
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Condensed π-molecular arrangement for –C2H4SO3− armed naphthalenediimide†
Ryo
Ide
a,
Ayumi
Kawasaki
a,
Takashi
Takeda
 abc,
Shun
Dekura
ab,
Norihisa
Hoshino
d,
Wakana
Matsuda
e,
Shu
Seki
e and
Tomoyuki
Akutagawa
*ab
abc,
Shun
Dekura
ab,
Norihisa
Hoshino
d,
Wakana
Matsuda
e,
Shu
Seki
e and
Tomoyuki
Akutagawa
*ab
aGraduate School of Engineering, Tohoku University, Sendai 980-8579, Japan
bInstitute of Multidisciplinary Research for Advanced Materials (IMRAM), Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai 980-8577, Japan. E-mail: akutagawa@tohoku.ac.jp
cFaculty of Science, Shinshu University, 3-1-1 Asahi, Matsumoto 390-8621, Japan
dFaculty of Engineering, Niigata University, Niigata 950-2102, Japan
eGraduate School of Engineering, Kyoto University, Kyoto, 615-8510, Japan
First published on 11th January 2024
Abstract
A highly condensed packing structure with π-molecules is important to achieve high carrier transport properties. To realize condensed π-molecular arrangements, we focused on electrostatic crystal lattices consisting of cation–anion pairs and further designed an anionic N,N′-bis(ethyl sulfonate)-naphthalenediimide (ESNDI2−), which introduces structural flexibility at two –C2H4SO3− side arms. The size of the counter cation (M+) was varied from Na+, K+, Rb+, and Cs+ to design a precise crystal lattice of (M+)2(ESNDI2−)·(H2O)n salts, which is divided into hydrated salts (M+ = Na+ and K+) and anhydrous salts (M+ = K+, Rb+, and Cs+). Salt for M+ = K+ is a boundary between hydrated and anhydrous crystals, and exhibited reversible H2O adsorption–desorption behavior at 298 K. In addition, the K+ salts showed transient conductivity (ϕΣμ) switching behavior associated with the H2O adsorption–desorption cycle. The anhydrous salts for M+ = K+, Rb+, and Cs+ had isomorphous crystal structures, with the appearance of a two-dimensional (2D) herringbone arrangement in ESNDI2−. The condensed packing arrangement and highest ϕΣμ value were observed in (Rb+)2(ESNDI2−) salt. In these anhydrous salts, crystalline domains were easily formed using the simple casting method on the substrate surface. The temperature- and frequency-dependence of the dielectric constant indicated the presence of thermally induced structural fluctuations in these ionic crystal lattices. In hydrated crystals, thermal motions of Na+, K+, and H2O were dominant, whereas thermal fluctuations in the rigid M+⋯−O3S– Coulomb lattice were observed in anhydrous crystals as Debye-type relaxation. Precise structural control of the electrostatic crystal lattice determines the optimization conditions for achieving condensed π-molecular arrangement.
Introduction
The crystal structures formed by organic molecules are governed by diverse intermolecular interactions compared to inorganic materials.1–3 Even simple aromatic hydrocarbons composed of carbon and hydrogen atoms, such as naphthalene,4 pyrene,5 perylene,6 and coronene,7 undergo π–π and C–H interactions, reflecting their molecular shapes, and various packing patterns such as π-stacking and herringbone arrangements are observed.8–10 Intermolecular interactions acting on organic π-molecules have a wide range of energy scales (1–100 kJ mol−1) and directionality, including electrostatic, hydrogen-bonding, charge-transfer, and van der Waals interactions.11–14 The essential feature of organic materials is the high degree of chemically design freedom for the crystal lattice by focusing on intermolecular interactions.Higher dimensionality of intermolecular interactions is advantageous for high carrier transport properties, which improve in the order of three-dimension (3D) > two-dimension (2D) > one-dimension (1D).15–18 While most inorganic materials are formed by 3D chemical bonds, planar π-electronic organic molecules tend to form 1D π-stacking structures, which exhibit instability to external factors such as impurities and lowering of temperature.19,20 Many organic materials composed of π-planes need to have a specific molecular design to form a 2D molecular arrangement. This is an important design strategy for achieving high carrier transport properties. One chemical solution to form a 2D electronic structure with high carrier transport properties is to introduce heavy atoms such as S and Se with large polarization into the π-plane composed of carbon and hydrogen atoms.21–28 This is a molecular design that has been used with great success in molecular systems such as bis(ethylenedithiolo)-tetrathiafulvalene (BEDT-TTF) and [1]benzothieno[3,2-b][1]benzothiophene (BTBT).27–30 There are also diverse molecular systems such as phthalocyanine with a wide π-plane and C60 with 3D intermolecular interactions can be realized, and various approaches have been proposed to control the strength and dimensionality of the intermolecular interactions.31–36
Electrostatic interactions with energies above 100 kJ mol−1 can be used to control the arrangement of organic π-molecules and increase intermolecular interactions.37–41 Recently, an approach for controlling the arrangement of π-planar cations and anions was reported by Maeda et al. to develop organic materials in which the Coulomb interaction can be controlled.39–41 We have designed supramolecular cations encapsulated with crown ethers to reduce the Coulomb interactions on anions by shielding positive charges, and have designed a variety of anion arrangements for π-electronic molecules.42–46 By screening the positive and negative charges, various types of molecular arrangements similar to those of van der Waals crystals can be realized.
π-Planar naphthalene diimide (NDI) molecules exhibit n-type semiconducting properties and are used to fabricate a variety of functional organic materials due to their electronic functions such as carrier transport, molecular adsorption, magnetic and chromic properties.47–58 Various molecular designs for the nitrogen sites at the end of the NDI molecule are easily possible, and a variety of molecules can be prepared. Therefore, the NDI system provides interesting research targets in terms of electronic function design and dense packing of π-molecules. Notably, N,N′-bis(cyclohexyl)-NDI crystals exhibit high electron mobility due to their 2D molecular arrangement.58 For similar n-type organic semiconducting perylene diimide (PDI) derivatives, various substituents can be also introduced to the terminal nitrogen atom, and the molecular arrangements from nanoscale to mesoscale, including supramolecular polymers, have been reported. Since the size of the π-electron system differs between NDI and PDI π-molecules, the substituent introduced to the nitrogen sites has a different effect on the molecular arrangement and packing.59–63 The simple NDI π-core facilitates detailed studies on the design of the molecular arrangement.
Focusing on effective electrostatic interactions in crystals, we have been studying the control of molecular arrangement of n-type organic semiconducting anionic NDI derivatives, and the fabrication of functional materials to design crystal lattices with high chemical stability.64–68 An NDI derivative with benzenesulfonate groups (BSNDI2−) showed high thermal stability with the crystal reaching a decomposition temperature around 850 K with alkali metal cations such as Na+ and K+.64 However, the structurally rigid benzenesulfonate group allowed only rotational freedom around the N–C6H4SO3− bond, resulting in a low degree of freedom in molecular conformation, which did not lead to a suitable electron transport 2D layer with a condensed π-molecular arrangement. To solve the above problem, the NDI π-core with a terminal flexible –C2H4COO− group, which improves the molecular conformational freedom, was combined with M+ = Li+, Na+, K+, Rb+, and Cs+ to form cationic-anion salts.67,68 As a result, the high structural stability of the Coulomb crystal lattice and control of the electronic transport properties in conjunction with the H2O adsorption–desorption behavior were confirmed. It is important to investigate in detail what kind of anionic structural units can realize the condensed NDI array with the 2D properties, high thin film formation ability, and high electron mobility from the viewpoints of molecular structure, crystal structure, and control of physical properties.
Recently, an NDI derivative with the –C2H4PO3H− group (EPNDI2−) was used for the proton conducting crystal via a hydrogen-bonding network between –PO3H− and NH4+ in (NH4+)2(EPNDI2−) salt.69,70 In contrast, the formation of pyrophosphate bonds (–PO2–O–PO2–) through the thermal polymerization reaction between the –PO3H− units of the adjacent EPNDI2− was observed in the (Na+)2(EPNDI2−) crystals. The unit introduced at the terminal nitrogen site of the NDI skeleton plays an important role in the physical properties and the condensed arrangement of NDI. In this study, we focused on ESNDI2− with a flexible ethyl sulfonate (–C2H4SO3−) group for structural flexibility and prepared (M+)2(ESNDI2−) salts with alkali metal ions of M+ = Na+, K+, Rb+, and Cs+ (Scheme 1). In the previous BSNDI2− salts, only rotational degree of freedom around the N–C4H4SO3− bond is allowed, whereas in ESNDI2−, the conformational freedom of the ethyl sulfonate group is enhanced and the various molecular arrangement of the NDI π-core is observed. Although the crystal structure and carrier generation by photoirradiation of the ESNDI2− molecule combined with M+ = Na+, K+, Rb+, Mg2+, and Al3+ salts have been reported,71–75 there has been no comparative study on the molecular arrangements, formation of condensed π-packing structures, and electron transport properties when the size of M+ is systematically changed. We focused on (M+)2(ESNDI2−) salts with M+ = Na+, K+, Rb+, and Cs+ and attempted to investigate changes in the molecular arrangement and electron transport properties in a series of crystals in terms of the appearance of condensed π-molecular arrangements. Hydrated crystal (Na+)2(ESNDI2−)·(H2O)4.0 (1) and anhydrous crystal (Na+)2(ESNDI2−) (1′) were obtained, whereas hydrous (K+)2(ESNDI2−)·(H2O)1.0 (2) and anhydrous crystal (K+)2(ESNDI2−) (2′) were also obtained. The reversible adsorption–desorption cycle of H2O was confirmed through the sorption isotherms and the reversible conversion between hydrated and anhydrous crystals was possible in a single crystal state for M+ = K+. For Rb+ and Cs+ salts, only the anhydrous crystals (Rb+)2(ESNDI2−) (3) and (Cs+)2(ESNDI2−) (4) were obtained. We examine the molecular arrangement, thin film formation ability, dielectric response, and electron transport properties of the series of crystals 1–4.
