
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceKelvin probe force microscopy on patterned large-area biofunctionalized surfaces: a reliable ultrasensitive platform for biomarker detection†
Cinzia
Di Franco‡
 *a,
Matteo
Piscitelli‡
ab,
Eleonora
Macchia
cd,
Cecilia
Scandurra
e,
Michele
Catacchio
e,
Luisa
Torsi
*e and
Gaetano
Scamarcio
*ab
*a,
Matteo
Piscitelli‡
ab,
Eleonora
Macchia
cd,
Cecilia
Scandurra
e,
Michele
Catacchio
e,
Luisa
Torsi
*e and
Gaetano
Scamarcio
*ab
aCNR – Institute of Photonics and Nanotechnologies, Via Amendola 173, 70126 Bari, Italy. E-mail: cinzia.difranco@cnr.it
bDipartimento Interateneo di Fisica ‘‘M. Merlin’’, Università degli Studi di Bari Aldo Moro, 70126 Bari, Italy. E-mail: gaetano.scamarcio@uniba.it
cDipartimento di Farmacia-Scienze del Farmaco, Università degli Studi di Bari Aldo Moro, 70126 Bari, Italy
dThe Faculty of Science and Engineering, Åbo Akademi University, 20500 Turku, Finland
eDipartimento di Chimica, Università degli Studi di Bari Aldo Moro, Via E. Orabona 4, 70125 Bari, Italy. E-mail: luisa.torsi@uniba.it
First published on 20th November 2023
Abstract
Kelvin probe force microscopy (KPFM) allows the detection of single binding events between immunoglobulins (IgM, IgG) and their cognate antibodies (anti-IgM, anti-IgG). Here an insight into the reliability and robustness of the methodology is provided. Our method is based on imaging the surface potential shift occurring on a dense layer of ∼5 × 107 antibodies physisorbed on a 50 μm × 90 μm area when assayed with increasing concentrations of antigens in phosphate buffer saline (PBS) standard solutions, in air and at a fixed scanning location. A comprehensive investigation of the influence of the main experimental parameters that may interfere with the outcomes of KPFM immune-assay is provided, showing the robustness and reliability of our approach. The data are supported also by a thorough polarization modulation infrared reflection–absorption spectroscopy (PM-IRRAS) analysis of the physisorbed biolayer, in the spectral region of the amide I, amide II and amide A bands. Our findings demonstrate that a 10 min incubation in 500 μL PBS encompassing ≈ 30 antigens (100 zM) triggers an extended surface potential shift that involves the whole investigated area. Such a shift quickly saturates at increasing ligand concentration, showing that the developed sensing platform works as an OFF/ON detector, capable of assessing the presence of a few specific biomarkers in a given assay volume. The reliability of the developed methodology KPFM is an important asset in single molecule detections at a wide electrode interface.
Introduction
Kelvin probe force microscopy (KPFM) is a powerful analytical technique that allows surface potential (SP) mapping with high spatial resolution by measuring the contact potential difference (CPD) between the metal coated tip of an atomic force microscope (AFM) operated in tapping mode and the sample surface.1 It allows the simultaneous high-resolution morphological imaging of the sample surface and the mapping of the electrostatic configuration, originating from the local distribution of surface dipoles. While KPFM is inherently a non-invasive technique, it is extremely sensitive to changes occurring in the top-most atomic layer structures, as caused by terminal group modifications, molecular bindings or deposition of adlayers.2–4Several reports have shown the possibility to use KPFM to study biorecognition processes on biofunctionalized surfaces. For example, DNA either immobilized or physisorbed on Au coated Si substrates has been detected after exposure to dilutions in the μM range.2,5–7 ATP-binding on protein kinase has been detected with sensitivity of 10 pM and DNA-bound Hg+ ions with sensitivity in the fM range.8,9 KRAS and EGFR oncogene mutant DNA, bonded to Au nanoparticles, have been revealed with sensitivity in the pM range.10 Recently, we have demonstrated that KPFM can be used to assess the surface potential shift11 triggered by affinity binding on the biofunctionalized gate of a single molecule with a large transistor (SiMoT) sensor12,13 incubated with ligand concentrations as low as 10−19 mole L−1![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) 102 zM, corresponding to the ultimate limit of single molecule detection.
102 zM, corresponding to the ultimate limit of single molecule detection.
This work is focused on the development and optimization of a KPFM based sensing platform for the detection of immunoglobulin biomarkers on large-area biofunctionalized surfaces. Specifically, we demonstrate the detection of a few immunoglobulins (IgM, IgG) by mapping the surface potential of Au/anti-immunoglobulin M or G (anti-IgM, anti-IgG) interfaces with unprecedented sensitivity in the zeptomole range. Our method is based on the comparison of KPFM images taken in air at a fixed scanning location across the Au/biolayer interface, after 10 minute long sequential incubations at increasing concentrations of analyte in phosphate buffer saline (PBS) standard solutions followed by a drying procedure. This procedure inherently causes reproducibility issues, mainly due to surface contamination by adventitious carbon.14,15 Hence, we have carried out a comprehensive investigation of the influence of the main experimental parameters that may interfere with the outcome of a conventional KPFM analysis, showing the efficacy and reliability of a method based on the simultaneous SP mapping of both the bare and the bio-functionalized Au areas. Our findings reveal that the formation of the immune-complex reliably triggers an extended SP shift due to a surface amplification effect. This approach paves the way for a fast, reliable, and qualitative analysis of biomolecular interactions.
