
Methyl red based metal–organic frameworks for the selective and tuneable sensing of ammonia gas†
Craig N. G.
Weir
,
Rodney J.
Blanchard
,
Amanda P.
Parsons
,
Gauthaman
Kalarikkandy
and
Michael J.
Katz
 *
*
Department of Chemistry, Memorial University of Newfoundland, St. John's, Newfoundland and Labrador A1B 3X7, Canada. E-mail: mkatz@mun.ca
First published on 16th November 2023
Abstract
This work examines a family of MOFs that are colourimetric sensors to acid and base vapours. With the strongest colour change associated with strong base vapours such as ammonia, this material can act as an ammonia sensor for applications in health and safety. A methyl red based linker is synthesized and incorporated into a UiO-66 based metal–organic framework (MOF). Using terephthalic acid and the methyl red based linker, four different methyl red MOF compositions were synthesized and characterized. The degree of incorporation ranged from 10–100%. The as-synthesized MOF was an red-orange colour with the colour depending on the amount of methyl red based linker incorporated. Exposure of the MOF to acid vapour produced an irreversible, with respect to time, darker red colour. Exposing the MOF to ammonia vapour produced a stark and reversible, with respect to time, colour change to yellow. No other base vapours were able to elicit this response making this MOF selective for ammonia vapour. The sensitivity of the MOF to ammonia is proportional to the degree of methyl red linker incorporation. We further incorporate the MOFs into a mixed matrix membrane using polyvinylidene fluoride as the polymer host. As with the parent MOFs, the membranes demonstrated a strong colourimetric response to ammonia. These materials have potential applications in future shipping vessels that will utilize ammonia as a fuel source.
Introduction
With the aim to achieve net-zero emissions by 2050,1,2 ammonia has become a promising future fuel in shipping vessels.3 If ammonia is produced from renewable sources (green-ammonia),4,5 then it becomes a zero-carbon fuel. Furthermore, under ideal combustion conditions, ammonia is converted to water vapour and nitrogen gas.3 This makes ammonia an ideal fuel source. Given ammonia's potential, it is no surprise that the Danish funding agency has established a consortium to develop the first ammonia-fuelled engines by 2024.Despite the advantages of ammonia, one of the primary challenges with ammonia as a fuel source is the health and safety concerns associated with ammonia exposure. Long term exposure to ammonia gas leads to desensitization making it easy for individuals to fail to identify leaks. Meanwhile, exposure to ammonia can lead to irritation and damage of the respiratory system as well as chemical burns on the skin and eyes. Environmentally, release of ammonia gas can result in adverse effect to oceans and the marine aquaculture. With that in mind, it is important to develop materials capable of selectively sensing ammonia gas at various partial pressures.
Metal–organic frameworks (MOFs) are a class of porous materials made from inorganic nodes (e.g., metal cations, pairs of metal cations, metal–oxo/hydrox/aquo-based clusters, etc.) and organic bridging ligands (linkers) possessing two or more functional groups that are designed to bridge nodes to one another (e.g., carboxylates, pyridines, imidazoles). Judicious combination of nodes and linker allow researchers to tailor the pore size and pore aperture of the 3D structure.6–8 To impart further properties to the MOF, additional functional groups (e.g., amines, hydroxides, catalysts, etc.) can be covalently attached to the linker(s) such that the pore is decorated by these groups.9–11 Given the multitude of nodes and linkers, MOFs have been featured in gas and vapour storage,12–15 catalysis,13,16,17 drug delivery,18–20 and chemical sensing.21–23
With our ongoing interests in developing MOFs for environmental applications such as storage,24 separation,25,26 and reactivity of gases,27 the present work focuses on sensing ammonia gas. As illustrated in Fig. 1, if a methyl red based linker (MRL)28–30 can be incorporated into a MOF, then the solid sorbent would respond to the pKa of a guest gas. In its red hydrazone/quinone form, methyl red could react with a basic gas such as ammonia. This would turn the red, acidic, form of the indicator into the azo, yellow, form of the indicator. Thus, for an ammonia sensing MOF, the MOF has to be acidic enough to stabilize the hydrazone/quinone form. With that in mind, this work incorporates MRL into UiO-66, Zr6μ3-O4(μ3-OH)4(BDC)6 (BDC: benzenedicarboxylate) by using a mixed linker approach with both BDC and MRL. Given UiO-66 is synthesized under highly acidic conditions31 and the protons on the μ3-OH have been shown to be acidic,32 this should be sufficient to result in the formation of the hydrazone/quinone form of the MOF. The present work demonstrates the sensing capabilities of MRL containing MOFs with particular focus on ammonia sensing. Given the ease in which the ratio of linkers can be synthetically tuned, the detection limit of the sensor can be optimized via the degree of MRL vs. BDC incorporation; lower methyl red incorporation is expected to result in lower detection limits. With focus on applications, we further demonstrate that these MOFs can be incorporated into mixed matrix membranes.