 | ||
| Scheme 1 Molecular structures of ESNDI2− and its salts with M+ = Na+, K+, Rb+, and Cs+ in this study. | ||
Results and discussion
Thermal stability
The thermal stability of crystals 1–4 was evaluated using TG measurements (Fig. S1, ESI†). Salts 1 and 2 were stably obtained by hydrated single crystals containing four H2O molecules and one H2O molecule as crystallization solvents, whereas salts 3 and 4 were isolated as anhydrous crystals. Crystal 1 exhibits a weight loss of 12.1% upon crystal heating from 353 to 393 K, and changes to anhydrous crystal 1′ due to the desorption of four H2O molecules (calculated weight of 12.0%). The hydrated crystal 2 also forms a dehydrated crystal 2′ with one H2O molecule desorbed, with a weight loss of about 2.71% around 370 K. The decomposition temperatures of dehydrated salts 1′, 2′, 3, and 4 were 713, 749, 749, and 752 K, respectively, indicating high thermal stability. Salts 1′, 2′, 3, and 4 showed high thermal stability, whereas the van der Waals crystal, N,N′-bis(phenyl)-NDI, showed a melting point at 593–595 K,81 indicating that M+⋯−O3S– electrostatic Coulomb interactions between the cation and anion were effective in improving thermal stability. The phase transition behavior of salts 1′, 2′, 3, and 4 after dehydration was also evaluated using DSC measurements (Fig. S2, ESI†), and there was no phase transition peak and a single crystalline phase in a wide temperature range of 200–600 K.Crystal structures
The crystal structures of salts 1·4.0(H2O) and 3 have been reported previously.71–75 However, there is no evaluation of the dimensionality of the electronic structure and electron transport properties. No systematic changes in the molecular arrangement have been reported when cations are increased in size from Na+, K+, Rb+, to Cs+. Herein, we attempted to focus on the dimensionality change of the electronic structure of crystals 1–4.76,77 In crystal 2, the desorption of H2O molecules was observed in the single crystalline state upon temperature increase, and the molecular arrangements before and after the H2O desorption were evaluated in single crystals 1 at 100 K and anhydrous 1′ at 300 K.The molecular arrangements of crystals 1–4 were similar to each other, although the number of crystallographically independent species was different. In particular, anhydrous crystals 2′, 3, and 4 had similar lattice constants and similar molecular arrangements (Fig. S1, ESI†). For crystals 1 and 2, the presence of H2O molecules was necessary to form a closest-packing structure. Fig. 1 shows the crystal structure of salt 4 at 100 K. The crystal structure of salt 4 is identical to that of salt 3 at 300 K as previously reported.71 One ESNDI2− and two Cs+ are the crystallographically independent structural units, and the electronic conducting layer consisting of the NDI backbone of ESNDI2− was observed in the bc plane. The cation–anion electrostatic inorganic layer in the bc plane composed of Cs+⋯−O3S– and NDI layers was alternatively elongated along the a-axis. The electrical conducting NDI layer in the bc plane is in a herringbone arrangement, which is expected to form a 2D electronic structure.
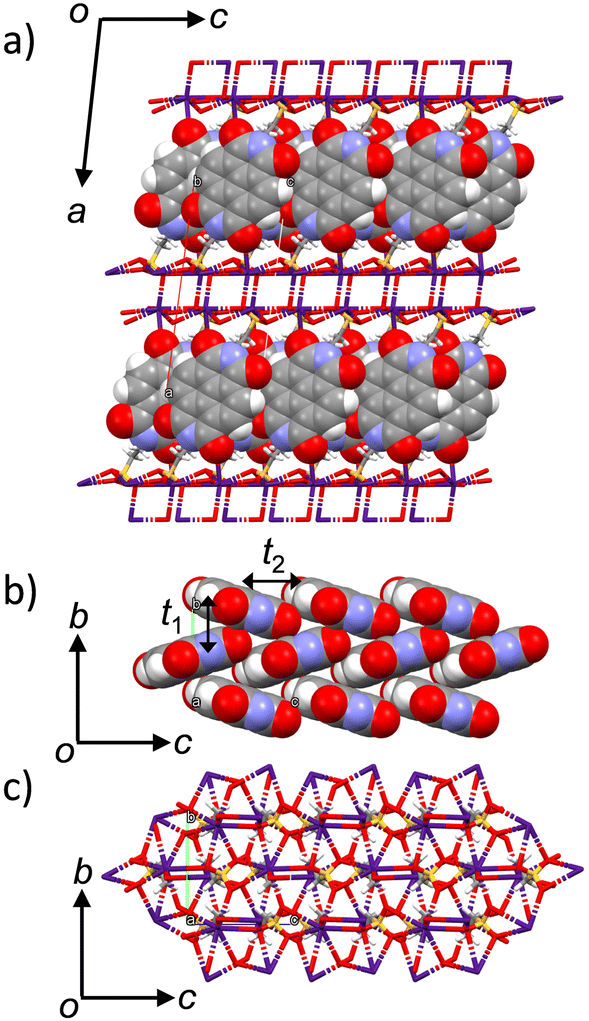 | ||
| Fig. 1 Crystal structure of salt 4 at 100 K. (a) Unit cell viewed along the b-axis using a CPK representation of DNI units. (b) The 2D herringbone NDI arrangement in the bc plane with t1 and t2 interactions. (c) The 2D Cs+⋯−O3S– electrostatic networks in the bc plane. | ||
Electronic structures
Using the extended Hückel calculation, the transfer integral (t, meV) of the LUMOs between the NDI layers of salt 4 was calculated to be t1 = −21.0 meV along the b-axis and t2 = −18.6 meV along the c-axis, suggesting the formation of a 2D electronic structure (Fig. S3, ESI†).78 Calculation of the LUMO band of salt 4 using the tight-binding approximation yields a closed 2D Fermi surface assuming a partially occupied state of Fermi energy EF = 0.15, confirming the formation of a 2D electronic structure (Fig. S4, ESI†). In the inorganic layer in the bc plane, two –SO3− groups and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O oxygen of ESNDI2− formed a hepta-coordinated structure around Cs+, forming a cation–anion electrostatic network layer. The average distance (dCs–O) between the oxygen atoms of the –SO3− group and Cs+ was 3.154 Å, whereas the dCs-O between the oxygen atom of CO and Cs+ was 3.279 Å.
O oxygen of ESNDI2− formed a hepta-coordinated structure around Cs+, forming a cation–anion electrostatic network layer. The average distance (dCs–O) between the oxygen atoms of the –SO3− group and Cs+ was 3.154 Å, whereas the dCs-O between the oxygen atom of CO and Cs+ was 3.279 Å.
The electrical conducting NDI arrangements in crystals 1–4 exhibited a herringbone pattern that could be represented by the ratio of t1 and t2 interactions, wherein the LUMO band changed depending on the cation size (Fig. S3, ESI†). In crystal 1, the intermolecular interactions of t1 = −0.69 meV and t2 = +5.24 meV were observed along the b- and the c-axis, respectively, wherein these values were smaller than those found in salt 4. The negligible magnitude of t1 interaction along the b-axis of salt 1 forms a 1D electronic structure, in which the LUMO band showed a narrow bandwidth of less than 0.005 eV at the Γ–S energy trace (Fig. S5, ESI†). Assuming the Fermi surface of salt 1 at EF = 0.15, the formation of a 1D electronic structure is confirmed. In salt 2, the intermolecular interactions of t1 = −2.20 meV and t2 = 18.9 meV were observed along the a- and the b-axis, respectively, by changing cations from Na+ to K+, which also changed the number of crystallization H2O molecules from four to one. The intermolecular interaction between the LUMOs increased 3 times due to the replacement the cation from Na+ to K+ in salt 2. Additional t3 = 3.20 meV is also observed along the a + b axis. The structure of the LUMO band indicates the presence of a 1D Fermi surface (Fig. S6b, ESI†), assuming EF = 0.15. When crystal 2 changed to anhydrous 2′, significant enhancement of intermolecular interactions is observed with t1 = −27.2 meV along the b-axis and t2 = −19.4 meV along the c-axis. The resulting LUMO band showed the presence of a circular 2D Fermi surface assuming EF = 0.15 (Fig. S6a, ESI†). The crystal change from hydrated salt 2 to anhydrous salt 2′ modulated the closest-packing structure and transformed the molecular arrangement and electronic structure of the NDI π-core from a 1D to 2D pattern. In salt 3, an almost equivalent intermolecular interaction of t1 = −25.1 meV along the b-axis and t2 = −21.7 meV along the b-axis was observed, where the LUMO band assuming EF = 0.15 indicated the presence of a 2D electronic structure (Fig. S7, ESI†). The t values in salt 4 were smaller than those in salt 3, suggesting the best-fit cation size of Rb+ in the condensed NDI π-arrangement.
