
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Uncovering cation disorder in ternary Zn1+xGe1−x(N1−xOx)2 and its effect on the optoelectronic properties†
Zhenyu
Wang
 ab,
Daniel M.
Többens
a,
Alexandra
Franz
a,
Stanislav
Savvin
c,
Joachim
Breternitz‡
*a and
Susan
Schorr
*ab
ab,
Daniel M.
Többens
a,
Alexandra
Franz
a,
Stanislav
Savvin
c,
Joachim
Breternitz‡
*a and
Susan
Schorr
*ab
aHelmholtz-Zentrum Berlin für Materialien und Energie, Department Structure and Dynamics of Energy Materials, Hahn-Meitner-Platz 1, 14109 Berlin, Germany. E-mail: joachim.breternitz@fh-muenster.de; Susan.Schorr@helmholtz-berlin.de
bFreie Universität Berlin, Department Geosciences, Malteserstraße 74-100, 12249 Berlin, Germany
cInstitut Laue-Langevin, Avenue des Martyrs, 38000 Grenoble, France
First published on 14th December 2023
Abstract
Ternary nitride materials, such as ZnGeN2, have been considered as hopeful optoelectronic materials with an emphasis on sustainability. Their nature as ternary materials has been ground to speculation of cation order/disorder as a mechanism to tune their bandgap. We herein studied the model system Zn1+xGe1−x(N1−xOx)2 including oxygen – which is often a contaminant in nitride materials – using a combination of X-ray and neutron diffraction combined with elemental analyses to provide direct experimental evidence for the existence of cation swapping in this class of materials. In addition, we combine our results with UV-VIS spectroscopy to highlight the influence of disorder on the optical bandgap.
Introduction
The general suitability of nitride materials for optoelectronic applications has long been demonstrated1 and even culminated in the nobel prize for (In,Ga)N based blue LED materials – the famous III–V materials.2–4 Besides their application as LEDs, a number of studies also evaluated the potential of nitride materials for photovoltaic applications.5 Growing concern, however, is associated with the use of scarce and toxic elements in photovoltaic applications,6 as their use will become more and more widespread. Particularly, indium is one of the scarcest elements7 and its use would hence potentially impede their widespread application. With the choice of trivalent cations that can replace In3+ being limited, a strategy that has been proven fruitful is the replacement of the trivalent cations with equimolar amounts of divalent and tetravalent cations. A similar strategy has been used to replace In3+/Ga3+ in Cu(In,Ga)S2 (CIGS) with Zn2+ and Sn4+ to arrive at Cu2ZnSnS4 (CZTS)8–10 as well as in double perovskites, such as Cs2AgInBr6, where Ag+ and In3+ replace the toxic Pb2+ in lead halide perovskites.11,12For nitrides, a combination of divalent Zn2+ and a tetravalent cation of group 14 (Si4+, Ge4+, and Sn4+) has been proven to yield materials with a suitable optical bandgap and stability that can be produced in the form of thin-films and bulk materials with the general formula ZnMN2 (M = Si, Ge, Sn).13–15 The transition from binary to ternary nitride materials not only allows the more traditional bandgap tuning mechanism through alloying of different tetravalent cations – similar to Al, Ga, In alloying in III–V's – but the particular arrangement of different cation types influences the bandgap, too.16–25 While a compound with fully statistical distribution of the cations would crystallise isostructurally to (In,Ga)N in the wurtzite-type structure in the hexagonal space group P63mc,26 this crystal structure type does not allow for ordering in the crystal structure, since there is only one crystallographic position for the cations.27 Instead, fully or partially ordered compounds crystallise in the β-NaFeO2-type structure in the orthorhombic space group Pna21, a subgroup of P63mc.27–29 Herein, the cation and anion positions are split in two distinct crystallographic sites on the general Wyckoff position 4a and hence allow a great degree of freedom in the crystal structure.
A number of theoretical works have studied the influence of cation ordering on the bandgap of the material,20,23,30 showing a strong trend to bandgap narrowing when cation disorder is introduced. Such trends were also observed experimentally and attempts to rationalise the degree of disorder based on the lattice constants have proven some success.14,31,32 However, there are two fundamental problems when studying the order phenomena in these materials: (a) Zn2+ and Ge4+, the cations in the best studied material of this series, are isoelectronic and hence hardly distinguishable using standard X-ray diffraction (XRD) techniques and (b) the accidental or voluntary introduction of oxygen into the compounds appears to cause a similar phenomenon of narrowing the optical bandgap.26,33 We could recently show that compounds made through ammonolysis of Zn2GeO4 follow the general formula Zn1+xGe1−x(N1−xOx)2 within the technologically relevant reaction region.33 While neither of the pairs Zn2+/Ge4+, and O2−/N3− can be distinguished confidently using standard XRD, the neutron scattering lengths of the elements (bZn = 5.68 fm, bGe = 8.185 fm, bO = 5.803 fm, and bN = 9.36 fm)34 allow a clear distinction between the elements and hence permit the unambiguous quantification of order/disorder in Zn1+xGe1−x(N1−xOx)2.
