientation, evolving from perpendicular with bare substrates to, notably, flat-lying with CuI underlayers – which is acknowledged to improve efficiency of charge generation and transfer – while PTCDA underlayers do not affect orientation.48 In turn, the geometrical configuration of the donor/acceptor interfaces is known to significantly impact the optoelectronic properties of the processed structures, as for instance the rates of exciton dissociation and charge recombination in solar cells based on pentacene – fullerene heterojunctions, or the nature of optical excitation – as modelled for pentacene – TiO2 interfaces.9,49,50 It is therefore important to screen and discuss such structural aspects for our ITO – pentacene structures, which is done in the next section.
3.2. Topographic and structural properties
The morphology and topography of neat plasmonic ITO, neat pentacene (PEN), and plasmonic ITO – pentacene (PEN–ITO) bilayers, are established from top-view and cross-section SEM as well as tapping mode AFM measurements (Table 1 and Fig. 1). Plasmonic ITO films (Fig. 1a) consist of densely packed nanocrystals with an average lateral grain size of 19 ± 11 nm, while pentacene films (Fig. 1b) consist of heterogeneously shaped grains with an average grain size of 125 ± 60 nm. In both cases, the neat films are observed to be highly continuous, homogeneous, and pinhole-free. Roughness analysis indicates smooth ITO layers – with an average Rq value of 0.9 ± 0.1 nm – and of quite rougher pentacene films – with Rq of 12.9 ± 0.2 nm. When deposited onto ITO, pentacene layers retain their high degree of coverage and morphology (Fig. 1c), with an average lateral grain size of 102 ± 47 nm. The size is similar to the layers grown onto Si substrates within error. The roughness (Rq of 10.7 ± 1.7 nm) is not affected by the underlying layer within error, although morphology is less heterogeneous with some grains protruding from the interface. Complementary optical microscopy images and photographs of samples are shown in Fig. S2 (ESI†).| Sample | Roughness Rq (nm) | Lateral grain size (nm) | Out-of-plane grain sizea (nm) |
|---|---|---|---|
| a Scherrer analysis is performed for plasmonic ITO on the (222) reflection of ITO at 2θ = 30.7°, and for PEN and PEN–ITO on the (001) reflection of pentacene at 2θ = 5.8°. | |||
| ITO | 0.9 ± 0.1 | 19 ± 11 | 7 ± 1 |
| PEN | 12.9 ± 0.2 | 125 ± 60 | 76 ± 2 |
| PEN–ITO | 10.7 ± 1.7 | 102 ± 47 | 31 ± 1 |
 | ||
| Fig. 1 Cross-section (i) and top-view (ii) SEM images, and (iii) AFM images of (a) plasmonic ITO, (b) PEN, and (c) PEN–ITO deposited onto silicon substrates. Scale bars are set at 200 nm for cross-section SEM images, 1 μm for top-view SEM images and 2 μm for AFM images. | ||
X-ray diffraction (XRD) supports the interpretation of pentacene molecules being differently packed on bare and plasmonic ITO-covered substrates. The XRD pattern of neat pentacene (Fig. 2, blue curve) highlights the main characteristic (00l) planes from the thin film phase (CCDC 665900)51 at 2θ = 5.8° (001), 11.6° (002), 17.3° (003), 23.2° (004) and 29.0° (005). In good agreement with previous works45–48 where 0.5 Å s−1 rates were also used for pentacene growth by OMBD, this indicates a highly-ordered pentacene molecular layer consisting of two polymorphs – a thin film phase with its dominant (001) peak at 2θ = 5.8°, plus a secondary bulk phase with a weak (001) signal at 2θ = 6.1° – with molecules being aligned in a nearly perpendicular orientation to the substrate surface. An out-of-plane grain size of 76 ± 2 nm is further determined using the Scherrer equation on the (001) plane, with a FWHM of 0.1°/0.002 radians (Table 1). In comparison, a pentacene film deposited onto plasmonic ITO results in almost suppressed scattering from the (001) plane (Fig. 2, purple curve and inset) and strongly reduced out-of-plane grain sizes (31 ± 1 nm, with FWHM increasing to 2θ = 0.2°/0.004 radians). These observations imply less crystalline order, with fewer molecules aligning perpendicular when deposited onto plasmonic ITO. The XRD signature of the bilayer structure also shows a small contribution at 2θ = 30.7° (crystallite size of 7 ± 1 nm), which corresponds to the (222) reflection of body-centered cubic indium oxide In2O3 (reference pattern JCPDS File No. 06-0416),52 and unchanged following Sn-doping, which is also characteristic of the neat ITO (Fig. 2, red curve).31,35
 | ||
| Fig. 2 XRD patterns of PEN film deposited onto silicon (blue curve), PEN film deposited onto plasmonic ITO layer covered silicon (PEN–ITO; purple curve), and plasmonic ITO layer deposited onto silicon (red curve). The diffraction scans are stacked at regular intervals of 2000 counts. The inset presents the close up (2θ range of 5.0–7.0°) on the (001) plane of pentacene in PEN–ITO. ° symbol highlights the presence of the (222) reflection of ITO. | ||
In all generality, such a molecular orientation change is governed by the balance between the electrostatic forces between the layer and the substrate, and the intermolecular quadrupolar interactions.48 Specifically, the pentacene herringbone arrangement is driven by quadrupolar interactions across the dimer pair. When deposited onto a negatively charged surface, such as CuI, the pentacene molecules undergo a transition from a standing orientation to a flat-lying orientation, whilst maintaining the herringbone molecular arrangement. A similar principle can be extended and postulated for the present pentacene–ITO bilayer system. The polarity of the ITO surface can be assumed to be varied with clusters of positive and negative charges randomly distributed.53 In the areas of negative charges, a similar interaction to that observed in the pentacene/CuI bilayer system can be expected. On the other hand, in areas of positive charges, the intermolecular interactions between the pentacene molecules are dominant and the molecules can maintain their thermodynamically preferred standing arrangement. The varied distribution of the interactions between the pentacene molecules and ITO surface presumably results in a mixture of pentacene molecules aligned parallel and perpendicular to the ITO surface.