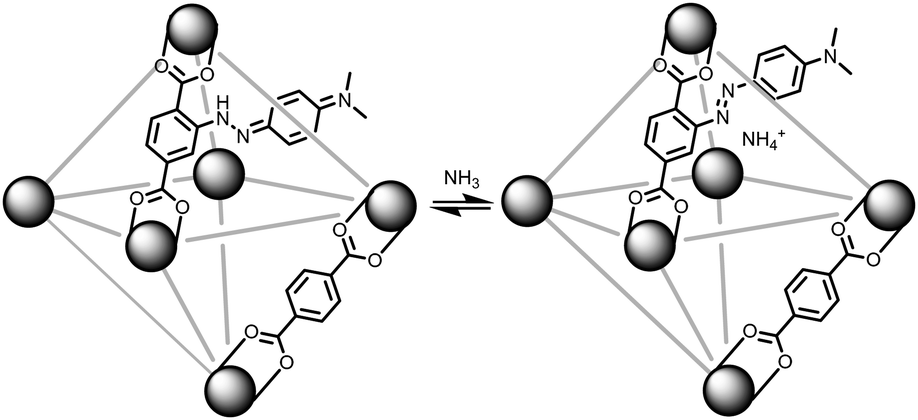 | ||
| Fig. 1 (left) MR-MOF formed by the reaction of H2MRL, H2BDC, and ZrCl4. The MRL linker exists in the hydrazone/quinone (red) form. (right) When MR-MOF is exposed to ammonia gas, the MRL linker is converted to the azo (yellow) form. | ||
Experimental
All chemicals used were purchased from commercial sources and no further purification was performed. PVDF was supplied free of charge by Arkema, under the name Kynar 761.Gas adsorption analyses were carried out on a Micromeritics Tristar II 3020 instrument. Samples were maintained at 77 K using a liquid nitrogen dewar and an isothermal jacket to ensure a constant temperature over the measurement time. Data analysis was carried out using the Micromeritics MicroActive software package (version 4.02). Prior to gas adsorption analysis, samples were activated on a Micromeritics SmartVac Prep instrument. Initially, samples were heated at 150 °C while reducing the pressure (5 mmHg min−1). The samples were held at this temperature for 30 min. Afterwards, samples were heated at 150 °C for 600 h. Samples were subsequently back filled with nitrogen gas prior to analysis.
Vapour exposure was carried out as described previously.33 Briefly, the MOF (powder or film) was placed in a vial or Petri dish. A filter paper was placed in the vial/dish with a few drops of analyte (e.g., hydrochloric acid solution, ammonium hydroxide solution) to generate a vapour pressure of the analyte (i.e., hydrochloric acid or ammonia gas). The filter paper did not contact the powder/film thereby ensuring that the vapour and not the liquid was responsible for the chemical reactivity.
UV-Visible absorption was carried out on an Agilent Cary 100 UV-Vis spectrophotometer instrument equipped with an integrating sphere.
Infrared spectroscopy was carried out on a Bruker INVENIO-R infrared spectrometer equipped with a Transit Platinum ATR sample holder with a DTGS detector.
Solution-phase 1H-NMR was carried out on a Bruker AVANCE 500 MHz spectrometer equipped with an inverse probe. Solid-state 13C data were collected on a Bruker Avance II 600 spectrometer, equipped with a SB Bruker 3.2 mm MAS triple-tuned probe operating at 600.29 MHz for 1H and 150.96 MHz for 13C. Chemical shifts were referenced to tetramethylsilane (TMS) using adamantane as an intermediate standard for 13C. The samples were spun at 20 kHz. Cross-polarization (CPMAS) spectra were collected with a Hartmann–Hahn match at 62.5 kHz and 100 kHz 1H decoupling, with a contact time of 2 ms, a recycle delay of 3 s, and 2k scans.
Synthesis of 2-[2-[4-(dimethylamino)phenyl]diazenyl]-1,4-benzenedicarboxylic acid (H2MRL)
The synthesis of H2MRL was based off previous work with minor adjustments.34 To a 25 mL round bottom flask was added 0.200 g (1.10 mmol) of 2-aminoterephthalic acid, 2.00 mL of deionized water, and 0.2 mL of 12 M HCl. The flask was cooled below 5 °C using an ice bath. Once cooled, 0.220 g (3.19 mmol) of NaNO2 dissolved in 0.2 mL of water was added and the solution was stirred for 5 min. Afterwards, 0.436 mL (3.44 mmol) of dimethylaniline was added to the solution. The addition of dimethylaniline caused an immediate colour change from red/orange to a dark red/brown colour. The solution was stirred for an additional 15 min before 0.420 g (5.12 mmol) of sodium acetate dissolved in 0.2 mL of water was added. The solution was stirred for 1 h before being placed into a fridge overnight. The following day, the dark red precipitate was collected by suction filtration and washed 3 times with 15 mL of hexanes to remove leftover dimethylaniline. The solid was further washed 3 times with 15 mL of methanol to remove leftover sodium acetate/acetic acid. A total of 0.300 g (0.958 mmoles), an 86% yield, was isolated. The 1H-NMR was collected on a Bruker Avance III 300 MHz spectrometer in DMSO-d6: δ/ppm 3.08 (s, 6H), 6.85 (d, J = 7.7 Hz, 2H), 7.72 (d, J = 7.9 Hz, 1H), 7.78 (d, J = 7.7 Hz, 2H), 7.95 (d, J = 7.9 Hz, 1H), 8.11 (s, 1H).Synthesis of MR-MOF
The synthesis of MR-MOF is based off of a previously published synthesis for UiO-66.31 Different degrees of MRL were incorporated by adjusting the H2MRL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2BDC ratio. Table 1 indicates the masses used in this work. For clarity, we include the synthesis of MR-MOF with 25% H2MRL and 75% H2BDC.