Herein, we present a systematic approach to study the cation order in Zn1+xGe1−x(N1−xOx)2 directly using powder neutron diffraction (PND). By studying the trilogy of chemical compositions, optical properties and cation order, we are able to deconvolute the effects of oxygen and intrinsic disorder on the bandgap for the first time and show that both effects – although resulting in similar trends – are unrelated to each other.
Experimental section
Samples of Zn1+xGe1−x(N1−xOx)2 with varying compositions were prepared using a method described earlier.26,33,35X-ray fluorescence (XRF) measurements of the metals were used to determine the overall chemical composition of the samples. The calculation was based on the general formula, since we could show that this approach yields in reliable overall compositions.
UV-Vis measurements (UV-VIS) were performed using a PerkinElmer Lambda 750S spectrometer using a praying mantis diffuse reflectance sample holder and a 100 mm integrating sphere as the detector. The data interpretation was conducted through Kubelka–Munk treatment36 combined with a Tauc-plot.37 An exponent for a direct allowed bandgap was used in the Tauc-plot.38
Powder X-ray diffraction (XRD) was performed using a Bruker D8 advance powder diffractometer with Ni-filtered Cu-Kα radiation (λ = 1.5418 Å) in the range of 15° ≤ 2θ ≤ 140° with a step width of 0.02° and LaB6 as the internal standard.
Powder neutron diffraction (PND) was performed using either the E9 powder diffractometer39 at the BERII research reactor of Helmholtz-Zentrum Berlin or the D2B powder diffractometer at the Institut Laue-Langevin.40
Anomalous X-ray diffraction data for Rietveld refinement were collected at the KMC-2 Diffraction station at KMC-2 beamline,41 BESSY II, Berlin, Germany. Samples were mounted in symmetric reflection geometry using a zero-background silicon sample holder and an area sensitive gas detector (Vantec 2000, Bruker AXS). The instrumental resolution function was determined from a sample of LaB6. A preliminary energy-dependent scan of one sample determined the positions of the absorption edges as established by the increase of the fluorescence background as 9660(2) eV and 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 102(2) eV. These values did not deviate significantly from the literature values of 9659 eV (Zn–K) and 11103 eV (Ge–K).42
102(2) eV. These values did not deviate significantly from the literature values of 9659 eV (Zn–K) and 11103 eV (Ge–K).42
Full powder diffraction sets were collected at energies of 8048 eV (λ = 1.5406 Å, equivalent to Cu Kα1), 9649 eV (below Zn–K absorption edge), and 11093 eV (below Ge–K edge).
Rietveld refinements were performed using Fullprof-Suite.43 For the neutron diffraction samples, both XRD and NPD were refined simultaneously for each sample in order to combine the accuracy of lattice constants and contrast between cations and anions from XRD with the contrast of the cation pair Zn2+/Ge4+ and the anion pair O2−/N3−. The background was modelled by linear interpolation between positions with little contribution from Bragg peaks. Symmetric Thompson–Cox–Hastings pseudo-Voigt functions were used for the peak shape. Anisotropic domain size broadening according to Scherrer was refined by symmetry-adapted spherical harmonics up to second order. Further details on the anomalous diffraction refinement procedure may be found in the ESI,† in particular the form factors used for N3− and O2− anions.44
Types of cation disorder in Zn1+xGe1−x(N1−xOx)2
To signify disordered cations clearly, we use notation in the form of ZnGe for zinc residing at the germanium position, GeZn for germanium residing at the zinc position and ZnZn and GeGe for the respective atoms residing at their conventional crystallographic position (Fig. 1). An ideal, fully ordered ZnGeN2 would show the anti-sites GeGe and ZnZn. | ||
Fig. 1 Schematic of the cation occupation of the two cation sites of the β-NaFeO2 structure type. The left-hand case has no intrinsic disorder and a certain degree of extrinsic disorder (ZnGe), while the right-hand case depicts the same composition with extrinsic (ZnGe) and intrinsic (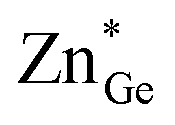 and and  ) disorders. ) disorders. | ||
Two distinct and independent types of disorder must be regarded in the zinc germanium oxide nitride materials as produced from the ammonolysis of Zn2GeO4. As a consequence of the reaction mechanism, no sub-stoichiometric amounts of cations or anions are expected, i.e. the ratio of cations to anions is always 1:1.33 This fact, on the other hand, in combination with the decrease in overall anion charge when the oxygen amount increases means that all oxide nitrides are rich in Zn resulting in Zn/Ge > 1. As a consequence, a certain amount of Zn that is commensurate with the oxygen content necessarily resides at the Ge position, as there is more than one equivalent of Zn in such a compound, while it contains less than one equivalent of Ge. The amount of ZnGe in an otherwise completely ordered sample (notably GeZn = 0) would hence correspond to the extrinsic disorder, since it is a direct consequence of the composition (Fig. 1, left).