Cation dependence for packing structures
In the inorganic electrostatic layer of salt 1, oxygen atoms of two –SO3−, four H2O, and oneO of ESNDI2− were hepta-coordinated to Na+ in a pentagonal bipyramidal coordination geometry, forming an electrostatic network in the ab plane, where one Na+ was coordinated in an equatorial plane of two –SO3− groups and one O in a pentagonal configuration with the average distance of dNa–O = 2.395 Å. In addition, the two H2O molecules coordinated up and down concerning Na+, and the average Na+⋯O distance of dNa–O = 2.460 Å was almost the same as pentagonal coordination. In salt 2, the two –SO3− groups, one H2O, and one O of ESNDI2− formed a pseudo-dodecahedral-shaped octa-coordinated structure around K+, forming an electrostatic network in the ab plane. The interaction between the O group and K+ was observed at dK–O = 2.967 Å, whereas that between the –SO3− oxygen atom and K+ was observed at an average distance of dK–O = 2.883 Å. In the anhydrous salt 2′, a clear change in the molecular arrangement of the NDI π-core was observed, whereas no significant change in the coordination structure around K+ was observed. The oxygen atoms of –SO3− and the O of the ESNDI2− formed seven-coordinated geometry to the Rb+ salt, forming a multipoint electrostatic interaction network, with dRb–O = 3.180 Å for O and dRb–O = 2.970 Å for –SO3−. In all salts, two –SO3− groups were coordinated to M+, connecting the adjacent NDI layers and forming a Coulomb crystal lattice.
Fig. 2 summarizes the M+⋯−O3S– arrangements in the inorganic network layers and the t values of NDI for salts 2′, 3, and 4. The cation M+ forms a coordination structure through electrostatic interactions with two –SO3− groups (Fig. 2a). Therefore, a much larger size of M+ increases the average distance between –SO3− groups. Flexible –C2H4SO3− groups of the ESNDI2− molecule have a high degree of conformational freedom. An increase in M+ size alters the intermolecular interactions of the NDI π-core responsible for carrier transport property, leading to conformational changes that form a closest-packing structure. Fig. 2b summarizes the S⋯S distances and angles between the three S atoms of the neighboring –SO3− groups of ESNDI2− molecules for M+ = K+, Rb+, and Cs+, where the S⋯S distances dS–S increase in the order of Cs+ (dS–S = 7.75 Å) > Rb+ (dS–S = 7.56 Å) > K+ (dS–S = 7.43 Å) with increasing M+ size, whereas the S⋯S⋯S (fS–S–S) angle between the three S atoms decreases in the order fS–S–S = 51.7° (K+) > 49.0° (Rb+) > 44.3° (Cs+). The t values reached a maximum at t1 = 25.1 and t2 = 21.7 meV for M+ = Rb+, and the change of M+ size from Rb+ to Cs+ reduced to t1 = 21.0 and t2 = 18.6 meV, where the intermolecular interactions between the NDI π-cores were suppressed. Regarding the dimensionality of the intermolecular interactions between NDIs, the dimensionality tended to improve with increasing M+ size, with t2/t1 values of 0.89 (Cs+) > 0.87 (Rb+) > 0.71 (K+). However, the increase in dimensionality and the magnitude of the t value tend to be in conflict, and it can be concluded that the Rb+ salt was the best condition for obtaining the 2D electronic structure.
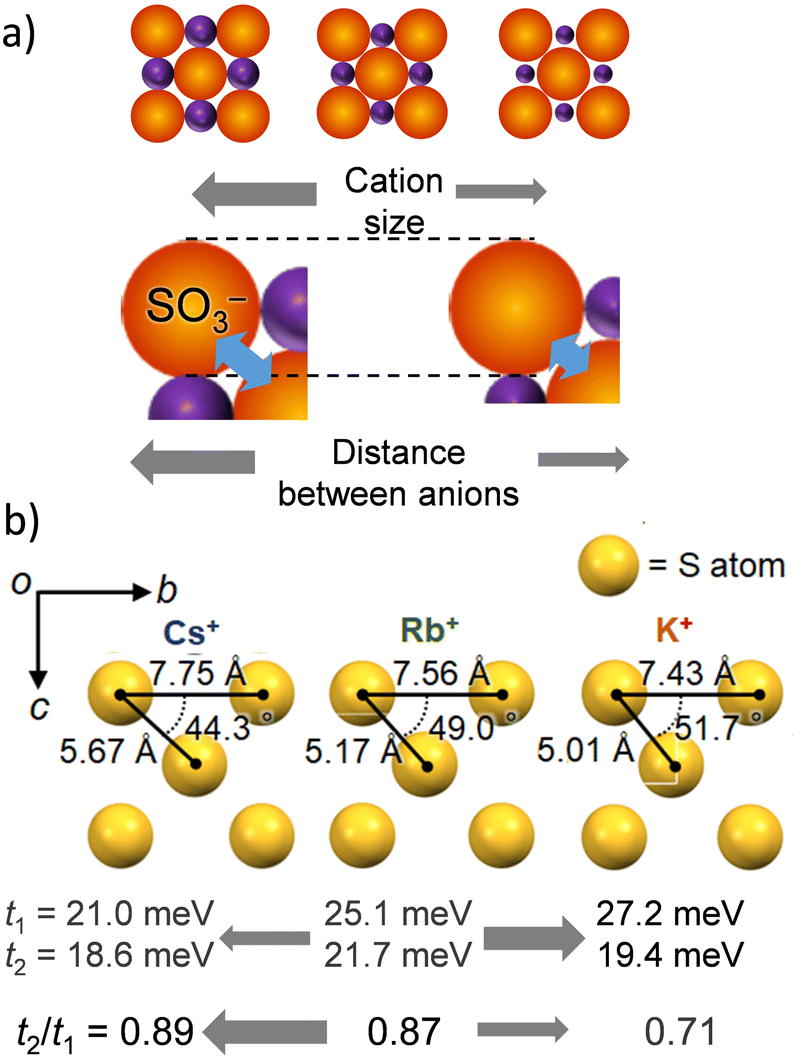 | ||
| Fig. 2 Change in the inorganic M+⋯−O3S– electrostatic layer from M+ = K+, Rb+ to Cs+. (a) Modulation of the M+⋯−O3S– electrostatic inorganic layer with increasing M+ size. The orange and purple spheres represent –SO3− and M+, respectively. (b) S⋯S interatomic configuration and t1 and t2 values for M+ = K+, Rb+, and Cs+. The M+ dependent t1, t2, and their ratio t2/t1 are also shown in the bottom. | ||
Structural transformation from 2 to 2′
Single crystals 2 can undergo a structural change from monohydrate 2 to anhydrous crystal 2′ while retaining its single crystalline state. Single crystal X-ray structural analyses of both states reveal how the molecular arrangement changes with the H2O adsorption–desorption cycle. H2O molecules incorporated in the inorganic K+⋯−O3S– network are desorbed from the crystal lattice by thermal annealing, changing the rearrangement of K+ and –SO3− and modulating the molecular arrangement in the NDI layer responsible for electron transport property (Fig. 3a). Fig. 3b shows the change in intermolecular interactions in the 2D layer of NDIs using t values. Intermolecular interactions of t1 = +18.9, t2 = −3.2, and t3 = +2.20 meV are observed in salt 2, and the small magnitude of the latter two t values forms a 1D electronic structure in which the t1 value along the a-axis is essentially dominant (Fig. 3b). After H2O desorption by thermal annealing, the π-stacked NDI arrangement tilts toward the molecular long axis and changes to a herringbone arrangement. The desorption of H2O molecules from single crystal 2 exhibits a drastic effect on the electronic structure.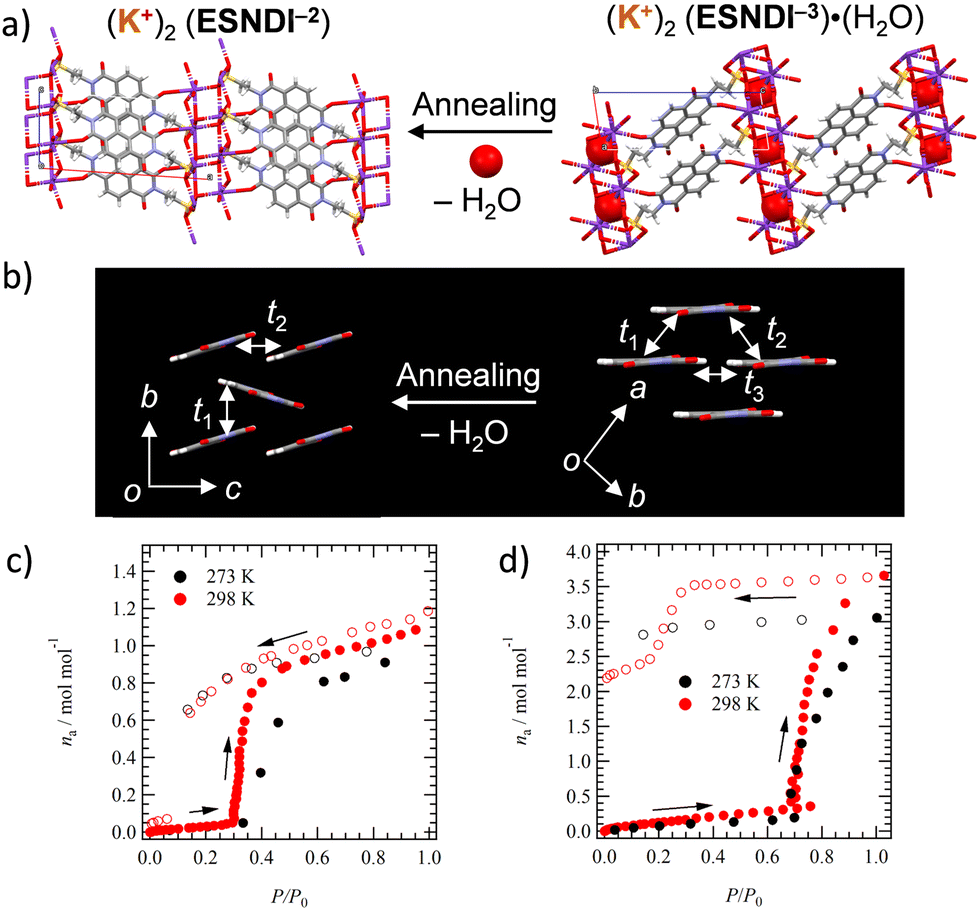 | ||
| Fig. 3 Structural changes associated with the H2O adsorption–desorption cycle. (a) Change in the molecular arrangement in salts 2 and 2′. (b) Change in intermolecular interactions between NDI π-cores in salts 2 and 2′. Adsorption–desorption isotherms of (c) salt 2′ at 273 and 298 K for H2O, and (d) salt 1′ at 273 and 298 K for H2O. | ||
H2O adsorption–desorption of salts 1 to 2