Materials and methods
Materials
HPLC-grade water, hydrogen peroxide 30%, VLSI grade, and sulfuric acid 96% VLSI Selectipur grade were purchased from Avantor. Polyclonal antibodies: anti-human immunoglobulin M (anti-IgM), anti-human immunoglobulin G (anti-IgG), human IgM (∼950 kDa), human IgG (∼150 kDa) (Sigma–Aldrich), were used without additional purifications. Phosphate-buffered saline (PBS, Sigma-Aldrich) solution had ionic strength of 162 mM, pH 7.4, at room temperature.Biofunctionalization protocol
The biofunctionalized surfaces were prepared starting from 10 mm × 5 mm substrates cut from As-doped silicon wafers with a 300 nm SiO2 layer on top. The substrates were preliminary cleaned by ultrasonic baths in acetone and 2-propanol for 10 min and dried with nitrogen. Afterwards, a 5 nm thick layer of Ti and a 50 nm Au layer were deposited by electron-beam evaporation. Before the biofunctionalization, each substrate was cleaned in a piranha solution (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of sulfuric acid and hydrogen peroxide), rinsed with HPLC water, and dried with nitrogen. The flowchart of the gold biofunctionalization protocol is sketched in Fig. S1 (ESI†). A suitable polymeric mask shadowed one half of the gold substrate. Subsequently, the sample was submerged in a PBS solution containing 0.1 mg mL−1 of anti-IgM or anti-IgG for 150 minutes at room temperature. The substrates were then extensively rinsed sequentially in PBS and in HPLC water and dried by spinning at 3000 rpm for 60 s before any characterization. The surface coverage is 1.2 × 1012 cm−2 antibodies as measured by surface plasmon resonance experiments.11
:1 mixture of sulfuric acid and hydrogen peroxide), rinsed with HPLC water, and dried with nitrogen. The flowchart of the gold biofunctionalization protocol is sketched in Fig. S1 (ESI†). A suitable polymeric mask shadowed one half of the gold substrate. Subsequently, the sample was submerged in a PBS solution containing 0.1 mg mL−1 of anti-IgM or anti-IgG for 150 minutes at room temperature. The substrates were then extensively rinsed sequentially in PBS and in HPLC water and dried by spinning at 3000 rpm for 60 s before any characterization. The surface coverage is 1.2 × 1012 cm−2 antibodies as measured by surface plasmon resonance experiments.11
Kelvin probe force microscopy
KPFM measurements were performed using an AFM system (mod. NTEGRA Spectra, NT-MDT, Moscow, Russia) equipped with a platinum-iridium coated tip FMG01/Pt (TipsNano) with an apex size of 35 nm, a resonance frequency f = 77.7 kHz, and a quality factor Q = 242. The sample was grounded using a metallic clamp. Morphology, phase, and surface potential images have been acquired simultaneously using the two-pass mode in which each line is sequentially scanned 2 times. In the first pass, the morphology is recorded in semi-contact mode, while the AFM cantilever is mechanically excited at frequency f. In the second pass, the tip is retraced at a set lift height (hlift) from the sample surface to measure the surface potential. During the second pass the cantilever is no longer excited mechanically but electrically by applying Vtip. During this phase, the tip and the sample behave like the opposing plates of a capacitor. Because of their different work functions, a force between the plates exists, which can be modulated by applying a voltage Vtip to the tip.1 In the vibrating capacitor model, Vtip consists of a constant (VDC) and an alternated (VAC) component with a frequency matching the cantilever resonant one. The force felt by the vibrating capacitor is: | (1) |
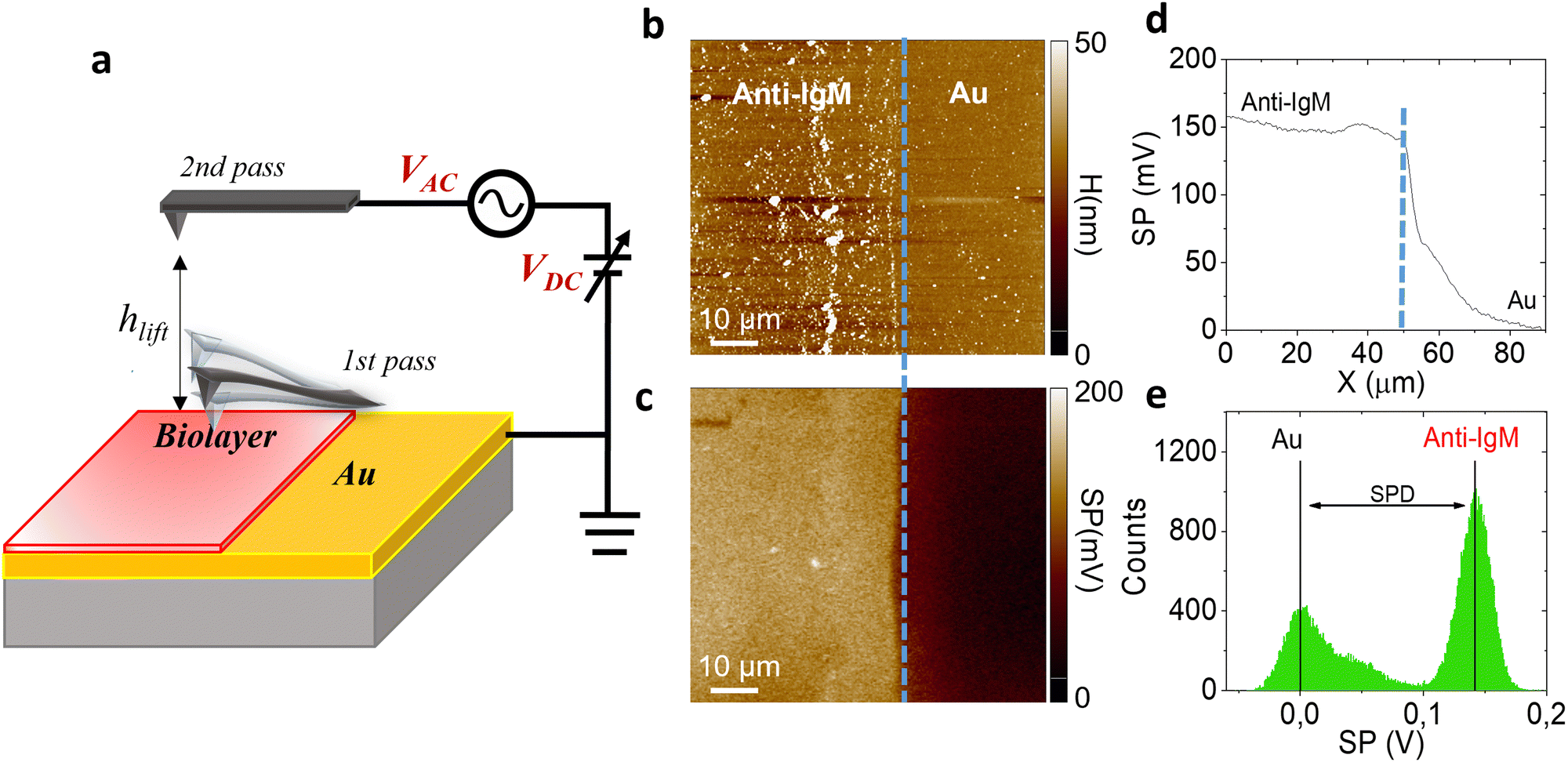 | ||
| Fig. 1 (a) Schematics of two pass-mode Kelvin probe force microscopy. Each line is sequentially scanned two times. In the first pass the morphology is recorded via semi-contact AFM mode, in the second pass the tip is re-traced at a set lift-height (hlift) from the sample surface to detect the surface potential. (b) Representative morphology (90 μm × 90 μm area) of a patterned anti-IgM layer at the edge between the gold area and the protein region. A sharp edge between the bio-functionalized (leftmost side) area and gold (rightmost side) region is visible, highlighted by the dashed blue line. (c) Representative surface potential (SP) image. (d) Average surface potential profile. (e) Histogram of the surface potential distribution. | ||