On the other hand, one could imagine a situation, where a Zn atom swaps sites with a Ge atom, thus a pair of ZnGe and GeZn are formed simultaneously. We will refer to this as intrinsic disorder (named  and
and  ), since it is independent of the chemical composition and can exist, in principle, at any oxygen level in any compound (Fig. 1, right). This latter is also the kind of disorder that has been investigated computationally in terms of its influence on the bandgap. Complete disorder in stoichiometric ZnGeN2 – i.e. without any extrinsic disorder – is reached when ZnGe = GeZn = 0.5 is observed. In this case, both elements are statistically distributed over both cation positions, rendering them indistinguishable from each other.
), since it is independent of the chemical composition and can exist, in principle, at any oxygen level in any compound (Fig. 1, right). This latter is also the kind of disorder that has been investigated computationally in terms of its influence on the bandgap. Complete disorder in stoichiometric ZnGeN2 – i.e. without any extrinsic disorder – is reached when ZnGe = GeZn = 0.5 is observed. In this case, both elements are statistically distributed over both cation positions, rendering them indistinguishable from each other.
Refinement strategy for neutron powder diffraction
As a consequence of the group–subgroup relationship between the wurtzite type structure and the β-NaFeO2-type structure, the differences in the diffraction patterns mostly occur in split reflections and only weak supplementary reflections for the latter crystal structure. Since differences in stoichiometry and cation disorder cause a quasi-continuous change between the wurtzite-type structure and the β-NaFeO2-type structure, there are cases where the split reflections, which are significant for ordered or partially ordered materials, overlap to a large degree. In such cases, the exact peak positions – indicative for the lattice constants – and the exact intensities – indicative for the atomic arrangement – are not straightforward to separate due to a strong peak overlap. In order to ease this problem, we have combined neutron and X-ray powder diffraction measurements in a simultaneous refinement (see Fig. 2 for a representative example). Hereby, we profit from the typically much narrower peaks in X-ray diffraction (due to a better instrumental resolution) for the exact determination of the reflection positions and the distinction between isoelectronic atoms in neutron diffraction. | ||
| Fig. 2 Rietveld plots of Zn1.032Ge0.968(N0.968N0.032)2 using neutron diffraction and X-ray diffraction (inset). Experimental values: red dots, calculated values: black line, difference: blue line and calculated reflection positions: green stacks. The calculated positions are given for the title phase (top) and LaB6 (bottom) was used as the internal standard. | ||
Still, the complex convolution of chemical compositions and four atomic sites on general positions makes such a refinement prone to errors and potentially unstable. Therefore, we introduced our knowledge of the chemical composition as a constraint on the composition into the refinement. We were recently able to elucidate the reaction mechanism for the later stages of the ammonolysis reaction and can therefore determine the chemical composition from XRF measurements with good confidence.33 Two important conclusions can be drawn from the reaction model: the chemical composition of the full compound is only dependent on one variable x and the crystallographic sites are fully occupied, since the cation-to-anion ratio in Zn1+xGe1−x(N1−xOx)2 is always 1:1. An important drawback is the fact that the results for multiple phase samples, in which traces of decomposition products, such as Ge and Ge3N4, are present cannot be treated in this way, since the chemical composition of the whole sample is no longer indicative for the ternary oxide nitride phase. Using this strategy, we have achieved stable refinements throughout the series of single-phase products studied. Fig. 3 shows exemplarily the resulting cation distribution for a series of Zn1+xGe1−x(N1−xOx)2 compounds. It has to be noted that there is always some Ge on the Zn site.
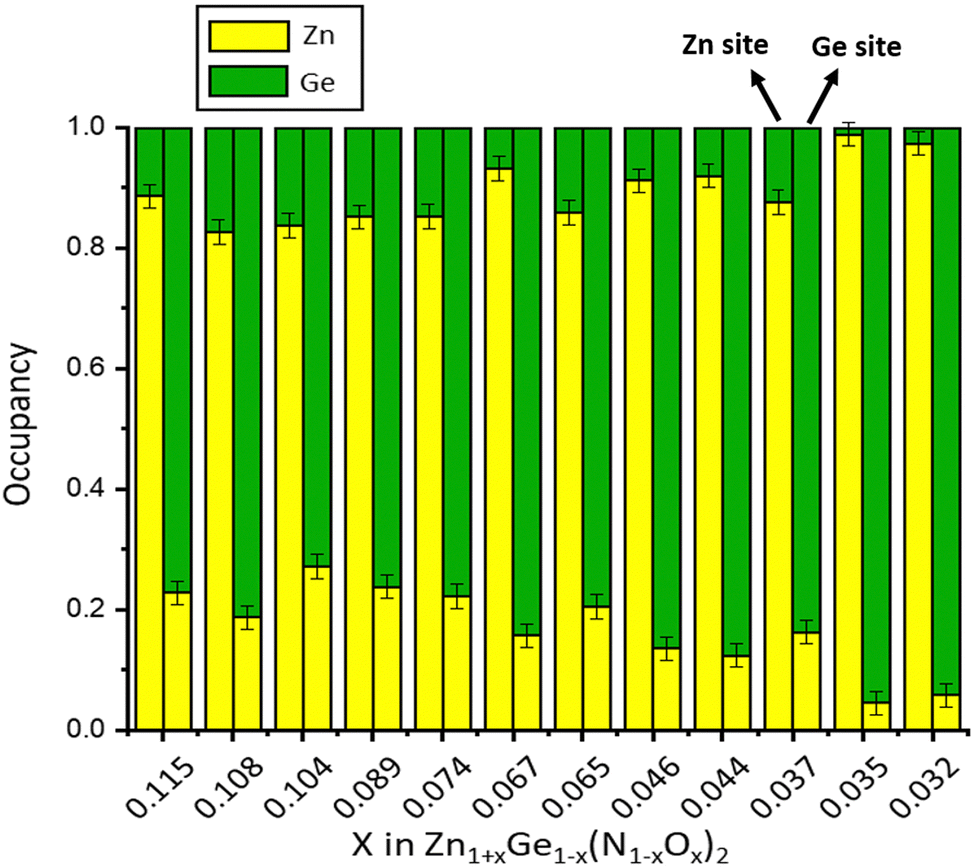 | ||
| Fig. 3 Cation distributions determined by the simultaneous refinement of neutron and X-ray powder diffraction data for a series of Zn1+xGe1−x(N1−xOx)2 compounds. The x-axis shows the oxygen content, and the y-axis shows the cation site occupancy. | ||
Structural studies of Zn1+xGe1−x(N1−xOx)2
Are extrinsic and intrinsic disorders correlated?