We then consider Raman spectroscopy to strengthen our understanding of the structural order and molecular orientation of pentacene. Related literature54 has shown that the intensity of C–H bending modes at the extremity of the pentacene molecules (1157 and 1180 cm−1; Fig. 3a) relative to the intensity of C–C ring stretching modes along the short-axis of pentacene molecules (1353 and 1374 cm−1; Fig. 3b) could be used to determine the degree of deviation from equilibrium packing. Presently, the slight increase of I1157/I1374 for pentacene on plasmonic ITO (I1157/I1374 = 0.67) compared to a bare substrate (I1157/I1374 = 0.60) is therefore indicative of increased (001) d-spacing and more lattice strain. Additionally, the increase of the C–CLong peak (1596 cm−1; Fig. 3c) is indicative of pentacene molecules transitioning from perpendicular to flat-lying orientation.
 | ||
| Fig. 3 Raman spectra measured over (a) 1135–1200 cm−1, (b) 1335–1395 cm−1 and (c) 1550–1675 cm−1 ranges of plasmonic ITO layer (red curve), PEN film (blue curve), and PEN film deposited on plasmonic ITO layer (PEN–ITO; purple curve). Data are normalized to the intensity of the C–CShort stretch at 1374 cm−1. Arrows highlight the evolution of PEN–ITO signals vs. PEN ones. | ||
Complementary ATR-FTIR spectroscopy analysis further confirms these trends. The two bands observed at 904 and 1296 cm−1 for neat pentacene layers (see spectra on Fig. S3a and b, blue curve, ESI†) are associated to the molecular Z and X modes, i.e. vibrational modes along the Z- and X- short-axes, respectively (see inset with pentacene's molecular structure), while the characteristic bands of the Y mode (along the Y- long-axis) at 1443 and 1498 cm−1 are actually missing; based on literature,55,56 these observations can be correlated to pentacene molecules mostly standing upright on the surface. When pentacene molecules are deposited onto plasmonic ITO layers (purple curve on Fig. S3a and b, ESI†), notably, the intensity of the 904 cm−1 band (Z mode) significantly decreases, the 1296 cm−1 band (X mode) remains noticeable, and a supplementary band slightly appears by 1443 cm−1 (attributed to the Y mode). Such an “heterogeneous” signature can therefore be attributed to less organized and more randomly oriented pentacene molecules.
3.3. Optoelectronic properties
Fig. 4 shows optical spectra of neat pentacene, neat plasmonic ITO and ITO–pentacene bilayers for both VIS and NIR ranges. Absorptance data are presented, being obtained from total transmittance and total reflectance measurements (presented in Fig. S4a, ESI†) as A(%) = 1 − T(%) − R(%); additional optical constants being further extracted from the VIS-NIR spectra, such as absorption coefficient α, extinction coefficient k and refractive index n, are reported and discussed in ESI† (see Fig. S4b–d and complementary analysis). In VIS (Fig. 4a), neat pentacene films contain four characteristic peaks: the lowest singlet transition (S0 → S1) is observed through two signals attributed to Davydov splitting (DS),57 with the lower level (1) at 667 nm (1.86 eV) and the upper level (2) at 632 nm (1.96 eV); peaks (3) and (4) at 584 nm (2.12 eV) and 545 nm (2.27 eV) correspond to pentacene–pentacene charge-transfer (CT) transitions (S0 → CT).10 Neat ITO layers also present their characteristic signatures with a strong LSPR peak in NIR (Fig. 4b) centered here around 2.15 μm (0.57 eV) together with VIS transparency, corresponding to literature (notably by D. J. Milliron et al.).31,58 The ITO–pentacene bilayers combine the optical responses of pentacene in VIS and of ITO in NIR. Band gaps of ITO and pentacene, calculated from Tauc plots (Fig. S5, ESI†), are similar in neat and bilayer configurations: 1.75 ± 0.05 eV for pentacene, 4.05 ± 0.05 eV for ITO. | ||
| Fig. 4 (a) VIS and (b) NIR absorptance spectra of PEN film deposited onto quartz (blue curve), plasmonic ITO layer deposited onto quartz (red curve) and PEN film deposited onto ITO layer covered quartz (PEN–ITO; purple curve). | ||
Differences are observed when comparing the optical curves of the bilayers with the individual films. Most strikingly, in the NIR, the LSPR peak maximum wavelength blueshifts from 2.15 μm (0.57 eV) for ITO to 2.11 μm (0.59 eV) for ITO–pentacene, while the LSPR absorption intensity increases by ∼41% (based on integrated area). As further described in full details in Section 4 “Discussion” below, these variations can be directly correlated to an increase of free carrier concentration in ITO, being a consequence of a charge injection process that presumably occurs following photoexcitation of pentacene and exciton separation at the interface. Optical changes are also observed in VIS, where the electronic transitions of pentacene are obviously impacted by the presence of plasmonic ITO: the relative intensity of peak (2) to peak (1) slightly decreasing (0.81 to 0.78), together with a redshift [blueshift] of peak (1) [(2)] from 667 [632] to 668 [628] nm, resulting in a slight increase of DS to 0.12 eV vs. 0.11 eV with single pentacene layers. The shift of peak (2) toward higher photon energies (shorter wavelength) and the variation of the relative intensities of the two Davydov components, i.e. peaks (1) and (2), have been attributed to a change in the polarizability of the local environment of pentacene,7,10,45,59 which may be due in our case to the presence of the plasmonic ITO layer, but also to enhanced intermixing and electronic coupling occurring between adjacent perpendicular (in majority) and flat-lying (in minority) pentacene molecules. These changes of intensity and DS are thus indicative of stronger intermolecular coupling and π–π interactions within the pentacene layer, pointing out a larger degree of charge transfer character in the lower-lying excited state of pentacene when deposited onto plasmonic ITO as compared to the single pentacene layer, as well as a globally slower charging rate.5,10 However, it remains unclear at this stage whether these variations could result from the “destructuration” of the pentacene molecules provoked by the underlying ITO layer, and/or from any enhanced acceptor effect being caused by the optoelectronic characteristics of ITO, i.e. its plasmonic nature.