The stability of anhydrous salts 1′ and 2′ under atmospheric conditions was evaluated. Salt 1′ was re-adsorbed using H2O by leaving it in H2O vapor overnight, and its H2O adsorption amount was evaluated using TG measurements. The salt 1′ exposed to distilled H2O vapor by allowing it to stand overnight in a vial at 298 K showed a weight loss of 12.1% equivalent to 4.0 molecules of H2O in the TG chart (iii in Fig. S8, ESI†), which was in agreement with the weight loss (12.1%) of hydrated crystal 1 (i in Fig. S8, ESI†). In contrast, the TG chart for the 1′ salt with H2O adsorbed in air showed no weight loss, indicating that salt 1′ does not re-adsorb H2O in the air (ii in Fig. S8, ESI†). Similar measurements were examined for salt 2′. Sample 2′ exposed to distilled H2O vapor showed a weight loss of 2.71% corresponding to 0.92 amount of H2O molecules (ii in Fig. S9, ESI†), and H2O re-adsorbed powder 2′ showed a weight loss of 2.11% corresponding to 0.71 molecules of H2O. Powder 2′ was confirmed to exhibit H2O re-adsorption behavior even under atmospheric conditions (i in Fig. S9, ESI†), which is different from the behavior of salt 1′.The adsorption and desorption process of H2O on crystal 2′ was investigated through the measurement of sorption isotherms at 273 and 298 K (Fig. 3c). In anhydrous crystal 2′, a rapid increase in adsorption amount (na, mol mol−1) was observed around relative pressure P/P0 = 0.3 with increasing H2O pressure, confirming a gate-opening H2O adsorption mechanism. When P/P0 is decreased, a stable state of na = 1.0 is observed around P/P0 = 0.1, and a hysteresis behavior for the H2O adsorption–desorption process is confirmed. Thus, crystal 2′ can be reversibly converted to a monohydrated crystal by changing the relative pressure of H2O. A similar change is observed for salt 1′ at a relative pressure of P/P0 = 0.7, whereas the adsorption of H2O at P/P0 = 1.0 for crystal 1′ at 298 and 273 K is na = 3.5 and 3.0, respectively, whose amount is lower than that of the four H2O molecules found in salt 1. When the relative pressure was decreased from P/P0 = 1.0, the hydrated state remained stable and no reversible change to na = 0 was observed.
Dielectric property
To evaluate the dynamics of polar structural units in salts 1–4, the temperature (T) and frequency (f) dependent dielectric responses were evaluated in the AC impedance method using a compressed pellet under nitrogen flow. Fig. 4a and b show the T- and f-dependent real part dielectric constant ε1 for salts 2 and 2′.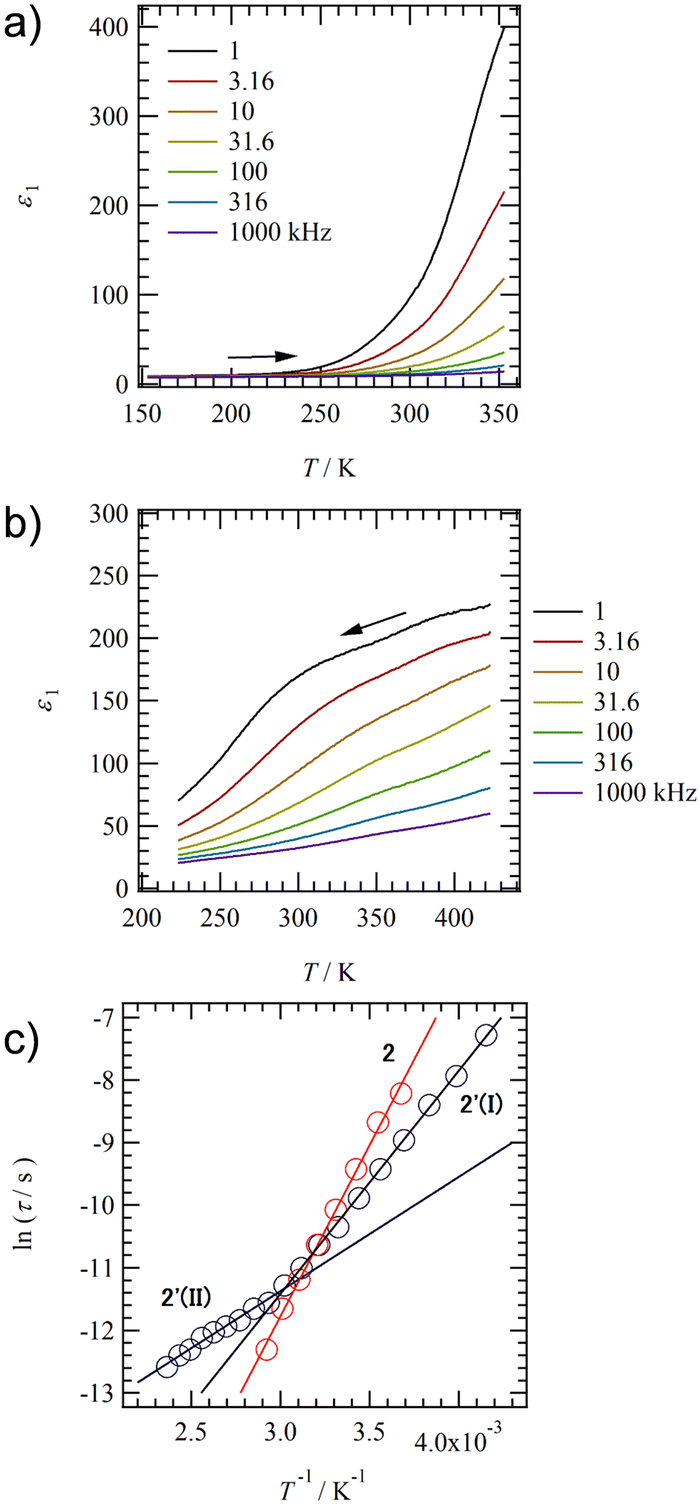 | ||
| Fig. 4 Dielectric response of hydrated salt 2 and anhydrous slat 2′ using the compressed pellets. T- and f-dependent ε1 of (a) salt 2 and (b) salt 2′. (c) ln(τ) − T−1 plots of salts 2 and 2′. A crossover between two types of relaxation processes I at T > 330 K and II at T < 330 K is observed in anhydrous salt 2′ around 330 K. | ||
The ε1 and imaginary part dielectric constant (ε2) of salt 2 are observed at ε1 ∼10 and ε2 ∼20 at 220 K, respectively, and these values increase significantly to ε1 ∼400 and ε2 ∼4000 at 350 K (Fig. S10a, ESI†). Since neutral NDI derivatives without ionic pairs or a crystallization solvent do not exhibit a dielectric response,64 the dielectric response found in salt 2 is presumably due to the dynamic behavior of the polar H2O molecule or the ionic component such as K+ and K+⋯−SO3–. A monotonic increase in ε1 and ε2 with increasing T is observed for salt 2. In contrast, ε1 in anhydrous salt 2′ shows a broad peak that depends on T- and f-values spanning the range of 200 < T < 350 K and 1.0 < f < 10 kHz (Fig. 4b). The T-dependence of the Cole–Cole plots for hydrated salt 2 and anhydrous salt 2′ (Fig. S10 and 11, ESI†) showed a semicircular trace in both T-ranges, indicating the presence of dielectric relaxation of a Debye-type single-component. For salt 2, the K+ conductivity (σK+) obtained from the Cole–Cole plot (Fig. S10, ESI†) showed σK+ = 1.6 × 10−7 S cm−1 at 350 K and Ea = 466 meV. Next, an ln(τ) − T−1 plot was constructed from the Cole–Cole plot of the anhydrous salt 2′, and Ea was calculated for dielectric relaxation. The ln(τ) − T−1 plot shows a slope change around 330 K, and a crossover phenomenon from Ea(I) = 310 meV at T < 330 K to Ea(II) = 160 meV at T > 330 K appears. The dielectric relaxation of anhydrous salt 2′ is considered to originate from the thermal fluctuation of the ionic crystal lattice because the Ea value at T > 330 K is about half of the value at T < 330 K and the Ea is more than 200 meV lower than that of the hydrated salt 2. In general, cations such as Na+ and K+ in the ionic crystals are more strongly bound to the crystal lattice than Li+.79,80 However, K+ coordinated with H2O suppressed the anion–cation electrostatic interaction and showed ionic conductivity at 300 K.80 In salt 2, the coordination of H2O to K+ shielded the positive charge of K+, resulting in K+ conduction. In contrast, ionic conduction in salt 2′ was suppressed by the strong electrostatic K+⋯−O3S– interaction, and a dielectric response due to thermal fluctuations at K+⋯−O3S– bond was observed. The crossover phenomenon is considered to be caused by the T-dependent change in the coordination environment of –SO3− and O concerning K+.