As shown in Fig. 4, the oxygen content of the synthesised Zn1+xGe1−x(N1−xOx)2 compounds can be tuned by the synthesis conditions: reaction time (t) and reaction temperature (T). | ||
| Fig. 4 Oxygen content x in Zn1+xGe1−x(N1−xOx)2 compounds in dependence on the synthesis conditions (reaction time t and reaction temperature). The different reaction temperature series are colour-coded. | ||
If intrinsic (cation swapping) and extrinsic (compositional) disorders would go hand-in-hand, there should be a clear trend in the plot of intrinsic disorder  vs. the oxygen content. The latter corresponds to extrinsic disorder, i.e. ZnGe, that is commensurate with the oxygen content. It is evident on the first glance that there is no such correlation in the data (Fig. 5).
vs. the oxygen content. The latter corresponds to extrinsic disorder, i.e. ZnGe, that is commensurate with the oxygen content. It is evident on the first glance that there is no such correlation in the data (Fig. 5).
 | ||
Fig. 5 Plot of the intrinsic disorder  versus oxygen content x for Zn1+xGe1−x(N1−xOx)2 compounds synthesized at different temperatures. versus oxygen content x for Zn1+xGe1−x(N1−xOx)2 compounds synthesized at different temperatures. | ||
While there appears to be a general trend in the sense that the high oxygen content, which corresponds to high extrinsic disorder, tends to go hand in hand with high intrinsic disorder, and the samples are pretty scattered, signifying that there is no causal relationship between both. Also, there are Zn1+xGe1−x(N1−xOx)2 compounds with the same oxygen content x but different values of intrinsic disorder 
This is a most significant point, since it not only proves that both types of disorder exist, but also that they are not correlated to each other. Therefore, both effects can be regarded independent of each other and we look at the relationship between the intrinsic disorder and the reaction conditions in order to explore, whether the intrinsic disorder can be tuned through the reaction conditions. Still, one has to bear in mind that for the oxide nitride system, both effects exist at the same time, and hence need to be regarded simultaneously. This influences the trends and a strictly mathematical trend can only be expected for either, if the other does not influence this specific parameter.
Order parameter
The Zn1+xGe1−x(N1−xOx)2 compounds synthesised crystallize either in the β-NaFeO2-type structure and show different degrees of cation disorder (as shown in Fig. 5) or in the wurtzite-type structure with a statistic distribution of the cations and hence full cation disorder. To describe the different levels of cation disorder, an order parameter has been introduced. The order parameter is equal to 1 for full order (no intrinsic disorder, i.e. ) and equal to 0 for full disorder
) and equal to 0 for full disorder  and is calculated according to
and is calculated according to | (1) |
 describes the intrinsic disorder and Ge is the Ge-content in Zn1+xGe1−x(N1−xOx)2. Different levels of cation disorder correspond to different values of the order parameter and can be tuned by the synthesis conditions (see Fig. 6).
describes the intrinsic disorder and Ge is the Ge-content in Zn1+xGe1−x(N1−xOx)2. Different levels of cation disorder correspond to different values of the order parameter and can be tuned by the synthesis conditions (see Fig. 6).