:H2BDC ratio. Table 1 indicates the masses used in this work. For clarity, we include the synthesis of MR-MOF with 25% H2MRL and 75% H2BDC.
| MR-MOF | H2BDC | H2MRL |
|---|---|---|
| a The number represents the percent of H2MRL vs. total linker used during the synthesis of MR-MOF. | ||
| MR-MOF-25a | 0.0934 g (0.563 mmol) | 0.0587 g (0.187 mmol) |
| MR-MOF-50a | 0.0623 g (0.375 mmol) | 0.1174 g (0.375 mmol) |
| MR-MOF-75a | 0.0311 g (0.188 mmol) | 0.1761 g (0.5625 mmol) |
| MR-MOF-100a | — | 0.2348 g (0.75 mmol) |
In a 20 mL Durex jar, 0.0587 g (0.187 mmol) of H2MRL, 0.0934 g (0.563 mmol) of terephthalic acid (H2BDC) and 0.123 g (0.528 mmol) of ZrCl4 were combined with 15 mL of N,N-dimethylformamide (DMF) and 1.00 mL of 12 M HCl. The contents of the jar were sonicated for 20 min before being placed in an 80 °C oven overnight. The following day, the contents of the Durex jar were cooled down, centrifuged (5000 rpm, 5 min) and the solution decanted. The remaining dark red solid was suspended in 15 mL of DMF and subsequently centrifuged (5000 rpm, 5 min). The process was repeated a total of three times with DMF and subsequently a minimum of three times with methanol, or until the filtrate was colourless. Afterwards, the solid was allowed to dry before further use. To determine the degree of linker incorporation, the MOF was dissolved with 1–2 drops of sulfuric acid followed by the addition of DMSO-d6, and then the 1H NMR measured on a Bruker Avance III 300 MHz NMR spectrometer using a solvent suppression routine to reduce the effect of the sulfuric acid on the spectra.
Synthesis of mixed-matrix membranes of MR-MOF in PVDF (MR-MMM)
The synthesis of MR-MMM was based on our previous work.25MR-MMM-25 was synthesized by first mixing 20 mg of MR-MOF-25 in a 20 mL vial with 2 mL of DMF overnight. In a second 20 mL vial, 0.100 g of polyvinylidene fluoride (PVDF) was stirred in 2 mL of DMF overnight. The following day, both mixtures were combined and stirred for 3 hours before being transferred to a circular mould (5.08 cm diameter, 0.24 mm depth) and placed into an 80 °C oven overnight. The following day the mould was removed from the oven and the dark red film was removed from the mould.
MR-MMM-50, MR-MMM-75, and MR-MMM-100 were synthesized using the same procedure as MR-MMM-25 but using 10 mg of MR-MOF-50 and MR-MOF-75, or 1 mg of MR-MOF-100. The lower degree of incorporation was due to the larger degree of MRL incorporation (Table 2).
| Ratio of BDC and MRL | BET surface area (m2 g−1) | |||
|---|---|---|---|---|
| MR-MOF | Added during synthesis | Determined by NMRa | As synthesized | After five cycles of ammonia exposurec |
| a Doesn’t account for any changes in the number of defects. b Represents the error. c Exposures were sufficient to convert 50 mg of MR-MOF yellow; this represents a worse-case scenario. | ||||
| MR-MOF-25 | 75:25 |
90:10(3b) |
1270 | 750 |
| MR-MOF-50 | 50:50 |
82:18(3b) |
820 | 620 |
| MR-MOF-75 | 25:75 |
65:35(3b) |
810 | 360 |
| MR-MOF-100 | 0:100 |
— | 530 | 30 |
Results and discussion
Characterization of MR-MOF
The methyl red containing linker (H2MRL) was synthesized via the diazotization reaction of 2-aminoterephthalic acid followed by coupling with dimethylaniline. With the 1H NMR spectrum confirming the synthesis of H2MRL (Fig. S1 in the ESI†), we incorporated different amounts of H2MRL and H2BDC with ZrCl4 to form four MRL-containing UiO-66 derivatives (MR-MOF-25, MR-MOF-50, MR-MOF-75, and MR-MOF-100; the number represents the molar% MRL used in the synthesis). Independent of the degree of MRL incorporation, MR-MOF had a red-orange colour that appeared to be proportional to the amount of MRL incorporated (Fig. 3 and 4).MR-MOF was characterized as illustrated in Fig. 2. The powder X-ray diffractograms (PXRD) of MR-MOF are consistent with the fcu topology associated with UiO-66 (Fig. 2a). The PXRD of MR-MOF-100 appears notably broader than the other derivatives. This suggests that there is likely a steric limit associated with MRL incorporation that results in a larger mosaicity or smaller particle size.