We then turn to ultrafast transient absorption spectroscopy (TAS) to investigate the specific nature and extent of electronic interactions in response to photoexcitation. In the 100 fs to 5 ns range, TAS signatures in VIS are similar for the ITO–pentacene bilayers compared to neat pentacene (Fig. 5a): two ground-state bleaching bands are noticed at 585 and 680 nm, in addition to three positive signals – two sharp bands at 620 and 650 nm related to heating effects,60 and a broad one above 720 nm (up to ∼1100 nm) related to triplet transitions – which are consistent with the literature.10,61 The triplet signal is monitored in NIR (Fig. 5b), showing a redshift (see the arrow in Fig. 5b-iii) from 915 nm (1.36 eV) in neat pentacene to 950 nm (1.30 eV) in ITO–pentacene bilayers. This spectral evolution, together with an increased intensity of a corresponding high-energy feature at 450–500 nm also noticed here (see arrow in Fig. 5a-iii), can be assigned to the formation of pentacene's radical cation,61 which might be suggestive of electron injection into the plasmonic ITO. That said, this hypothesis is nuanced from a global fitting analysis of the 840–1400 nm transient spectra of neat pentacene and ITO–pentacene, being performed to extract the time constant of the peak formation (Fig. S6, ESI†). Corresponding results indicate the presence of a signal rise at 945 nm (time constant 770 ± 360 fs) for pentacene and redshifted to 965 nm (time constant 869 ± 410 fs) for ITO–pentacene.
 | ||
| Fig. 5 (a) VIS and (b) NIR transient absorption spectroscopy data measured (excitation at 545 nm) on quartz samples covered with a PEN film (blue curve), ITO layer (red curve), and PEN film deposited onto the ITO layer (PEN–ITO, purple curve); (i) PEN and (ii) PEN–ITO spectra recorded between 100 fs and 5 ns probe times, with 525–556 nm range left blank due to artefact from excitation pulse scattering signal; (iii) ITO, PEN and PEN–ITO spectra recorded at a 10 ps probe time. Arrows highlight the evolution of PEN–ITO signal vs. PEN one. | ||
To determine the possibility for charge injection into ITO, transient absorption measurements in the microsecond timescale are further conducted. The kinetic traces (ΔOD vs. time, Fig. 6) from ITO LSPR absorption at 1600 nm (0.77 eV) supports the assumption of electron transfer from pentacene: this interpretation is based on the detection of the positive plasmonic signal after pentacene excitation only in ITO–pentacene bilayers but not in neat ITO nor neat pentacene films, all probed under the same conditions. Interestingly, this feature decays over an overall ∼50 μs timescale, which is much slower than the triplet-influenced hole charging process evoked earlier. This remains to be further investigated, especially since the analysis of the pentacene kinetic traces in VIS through samples probing at 665 nm (1.86 eV), i.e. pentacene absorption (see Fig. S7, ESI†), does not highlight any significant difference between neat pentacene and ITO–pentacene bilayers.
 | ||
| Fig. 6 Transient absorption dynamics of PEN film (blue curve), plasmonic ITO layer (red curve) and PEN film deposited onto the ITO layer (PEN–ITO, purple curve), all deposited on quartz substrates and measured at a probe wavelength of 1600 nm. | ||
4. Discussion
An energy diagram level of the ITO–pentacene structures is presented in Fig. 7. It represents the photoexcited state generation in pentacene and further charge injection into plasmonic ITO, being at the basis of the electronic and optical changes mechanisms taking place in the bilayers and leading to an increase of NIR absorption; notably, a ground-state charge transfer does not seem plausible here, as it would necessitate a higher energy pentacene S0 level than the EF of ITO.62 | ||
| Fig. 7 Hypothetical energy level diagram of ITO–pentacene structures. Evac stands for vacuum level; S0, S1 and T1 stand for the ground-state singlet, excited singlet and triplet levels in pentacene, respectively; WF, EF, EC, EV and Eg stand for work function, Fermi level, conduction band minimum, valence band maximum and band gap value in ITO, respectively, with corresponding energy values established from the literature.10,63–66 S1–S0 and Eg band gap values are retrieved experimentally from UV-VIS-NIR spectrometry (see Fig. 4 and Fig. S5, ESI†). | ||
Considering first the charge generation/transfer processes in pentacene, they are reported to primarily occur within up to 2–10 ns after photoexcitation, based upon the state-of-the-art on “model” donor–acceptor systems such as pentacene-fullerene structures.10 First, the absorption of VIS photons in pentacene leads to the photogeneration of singlet excitons (S0 → S1). These can then undergo either direct dissociation, which typically takes place within 1–100 ps timescales (even longer if the whole charging process is considered up to proper transfer towards and collection by the acceptor material), or singlet fission – which would theoretically occur within ultrafast kinetics of 80 fs and with up to 200% yield – so as to generate triplet excitons (see dashed arrows in Fig. 7) that would then be capable of dissociating and similarly inducing charge transfer towards the acceptor material.21,67–69
However, the data presently collected on the PEN–ITO samples do not allow at this stage to rule on the exact decay mechanism of singlet exciton in pentacene being interfaced with plasmonic ITO. This is due to the limit in the timescale resolution of the spectroscopic tools used (typically of 100 fs) as well as the intrinsically ultrafast kinetics of the singlet fission process (typically on a few tens-to-hundreds fs timescale for pentacene-based materials5,59,67). These aspects remain therefore to be further probed.