 | ||
| Fig. 6 Order parameter in dependence on the synthesis conditions reaction time t and reaction temperature. The different reaction temperature series are colour-coded. | ||
Influences on the lattice constants
While the hexagonal crystal system of the wurtzite-type structure restricts the lattice parameters a and b to be equal, this restriction is lifted in the orthorhombic β-NaFeO2-type structure. The distortion of the lattice in the latter case can, therefore, be conveniently described by the ratio of these lattice constants. Since a rather complex change of the unit cells occurs in this group–subgroup relationship,27 we first calculate pseudo-hexagonal lattice constants from the orthorhombic lattice constants as demonstrated previously.33 The lattice distortion a is calculated according to | (2) |
Again, there is an overall trend between the lattice distortion and both extrinsic and intrinsic disorders, but this trend is not straightforward enough to allow the prediction of any variable on the basis of another (see Fig. 7 and 8). This is most probably due to the fact that both disorder effects affect the lattice distortion in a similar manner.
 | ||
Fig. 7 Plot of the lattice distortion α versus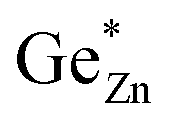 . . | ||
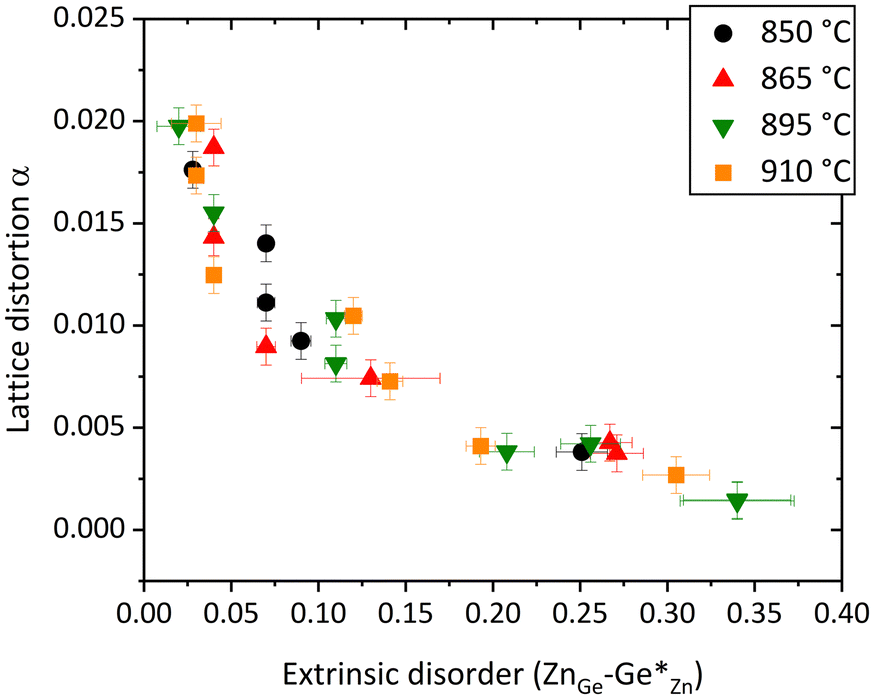 | ||
Fig. 8 Plot of the lattice distortion α versus the extrinsic disorder 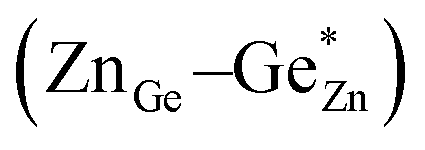 . . | ||
Intrinsic disorder and reaction conditions
Theoretical studies agree that the ordered state is energetically more favourable than any disordered state in this system but with different energy differences.23,25,31 At a first glance, one could therefore assume that the higher thermal energy at higher temperatures would provoke a higher degree of disorder in the final product, but this is not the case in our experiments. Instead, there is a general trend that samples with comparable reaction times exhibit less disorder when they are produced at higher temperatures (Fig. 6). The higher thermal activation allows the overcoming of the activation energy for cation swapping and the energetic difference between the ordered and disordered states appears to be high enough to lock the cations in their energetically favoured environment.Furthermore, this trend is even clearer for the dwelling times. Longer times at the same reaction temperature produce smaller levels of intrinsic disorder. The combination of the reaction time and the reaction temperature therefore allows directing the reaction into one particular composition. We found that the reaction conditions producing the smallest amount of intrinsic disorder in our experiments are 865 °C and 18.3 h of dwelling times with  . This sample is, within the experimental error, completely ordered Zn1+xGe1−x(N1−xOx)2. It is worth noting that this sample is also with the lowest extrinsic disorder, but has a nominal composition of Zn1.035(9)Ge0.965(9)(N0.965(9)O0.035(9))2 and is hence not completely oxygen free. Since traces of oxygen are very often found in nitride materials, it is an important information that even with such small amounts of oxygen, the material can be virtually fully ordered.
. This sample is, within the experimental error, completely ordered Zn1+xGe1−x(N1−xOx)2. It is worth noting that this sample is also with the lowest extrinsic disorder, but has a nominal composition of Zn1.035(9)Ge0.965(9)(N0.965(9)O0.035(9))2 and is hence not completely oxygen free. Since traces of oxygen are very often found in nitride materials, it is an important information that even with such small amounts of oxygen, the material can be virtually fully ordered.