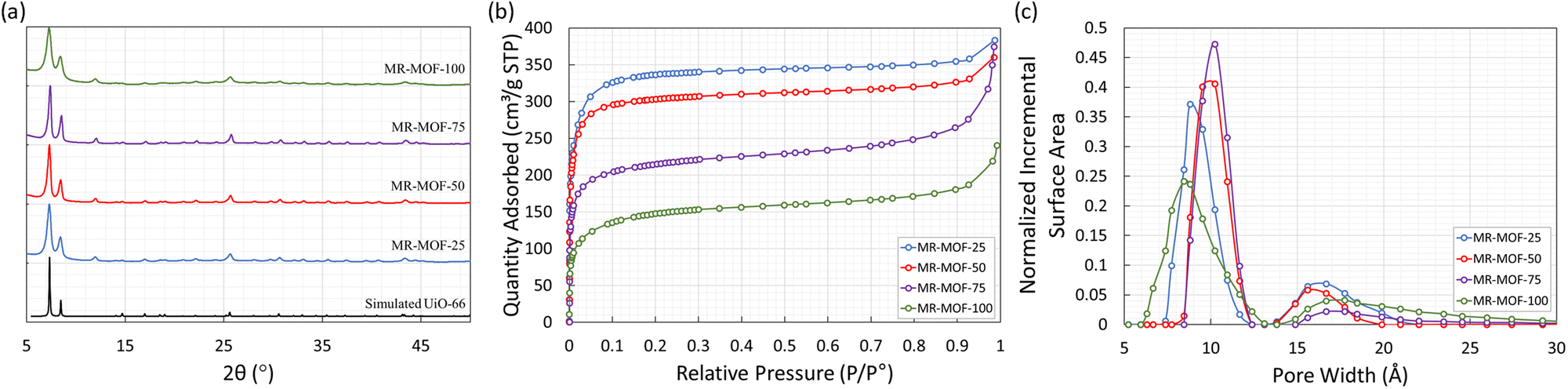 | ||
| Fig. 2 (a) Powder X-ray diffractograms of MR-MOF and the simulated diffractogram for UiO-66. (b) Nitrogen gas adsorption isotherms, measured at 77 K for MR-MOF. (c) BET surface area normalized pore size distributions for MR-MOF determined using the Tarazona NLDFT (Esf = 30.0 K) model. The standard deviations for the fits are 3.41091 (MR-MOF-25), 4.18722 (MR-MOF-50), 3.23237 (MR-MOF-75), and 0.63011 cm3 g−1 (MR-MOF-100). | ||
The nitrogen gas adsorption isotherms were measured at 77 K and are shown in Fig. 2b with the nitrogen accessible Brunauer–Emmett–Teller surface areas (BET-SA) summarized in Table 2. For MR-MOF, the BET-SA decreases with increasing MRL incorporation. MR-MOF-25 has the highest BET-SA of 1270 m2 g−1 with MR-MOF-100 being as low as 530 m2 g−1. The BET-SA for all four MR-MOF derivatives were lower than the ca. 1500 m2 g−1 associated with UiO-66 synthesized via this procedure.35 However, these BET surface areas are consistent with derivatives of UiO-66.31,35 To further explore MR-MOF, the NLDFT pore size distributions (PSD) were calculated using the Tarazona model (Fig. 2c). For MR-MOF-25–MR-MOF-75, the PSDs are more similar than different. As more MRL is incorporated, the 10 Å pore shifts to lower pore width; this is consistent with the sterics associated with the MRL linker, which decreases the overall pore size. The pores centered at 16 Å are associated with defect-based pores.36 Incorporating additional MRL doesn’t drastically affect the defects in the MOF. MR-MOF-100 shows the biggest difference in the PSDs with the PSDs being broader than the other derivatives. This is consistent with the breadth differences in the PXRD of MR-MOF-100 (Fig. 2a).
To further characterize the materials, the four MOFs were dissolved in sulfuric acid, diluted with DMSO-d6 and their 1H-NMR spectra measured to determine the ratio of BDC to MRL linker (Table 2). As shown in Table 2, the degree of MRL incorporation is consistently lower than the mole ratio used in the synthesis. This is likely due to the steric profile of the MRL linker limiting the degree of incorporation. This is, once again, consistent with the broader PXRD (Fig. 2a) and the broader PSDs (Fig. 2c) of MR-MOF-100 relative to the other derivatives. This also demonstrates the importance of the BDC linker as a structural supporting group in MR-MOF. The synthetic success of MR-MOF-100 also demonstrates that higher MRL incorporations should be possible, but may not be necessary (see below), by adjusting the H2MRL:H2BDC ratio.