Fig. 5a shows the T- and f-dependent ε1 behaviour of salt 3. The ε1 value of salt 3 exhibited a broad peak over a wide measurement T- and f-ranges of 223 < T < 350 K and 1.0 < f < 31.6 kHz, respectively, showing similar behaviour to those of salt 2′. The crystal structure of anhydrous salt 2′ was similar to that of salt 3, suggesting the same dielectric relaxation mechanism of a Coulomb crystal lattice composed of M+⋯−O3S– for M+ = K+ and Rb+. The T-dependent Cole–Cole plot for salt 3 showed a semicircular trace and used the peak top fp values to deduce an ln(τ) − T−1 plot to obtain the Ea value (Fig. 5d and Fig. S14, ESI†). A crossover of Ea values obtained from dielectric relaxation processes I to II with temperature change was already observed for anhydrous salt 2′, while Ea = 256 meV explained the dipole relaxation process of salt 3 in all measured T-ranges. The Ea value of salt 3 has the same origin as the dielectric relaxation process I at the low-T region of salt 2 (Ea = 160 meV), which is considered to undergo the thermal fluctuation of the Rb+⋯−O3S– Coulomb lattice. The larger Ea value for salt 3 than for anhydrous salt 2′ is the result of the much heavier Rb+⋯−O3S– lattice than the K+⋯−O3S– lattice, which needs more thermal energy.
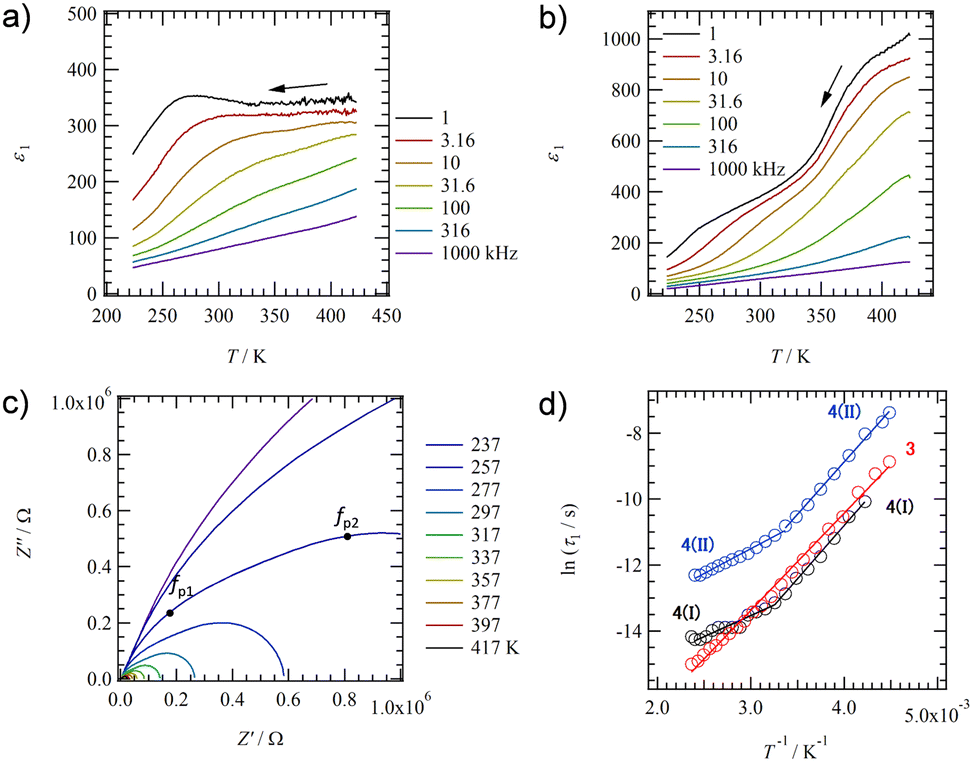 | ||
| Fig. 5 Dielectric response of salts 3 and 4 using the compressed pellets. T- and f-dependent ε1 of (a) salt 3 and (b) salt 4. (c) T-dependent Cole–Cole plots of salt 4 with two f-peaks at fp1 and fp2. (d) ln(τ) − T−1 plots of salts 3 and 4. A crossover between two types of relaxation process I at T > 310 K and process II at T < 310 K is observed in salt 4. | ||
Fig. 5b shows the T- and f-dependent ε1 behaviour for salt 4 (Fig. S15, ESI†). Two dielectric relaxation processes were observed in salt 4 as a broad peak at 223 < T < 350 K and 1.0 < f < 31.6 kHz on the low-T side (relaxation I) and a broad peak at 350 < T < 423 K and 1.0 < f < 316 kHz on the high-T side (relaxation II). This was similar to the behaviour of anhydrous salts 2′ and 3, and more clearly two different relaxation processes were observed in salt 4. These three anhydrous salts of 2′, 3, and 4 exhibited similar dielectric responses because they have the same molecular arrangement. The T-dependent Cole–Cole plot for salt 4 shows two overlapping semicircular traces, and the presence of two types of dielectric relaxation (I and II) can be also observed (Fig. 5c). Peak top frequencies of fp1 and fp2 were determined for the two overlapping semicircles in the Cole–Cole plot at each T, forming two ln(τ) − T−1 plots (Fig. 5d). For Relaxation I on the low-T side, a crossover from Ea = 121 meV at T > 310 K to 269 meV at T < 310 K was observed around 310 K. For relaxation II, a crossover from Ea = 110 meV at T > 310 K to 279 meV at T < 310 K is also observed around 310 K. Since the activation energies of relaxations I and II are similar to each other and the T-range of the crossover from a low to high-T region is also similar to each other, the motional freedom of dipole moments is interconnected to each other. A similar dielectric relaxation process has been observed for (M+)2(PCNDI2−) salts for M+ = Rb+ (Ea1 = 46.8 meV and Ea2 = 62.8 meV) and Cs+ (Ea1 = 89.6 meV and Ea2 = 90.9 meV), which arise from two relaxation processes involving the thermally activated Rb+⋯−OOC– Coulomb lattice.68 The dielectric relaxation of salt 4 is also attributed to the similar thermal fluctuations of the electrostatic bonding Cs+⋯−O3S– lattice, and the crossover process is due to the presence of –SO3− and O groups in different coordination environments around Cs+. The Ea value at the low-T side of salt 4 (Ea = 279 meV) is larger than that of salt 3 (Ea = 256 meV), which is due to the heavy crystal lattice of Cs+⋯−O3S–.
Thin film formation
To evaluate the film formation properties of salt 1–4, thin films were prepared on a Si substrate with a SiO2 layer of 200 nm using the drop-casting method, and the surface structure was observed via polarized optical microscopy (POM) performed in a cross-Nicole optical arrangement and via atomic force microscopy (AFM). POM images of the salt 1 thin film showed the formation of fibrous domain structures, whereas those of salts 2–4 thin films indicated the formation of polycrystalline thin films consisting of dense plate-like crystalline domains (Fig. S16, ESI†). Fig. 6 shows an AFM image of the salts 2–4 thin films. The salt 1 thin film consists of fibrous domains with heights ranging from several tens to several hundreds of nanometers, and the formation of a uniform thin film domain was not observed (Fig. S17, ESI†). In contrast, the formation of a flat crystalline domain was observed in the salt 2–4 thin film, with domain sizes ranging from 200 to 400 nm. In these thin films, a striped surface structure with overlapping crystalline domains was observed, and the step-terrace between the crystalline terraces was estimated to be about 1.4–1.8 nm in height from the cross-sectional profile analysis (lower in Fig. 6). The step height corresponds to the length of the a-axis obtained in the single crystal X-ray structure, and the domains had the step-terrace structure characteristic of single crystalline domains. Salts 2–4 can be used to fabricate polycrystalline thin films consisting of single crystalline domains on Si substrates using a simple drop-casting method and exhibit excellent film-forming properties. | ||
| Fig. 6 AFM images of thin films for salts (a) 2, (b) 3, and (c) 4 with a scan area of 500 × 500 nm2. The lower part includes the cross-sectional image along the upper red line. | ||
The correspondence between thin films fabricated on quartz substrates and crystal structures obtained from single crystal structure analysis was examined from the XRD measurements of thin films 1–4. Fig. 7a summarizes the XRD patterns of crystals and thin films for salt 2. The XRD pattern calculated using the single crystal X-ray structural analysis of 2 at 100 K (pattern i in Fig. 7a) shows good agreement with the XRD pattern of thin film 2, confirming high crystallinity even in the thin film state. Diffraction peaks at d001 = 15.8 Å, d002 = 7.90 Å, and d003 = 5.25 Å were observed at 2θ = 5.59, 11.2, and 16.9°, respectively, corresponding to reflections corresponding to the layered structures, which are consistent with the layer period of c = 15.7629(3) Å for the single crystal at 100 K. Anhydrous thin film 2′ was prepared by heating hydrated thin film 2 to 393 K. The XRD patterns of thin film 2′ were observed around 2θ = 5.08, 10.2, 20.5, and 25.7° at d100 = 17.4, d200 = 8.67, d400 = 4.33, and d500 = 3.47 Å. These are in good agreement with the XRD pattern obtained from the simulation of single crystal 2′ with a = 17.381(15) Å. Therefore, ESNDI2− molecules in salts 2 and 2′ are aligned perpendicular to the molecular long axis on the substrate while maintaining crystallinity during the reversible H2O adsorption–desorption cycle.