Cumulating the information gained under different reaction conditions allows an experimental window to be defined, in which a variable degree of disorder can be achieved (Fig. 9). It is evident that the tuneable time window is much smaller at higher temperatures than at lower temperatures. Using higher temperatures may therefore be a key to achieving full order efficiently, while lowering the temperature allows for finer tuning of the degree of disorder. The determination of the reaction window is therefore highly beneficial for the directed conduction of the reaction in further experiments.
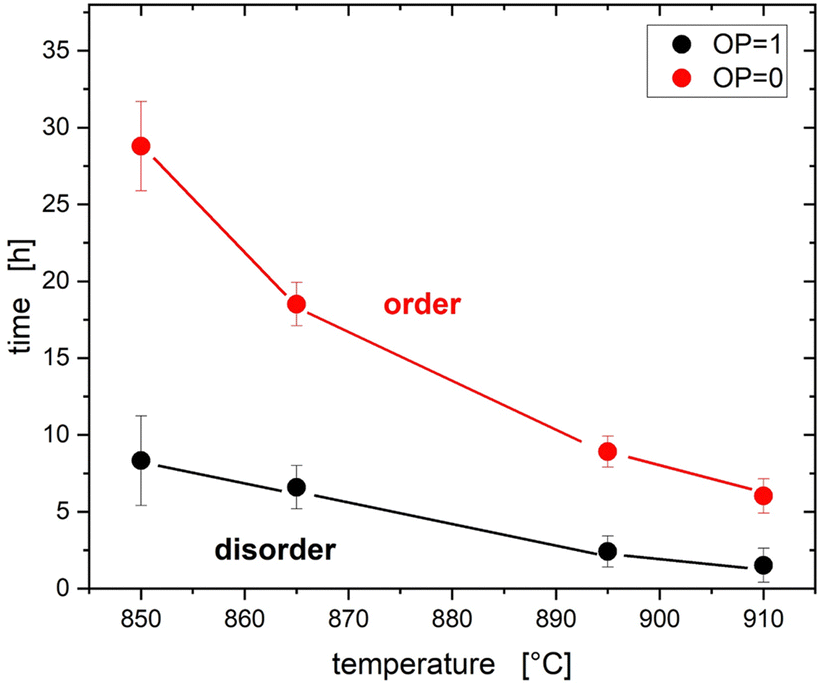 | ||
| Fig. 9 Overview of intrinsic disorder and reaction conditions. Between the lines for full disorder (OP = 1) and order (OP = 0) the level of cation disorder can be tuned by the reaction conditions. | ||
Bandgap energy trends
The bandgap energy values tend to become the lower, the higher the oxygen content and the extrinsic cation disorder is (Fig. 10). This trend is similar to the previous data acquired using different synthesis methods, with varying degrees of oxygen composition.45 As outlined, however, the chemical composition is not sufficient to characterise the sample, since intrinsic and extrinsic disorders are not strictly correlated. All synthesized Zn1+xGe1−x(N1−xOx)2 compounds lie in a band gap energy range of 2.7–3.5 eV, which also is very much in line with previous studies. | ||
| Fig. 10 Oxygen content x in Zn1+xGe1−x(N1−xOx)2versus the optical bandgap Eg. Open symbols refer to Zn1+xGe1−x(N1−xOx)2 with full cation disorder. The star marks the Zn1+xGe1−x(N1−xOx)2 compound with the lowest degree of cation disorder. The triangle shows the theoretical value [23]. | ||
A key question in this study was the question, as to whether the intrinsic disorder really affects the bandgap energy in the way it was predicted theoretically. As with many of the other values, there are some degrees of scattering of values around the general trend that the bandgap energy decreases with the decreasing order parameter (Fig. 11). Therefore, the extrinsic disorder (Fig. 10) and intrinsic disorder (Fig. 11) both affect the optical bandgap in a similar manner, but independently of each other. Since both effects are heavily intertwined, the exact reaction conditions need to be precisely met, in order to reliably produce a distinct composition with a certain disorder and hence an exact bandgap.
 | ||
| Fig. 11 Order parameter versus optical bandgap Eg. The star marks the Zn1+xGe1−x(N1−xOx)2 compound with nearly full cation order. | ||
The situation becomes much clearer when considering the trend of the optical bandgap within time series at distinct temperatures (Fig. 12). A correlation is very clear in these series, which is in line with thermodynamic considerations outlined earlier. This indicates that the prolongation of the reaction time monotonically influences the intrinsic disorder rather than the extrinsic disorder. Therefore, the level of intrinsic disorder can be influenced straightforwardly through the choice of the reaction temperature. In combination with the choice of the appropriate reaction time, a targeted formulation of composition and intrinsic disorder can be achieved. However, since the reaction is strongly kinetically influenced, the concrete combinations will be highly dependent on the amount of the material produced and hence should be adjusted to the system in use.