Considering the optical behavior of ITO as plasmonic electrochromic material, it is important to note here that it implies no (eye-)visible color change but a tuning of NIR absorption, evolving towards more NIR-darkening when ITO carrier concentration is increased from electronic doping. Here, the tuning of ITO NIR absorption is aimed to be driven by the incorporation of the pentacene layer, allowing for an in situ electronic doping, i.e. without external electrical powering like in state-of-the-art electrochromic setups. At lab-scale, these classically consist of a 3-electrodes cell made of a conducting glass working electrode covered with the plasmonic ITO film, completed with reference and counter electrodes all immersed in a liquid electrolyte; tuning of ITO NIR absorption between dark and clear states is then driven by electrochemical bias between charging and discharging potentials, respectively.31,34–36,39,58
In the present study, the association of pentacene to plasmonic ITO is observed to already impact the optical properties of the “as deposited” state: as seen on Fig. 4b, PEN–ITO samples (purple curve) display a small blueshift of ITO LSPR peak in NIR together with increased absorption intensity, in comparison with neat plasmonic ITO (red curve). From these data being further correlated by TAS measurements (Fig. 5 and Fig. 6), the optical change occurring in PEN–ITO structures is obviously the result of an electron transfer process from pentacene into plasmonic ITO. The resulting electronic tuning of ITO absorbance can then be first described and quantified according to a Drude model,31 assuming free carrier concentration nc and LSPR resonance frequency ωLSPR, as being given in all generality by eqn (1) and (2):
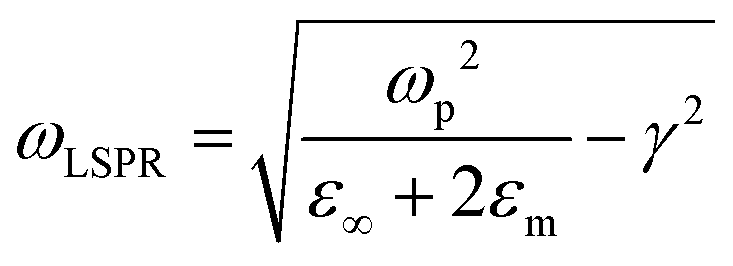 | (1) |
 | (2) |
With ωp being the bulk plasmon frequency of free carriers in the material (i.e. electrons);  the electron effective mass, i.e. 0.35 electron mass for ITO;69,70ε∞, εm and ε0 the dielectric constants of ITO in VIS range, of surrounding environment, and of free space (respectively); and γ the damping of the bulk plasmon. We have evaluated the value of ωp at 2.0 eV from a fit of the optical absorption of ITO nanoparticles in solution (see ESI,† Fig. S8) with an extended Drude model.71
the electron effective mass, i.e. 0.35 electron mass for ITO;69,70ε∞, εm and ε0 the dielectric constants of ITO in VIS range, of surrounding environment, and of free space (respectively); and γ the damping of the bulk plasmon. We have evaluated the value of ωp at 2.0 eV from a fit of the optical absorption of ITO nanoparticles in solution (see ESI,† Fig. S8) with an extended Drude model.71
The LSPR frequency given by eqn (1) is valid for an isolated nanoparticle in a homogeneous medium described by εm. However, in the present situation, the interactions between ITO nanoparticles within the film, and the presence of both quartz substrate underneath (εm = 4.0) and pentacene layer above (εm = 3.6–4.0), are all influencing the resonance wavelength. The first effect is modeled here by considering an effective dielectric function for the ITO film (according to Maxwell–Garnett theory), and the second effect is taken into account by considering εm > 1.72,73 The absorption in the NIR of the ITO film on the quartz substrate is qualitatively reproduced for εm = 2.0 (Fig. S9a, blue curve, ESI†). To describe the effect of the pentacene layer, the sole modification of εm without charge injection, i.e. with a free carrier density nc that does not increase (constant ωp), would lead to a redshift of the LPSR (Fig. S9a, orange curve vs. blue curve, ESI†), which is opposite to what is seen here. Instead, our observation of a small blueshift of the LSPR and an increase of absorption intensity suggests that the presence of the pentacene film not only provokes the increase of εm but also results in an increase of nc (increase of ωp) in ITO as a consequence of a charge injection process (Fig. S9b, orange and green curves vs. blue curve, ESI†). A good description of the modification of the absorption after the deposition of the pentacene layer is therefore obtained for ωp = 2.2 eV and εm = 2.4 (Fig. S9b, green curve, ESI†).
Based on this modeling approach, one can now evaluate the density of electrons nc (per nm−3) provoking the optical modulation of the LSPR peak between neat ITO and ITO–pentacene bilayers, as observed in Fig. 4b. From eqn (2), it can be established that:
| nc = 0.2174ωp2 | (3) |
Then, for a sphere of radius R (in nm), one obtains:
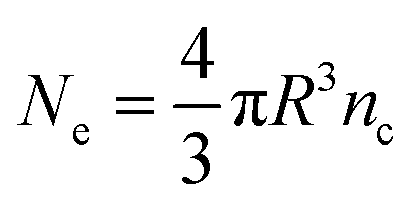 | (4) |
With Ne being the number of free carriers (electrons) contained in a spherical ITO nanoparticle.
Furthermore, a Δω (%) increase of ωp can be correlated to a Δnc (%) increase of nc as:
| 1 + Δnc = (1 + Δω)2 | (5) |
ω p of ITO is estimated at 2.0 eV, leading to n = 0.869 nm−3 and to Ne = 98 electrons, respectively, considering R = 3 nm. When pentacene is associated with ITO, ωp evolves to 2.2 eV (Fig. S9a, ESI†), leading to a Δω increase of ∼10% and a Δnc increase of ∼21% (see eqn (5)). A ΔNe of ∼20 electrons (98 × 21%) per ITO nanoparticle is therefore obtained, accounting for the optical tuning of ITO LSPR absorption resulting from the presence of the pentacene layer. Literature generally establishes a wide range of 80 to 270 electrons per nanoparticle to quantify the extent of electron transfer occurring from external electrochemical (charging) bias in “real” plasmonic electrochromic systems.74–77 Here, basing upon our previous works using similar configuration of plasmonic electrochromic ITO layers,35,36 we estimate a ΔNe of ∼31 electrons per ITO nanoparticle, obtained from a measured increase of ∼15% for Δω and increase of ∼32% for Δnc (see eqn (5)). In terms of electrons transfer, the in situ charging of ITO via pentacene resulting from the present association of the two compounds seems thus to account for 20/31 = ∼65% of the value corresponding to ex situ electrochemical charging, which stands as a very promising pioneering result.