Acid & base sensing of MR-MOF
With MR-MOF characterized, we turned our attention to its sensing capabilities (Fig. 3). Initially, we explored the colourimetric response of MR-MOF-25 to both acid (hydrochloric acid) and base (ammonia) vapour; a similar response was observed for all MR-MOF variants. The parent MR-MOF-25 is red-orange in colour. When exposed to hydrochloric acid vapours, the MOF turned a dark red colour. Open to air, with no additional acid vapours and even at elevated temperatures, the colour persisted indefinitely. When the parent MR-MOF-25 is exposed to ammonia vapours, the MOF becomes yellow. When exposed to an ammonia-free environment, MR-MOF-25 returns to the red-orange colour. The colourimetric response was sustained over five acid-to-base cycles with no obvious change in colourimetric response. This indicates that the MOF can sense both acids and bases in its parent form, which illustrates that the MRL linker exists in both azo and hydrazone/quinone forms (Fig. 1) inside the MOF. To better understand the response of MR-MOF to acid/base vapours, the IR and 13C-MAS spectroscopy were collected for MR-MOF-100 (Fig. S4 and S5, ESI†). | ||
| Fig. 3 Colourimetric response of MR-MOF-25 upon exposure to hydrochloric acid vapours, ammonia vapours, and after 1 and 24 h exposure to air (post ammonia exposure). | ||
The 13C-MAS spectra is illustrated in Fig. S5 (ESI†). The methyl region of MRL linker contains resonances at 25–50 ppm while the remaining MRL linker contains resonances at 100–200 ppm. The ratio of these two regions is approximately 2:14, consistent with the ratio of the carbon atoms found in the linker. The azo form of MR-MOF-100 is expected to contain one methyl resonance due to free rotation, albeit slow, about the Ar–N bond. For the hydrazone/quinone form, where there is no free rotation about the Ar![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, we expect to see two methyl resonances. As illustrated in Fig. S5b–f (ESI†), there are three peaks observed in the methyl region for the as-synthesized and acid-exposed form of MR-MOF-100. However, the relative intensities of these resonances are different. The 13C MAS for ammonia-exposed MR-MOF-100 (Fig. S5a and d, ESI†) contained one dominant methyl resonance (the most downfield resonance), consistent with the azo form of the linker. Returning to the acid-exposed MR-MOF-100, relative to the as-synthesized MR-MOF-100, the integration of the two upfield hydrazone/quinone resonances are higher than the azo form. This illustrates that MR-MOF is synthesized as a mixture of the two forms, and that the acid-exposed MR-MOF-100 contains more of the hydrazone/quinone form.
N bond, we expect to see two methyl resonances. As illustrated in Fig. S5b–f (ESI†), there are three peaks observed in the methyl region for the as-synthesized and acid-exposed form of MR-MOF-100. However, the relative intensities of these resonances are different. The 13C MAS for ammonia-exposed MR-MOF-100 (Fig. S5a and d, ESI†) contained one dominant methyl resonance (the most downfield resonance), consistent with the azo form of the linker. Returning to the acid-exposed MR-MOF-100, relative to the as-synthesized MR-MOF-100, the integration of the two upfield hydrazone/quinone resonances are higher than the azo form. This illustrates that MR-MOF is synthesized as a mixture of the two forms, and that the acid-exposed MR-MOF-100 contains more of the hydrazone/quinone form.
The IR spectra of the as-synthesized MR-MOF-100 and the base-exposed MR-MOF-100 look more similar than different, while the IR spectrum of the acid-exposed sample illustrated notable differences at 1400 and 1250 cm−1 (Fig. S4, ESI†). This indicates that the MRL in MR-MOF exists primarily in the azo form. This indicates that a sub-stoichiometric amount of ammonia (relative to the total MRL incorporation) is necessary to illicit a colourimetric response; this makes MR-MOF more sensitive to ammonia than initially postulated.
The IR spectrum of MR-MOF-100 after exposure to acid vapour persisted even after heating at 80 °C for 4 hours. We hypothesize that the hydrazone/quinone form either can’t release hydrochloric acid due to how the MR-MOF balances the charges of MRL, or the formation of hydrochloric acid vapours and the subsequent formation of the azo form of the linker is thermodynamically disfavoured relative to the hydrazone/quinone form of the linker. While we propose the latter is more likely, further studies are necessary to rule out the former.
While both acid and base are sensed, the base response is more drastic, and reversible. Furthermore, given the persistent dark red colour change of the acid-exposed MR-MOF-25, a starker change exists if MR-MOF is first exposed to acid. Attempts to elicit a similar colourimetric response from weaker acids/bases was unsuccessful. We observed that only strong acids and strong bases were able to produce a notable colourimetric response. It is worth noting that the parent linker H2MRL showed no response to base vapours. This illustrates the importance of the MOF for the sensing capabilities.
Ammonia sensing of MR-MOF
Given the stark colour change associated with ammonia exposure, we focused on the ammonia sensing capabilities of MR-MOF. To explore the sensitivity of MR-MOF, we placed all four variants equidistant from a filter paper containing ammonium hydroxide. The four MOFs were monitored as a function of time (Fig. 4). All four MR-MOF samples had a similar red-orange colour prior to ammonia vapour exposure, with the higher MRL loadings exhibiting a darker colour. Within a matter of seconds of ammonia exposure, MR-MOF-25 exhibits a change to a yellow colour. After approximately 30 s, MR-MOF-50 also changes to yellow. For MR-MOF-75, a small amount of red colour persists even after 120 s. For MR-MOF-100, the colour change is only slightly noticeable after 60 s with the MOF unable to completely change colour even after 300 s. To explore the role of water vapour on the colour change, we repeated the experiment by flowing the ammonia vapour through a calcium oxide column to remove the water. The colourimetric response to ammonia was still observed. This indicates that water is not necessary for the transformation from the hydrazone/quinone to the azo form of MR-MOF. | ||
| Fig. 4 Time-lapse of MR-MOF exposed to ammonia vapour. | ||
The inverse relationship between the degree of MRL incorporation and the sensitivity of the MOF is expected; the less MRL present, the fewer ammonia molecules are necessary to deprotonate all the red hydrazone/quinone form of MRL to the azo yellow form. These results indicate that MR-MOF can be tuned to respond to a specific partial pressure (mixing ratio) of ammonia. This allows MR-MOF to be optimized for different applications while still being porous to other gases.