 | ||
| Fig. 12 Trend of the optical bandgaps as a function of the dwelling times at different reaction temperatures. | ||
Conclusions
We first demonstrated the direct experimental evidence of cation swapping in the ternary nitride (oxide nitride) system Zn1+xGe1−x(N1−xOx)2, which was previously predicted computationally. Using a combination of X-ray and neutron diffraction, we showed that not only intrinsic cation disorder in the form of cation swapping exists in these compounds, but also a second form of disorder exists, which is not directly correlated with the first. This extrinsic disorder is directly related to the oxygen content and hence to the composition of the compound. Both disorder types influence the bandgap energy in similar, but unrelated ways. Using structural evidence, we therefore show for the first time that the predicted bandgap tuning mechanism through cation swapping truly exists in these materials. Further studies in the future will be revealing the individual contributions of the different types of disorder to ideally allow the individual tuning of both types independently of each other.Author contributions
Su.S. conceived the initial idea for the study. J.B., Su.S. and Z.W. developed the experimental plan and designed the experiments. Z. W. and J. B. conducted the synthesis experiments. J.B., Z.W., St.S., A.F. and Su.S. conducted the neutron diffraction experiments. Z.W. performed the data refinement. Su.S., J.B. and Z.W. performed the data interpretation. J.B. wrote the manuscript with input from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Z.W. would like to acknowledge the MatSEC graduate school of HZB for PhD studentship. Helmholtz-Zentrum Berlin, Institut Laue-Langevin and Paul-Scherrer Institut are acknowledged for the provision of beamtime under proposal numbers 182-07291-EF/R (HZB), 5-21-1126, EASY-601, EASY-602, EASY-1012, and EASY-1013 (ILL). The authors would like to thank Galina Gurieva for assistance with the neutron measurements.Notes and references
- T. Matsuoka, Int. J. Optomechatronics, 2015, 9, 1–8 CrossRef.
- P. Von Dollen, S. Pimputkar and J. S. Speck, Angew. Chem., Int. Ed., 2014, 53, 13978–13980 CrossRef CAS PubMed.
- I. Akasaki, Angew. Chem., 2015, 127, 7860–7873 CrossRef.
- J. Heber, Nat. Phys., 2014, 10, 791 Search PubMed.
- N. Lu and I. Ferguson, Semicond. Sci. Technol., 2013, 28, 074023 CrossRef.
- D. Shin, B. Saparov and D. B. Mitzi, Adv. Energy Mater., 2017, 7, 1602366 CrossRef.
- A. A. Yaroshevsky, Geochem. Int., 2006, 44, 48–55 CrossRef.
- D. M. Többens, G. Gurieva, S. Levcenko, T. Unold and S. Schorr, Phys. Status Solidi B, 2016, 253, 1890–1897 CrossRef.
- S. Schorr, G. Gurieva, M. Guc, M. Dimitrievska, A. Pérez-Rodríguez, V. Izquierdo-Roca, C. S. Schnohr, J. Kim, W. Jo and J. M. Merino, J. Phys. Energy, 2019, 2, 012002 CrossRef.
- L. E. Valle Rios, K. Neldner, G. Gurieva and S. Schorr, J. Alloys Compd., 2016, 657, 408–413 CrossRef CAS.
- J. Breternitz, S. Levcenko, H. Hempel, G. Gurieva, A. Franz, A. Hoser and S. Schorr, J. Phys. Energy, 2019, 1, 025003 CrossRef CAS.
- F. Igbari, Z.-K. Wang and L.-S. Liao, Adv. Energy Mater., 2019, 9, 1803150 CrossRef.
- N. C. Coronel, L. Lahourcade, K. T. Delaney, A. M. Shing and H. A. Atwater, Earth-Abundant ZnSnxGe1–xN2 Alloys as Potential Photovoltaic Absorber Materials. In 2012 38th IEEE Photovoltaic Specialists Conference; 2012; pp 003204–003207. https://doi.org/10.1109/PVSC.2012.6318259.
- A. L. Greenaway, C. L. Melamed, M. B. Tellekamp, R. Woods-Robinson, E. S. Toberer, J. R. Neilson and A. C. Tamboli, Annu. Rev. Mater. Res., 2021, 51, 591–618 CrossRef CAS.
- W. Sun, C. J. Bartel, E. Arca, S. R. Bauers, B. Matthews, B. Orvañanos, B.-R. Chen, M. F. Toney, L. T. Schelhas, W. Tumas, J. Tate, A. Zakutayev, S. Lany, A. M. Holder and G. Ceder, Nat. Mater., 2019, 18, 732–739 CrossRef CAS PubMed.
- T. D. Veal, N. Feldberg, N. F. Quackenbush, W. M. Linhart, D. O. Scanlon, L. F. J. Piper and S. M. Durbin, Adv. Energy Mater., 2015, 5, 1501462 CrossRef.
- N. Feldberg, J. D. Aldous, W. M. Linhart, L. J. Phillips, K. Durose, P. A. Stampe, R. J. Kennedy, D. O. Scanlon, G. Vardar, R. L. Field, T. Y. Jen, R. S. Goldman, T. D. Veal and S. M. Durbin, Appl. Phys. Lett., 2013, 103, 042109 CrossRef.
- J. J. Cordell, J. Pan, A. C. Tamboli, G. J. Tucker and S. Lany, Phys. Rev. Mater., 2021, 5, 024604 CrossRef CAS.