Fig. 1 shows the morphology (b) of a representative 90 μm × 90 μm KPFM substrate, along with the corresponding surface potential image (c). Notably, the morphology image and the SP map exhibit a sharp nanometric boundary, demarcating the bio-functionalized region (leftmost side) and the gold region (rightmost side). The anti-IgM biofunctionalized area consists of a uniform coating with a significant different contrast with respect to the adjacent gold region, with a SP value 150 ± 11 mV higher, as shown in Fig. 1d. The histograms of the SP distributions are given in Fig. 1e. We chose as an analytical parameter the surface potential difference (SPD) calculated between the peaks of the SP distributions corresponding to the anti-IgM and the Au areas, respectively. The immunological assays were performed by sequentially recording the SP images, at a fixed scanning location, after incubation for 10 min, in 0.5 mL of phosphate buffer saline (PBS) standard solutions of immunoglobulins (IgM and IgG) with concentrations ranging from 102 zM to 106 zM. After incubation in each of the PBS standard-solutions, the substrates were washed thoroughly with HPLC water to remove the unreacted ligands, dried by spinning and measured by KPFM.
Polarization modulation infrared reflection–absorption spectroscopy
Polarization modulation infrared reflection–absorption spectroscopy (PM-IRRAS) measurements were performed using a Nicolet iS50 Fourier transform spectrometer equipped with a research module containing a wire-grid polarizer, a photo-elastic polarization modulator (PEM) operating at 50 kHz, a variable angle sample holder and a liquid-nitrogen-cooled mercury–cadmium–telluride (MCT) detector. An air purging system kept the relative humidity below 17% and a low level of carbon dioxide in the system compartments.The incident and reflected IR beams propagate at angles of 82° with respect to the sample surface normal. The PEM phase was set to have a maximum PM-IRRAS signal in the spectral regions (1000–1800 cm−1) and (2600–3500 cm−1). Each spectrum was the average of 1000 scans with a resolution of 4 cm−1. Two PM-IRRAS reflective substrates were prepared depositing over a glass slide a 5 nm-thick Ti adhesion promoter layer and a 50 nm-thick Au layer, via electron beam evaporation. They were cleaned with a sequential ultrasonic bath in water, acetone, and 2-propanol for 10 minutes and dried under nitrogen flux. One substrate was further treated for 10 minutes in an ozone cleaner before submerging it into 0.1 mg mL−1 anti-IgM PBS (pH = 7.4, ionic strength = 162 mM) solution for 2 hours at 25 °C to allow the formation of a stable layer of physisorbed antibodies. Then, it was thoroughly rinsed with HPLC water, and dried using a spinner (1500 rpm, 3 minutes) before PM-IRRAS analysis. The PM-IRRAS spectra of the two bare substrates were first measured to check for the same starting chemical composition of their surface. A layer of anti-IgM was prepared on one substrate, as described above. Then, it was thoroughly rinsed with deionized water, dried by spinning and the PM-IRRAS spectrum recorded. Afterwards, both substrates were stored at 4 °C, submerged in PBS solution for 24 h. The day after they were rinsed with water and dried, the PM-IRRAS spectra were measured, and they were stored again in buffer solution at 4 °C. This protocol was repeated to measure the spectra of both the bare gold substrate and that of the protein layer after 48 h, 72 h, and 144 h. The baseline correction on the original PM-IRRAS signal was calculated in the ranges 950–1780 cm−1 and 2700–3500 cm−1 using a least-square third-order polynomial fitting method.16
Results and discussion
Experimental data and method optimization
The SP values extracted by KPFM are inherently affected by the surface conditions or experimental parameters. In particular, the strongest influence is due to the chosen tip-sample distance hlift during the second pass, or the surface contamination by adventitious carbon in ambient air.14,15 On the other hand, while the SP values may vary as a function of extrinsic parameters, the average value of the relative contact potential differences (SPD) between the bio-functionalized and the Au areas is less sensitive to the external conditions and can be reliably used as a suitable analytical quantity to assess the surface transformation induced by affinity binding events.To confirm this claim, we first consider the effect of varying hlift. Controlling the tip-sample distance involves carefully calibrating the AFM setup, minimizing thermal drift, and accounting for any vibration-induced perturbations. Fig. 2 shows the peak values in the SP histograms and the corresponding surface potential differences of a representative anti-IgM/Au substrate, as a function of the tip-sample distance. While for small distances SP and SPD vary substantially, for hlift > 200 nm they reach a constant value. Accordingly, all KPFM measurements have been performed setting hlift = 250 nm.