To explore the stability of MR-MOF to ammonia vapour, we exposed the MOF to five ammonia adsorption–desorption cycles. The amount of ammonia vapour used was sufficient to switch 50 mg of MR-MOF from red-orange to yellow. This likely represents a worse-case scenario due to the large amount of ammonium hydroxide needed to accomplish this task. As shown in Table 2 and Fig. S2 and S3 (ESI†), the surface areas for MR-MOF-25–MR-MOF-75 decrease but remain porous despite the aggressive treatment. For MR-MOF-100, the BET-SA can be considered 0 m2 g−1. This data further suggests that the BDC linker in MR-MOF-25–MR-MOF-75 acts as a structural supporting group. In the absence of this support, a high partial pressure of ammonia vapour causes the MOF to collapse.
Ammonia sensing in mixed matrix membranes of MR-MOF
To further explore the utility of MR-MOF, we incorporated MR-MOF into mixed matrix membranes (MMMs) with polyvinylidene fluoride (PVDF). PVDF was chosen because of its overall thermal stability, which may be necessary around ammonia-based engines. To explore the composites (MR-MMM) we measured the reflectance of MR-MMM before and after ammonia vapour (Fig. 5). The MMMs were stable over six months without any evidence of a reduced colourimetric response.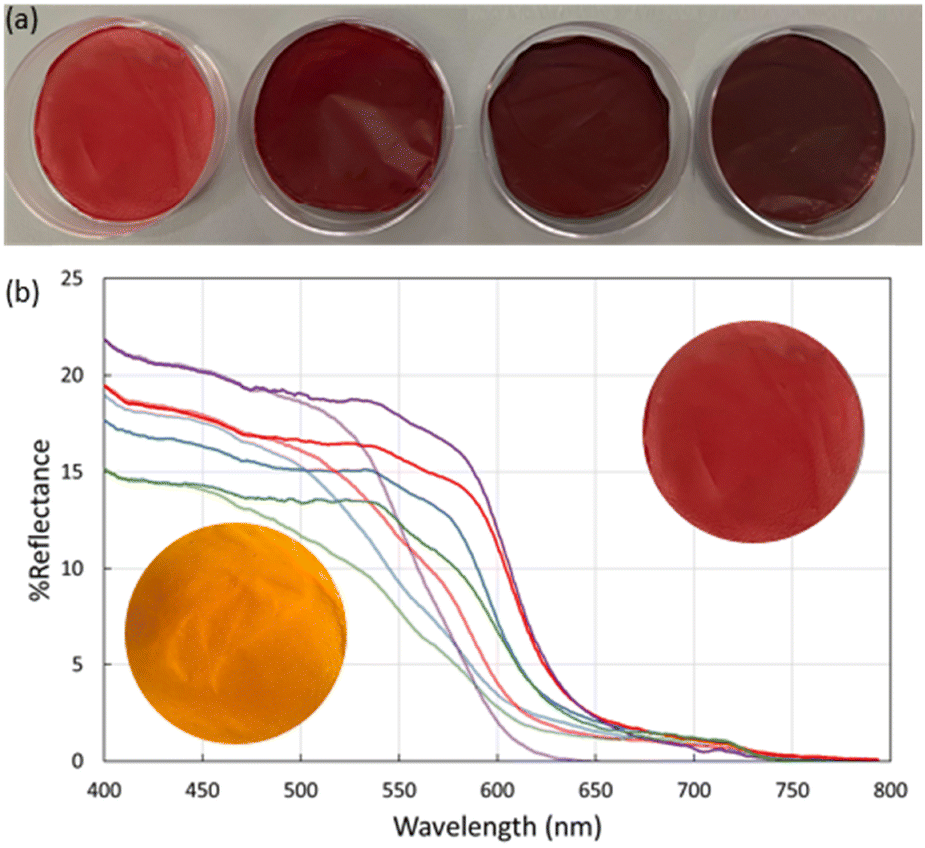 | ||
| Fig. 5 (a) MR-MMM samples of MR-MOF in a PVDF matrix. From left to right the MMMs contain MR-MOF-25 (20 mg), MR-MOF-50 (10 mg), MR-MOF-75 (10 mg), and MR-MOF-100 (1 mg) in 100 mg of PVDF. (b) Reflectance measurements of MR-MOF before (low-energy onset) and after (higher energy onset) ammonia exposure. Photographic inset shows MR-MMM-25 before and after ammonia exposure. MR-MMM-25 (blue), MR-MMM-50 (red), MR-MMM-75 (purple), and MR-MMM-100 (green). Traces are boxcar averaged. | ||
The as-synthesized MMMs are shown in Fig. 5a. MR-MMM-25 exhibits a bright red colour while the other samples are dark red and look more similar than different. Reflectance measurements (Fig. 5b) illustrate that all the parent MMMs show an absorption onset of ca. 650 nm. The maximum reflectance is related to the amount of MRL that is present in the sample. For example, MR-MMM-75 and MR-MMM-50 (Fig. 5b purple traces vs. blue traces) have the same w/w ratio of MOF in the MMM but MR-MMM-75 shows a higher reflectance. MR-MMM-25 (Fig. 5b blue trace) and MR-MMM-50 show similar reflectance due to a 50% decrease in MOF incorporation (w/w). Lastly, MR-MMM-100 only contains 1 mg of MOF and thus has the lowest maximum reflectance. This illustrates another degree of tunability for MR-MOF.