- E. W. Blanton, K. He, J. Shie and K. Kash, J. Cryst. Growth, 2017, 461, 38–45 CrossRef CAS.
- M. S. Haseman, M. R. Karim, D. Ramdin, B. A. Noesges, E. Feinberg, B. H. D. Jayatunga, W. R. L. Lambrecht, M. Zhu, J. Hwang, K. Kash, H. Zhao and L. J. Brillson, J. Appl. Phys., 2020, 127, 135703 CrossRef CAS.
- D. Skachkov, P. C. Quayle, K. Kash and W. R. L. Lambrecht, Phys. Rev. B, 2016, 94, 205201 CrossRef.
- D. Skachkov, A. P. Jaroenjittichai, L. Huang and W. R. L. Lambrecht, Phys. Rev. B, 2016, 93, 155202 CrossRef.
- P. C. Quayle, E. W. Blanton, A. Punya, G. T. Junno, K. He, L. Han, H. Zhao, J. Shan, W. R. L. Lambrecht and K. Kash, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 205207 CrossRef.
- A. Punya, T. R. Paudel and W. R. L. Lambrecht, Phys. Status Solidi C, 2011, 8, 2492–2499 CrossRef CAS.
- S. Lany, A. N. Fioretti, P. P. Zawadzki, L. T. Schelhas, E. S. Toberer, A. Zakutayev and A. C. Tamboli, Phys. Rev. Mater., 2017, 1, 035401 CrossRef.
- J. Breternitz, Z. Wang, A. Glibo, A. Franz, M. Tovar, S. Berendts, M. Lerch and S. Schorr, Phys. Status Solidi A, 2019, 216, 1800885 CrossRef.
- J. Breternitz and S. Schorr, Acta Crystallogr., Sect. A: Found. Adv., 2021, 77, 208–216 CrossRef CAS PubMed.
- P. C. Quayle, Acta Crystallogr., Sect. A: Found. Adv., 2020, 76, 410–420 CrossRef CAS PubMed.
- P. C. Quayle and J. Breternitz, Acta Crystallogr., Sect. A: Found. Adv., 2021, 77, 217–221 CrossRef CAS PubMed.
- W. R. L. Lambrecht, E. Alldredge and K. Kim, Phys. Rev. B, 2005, 72, 155202 CrossRef.
- C. L. Melamed, J. Pan, A. Mis, K. Heinselman, R. R. Schnepf, R. Woods-Robinson, J. J. Cordell, S. Lany, E. S. Toberer and A. C. Tamboli, J. Mater. Chem. C, 2020, 8, 8736–8746 RSC.
- C. L. Melamed, M. B. Tellekamp, J. S. Mangum, J. D. Perkins, P. Dippo, E. S. Toberer and A. C. Tamboli, Phys. Rev. Mater., 2019, 3, 051602 CrossRef CAS.
- Z. Wang, D. Fritsch, S. Berendts, M. Lerch, J. Breternitz and S. Schorr, Chem. Sci., 2021, 12, 8493–8500 RSC.
- V. F. Sears, Neutron News, 1992, 3, 26–37 CrossRef.
- R. Viennois, T. Taliercio, V. Potin, A. Errebbahi, B. Gil, S. Charar, A. Haidoux and J.-C. Tédenac, Mater. Sci. Eng. B, 2001, 82, 45–49 CrossRef.
- P. Kubelka and F. Munk, Z. Techn. Phys., 1931, 12, 593–601 Search PubMed.
- Y. Feng, S. Lin, S. Huang, S. Shrestha and G. Conibeer, J. Appl. Phys., 2015, 117, 125701 CrossRef.
- J. Tauc, Mater. Res. Bull., 1968, 3, 37–46 CrossRef CAS.
- A. Franz and A. Hoser, J. Large-Scale Res. Facil., 2017, 3, 103 CrossRef.
- A. W. Hewat, Materials Science Forum, 1986, vol. 9, pp. 69–80 Search PubMed.
- Helmholtz-Zentrum für Materialien und Energie, J. Large-Scale Res. Facil., 2016, 2, A49.
- E. A. Merritt, X-ray Anomalous Scattering, http://skuld.bmsc.washington.edu/scatter/, accessed June 2014 Search PubMed.
- J. Rodruigez-Carvajal, Fullprof.2k. 2012: ILL-JRC.
- P. C. Schmidt and A. Weiss, Z. Naturforsch. A, 1979, 4, 1471–1481 Search PubMed.
- R. R. Schnepf, J. J. Cordell, M. B. Tellekamp, C. L. Melamed, A. L. Greenaway, A. Mis, G. L. Brennecka, S. Christensen, G. J. Tucker, E. S. Toberer, S. Lany and A. C. Tamboli, ACS Energy Lett., 2020, 5, 2027–2041 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Table of experimental results. See DOI: https://doi.org/10.1039/d3tc02650g |
| ‡ Current address: FH Münster University of Applied Sciences, Stegerwaldstr. 39, 48565, Steinfurt, Germany. |
| This journal is © The Royal Society of Chemistry 2024 |