5. Conclusions and perspectives
This study addresses the practical design of hybrid ITO–pentacene bilayers in the context of developing novel plasmonic electrochromic systems. Multi-modal analysis through surface microscopy, X-ray diffraction and Raman spectroscopy reveals that plasmonic ITO modifies the molecular packing of the subsequently-deposited pentacene layers, decreasing the proportion of perpendicularly oriented molecules vs. flat-lying ones; this in turns impacts the capacities of charge production and transfer, as steady-state and time resolved optical spectroscopies highlight the photogeneration of electrons in pentacene, further inducing a modification of ITO LSPR and resulting in enhanced NIR absorption.At this stage, it remains to be established whether specific intermolecular and/or molecule–substrate interactions do take place within the ITO–pentacene bilayers, and if so, what are their exact nature and strength. Relevant literature on pentacene-based structures has highlighted the presence of driving forces such as hydrogen bonding, π–π interactions, dipolar interactions of attractive or repulsive nature, or surface polarity (as in the noticeable case of zinc oxide (ZnO) sublayers, shown to be highly impacted by the specific atomic termination, i.e. either Zn-(positive) or O-(negative)).48,78–80 Overall, further advanced surface characterization (which may imply (GI)SAXS/WAXS81 or angle-resolved polarized absorption measurements82) as well as in-depth theoretical simulations would help in further identifying, quantifying and rationalizing the dynamics of the interactions specifically occurring in the ITO–pentacene systems presented in this study. This will allow to further evaluate their impact in terms of optical and electronic properties, especially for clarifying the exact effect of various pentacene's molecular orientation and organization on the nature and efficiency of charge generation and transfer to ITO. Further optoelectronic characterization, both experimentally and numerically, is also needed in order to fully describe and understand how the charge transfer to plasmonic ITO takes place at the molecular level in pentacene, especially to determine whether it is driven by singlet fission and/or other photochemical processes. Such prospects are clearly anticipated as upcoming steps of complementary fundamental investigations.
Overall, continued optimization of deposition/interfacing approaches is also desirable towards more integrated, interconnected ITO–PEN structures, notably exploring thinner pentacene films (down to 10 nm, which is the typical minimal threshold for continuous pentacene films produced by OMBD). Alternative methodologies could also be implemented, for instance by infiltrating dissolved pentacene molecules into the porous network of ITO NCs through “dye-loading”, or by obtaining monolayers of pentacene molecules from other processing techniques than OMBD, including wet coating approaches. Exploration of alternative layer compositions may ultimately be undertaken, including other formulations of donor/acceptor materials.
From a broader perspective, the present study attests that the considered composition and design of materials bear high potential for the outcome of a novel generation of self-powered electrochromic systems. Ground-breakingly, these would allow autonomous charge supply and high spectral selectivity throughout the whole VIS-NIR solar range, while being designed following simplified structural architectures as heterojunction-like and therefore electrolyte-free configurations. In order to promote a capacity of dynamic switching between different optical states from in situ charging–discharging processes, one will notably have to consider how to carefully control the exact quantity of electrons being generated in pentacene, and then transferred to plasmonic ITO (in order to NIR-darken the systems), and how to regulate these processes “on demand” in order to make them progressive and reversible (NIR-bleach them back) as well as long-term cyclable. Therefore, as a prospective technological step, the present PEN–ITO bilayers should be further engineered into fully-functional electrochromic devices, first as lab-scale demonstrators then upscaled into “real” smart windows.
In practice, fabricating such devices will rely first on ITO and PEN layers being properly and progressively processed onto transparent conducting glass substrates, potentially following patterning approaches, before being covered with a metallic layer as top contact material.83–85 Similarly to benchmark works available in the literature,44,84 current–voltage measurements in the dark and under solar illumination as well as in situ spectro-electrochemical recordings should then be conducted in order to establish the performances of the obtained device architectures in terms of light photoconversion and of resulting VIS-NIR absorption. The extent of the generated photocurrent and the resulting optical contrast, switching kinetics and coloration efficiency, among other figures of merits of (photo)electrochromic systems,39,43,44 will have to be carefully determined and refined. These will certainly be influenced by multiple experimental parameters including the respective thicknesses of the ITO and PEN layers, the nature (Au, Ag, Pt…) and thickness of the top metallic layer, the device area – from mm2 to cm2 range, the intensity of solar illumination, etc.
Open questions for future fundamental researches will thus be to quantitatively determine whether this process actually produces significant NIR plasmonic absorption at solar intensities, and if it does, how to properly modulate it towards controllable, efficient NIR photoelectrochromism. From a more applied, technological point of view, such configurations of smart windows would not only be “self-powerable” but also of additional interest in terms of aesthetics of architectural buildings, as these materials would not be colorless but tinted in their native state (here in blue as a result of using pentacene – see Fig. S2, ESI†); in the meantime, they remain semi-transparent and not visually opaque thanks to the low film thickness. These novel intelligent glazing systems would then commute between a warm state, allowing for heating up the interior of buildings (NIR-transparent) while opacifying the sunlight (VIS-darkened), and a fully-dark state being opaque to both external light-VIS and heat-NIR. The specific use of these warm and dark modes of dynamic fenestration would therefore be of favorable use in cold climates and/or during winter days.86 Ultimately, such innovative technology of self-powered NIR-modulable electrochromic materials will certainly pave the way towards a reinforced lowering of energy consumption and waste in buildings from heating, air conditioning and artificial lighting, and further reducing global electricity demand.
Author contributions
Conceptualization: A. M. and S. H. Funding acquisition: A. M., R. C., and S. H. Investigation: A. M., D. K. K., J. W., E. B., and S. D. D. Methodology: A. M., D. K. K., S. D. D., and S. H. Project administration: A. M., R. C., and S. H. Validation: A. M., D. K. K., L. H., Y. O., S. D. D., R. C., and S. H. Visualization: A. M., D. K. K., J. W., E. B., and S. D. D. Writing – original draft preparation: A. M. Writing – review and editing: D. K. K., J. W., E. B., L. H., Y. O., S. D. D., R. C., and S. H. All authors have given approval to the final version of the manuscript.Data availability
All data that have been generated or analyzed during this study are included in this article and its ESI.† Additional information is available upon request from the corresponding author.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by A. M.'s WBI.WORLD postdoctoral fellowship awarded by Wallonie-Bruxelles International, with additional support from the GREEnMat laboratory of the University of Liège, and of the Department of Materials at Imperial College London. The authors acknowledge fellow team members for additional scientific, technical and administrative support. A. M. further thanks the University of Bordeaux – ICMCB for additional funding and support, especially Dr Alexandre Fargues for his assistance with optical measurements. D. K. K. is further grateful to the Department of Materials, Imperial College London for PhD funding. L. H. and Y. O. acknowledge funding from the Namur Institute of Structured Matter. E. B. is grateful to the EPSRC Centre for Doctoral Training in the Advanced Characterization of Materials (EP/S023259/1) for PhD funding. S. D. D. was funded through EP/V010913/1.References
- J. E. Anthony, Chem. Rev., 2006, 106, 5028 CrossRef CAS PubMed.