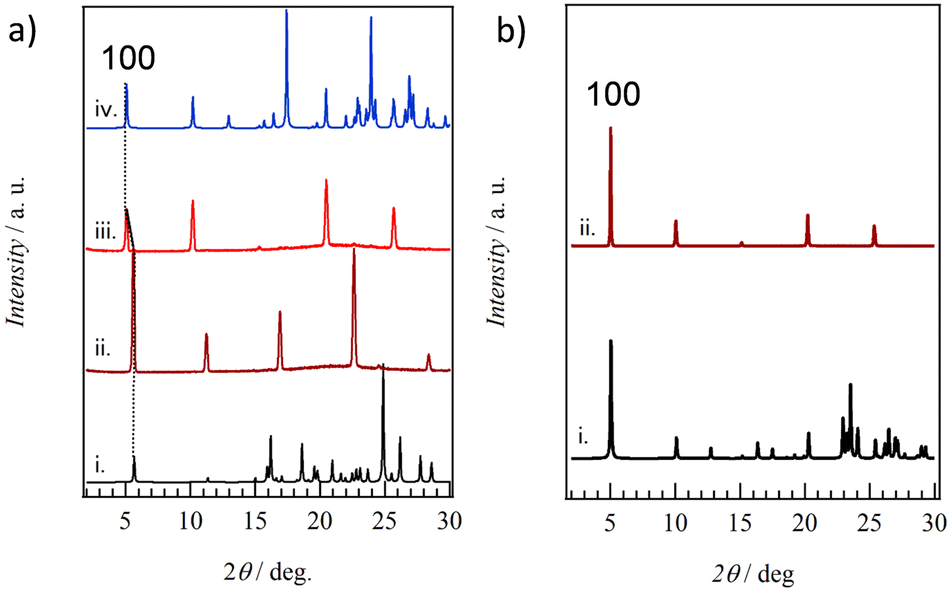 | ||
| Fig. 7 XRD patterns of thin films. (a) Salts 2 and 2′ of the simulation based on the single crystal X-ray structural analysis of 2 at 100 K (i), a thin film of 2 (ii), single crystal X-ray structural analysis of 2′ at 100 K (iii), and the thin film of 2′. (b) Salt 3 of the simulation based on single crystal X-ray structural analysis at 100 K (i) and thin film (ii). | ||
For hydrated single crystal 1, good agreement was also observed between the calculated XRD pattern and that of thin film 1, where diffraction peaks around 2θ = 6.37 and 12.7° corresponded to d002 = 13.9 Å and d004 = 6.97 Å, respectively, (Fig. S18, ESI†). Therefore, the molecular long axis of ESNDI2− was perpendicularly oriented on the substrate surface while retaining the layered structure of c = 27.7283(9) Å confirmed by the single crystal X-ray structural analysis of salt 1. The XRD pattern of anhydrous thin film 1′ dehydrated by heating at 393 K showed sharp reflections around 2θ = 6.77 and 13.6°, in good agreement with that of anhydrous powder 1′ dehydrated by heating. Both thin films 1 and 1′ showed clear Bragg reflections, confirming high crystallinity due to the H2O adsorption–desorption cycle.
Fig. 7b shows the simulated XRD pattern based on the single crystal X-ray structural analysis of 3 at 100 K and thin film state 3. Both are in good agreement, and reflections around 2θ = 5.03, 10.1, 15.1, 20.2, and 25.3°correspond to d100 = 17.6, d200 = 8.76, d300 = 5.87, d400 = 4.40, and d500 = 3.52 Å, respectively, for thin film 3. This periodicity is in good agreement with the lattice constant of a = 17.6025(4) Å in the crystal structural analysis of 3 at 100 K. Similar XRD patterns were also observed in thin film 4 (Fig. S19, ESI†), in good agreement with the XRD pattern calculated from single crystal 4 at 100 K. Periodicities of d300 = 5.95, d400 = 4.46, and d500 = 3.56 Å were observed, consistent with the lattice constant of a = 17.9643(4) Å based on the single crystal structural analysis of 4 at 100 K. Thin films 3 and 4 were confirmed to retain high crystallinity, consistent with the presence of crystalline domains as found via AFM, and were aligned perpendicular to the molecular long axis of ESNDI2− on the substrate surface.
Carrier transport property of thin films
The carrier transport properties of thin films 1–4 were evaluated using FP-TRMC measurements at 298 K in the air (Fig. 8).81,82 The same quartz substrate employed for the XRD measurements was used to confirm the crystallinity and structure of the thin films in the context of FP-TRMC measurements. ϕΣμ values for thin films 1, 1′, 2, 2′, 3, and 4 at 298 K obtained from ϕΣμ - t plots were 3.7 × 10−9, 4.4 × 10−9, 7.5 × 10−9, 4.4 × 10−8, 1.5 × 10−7, and 8.3 × 10−8 m2 V−1 s−1, respectively. While no difference in ϕΣμ values with and without H2O is observed in thin films 1 and 1′, a change in ϕΣμ values according to the existence of H2O is observed in thin films 2 and 2′ (Fig. 8b). ϕΣμ in anhydrous thin film 2′ is about 6 times larger than in hydrated 2, corresponding to the change in the electronic structure from 1D to 2D structure upon H2O desorption as shown by single crystal X-ray crystal structural analyses (Fig. 3b). The change in electronic transport properties due to the H2O adsorption–desorption cycle in thin film 2 was confirmed repeatedly, and a reversible H2O adsorption–desorption cycle and switching behavior of electronic transport properties at the adsorption isotherm were observed. Thin film 3 exhibits the highest ϕΣμ value in the series, which results from the 2D intermolecular interactions at t1 = 25.1 meV and t2 = 21.7 meV. The ϕΣμ value of 4 is lower than that of 3 for t1 = 21.0 meV and t2 = 18.6 meV, which represents the 2D electronic structure but results from low absolute t values of intermolecular interactions.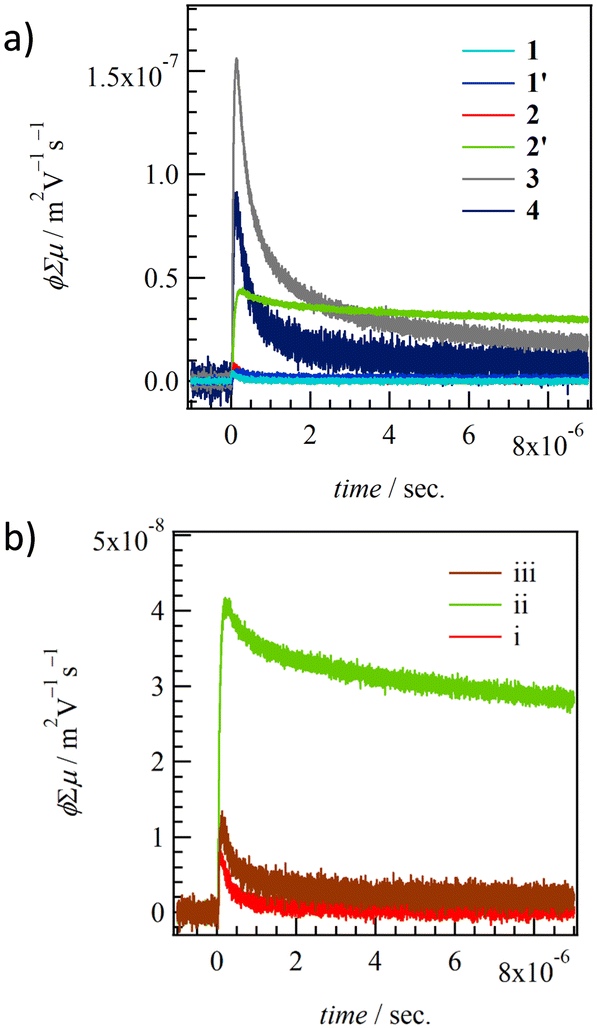 | ||
| Fig. 8 TRMC kinetics of (a) thin films for 1–4 and (b) H2O-dependent change of hydrated thin film 2 (i), anhydrous 2′ (ii), to H2O re-hydrated 2 (iii). | ||
Fig. 9 summarizes the ϕΣμ values of thin films 1–4 and (M+)2(BSNDI2−) at 298 K from the dimensionality of the electronic structure. The ϕΣμ values of thin films increase in proportion to the intermolecular interactions of the NDI π-core and the dimensionality of the electronic structure. Salt 3 (t2/t1 = 0.86) and salt 4 (t2/t1 = 0.89) have a 2D electronic structure, showing prominently higher ϕΣμ values. In particular, the ϕΣμ value of thin film 3 was 2.5 times higher than that of (Na+)2(BSNDI2−). The (M+)2(BSNDI2−) salt was linked through a phenylene spacer unit, which is rigid and has low structural degrees of freedom, whereas (M+)2(ESNDI2−) was linked through ethyl groups, which have high structural degrees of freedom. The latter unit enabled the closest-packing structure and the dense π-arrangement in molecular assembly. The high structural freedom for the H2O adsorption–desorption process also changes the dimensionality of the electronic structure while maintaining a high crystalline state.