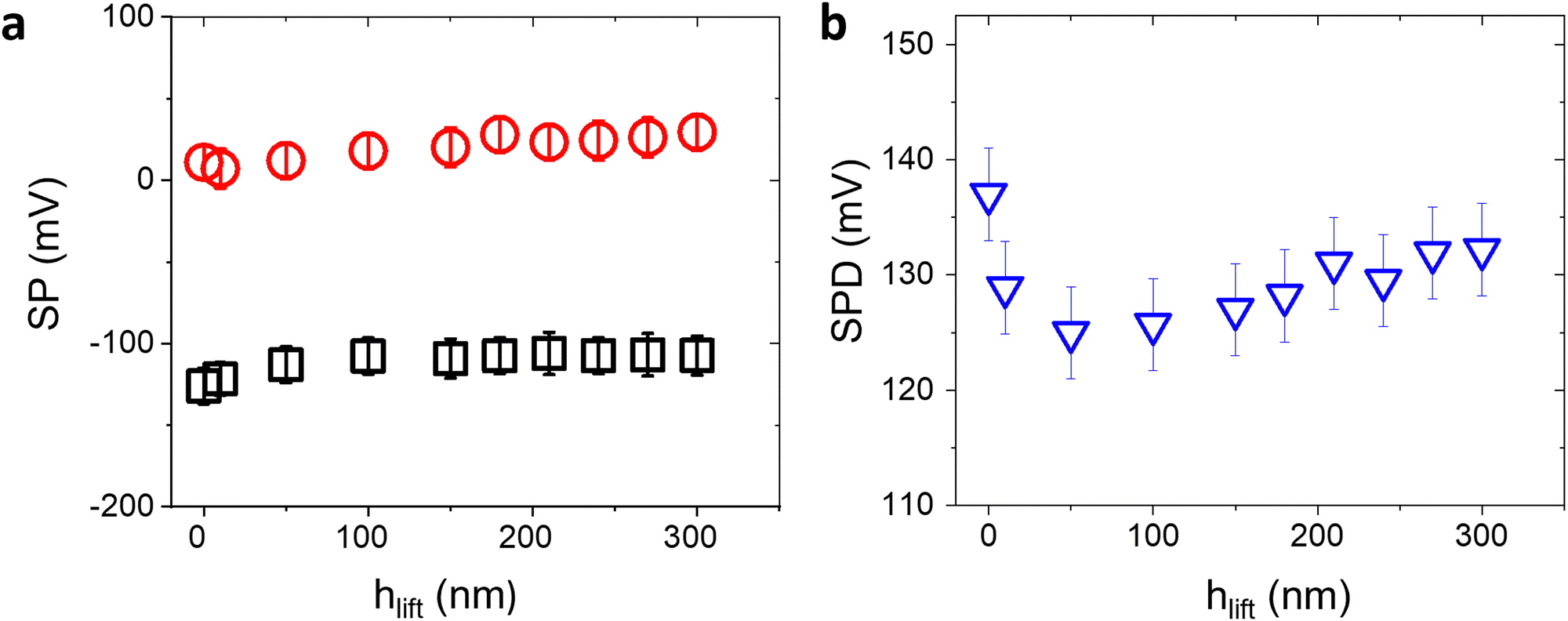 | ||
| Fig. 2 (a) Measured SP of Au (black square symbols) and anti-IgM areas (red circles) and SPD (b) as a function of the tip-sample distance in the 2nd pass of KPFM. The tip oscillation amplitude has been kept constant at 70 nm. The error bars have been calculated from the standard deviation of SP distributions. Increasing the tip-sample distance affects the SP values, however the SPD becomes approximately constant for hlift >200 nm. | ||
Surface contamination by hydrocarbons and carbo-oxides17 typically increases with storage time and becomes noticeable in a matter of minutes. With the aim of investigating the role of contaminants on the Au/anti-IgM substrates, we have plotted in Fig. 3 the measured SP and the corresponding SPD values as a function of time over several days. While the SP of both regions varies with time, probably due to the increased surface contamination by airborne adventitious carbon, it is worth noting that the SPD values remain approximately constant. Fig. 4b shows the time evolution of the PM-IRRAS spectra over a 6 day period. The largest modification occurs for the infrared spectrum of the bare gold substrate in which three bands centred at 1540 cm−1, 1660 cm−1 and 1740 cm−1, ascribed to the C–N, C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of carbonaceous molecules appear, respectively.18,19 The intensities of these bands monotonically increase with time. Also, the Amide I and Amide II bands measured on anti-IgM coated samples change with time, though to a lesser extent. These findings demonstrate the progressive contamination of substrates due to the procedure of storage, drying and exposure to ambient air, as described in the method paragraph. On the other hand, the contamination effects can be effectively tackled by considering the relative surface potential shift SPD, so that we can use KPFM measurements as a reliable analytical platform, as demonstrated in the following.
O stretching vibrations of carbonaceous molecules appear, respectively.18,19 The intensities of these bands monotonically increase with time. Also, the Amide I and Amide II bands measured on anti-IgM coated samples change with time, though to a lesser extent. These findings demonstrate the progressive contamination of substrates due to the procedure of storage, drying and exposure to ambient air, as described in the method paragraph. On the other hand, the contamination effects can be effectively tackled by considering the relative surface potential shift SPD, so that we can use KPFM measurements as a reliable analytical platform, as demonstrated in the following.