Upon exposure to ammonia vapours, the reflectance onset shifts to higher energy. This is consistent with the change in adsorption from red-orange to yellow for MR-MOF (Fig. 4). Of the four films explored in this work, the maximum reflectivity difference between the as-synthesized and ammonia-exposed MR-MMM occurs for MR-MMM-75. This illustrates that optimizing the ratio of MRL in the MOF and the amount of MR-MOF in MR-MMM is necessary to optimize the response characteristics of the material.
In comparison with MR-MOF, MR-MMM has a slower response time. This suggests that slow diffusion of ammonia through MR-MMM is occurring. The slower response kinetics of MR-MMM is beneficial in applications where a sustained leak needs to be detected over a periodic time. In principle, the response time can be adjusted by changing the polymer component of the MMM. Thus, the combination of the sensitivity of MR-MOF and the diffusion times of MR-MMM allow for a great deal of tuning for ammonia sensing.
Conclusions
Incorporating a linker based on methyl red into the UiO-66 framework allowed us to demonstrate that an ammonia sensing MOF can be easily synthesized and the colourimetric response tuned. With the upcoming utilization of ammonia as a fuel source, materials that can be implemented in quality control and safety are important. Given the wide range of colours and colour changes associated with MR-MOF and MR-MMM, we believe that the ideal ammonia response can be readily fashioned. For fast detection, low concentration detection, and recyclability, a low degree of MRL incorporation is ideal. For threshold detection, the acid-exposed MR-MOF, which starts as a dark red rather than a red-orange colour, exhibits a stronger colorimetric response and would be easier to see; this would also be ideal for instrumentation purposes. In areas where small leaks may be tolerated, a higher MRL incorporation and the acid-exposed form of the MOF would be ideal. Overall, the more sensitive material requires less MRL and start in the as-synthesized (red-orange) form, and the less sensitive material would require more MRL and start in the acid-exposed (red) form of the MOF. We are currently exploring the implementation of these materials into wearable sensors and sensors that can be incorporated around pipe fittings.Abbreviations
| H2BDC | Terephthalic acid |
| BET-SA | Brunauer–Emmett–Teller surface area |
| DMF | N,N-Dimethylformamide |
| DMSO-d6 | Hexadeuterodimethyl sulfoxide |
| H2MRL | 2-[2-[4-(Dimethylamino)phenyl]diazenyl]-1,4-benzenedicarboxylic acid |
| MOF | Metal–organic framework |
| MMM | Mixed matrix membrane |
| PVDF | Polyvinylidene fluoride |
| PSD | Pore size distribution |
| PXRD | Powder X-ray diffraction |
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Supervision and funding acquisition (MK), investigation (CW, AP), validation (RB, KG), visualization (all), writing – original draft (CW), writing – review & editing (all).Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. K. would like to acknowledge Natural Sciences and Engineering Research Council of Canada (NSERC) for funding this research through a Discovery Grant, a Department of National Defence (DND) supplement, and a Research Tools and Instruments (RTI) grant for the gas adsorption instrumentation utilized in this study. M. K. and C. W. would like to acknowledge Memorial University for a Seed grant. M. K. and G. K. would like to acknowledge MITACS for a Globalink Research Internship (GK). M. K. would also like to thank Dr Céline Schneider and the Memorial University CREAIT Network for their support with this work; Dr Schneider collected and interpreted the 13C-MAS spectra. M. K. would like to acknowledge the Canada Foundation for Innovation (CFI) for a John R. Evans Leaders Fund (JELF) with additional funding provided by the Department of Industry, Energy and Technology (IET) of the Government of Newfoundland. The CFI/IET funding supported the purchase of the Bruker INOVO-R spectrometer used in this work.Notes and references
- S. Fankhauser, S. M. Smith, M. Allen, K. Axelsson, T. Hale, C. Hepburn, J. M. Kendall, R. Khosla, J. Lezaun, E. Mitchell-Larson, M. Obersteiner, L. Rajamani, R. Rickaby, N. Seddon and T. Wetzer, Nat. Clim. Change, 2022, 12, 15–21 CrossRef.
- T. Sun, I. B. Ocko, E. Sturcken and S. P. Hamburg, Sci. Rep., 2021, 11, 22173 CrossRef CAS.
- D. Erdemir and I. Dincer, Int. J. Energy Res., 2021, 45, 4827–4834 CrossRef.
- N. Salmon and R. Bañares-Alcántara, Sustainable Energy Fuels, 2021, 5, 2814–2839 RSC.