- R. Ruiz, D. Choudhary, B. Nickel, T. Toccoli, K.-C. Chang, A. C. Mayer, P. Clancy, J. M. Blakely, R. L. Headrick, S. Iannotta and G. G. Malliaras, Chem. Mater., 2004, 16, 4497 CrossRef CAS.
- Y. Yunus, N. A. Mahadzir, M. N. Mohamed Ansari, T. H. Tg Abd Aziz, A. Mohd Afdzaluddin, H. Anwar, M. Wang and A. G. Ismail, Polymers, 2022, 14, 1112 CrossRef CAS PubMed.
- P. M. Zimmerman, Z. Zhang and C. B. Musgrave, Nat. Chem., 2010, 2, 648 CrossRef CAS PubMed.
- M. W. B. Wilson, A. Rao, J. Clark, R. S. S. Kumar, D. Brida, G. Cerullo and R. H. Friend, J. Am. Chem. Soc., 2011, 133, 11830 CrossRef CAS PubMed.
- M. W. B. Wilson, A. Rao, B. Ehrler and R. H. Friend, Acc. Chem. Res., 2013, 46, 1330–1338 CrossRef CAS PubMed.
- D. Beljonne, H. Yamagata, J. L. Brédas, F. C. Spano and Y. Olivier, Phys. Rev. Lett., 2013, 110, 226402 CrossRef CAS PubMed.
- A. J. Carrod, V. Gray and K. Börjesson, Energy Environ. Sci., 2022, 15, 4982 RSC.
- Y. Yi, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2009, 131, 15777 CrossRef CAS PubMed.
- A. Rao, M. W. B. Wilson, J. M. Hodgkiss, S. Albert-Seifried, H. Bässler and R. H. Friend, J. Am. Chem. Soc., 2010, 132, 12698 CrossRef CAS PubMed.
- W. Zukun, R. Wu, Z. Chen, L. Ye, H. Li and H. Zhu, J. Phys. Chem. A, 2020, 124, 4185 CrossRef CAS PubMed.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334 CrossRef CAS PubMed.
- T. Nishi, M. Kanno, M. Kuribayashi, Y. Nishida, S. Hattori, H. Kobayashi, F. von Wrochem, V. Rodin, G. Nelles and S. Tomiya, Appl. Phys. Lett., 2018, 113, 163302 CrossRef.
- N. Beaumont, J. S. Castrucci, P. Sullivan, G. E. Morse, A. S. Paton, Z.-H. Lu, T. P. Bender and T. S. Jones, J. Phys. Chem. C, 2014, 118, 14813 CrossRef CAS.
- C. P. Theurer, M. Richter, D. Rana, G. Duva, D. Lepple, A. Hinderhofer, F. Schreiber, P. Tegeder and K. Broch, J. Phys. Chem. C, 2021, 125, 23952 CrossRef CAS.
- B. Ehrler, M. W. B. Wilson, A. Rao, R. H. Friend and N. C. Greenham, Nano Lett., 2012, 12, 1053 CrossRef CAS PubMed.
- B. Ehrler, B. J. Walker, M. L. Böhm, M. W. B. Wilson, Y. Vaynzof, R. H. Friend and N. C. Greenham, Nat. Commun., 2012, 3, 1019 CrossRef PubMed.
- D. Jariwala, S. L. Howell, K.-S. Chen, J. Kang, V. K. Sangwan, S. A. Fillippone, R. Turrisi, T. J. Marks, L. J. Lauhon and M. C. Hersam, Nano Lett., 2016, 16, 497 CrossRef CAS PubMed.
- L. Ye, Y. Zhao, R. Xu, S. Li, C. Zhang, H. Li and H. Zhu, J. Am. Chem. Soc., 2023, 145, 26257 CrossRef CAS PubMed.
- J. N. Schrauben, Y. Zhao, C. Mercado, P. I. Dron, J. L. Ryerson, J. Michl, K. Zhu and J. C. Johnson, ACS Appl. Mater. Interfaces, 2015, 7, 2286 CrossRef CAS PubMed.
- N. A. Pace, D. H. Arias, D. B. Granger, S. Christensen, J. E. Anthony and J. C. Johnson, Chem. Sci., 2018, 9, 3004 RSC.
- T. Banerjee, S. P. Hill, M. A. Hermosilla-Palacios, B. D. Piercy, J. Haney, B. Casale, A. E. DePrince, M. D. Losego, V. D. Kleiman and K. Hanson, J. Phys. Chem. C, 2018, 122, 28478 CrossRef CAS.
- E. Sundin, R. Ringström, F. Johansson, B. Küçüköz, A. Ekebergh, V. Gray, B. Albinsson, J. Mårtensson and M. Abrahamsson, J. Phys. Chem. C, 2020, 124, 20794 CrossRef CAS.
- Y. Yu, S.-C. Chien, J. Sun, E. C. Hettiaratchy, R. C. Myers, L.-C. Lin and Y. Wu, J. Am. Chem. Soc., 2019, 141, 8727 CrossRef CAS PubMed.
- T. Minami, Semicond. Sci. Technol., 2005, 20, S35 CrossRef CAS.
- K. Ellmer, Nat. Photonics, 2012, 6, 809 CrossRef CAS.
- D. Kim, A. W. H. Lee, J. I. Eastcott and B. D. Gates, ACS Appl. Nano Mater., 2018, 1, 2237 CrossRef CAS.