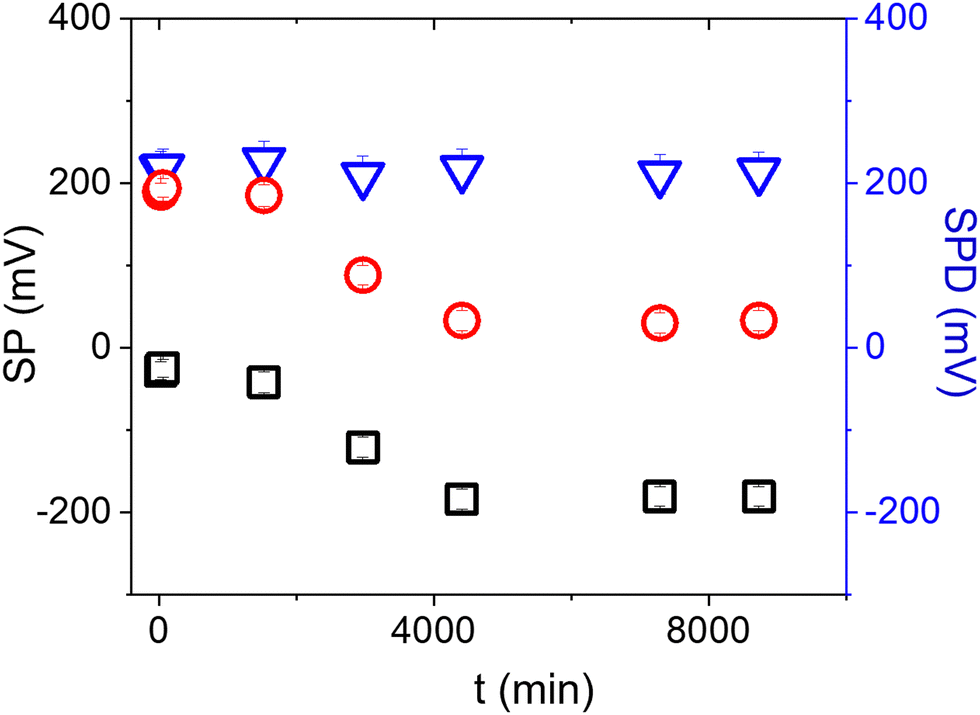 | ||
| Fig. 3 Time variation of the surface potential measured on the Au (black squares) and the anti-IgM (red circles) areas of an Au/anti-IgM patterned substrate. The SPD values are plotted as blue triangles. The tip-sample distance has been set at 250 nm. The error bars have been calculated from the standard deviation of the distributions. | ||
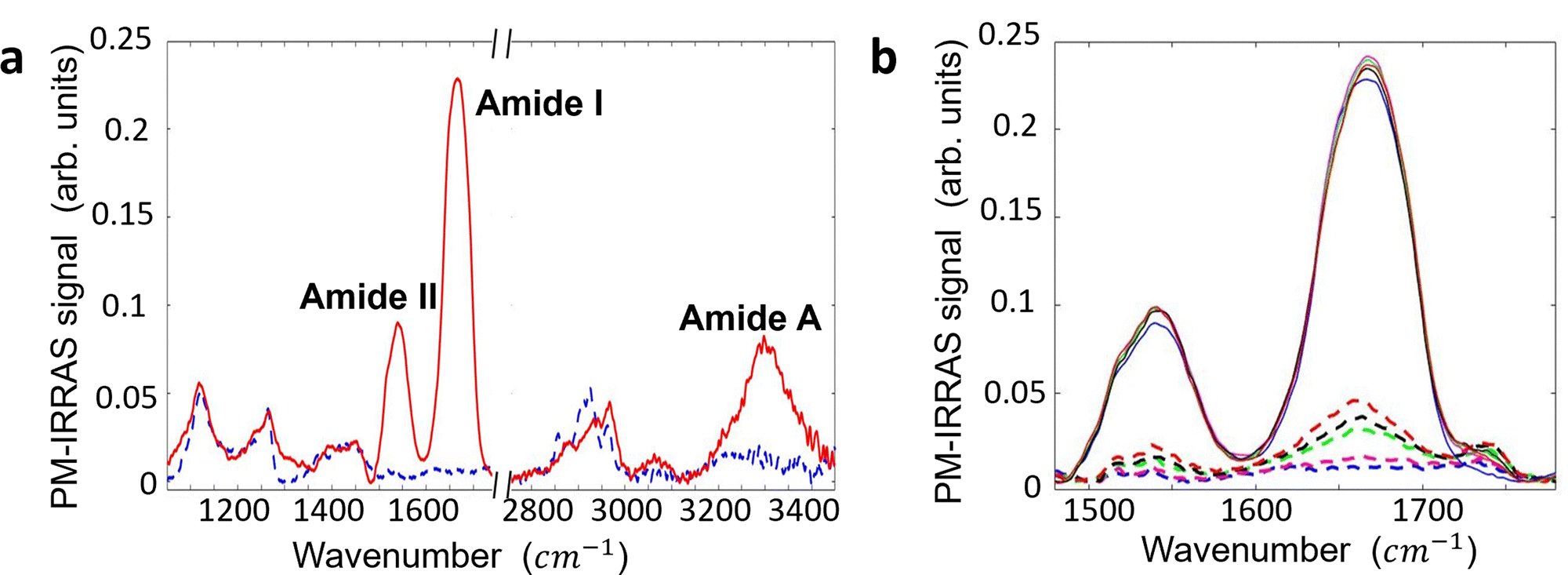 | ||
| Fig. 4 (a) PM-IRRAS spectra of the anti-IgM layer physisorbed on the Au substrate (solid red line) and bare gold sample (dashed blue line). Background subtraction was accomplished by dividing the recorded PM-IRRAS signal with a third-order polynomial curve. It has been obtained using a modified least square minimization algorithm implemented in MATLAB. The spectrum of the anti-IgM sample shows characteristic features of a protein layer, including: (i) Amide I (1600–1710 cm−1), (ii) Amide II (1500–1570 cm−1), and (iii) Amide A (3200–3400 cm−1). (b) PM-IRRAS spectra of the anti-IgM layer (solid lines) and Au substrate (dashed lines) over a span of 6 days. The initial spectra of both pristine samples are shown as blue lines. Subsequent spectra were recorded after 24 hours (represented by solid and dashed violet lines), 48 hours (solid and dashed green lines), 72 hours (solid and dashed black lines), and 144 hours (solid and dashed red lines). Throughout the 6-day period, the samples were stored in a buffer solution of HEPES with a pH of 7.4 and an ionic strength of 150 mM, in a refrigerated environment, between successive measurements. As the measurement time progressed, new spectral features emerged on both the bare gold and protein spectra, centered at approximately 1540 cm−1, 1660 cm−1, and 1740 cm−1. These spectral changes are attributed to the adsorption of adventitious layers on the sample surfaces. | ||
Method validation
Two antigen/antibody couples were used to validate our sensing approach, as well as to test its performance, along with levels of noise and limit of detections (LOD). The selected systems were the IgM/anti-IgM and IgG/anti-IgG couples. We have used KPFM to assess the occurrence of affinity binding interactions occurring at the biofunctionalized substrate after the incubation with increasing concentration of antigens in PBS standard solutions. The SP images were iteratively recorded at the same 90 μm × 90 μm area, across the interface between the Au region and the antibody one. Fig. 5 shows the SPD variation (ΔSPD) when a pristine Au/biolayer substrate is exposed to increasing concentrations of specific antigens (recognition experiments) or un-binding ligands (negative controls). The IgM/anti-IgM findings are given in Fig. 5a, and the corresponding KPFM images and histograms of the SP distributions are shown in Fig. S2 (ESI†). Taking the average value (ΔSPD = −15 mV) and the standard deviation (SDNC = 21.5 mV) of the negative control data as a reference, we note that the recognition events cause