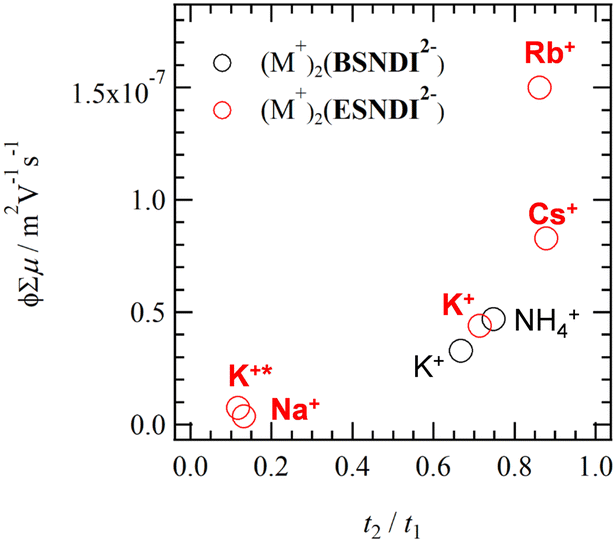 | ||
| Fig. 9 ϕΣμ – t2/t1 plots of thin films 1–4 (red circle) and (M+)2(ESNDI2−) (black circle). Points K+ and K+* correspond to thin films 2′ and 2, respectively. | ||
Conclusions
In ionic n-type organic semiconductors, the effects of structural flexibility of the spacer unit and the size of the counter cation (M+) on the packing structure and physical properties were investigated in (M+)2(ESNDI2−)·(H2O)n salts. Salts 1 (M+ = Na+) and 2 (M+ = K+) containing four molecules and one H2O molecule, respectively, as crystal solvents were obtained, whereas anhydrous crystals were obtained as single crystals for M+= K+, Rb+, and Cs+. Around a boundary, a single-crystal-to-single-crystal transformation with adsorption–desorption of H2O was observed in salt 2 for M+ = K+. For the electrostatic crystal lattice consisting of M+⋯−O3S–, the cation sizes of Na+ and K+ were small and H2O molecules were introduced to fill the crystalline space. In contrast, no crystalline space for H2O to be introduced was confirmed in M+ = Rb+ and Cs+, forming a closest-packing structure with a 2D NDI π-arrangement. As a result, the highest transient conductivity ϕΣμ was observed in the Rb+ salt. The ϕΣμ decreased in the order, salts 3 (Rb+) > 4 (Cs+) > 2′ (K+) > 2 (K+) ≈ 1 (Na+) ≈ 1′ (Na+), and reversible conductivity switching was observed for 2 and 2′. Salts with 2D intermolecular interactions form crystalline domains on the substrate surface, which leads to a high ϕΣμ value. Thermal fluctuations in the M+⋯−O3S– lattice and ionic conductivity for Na+ were observed via dielectric spectroscopy. The anhydrous salts 2′, 3, and 4 exhibited isomorphic crystal structures and showed the formation of a 2D electronic structure. Among them, salt 3 exhibited the lowest dipole fluctuations near room temperature, which achieved a condensed molecular arrangement and a high ϕΣμ value. The designed structural flexibility of the spacer unit of –C2H4SO3− in ESNDI2− compared to BSNDI2− is considered to provide more freedom for the realization of the condensed π-arrangement. A high degree of structural freedom is an important design parameter that both improves film formation and increases the packing density of the π-arrangement, which in turn affects the thermal stability and electron transport properties of n-type semiconductor materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on KAKENHI (Grant Numbers: JP20H05865, 22K19004, and 20H05862), the Japan Science and Technology Agency, Core Research for Evolutional Science and Technology (Grant Number: JPMJCR18I4), and the “Crossover Alliance to Create the Future with People, Intelligence and Material” project supported by the Ministry of Education, Culture, Sports, Science, and Technology.Notes and references
- A. I. Kitaigorodsky, Molecular Crystals and Molecules, Academic Press, New York, 1973 Search PubMed.
- J. D. Dunitz and A. Gavezzotti, Chem. Soc. Rev., 2009, 38, 2622–2633 RSC.
- A. J. Edwards, C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, Faraday Discuss., 2017, 203, 93–112 RSC.
- S. C. Abrahams, J. M. Robertson and J. G. White, Acta Cryst, 1949, 2, 233–238 CrossRef CAS.
- A. Camerman and J. Trotter, Acta Cryst, 1965, 18, 636–643 CrossRef CAS.
- D. M. Donaldson, J. M. Robertson and J. White, Proc. R. Soc. London, Ser. A, 1953, 220, 366–376 Search PubMed.
- E. Fawcett and J. Trotter, Proc. R. Soc. London, Ser. A, 1966, 289, 311–321 Search PubMed.
- M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 126, 4318–4328 CrossRef CAS PubMed.
- A. Guijarro, J. A. Vergés, E. San-Fabián, G. Chiappe and H. Louis, Chem. Phys. Chem., 2016, 17, 3548–3557 CrossRef CAS PubMed.
- C. Wang, D. Hashizume, M. Nakano, T. Ogaki, H. Takenaka, K. Kawabata and K. Takimiya, Chem. Sci., 2020, 11, 1573–1580 RSC.
- T. Akutagawa, Mater. Chem. Front., 2018, 2, 1064–1073 RSC.
- T. Akutagawa, Bull. Chem. Soc. Jpn., 2021, 94, 1400–1420 CrossRef CAS.
- M. Stein and M. Heimsaat, Crystals, 2019, 9, 665 CrossRef CAS.
- S. P. Thomas, P. R. Spackman, D. Jayatilaka and M. A. Spackman, J. Chem. Theory Comput., 2018, 14, 1614 CrossRef CAS PubMed.
- C. Rolin, E. Kang, J.-H. Lee, G. Borghs, P. Heremans and J. Genoe, Nat. Commun., 2017, 8, 14975 CrossRef CAS PubMed.
- P. J. Skabara, J.-B. Arlin and Y.-H. Geerts, Adv. Mater., 2013, 25, 1948–1954 CrossRef CAS PubMed.
- S. Giannini and J. Blumberger, Acc. Chem. Res., 2022, 55, 819–830 CrossRef CAS PubMed.
- N. Kasuya, J. Tsurumi, T. Okamoto, S. Watanabe and J. Takeya, Nature Mater., 2021, 20, 1401–1406 CrossRef CAS PubMed.
- J. C. Scott, Semiconductor and Semimetals. Highly Conducting Quasi-One-Dimensional Organic Crystals, ed. E. Conwell, Academic Press, New York, 1988, p. 385 Search PubMed.
- P. A. Lee, T. M. Rice and P. W. Anderson, Phys. Rev. Lett., 1973, 31, 462–465 CrossRef CAS.
- J. Shinagawa, Y. Kurosaki, F. Zhang, C. Parker, S. E. Brown and D. Jérome, Phys. Rev. Lett., 2007, 98, 147002 CrossRef CAS PubMed.
- T. Okamoto, M. Mitani, C. P. Yu, C. Mitsui, M. Yamagishi, H. Ishii, G. Watanabe, S. Kumagai, D. Hashizume, S. Tanaka, M. Yano, T. Kushida, H. Sato, K. Sugimoto, T. Kato and J. Takeya, J. Am. Chem. Soc., 2020, 142, 14974–14984 CrossRef CAS PubMed.
- W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113–6127 RSC.
- M. Mas-Torrent and C. Rovira, J. Mater. Chem., 2006, 16, 433–436 RSC.
- F. G. Brunetti, J. L. López, C. Atienza and N. Martín, J. Mater. Chem., 2012, 22, 4188–4205 RSC.
- M. Bendikov and F. Wudl, Chem. Rev., 2004, 104, 4891–4945 CrossRef CAS PubMed.
- T. Yamamoto and K. Takimiya, J. Am. Chem. Soc., 2007, 129, 2224–2225 CrossRef CAS PubMed.
- H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS PubMed.
- G. Saito, T. Enoki, K. Toriumi and H. Inokuchi, Solid State Commun., 1982, 42, 557–560 CrossRef CAS.
- G. Saito and Y. Yukihiro, Bull. Chem. Soc. Jpn., 2007, 80, 1–137 CrossRef CAS.
- O. A. Melville, B. H. Lessard and T. P. Bender, ACS Appl. Mater. Interfaces, 2015, 7, 13105–13118 CrossRef CAS PubMed.
- T. Mori, Bull. Chem. Soc. Jpn., 2016, 89, 973–986 CrossRef CAS.
- H. Hasegawa, T. Naito, T. Inabe, T. Akutagawa and T. Nakamura, J. Mater. Chem., 1998, 8, 1567–1570 RSC.
- T. Inabe and H. Tajima, Chem. Rev., 2004, 104, 5503–5534 CrossRef CAS PubMed.