- G. Li, Z. Ma, J. Zhao, J. Zhou, S. Peng, Y. Li and B. Wang, Clean Energy, 2023, 7, 116–131 CrossRef.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS.
- D. Zhao, D. J. Timmons, D. Yuan and H.-C. Zhou, Acc. Chem. Res., 2011, 44, 123–133 CrossRef CAS PubMed.
- A. Kirchon, L. Feng, H. F. Drake, E. A. Joseph and H.-C. Zhou, Chem. Soc. Rev., 2018, 47, 8611–8638 RSC.
- D. Kim, M. Kang, H. Ha, C. S. Hong and M. Kim, Coord. Chem. Rev., 2021, 438, 213892 CrossRef CAS.
- D. K. Yoo, B. N. Bhadra and S. H. Jhung, J. Hazard. Mater., 2021, 403, 123655 CrossRef CAS.
- S. Ali Akbar Razavi and A. Morsali, Coord. Chem. Rev., 2019, 399, 213023 CrossRef CAS.
- W. Fan, X. Zhang, Z. Kang, X. Liu and D. Sun, Coord. Chem. Rev., 2021, 443, 213968 CrossRef CAS.
- S. P. Shet, S. Shanmuga Priya, K. Sudhakar and M. Tahir, Int. J. Hydrogen Energy, 2021, 46, 11782–11803 CrossRef CAS.
- C. Petit, Curr. Opin. Chem. Eng., 2018, 20, 132–142 CrossRef.
- H. Lin, Y. Yang, Y.-C. Hsu, J. Zhang, C. Welton, I. Afolabi, M. Loo and H.-C. Zhou, Adv. Mater., 2023, 2209073 CrossRef.
- S. Ma, W. Han, W. Han, F. Dong and Z. Tang, J. Mater. Chem. A, 2023, 11, 3315–3363 RSC.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- S. Mallakpour, E. Nikkhoo and C. M. Hussain, Coord. Chem. Rev., 2022, 451, 214262 CrossRef CAS.
- J. Cao, X. Li and H. Tian, Curr. Med. Chem., 2020, 27, 5949–5969 CrossRef CAS PubMed.
- H. D. Lawson, S. P. Walton and C. Chan, ACS Appl. Mater. Interfaces, 2021, 13, 7004–7020 CrossRef CAS.
- B. Yan, J. Mater. Chem. C, 2019, 7, 8155–8175 RSC.
- S. Kamal, M. Khalid, M. S. Khan and M. Shahid, Coord. Chem. Rev., 2023, 474, 214859 CrossRef CAS.
- C. Jia, T. He and G.-M. Wang, Coord. Chem. Rev., 2023, 476, 214930 CrossRef CAS.
- M. C. Lawrence and M. J. Katz, J. Phys. Chem. C, 2022, 126, 1107–1114 CrossRef CAS.
- M. Basafa, A. P. Parsons, E. K. Berdichevsky, M. C. Lawrence and M. J. Katz, ACS Appl. Energy Mater., 2023, 6, 9170–9178 CrossRef CAS.
- B. J. Furlong and M. J. Katz, J. Am. Chem. Soc., 2017, 139, 13280–13283 CrossRef CAS PubMed.
- D. T. McGrath, M. D. Ryan, J. J. MacInnis, T. C. VandenBoer, C. J. Young and M. J. Katz, Chem. Sci., 2019, 10, 5576–5581 RSC.
- J. J. Gassensmith, H. Furukawa, R. A. Smaldone, R. S. Forgan, Y. Y. Botros, O. M. Yaghi and J. F. Stoddart, J. Am. Chem. Soc., 2011, 133, 15312–15315 CrossRef CAS.
- Y. Fu, K. Zhang, Y. Zhang, Y. Cong and Q. Wang, Chem. Eng. J., 2021, 412, 128722 CrossRef CAS.
- M. A. Nasalevich, M. G. Goesten, T. J. Savenije, F. Kapteijn and J. Gascon, Chem. Commun., 2013, 49, 10575–10577 RSC.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449–9451 RSC.
- R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2016, 4, 1479–1485 RSC.
- M. J. Katz, T. Ramnial, H.-Z. Yu and D. B. Leznoff, J. Am. Chem. Soc., 2008, 130, 10662–10673 CrossRef CAS PubMed.
- T. S. Basu Baul, D. Dutta, A. Duthie and M. F. C. Guedes da Silva, Inorg. Chim. Acta, 2017, 455, 627–637 CrossRef CAS.
- J. Winarta, B. Shan, S. M. McIntyre, L. Ye, C. Wang, J. Liu and B. Mu, Cryst. Growth Des., 2020, 20, 1347–1362 CrossRef CAS.
- W. Liang, C. J. Coghlan, F. Ragon, M. Rubio-Martinez, D. M. D'Alessandro and R. Babarao, Dalton Trans., 2016, 45, 4496–4500 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR of H2MRL as well as gas adsorption (isotherms and pore sizes) for the MR-MOF samples after five ammonia adsorption/desorption cycles are available. All published isotherms are also available in adsorption information file (AIF) format. See DOI: https://doi.org/10.1039/d3tc03120a |
| This journal is © The Royal Society of Chemistry 2024 |