an average ΔSPD shift of −74 ± 9 mV, which is significantly larger than the shift at the limit of detection, taken as 3 SDNC. Hence, with 99.7% confidence, we ascribe the observed ΔSPD shift to the extended modification of the surface dipole distribution in the anti-IgM recognition layer, which is triggered by a relatively small number of affinity binding events. Notably, the surface potential change occurs in the entire scanned surface, which is encompassed by ∼6 × 107 molecules, whereas the number of IgM ligands within the 500 μL incubation volume varies in the range of 3 × 101–3 × 105. The nominal numbers of ligands hosted at each concentration are estimated as C·V·NA, where C is the analyte concentration, V is the volume of the standard PBS solution in which the sample is incubated, and NA is the Avogadro number. The error associated with the sampling procedure can be estimated according to the Poisson's distribution. The KPFM assay performance was tested also in the recognition process between human-IgG and its cognate antibody anti-IgG. To this end, a patterned anti-IgG/Au sample was used. The representative morphology and the corresponding average profile are reported in Fig. S4 (ESI†).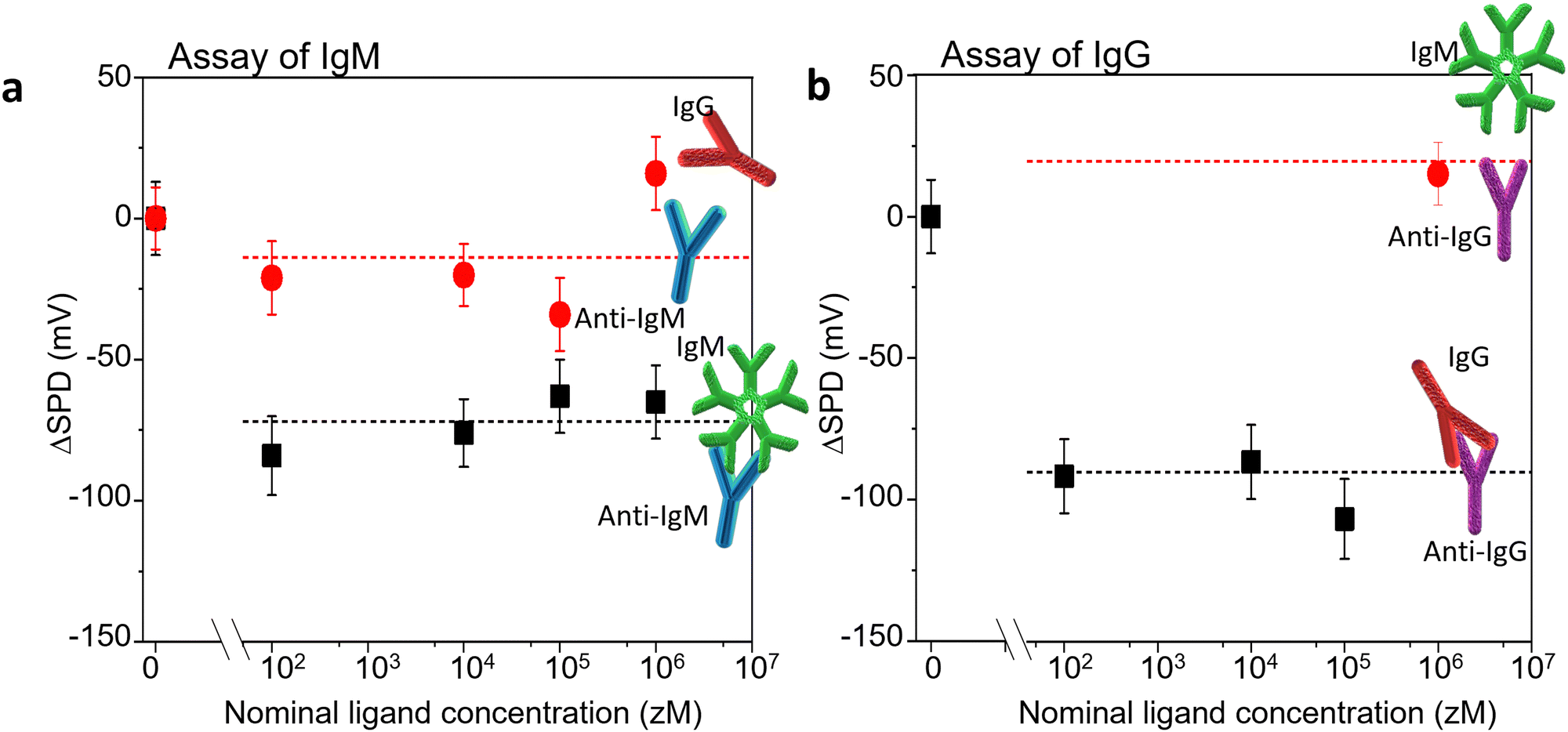 | ||
| Fig. 5 The SPD shift (ΔSPD) measured on the Au biofunctionalized substrate, as a function of the ligand concentration as compared to the SPD measured on pristine Au/biolayer substrates (reference). (a) Assay of IgM. Black squares: incubations with standard solutions of IgMs. Red circles: IgG incubations (negative control). The proteins are assayed from standard solutions in phosphate saline buffer solution (PBS, pH= 7.4, ionic strength: 162 mM), starting from the more diluted solution. The analytical parameter is the SPD shift (ΔSPD) at different analyte concentrations as compared to the SPD measured on the pristine sample exposed to a PBS solution (baseline). The values have been calculated from the peak values of the SP distributions. The binding and the negative control experiments have been performed on different anti-IgM/Au samples. The formation of the anti-IgM/IgM complex triggers an overall decrease by −74 mV of the investigated anti-IgM surface potential area. (b) Assay of IgG. Black squares: incubations with standard solutions of IgGs. Red circle: IgM 1 fM in PBS (negative control). The affinity binding and the negative control experiments have been performed on the same anti-IgG/Au sample. Upon binding, an overall decrease by −90 mV of the investigated anti-IgG surface potential was found. The error bars are taken as one standard deviation. | ||