- M. K. Gish, E. K. Raulerson, R. T. Pekarek, A. L. Greenaway, K. J. Thorley, N. R. Neale, J. E. Anthony and J. C. Johnson, Chem. Sci., 2021, 12, 11146 RSC.
- B. H. Farnum, Z. A. Morseth, M. K. Brennaman, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 15869 CrossRef CAS PubMed.
- M. Kanehara, H. Koike, T. Yoshinaga and T. Teranishi, J. Am. Chem. Soc., 2009, 131, 17736 CrossRef CAS PubMed.
- G. Garcia, R. Buonsanti, E. L. Runnerstrom, R. J. Mendelsberg, A. Llordes, A. Anders, T. J. Richardson and D. J. Milliron, Nano Lett., 2011, 11, 4415 CrossRef CAS PubMed.
- B. M. Crockett, A. W. Jansons, K. M. Koskela, D. W. Johnson and J. E. Hutchison, ACS Nano, 2017, 11, 7719 CrossRef CAS PubMed.
- J. Qu, C. Livache, B. Martinez, C. Gréboval, A. Chu, E. Meriggio, J. Ramade, H. Cruguel, X. Z. Xu, A. Proust, F. Volatron, G. Cabailh, N. Goubet and E. Lhuillier, ACS Appl. Nano Mater., 2019, 2, 1621 CrossRef CAS.
- T. E. Williams, C. M. Chang, E. L. Rosen, G. Garcia, E. L. Runnerstrom, B. L. Williams, B. Koo, R. Buonsanti, D. J. Milliron and B. A. Helms, J. Mater. Chem. C, 2014, 2, 3328 RSC.
- A. Maho, L. Comeron Lamela, C. Henrist, L. Henrard, L. H. G. Tizei, M. Kociak, O. Stéphan, S. Heo, D. J. Milliron, B. Vertruyen and R. Cloots, Sol. Energy Mater. Sol. Cells, 2019, 200, 110014 CrossRef CAS.
- A. Maho, C. A. Saez Cabezas, K. A. Meyertons, L. C. Reimnitz, S. Sahu, B. A. Helms and D. J. Milliron, Chem. Mater., 2020, 32, 8401 CrossRef CAS.
- O. Mashkov, J. Körfer, A. Eigen, A.-A. Yousefi-Amin, N. Killilea, A. Barabash, M. Sytnyk, N. Khansur, M. Halik, K. G. Webber and W. Heiss, Adv. Eng. Mater., 2020, 22, 2000112 CrossRef CAS.
- K. Wang, Q. Meng, Q. Wang, W. Zhang, J. Guo, S. Cao, A. Y. Elezzabi, W. W. Yu, L. Liu and H. Li, Adv. Energy Sustainable Res., 2021, 2, 2100117 CrossRef CAS.
- E. L. Runnerstrom, A. Llordés, S. D. Lounis and D. J. Milliron, Chem. Commun., 2014, 50, 10555 RSC.
- P. Pattathil, R. Giannuzzi and M. Manca, Nano Energy, 2016, 30, 242 CrossRef CAS.
- Y. Jin, L. Zhou, J. Liang and J. Zhu, Adv. Photonics, 2021, 3, 044002 CAS.
- A. Kolay, D. Maity, H. Flint, E. A. Gibson and M. Deepa, Sol. Energy Mater. Sol. Cells, 2022, 239, 111674 CrossRef CAS.
- A. Cannavale, F. Martellotta, F. Fiorito and U. Ayr, Energies, 2020, 13, 1929 CrossRef CAS.
- G. Syrrokostas, G. Leftheriotis and S. N. Yannopolous, Renewable Sustainable Energy Rev., 2022, 162, 112462 CrossRef CAS.
- D. Lubert-Perquel, E. Salvadori, M. Dyson, P. N. Stavrinou, R. Montis, H. Nagashima, Y. Kobori, S. Heutz and C. W. M. Kay, Nat. Commun., 2018, 9, 4222 CrossRef PubMed.
- D. Lubert-Perquel, A. A. Szumska, M. Azzouzi, E. Salvadori, S. Ruloff, C. M. W. Kay, J. Nelson and S. Heutz, J. Phys. Chem. Lett., 2020, 11, 9557 CrossRef CAS PubMed.
- D. Lubert-Perquel, D. K. Kim, P. Robaschik, C. W. M. Kay and S. Heutz, J. Mater. Chem. C, 2019, 7, 289 RSC.
- D. K. Kim, D. Lubert-Perquel and S. Heutz, Mater. Horiz., 2020, 7, 289 RSC.
- A. V. Akimov, A. J. Neukirch and O. V. Prezhdo, Chem. Rev., 2013, 6, 4496 CrossRef PubMed.
- M. P. Ljungberg, O. Vänskä, P. Koval, S. W. Koch, M. Kira and D. Sánchez-Portal, New J. Phys., 2017, 19, 033019 CrossRef.
- S. Schiefer, M. Huth, A. Dobrinevski and B. Nickel, J. Am. Chem. Soc., 2007, 129, 10316 CrossRef CAS PubMed.
- H. E. Swanson, N. T. Gilfrich and G. M. Ugrinic, Natl. Bur. Stand. Circ. (U. S.), 1955, 5, 26 Search PubMed.
- A. L. Swint and P. W. Bohn, Langmuir, 2004, 20, 4076 CrossRef CAS PubMed.
- J. Xu, Y. Diao, D. Zhou, Y. Mao, G. Giri, W. Chen, N. Liu, S. C. B. Mannsfeld, G. Xue and Z. Bao, J. Mater. Chem. C, 2014, 2, 2985 RSC.
- Y. Hosoi, K. Okamura, Y. Kimura, H. Ishii and M. Niwano, Appl. Surf. Sci., 2005, 244, 607 CrossRef CAS.
- E. Z. Fratczak, P. Uznanski and M. E. Moneta, Chem. Phys., 2015, 456, 49 CrossRef CAS.
- R. Hesse, W. Hofberger and H. Bässler, Chem. Phys., 1980, 49, 201 CrossRef CAS.