- C. Gaul, S. Hutsch, M. Schwarze, K. S. Schellhammer, F. Bussolotti, S. Kera, G. Cuniberti, K. Leo and F. Ortmann, Nat. Mater., 2018, 17, 439–444 CrossRef CAS PubMed.
- Y. Zhang, I. Murtaza and H. Meng, J. Mater. Chem. C, 2018, 6, 3514–3537 RSC.
- C. F. J. Faul, Acc. Chem. Res., 2014, 47, 3428–3438 CrossRef CAS PubMed.
- D. Wu, R. Liu, W. Pisula, X. Feng and K. Műllen, Angew. Chem., Int. Ed., 2011, 50, 2791–2794 CrossRef CAS PubMed.
- Y. Haketa and H. Maeda, Chem. Commun., 2017, 53, 2894–2909 RSC.
- S. Sugiura, T. Kubo, Y. Haketa, Y. Hori, Y. Shigeta, H. Sakai, T. Hasobe and H. Maeda, J. Am. Chem. Soc., 2023, 145, 8122–8129 CrossRef CAS PubMed.
- H. Tanaka, Y. Kobayashi, K. Furukawa, Y. Okayasu, S. Akine, N. Yasuda and H. Maeda, J. Am. Chem. Soc., 2022, 144, 21710–21718 CrossRef CAS PubMed.
- T. Akutagawa, T. Hasegawa, T. Nakamura, S. Takeda, T. Inabe, K. Sugiura, Y. Sakata and A. E. Underhill, Chem. – Eur. J., 2001, 7, 4902–4912 CrossRef CAS PubMed.
- T. Akutagawa, K. Shitagami, S. Nishihara, S. Takeda, T. Hasegawa, T. Nakamura, Y. Hosokoshi, K. Inoue, S. Ikeuchi, Y. Miyazaki and K. Saito, J. Am. Chem. Soc., 2005, 127, 4397–4402 CrossRef CAS PubMed.
- K. Sambe, N. Hoshino, T. Takeda, T. Nakamura and T. Akutagawa, J. Phys. Chem. C, 2020, 124, 13560–13571 CrossRef CAS.
- K. Sambe, N. Hoshino, T. Takeda, T. Nakamura and T. Akutagawa, Cryst. Growth Des., 2020, 20, 3625–3634 CrossRef CAS.
- K. Sambe, N. Hoshino, T. Takeda, T. Nakamura and T. Akutagawa, Cryst. Growth Des., 2021, 21, 5928–5942 CrossRef CAS.
- H. E. Katz, A. J. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y.-Y. Lin and A. A. Dodabalapur, Nature, 2000, 404, 478–481 CrossRef CAS.
- J. Penneau, B. J. Stallman, P. H. Kasai and L. L. Miller, Chem. Mater., 1991, 3, 791–796 CrossRef CAS.
- L. Zhao, D. Zhang, Y. Zhu, S. Peng, H. Meng and W. Huang, J. Mater. Chem. C, 2017, 5, 848–853 RSC.
- S.-L. Suraru and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 7428–7448 CrossRef CAS PubMed.
- N. Sakai, J. Mareda, E. Vauthey and S. Matile, Chem. Commun., 2010, 46, 4225–4237 RSC.
- M. Cui, R. Murase, Y. Shen, T. Sato, S. Koyama, K. Uchida, T. Tanabe, S. Takaishi, M. Yamashita and H. Iguchi, Chem. Sci., 2022, 13, 4902–4908 RSC.
- T. Tanabe, T. Sato, K. Fuku, S. Takaishi and H. Iguchi, ChemPlusChem, 2023, e202300140 CrossRef CAS PubMed.
- T. Ono, Y. Tsukiyama, S. Hatanaka, Y. Sakatsume, T. Ogoshi and Y. Hisaeda, J. Mater. Chem. C, 2019, 7, 9726–9734 RSC.
- Y. Takashima, V. M. Martínez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nat. Commun., 2011, 2, 168 CrossRef PubMed.
- A. Mizuno, Y. Shuku, M. M. Matsushita, M. Tsuchiizu, Y. Hara, N. Wada, Y. Shimizu and K. Awaga, Phys. Rev. Lett., 2017, 119, 057201 CrossRef.
- A. Mizuno, Y. Shuku, R. Suizu, M. Tsuchiizu and K. Awaga, CrystEngCom., 2021, 23, 5053–5059 RSC.
- D. Shukla, S. Nelson, D. C. Freeman, M. Rajeswaran, W. G. Ahearn, D. M. Meyer and J. T. Carey, Chem. Mater., 2008, 24, 7486–7491 CrossRef.
- M.-M. Ling, P. E. M. Gomez, M. Koenemann, J. Locklin and Z. Bao, Adv. Mater., 2007, 9, 1123 CrossRef.
- A. Nowak-Król and F. Würthner, Org. Chem. Front., 2019, 6, 1272–1318 RSC.
- O. Ogi, V. Stepanenko, K. Sugiyasu, M. Takeuchi and F. Würthner, J. Am. Chem. Soc., 2015, 137, 3300–3307 CrossRef PubMed.
- G. S. Perez, S. Dasgupta, W. Żuraw, R. F. Pineda, K. Wojciechowski, L. K. Jagadamma, I. Samuel and N. Robertson, J. Mater. Chem. A, 2022, 10, 11046–11053 RSC.
- R. S. Wilson-Kovacs, X. Fang, M. J. L. Hagemann, H. E. Symons and C. F. J. Faul, Chem. – Eur. J., 2022, 28, e202103443 CrossRef CAS PubMed.
- A. Kawasaki, T. Takeda, N. Hoshino, W. Matsuda, S. Seki and T. Akutagawa, J. Phys. Chem. C, 2019, 123, 15451–15457 CrossRef CAS.
- A. Kawasaki, T. Takeda, N. Hoshino, W. Matsuda, S. Seki and T. Akutagawa, Cryst. Grow. Des., 2020, 20, 1276–1284 CrossRef CAS.
- A. Kawasaki, T. Takeda, N. Hoshino, W. Matsuda, S. Seki, G. K. H. Shimizu and T. Akutagawa, ACS Appl. Mater. Interfaces, 2023, 15, 1661–1674 CrossRef CAS.
- H. Abe, A. Kawasaki, T. Takeda, N. Hoshino, W. Matsuda, S. Seki and T. Akutagawa, ACS Appl. Mater. Interfaces, 2020, 12, 37391–37399 CrossRef CAS PubMed.
- H. Abe, A. Kawasaki, T. Takeda, N. Hoshino, W. Matsuda, S. Seki and T. Akutagawa, J. Am. Chem. Soc., 2021, 143, 1046–1060 CrossRef CAS PubMed.
- R. Ide, A. Kawasaki, T. Takeda, S. Dekura, N. Hoshino, W. Matsuda, S. Seki and T. Akutagawa, J. Phys. Chem. C, 2023, 127, 16709–16720 CrossRef CAS.
- R. O. Macron, J. G. dos Santos, K. M. Figueiredo and S. Brochsztain, Langmuir, 2006, 22, 1680–1687 CrossRef PubMed.
- H. Ke, X.-M. Zhu, S.-M. Xie, P.-X. Ming and J. Z. Liao, Inorg. Chem. Front., 2022, 9, 2568–2574 RSC.
- X.-M. Zhu, Y. Jiang, S.-M. Xie, P.-X. Ming, C.-H. Zhang, H. Ke and J.-Z. Liao, Dyes Pigm., 2022, 207, 110747 CrossRef CAS.
- K. Ke, J.-Z. Liao and C.-Z. Lu, Acta Cryst, 2019, C75, 1128–1133 Search PubMed.
- J.-Z. Liao, J.-F. Chang, L. Meng, H.-L. Zhang, S.-S. Wanga and C. Z. Lu, Chem. Commun., 2017, 53, 9701–9704 RSC.
- J.-Z. Liao, S.-S. Wang, X.-Y. Wu, R. Yu, C.-Z. Lu and X.-L. Chen, Dalton Trans., 2018, 47, 1027–1031 RSC.
- Crystal Structure: Single Crystal Structure Analysis Software. Ver. 4.30, 2018. Rigaku Corporation and Molecular Structure Corporation.
- G. M. Sheldrick, SHELX2014 Programs for Crystal Structure Analysis; Universitat Göttingen: Göttingen, Germany, 2014.
- T. Mori, A. Kobayashi, Y. Sakaki, H. Kobayashi, G. Saito and H. Inokuchi, Bull. Chem. Soc. Jpn., 1984, 57, 627–633 CrossRef CAS.
- G. Greeuw and J. F. Verwey, J. Appl. Phys., 1984, 56, 2218–2224 CrossRef CAS.
- N. Yoshinari, S. Yamashita, Y. Fukada, Y. Nakazawa and T. Konno, Chem. Sci., 2018, 10, 587–593 RSC.
- S. Seki, A. Saeki, T. Sakurai and D. Sakamaki, Phys. Chem. Chem. Phys., 2014, 16, 11093–11113 RSC.
- Y. Tsutsui, G. Schweicher, B. Chattopadhyay, T. Sakurai, J.-B. Arlin, C. Ruzié, A. Aliev, A. Ciesielski, S. Colella, A. R. Kennedy, V. Lemaur, Y. Olivier, R. Hadji, L. Sanguinet, F. Castet, D. Beljonne, J. Cornil, P. Samorì, S. Seki and Y. H. Geerts, Adv. Matter., 2016, 28, 7106–7114 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2308337–2308340. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3tc04231f |
| This journal is © The Royal Society of Chemistry 2024 |