Fig. 5b shows the ΔSPD induced by the exposure of a pristine Au/anti-IgG substrate to increasing concentrations of IgG (affinity binding) ligands. A negative control experiment was performed by incubating the substrate with 1 fM IgM in PBS. No significant SPD change was found with respect to the baseline, confirming the high selectivity of the sensing platform. The same probe was sequentially incubated with increasing concentrations of IgG. A mean surface potential shift of −94 ± 10 mV was found, due to the immunorecognition events. The corresponding KPFM images and surface potential distributions are shown in Fig. S6 (ESI†). A qualitative agreement with the data of Fig. 5a is evident, confirming that KPFM imaging is a reliable and ultrasensitive platform for biomarker detection on large-area biofunctionalized surfaces.
It is worth noting that increasing the ligand concentration does not produce a further increase of the ΔSPD shift. This confirms the notion that the surface potential transition, locally triggered by a few affinity binding events, propagates over large areas, probably funnelled by the hydrogen bonding network.13 Accordingly, our sensing platform has the tendency to operate as an OFF/ON detector, capable of assessing the presence of a single specific biomarker in a given assay volume. This is analogous to similar findings obtained by us using a transistor-like detecting platform.20–25
Conclusion
We have developed and optimized an ultrasensitive detection platform based on KPFM and patterned surfaces biofunctionalized with dense physisorbed antibody layers, suitable for assessing the presence of a few ligands in an OFF/ON mode. We estimate that a small number of affinity binding events (30 ± 10 molecules) cause extended surface potential shifts over areas larger than hundreds of squared micrometres, hosting >107 highly packed capturing sites. This implies the existence of a propagation phenomenon of the electrostatic change occurring on a few capturing antibody sites. We postulated a domino-like surface phenomenon, ultimately affecting the densely packed neighbouring antibodies, and thus a single antibody–antigen binding occurrence could induce an extended electrostatic reorientation involving millions of antibody dipoles. While the detailed modelling of the observed amplification effect needs further studies, we have addressed here some reproducibility issues. PM-IRRAS analysis shows that adventitious contaminants actually affect the spectral features and chemical composition of both the biolayer and bare gold sample. Changes in the SP extracted by KPFM due to tip-sample distance, aging and storage effects cannot be neglected, nevertheless the simultaneous SP mapping of the bare and the bio-functionalized Au areas is a reliable tool to investigate the SP variation induced by antigen/antibody complex formation.Our findings show that a change of ∼−70 mV (∼−90 mV) is induced by the interaction of anti-IgM (anti-IgG) with the cognate IgM (IgG) ligands. The process is also proved to be irreversible, and thus no significant further surface potential change has been measured, if other affinity events occur. The presented data demonstrate a proof of concept and are relevant for future applications in the selective detection at the physical limit of immunoglobulin traces in real fluids.