- G. Garcia, R. Buonsanti, A. Llordes, E. L. Runnerstrom, A. Bergerud and D. J. Milliron, Adv. Opt. Mater., 2013, 1, 215 CrossRef.
- K. Broch, J. Dieterle, F. Branchi, N. J. Hestand, Y. Olivier, H. Tamura, C. Cruz, V. M. Nichols, A. Hinderhofer, D. Beljonne, F. C. Spano, G. Cerullo, C. J. Bardeen and F. Schreiber, Nat. Commun., 2018, 9, 954 CrossRef CAS PubMed.
- A. Rao, M. W. B. Wilson, S. Albert-Seifried, R. Di Pietro and R. H. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 195411 CrossRef.
- T. Sakanoue and H. Sirringhaus, Nat. Mater., 2010, 9, 736 CrossRef CAS PubMed.
- D. Shen, W.-C. Chen, M.-F. Lo and C.-S. Lee, Mater. Today Energy, 2021, 20, 100644 CrossRef CAS.
- L. Yang, M. Tabachnyk, S. L. Bayliss, M. L. Böhm, K. Broch, N. C. Greenham, R. H. Friend and B. Ehrler, Nano Lett., 2015, 15, 354 CrossRef CAS PubMed.
- R. Schlaf, H. Murata and Z. H. Kafafi, J. Electron Spectrosc. Relat. Phenom., 2001, 120, 149 CrossRef CAS.
- Y. Gassenbauer and A. Klein, J. Phys. Chem. B, 2006, 110, 4793 CrossRef CAS PubMed.
- P. Zhang, S. Zhao, H. Wang, J. Zhang, J. Shi, H. Wang and D. Yan, Adv. Elect. Mater., 2017, 3, 1700136 CrossRef.
- N. A. Pace, N. V. Korovina, T. T. Clikeman, S. Holliday, D. B. Granger, G. M. Carroll, S. U. Nanayakkara, J. E. Anthony, I. McCulloch, S. H. Strauss, O. V. Boltalina, J. C. Johnson, G. Rumbles and O. G. Reid, Nat. Chem., 2020, 12, 63 CrossRef CAS PubMed.
- P. J. Jadhav, P. R. Brown, N. Thompson, B. Wunsch, A. Mohanty, S. R. Yost, E. Hontz, T. Van Voorhis, M. G. Bawendi, V. Bulovic and M. Baldo, Adv. Mater., 2012, 24, 6169 CrossRef CAS PubMed.
- M.-S. Niu, X.-Y. Yang, C.-C. Qin, P.-Q. Bi, C.-K. Lyu, L. Feng, W. Qin, K. Gao and X.-T. Hao, Org. Electron., 2019, 71, 296 CrossRef CAS.
- I. Hamberg and C. G. Granqvist, J. Appl. Phys., 1986, 60, R123 CrossRef CAS.
- D. Mergel and Z. Qiao, J. Phys. D: Appl. Phys., 2002, 35, 794 CrossRef CAS.
- H. Sun, S. Ryno, C. Zhong, M. K. Ravva, Z. Sun, T. Körzdörfer and J.-L. Brédas, J. Chem. Theory Comput., 2016, 12, 2906 CrossRef CAS PubMed.
- T. C. Choy, Effective Medium Theory, Oxford University Press, Oxford, 2016 Search PubMed.
- A. Agrawal, I. Kriegel, E. L. Runnerstrom, F. Scotognella, A. Llordes and D. J. Milliron, ACS Photonics, 2018, 5, 2044 CrossRef CAS.
- C. R. Conti III, G. Quiroz-Delfi, J. S. Schwarck, B. Chen and G. F. Strouse, J. Phys. Chem. C, 2020, 124, 28220 CrossRef.
- B. Tandon, S. A. Shubert-Zuleta and D. J. Milliron, Chem. Mater., 2022, 34, 777 CrossRef CAS.
- S. A. Shubert-Zuleta, B. Tandon, B. J. Roman, X. Y. Gan and D. J. Milliron, Chem. Mater., 2023, 35, 3880 CrossRef CAS.
- C. Enengl, S. Enengl, M. Havlicek, P. Stadler, E. D. Glowacki, M. C. Sharber, M. White, K. Hingerl, E. Ehrenfreund, H. Neugebauer and N. S. Sariciftci, Adv. Funct. Mater., 2015, 42, 6679 CrossRef.
- N. Shioya, R. Murdey, K. Nakao, H. Yoshida, T. Koganezawa, K. Eda, T. Shimoaka and T. Hasegawa, Sci. Rep., 2019, 9, 579 CrossRef PubMed.
- S. Ikeda, Jpn. J. Appl. Phys., 2022, 61, 125504 CrossRef CAS.
- M. Hodas, P. Siffalovic, P. Nadazdy, N. Mrkyvkova, M. Bodik, Y. Halahovets, G. Duva, B. Reisz, O. Konovalov, W. Ohm, E. Majkova, A. Gerlach, A. Hinderhofer and F. Schreiber, ACS Appl. Nano Mater., 2018, 1, 2819 CrossRef CAS.
- D. T. James, J. M. Frost, J. Wade, J. Nelson and J.-S. Kim, ACS Nano, 2013, 7, 7983 CrossRef CAS PubMed.
- S.-I. Park, Y.-J. Quan, S.-H. Kim, H. Kim, S. Kim, D.-M. Chun, C. S. Lee, M. Tara, W.-S. Chu and S.-H. Ahn, Int. J. Precis. Eng. Manuf. - Green Technol., 2016, 3, 397 CrossRef.
- X. Jia, E. C. Baird, J. Blochwitz-Nimoth, S. Reineke, K. Vandewal and D. Spoltore, Nano Energy, 2021, 89, 106404 CrossRef CAS.
- M. Shrestha, G. K. Lau, A. K. Bastola, Z. Lu, A. Asundi and E. H. T. Teo, Appl. Phys. Rev., 2022, 9, 031304 CAS.
- A. Cots, S. Dicorato, L. Giovannini, F. Favoino and M. Manca, Nano Energy, 2021, 84, 105894 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc01154f |
| This journal is © The Royal Society of Chemistry 2024 |