Author contributions
C. D. F. and G. S. conceived the paper and designed the experiments. C. D. F. and M. P. performed the KPFM and PM-IRRAS measurements and analysis. C. S. and M. C. contributed to sample preparation and characterization, E. M. and L. T. contributed to the interpretation of the results and the funding project supervision. C. D. F., M. P. and G. S. wrote the manuscript that was approved by all authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The following projects are acknowledged for partial financial support: “A binary sensor with single-molecule digit to discriminate biofluids enclosing zero or at least one biomarker (NoOne)” ERC Starting Grant 2021 (GA 101040383); “Tecnologie portatili e protocolli innovativi per la diagnosi ultrasensibile di Xylella fastidiosa direttamente in piante e vettori” (1LIVEXYLELLA) Ministero dell’agricoltura, della sovranità alimentare e delle foreste – MIPAAF D.M. n.419161 del 13/09/2022; Centro di Innovazione Regionale Digital Assay, Regione PUGLIA DR n.702 del 08/11/2022; Italian network of excellence for advanced diagnosis (INNOVA), Ministero della Salute -code PNC-E3-2022-23683266 PNC-HLS-DA, CUP: C43C22001630001; Complementary National Plan PNC-I.1 “Research initiatives for innovative technologies and pathways in the health and welfare sector” D.D. 931 of 06/06/2022, DARE - DigitAl lifelong pRevEntion initiative, code PNC0000002, CUP: B53C22006420001, PNRR MUR project PE0000023-NQSTI; MUR – Dipartimenti di Eccellenza 2023–2027 – Quantum Sensing and Modelling for One-Health (QuaSiModO).References
- M. Nonnenmacher, M. O’Boyle and H. K. Wickramasinghe, Ultramicroscopy, 1992, 42–44, 268–273 CrossRef.
- D. C. Hansen, K. M. Hansen, T. L. Ferrell and T. Thundat, Langmuir, 2003, 19, 7514–7520 CrossRef CAS.
- G. Gauglitz, Anal. Bioanal. Chem., 2020, 412, 7043–7045 CrossRef CAS PubMed.
- M. Salerno and S. Dante, Materials, 2018, 11, 951–956 CrossRef.
- L. E. Cheran, M. Chacko, M. Zhang and M. Thompson, Analyst, 2004, 129, 161–168 RSC.
- L. E. Cheran, M. E. McGovern and M. Thompson, Faraday Discuss., 2000, 116, 23–34 RSC.
- L.-E. Cheran, S. Johnstone, S. Sadeghi and M. Thompson, Meas. Sci. Technol., 2007, 18, 567–578 CrossRef CAS.
- J. Park, S. Lee, K. Jang and S. Na, Biosens. Bioelectron., 2014, 60, 299–304 CrossRef CAS.
- J. Park, J. Yang, G. Lee, C. Y. Lee, S. Na, S. W. Lee, S. Haam, Y.-M. Huh, D. S. Yoon, K. Eom and T. Kwon, ACS Nano, 2011, 5, 6981–6990 CrossRef CAS.
- K. Jang, J. Choia, C. Park and S. Na, Biosens. Bioelectron., 2017, 87, 222–228 CrossRef CAS PubMed.
- C. Di Franco, E. Macchia, L. Sarcina, N. Ditaranto, A. Khaliq, L. Torsi and G. Scamarcio, Adv. Mater. Interfaces, 2022, 10, 2201829 CrossRef.
- E. Macchia, K. Manoli, B. Holzer, C. Di Franco, M. Ghittorelli, F. Torricelli, D. Alberga, G. F. Mangiatordi, G. Palazzo, G. Scamarcio and L. Torsi, Nat. Commun., 2018, 3223 CrossRef PubMed.
- E. Macchia, F. Torricelli, P. Bollella, L. Sarcina, A. Tricase, C. Di Franco, R. Österbacka, Z. M. Kovács-Vajna, G. Scamarcio and L. Torsi, Chem. Rev., 2022, 122, 4636–4699 CrossRef CAS PubMed.
- A. Liscio, V. Palermo, K. Müllen and P. Samorì, J. Phys. Chem. C, 2008, 112, 17368–17377 CrossRef CAS.
- N. Turetta, F. Sedona, A. Liscio, M. Sambi and P. Samorì, Adv. Mater. Interfaces, 2021, 10, 2100068 CrossRef.
- C. A. Lieber and A. Mahadevan-Jansen, Appl. Spectrosc., 2003, 57, 1363–1367 CrossRef CAS.
- P. Swift, Surf. Interface Anal., 1982, 4, 47–51 CrossRef CAS.
- A. Barth, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- V. A. Lorenz-Fonfria, Chem. Rev., 2020, 120, 3466–3576 CrossRef CAS.
- E. Macchia, Z. M. Kovács-vajna, D. Loconsole, L. Sarcina, M. Redolfi, M. Chironna, F. Torricelli, L. Torsi, D. Chimica and B. Aldo, Sci. Adv., 2018, 8, eabo0881 CrossRef.
- L. Sarcina, F. Viola, F. Modena, R. A. Picca, P. Bollella, C. Di Franco, N. Cioffi, M. Caironi, R. Österbacka, I. Esposito, G. Scamarcio, L. Torsi, F. Torricelli and E. Macchia, Anal. Bioanal. Chem., 2022, 414, 5657–5669 CrossRef CAS PubMed.
- E. Macchia, L. De Caro, F. Torricelli, C. Di Franco, G. F. Mangiatordi, G. Scamarcio and L. Torsi, Adv. Sci., 2022, 9, 2104381 CrossRef CAS PubMed.
- L. Sarcina, E. Macchia, G. Loconsole, G. D’Attoma, P. Bollella, M. Catacchio, F. Leonetti, C. Di Franco, V. Elicio, G. Scamarcio, G. Palazzo, D. Boscia, P. Saldarelli and L. Torsi, Adv. Sci., 2022, 9, e2203900 CrossRef.
- C. Scandurra, K. Björkström, L. Sarcina, A. Imbriano, C. Di Franco, R. Österbacka, P. Bollella, G. Scamarcio, L. Torsi and E. Macchia, Adv. Mater. Technol., 2023, 8, 2201910 CrossRef CAS.
- L. Sarcina, C. Scandurra, C. Di Franco, M. Caputo, M. Catacchio, P. Bollella, G. Scamarcio, E. Macchia and L. Torsi, J Mater. Chem. C Mater., 2023, 11, 9093–9106 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc03110a |
| ‡ Cinzia Di Franco and Matteo Piscitelli equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2024 |