DOI:
10.1039/D4TB00639A
(Paper)
J. Mater. Chem. B, 2024,
12, 10923-10933
A radical containing micellar probe for assessing esterase enzymatic activity with ultra-low field Overhauser-enhanced magnetic resonance imaging†
Received
26th March 2024
, Accepted 13th September 2024
First published on 16th September 2024
Abstract
The ability to track altered enzyme activity using a non-invasive imaging protocol is crucial for the early diagnosis of many diseases but is often challenging. Herein, we show that Overhauser magnetic resonance imaging (OMRI) can be used to monitor enzymatic conversion at an ultra-low field (206 μT) using a highly sensitive “off/on” probe with a nitroxide stable radical containing ester, named T2C12–T80. This TEMPO derivative containing probe forms stable electron paramagnetic resonance (EPR) silent micelles in water that are hydrolysed by esterases, thus yielding narrow EPR signals whose intensities correlate directly with specific enzymatic activity. The responsiveness of the probe to tumours, facilitated by increased esterase activity, was initially determined by comparing EPR signals measured upon incubation with 3T3 (healthy fibroblasts used as control), HepG2 (human hepatoma) and Hs766T (human pancreatic cancer cells) cell lysates and then with Hs766T and 3T3 living cells. Next, Overhauser MR images were detected on a phantom containing the probe and the esterases to show that the approach is well suited for being translated to the in vivo detection at the earth's magnetic field. Regarding detection sensitivity, ultra-low field OMRI (ULF-OMRI) is beneficial over OMRI at higher fields (e.g. 0.2 T) since Overhauser enhancements are significantly higher and the technique is safe in terms of the specific absorption rate.
Introduction
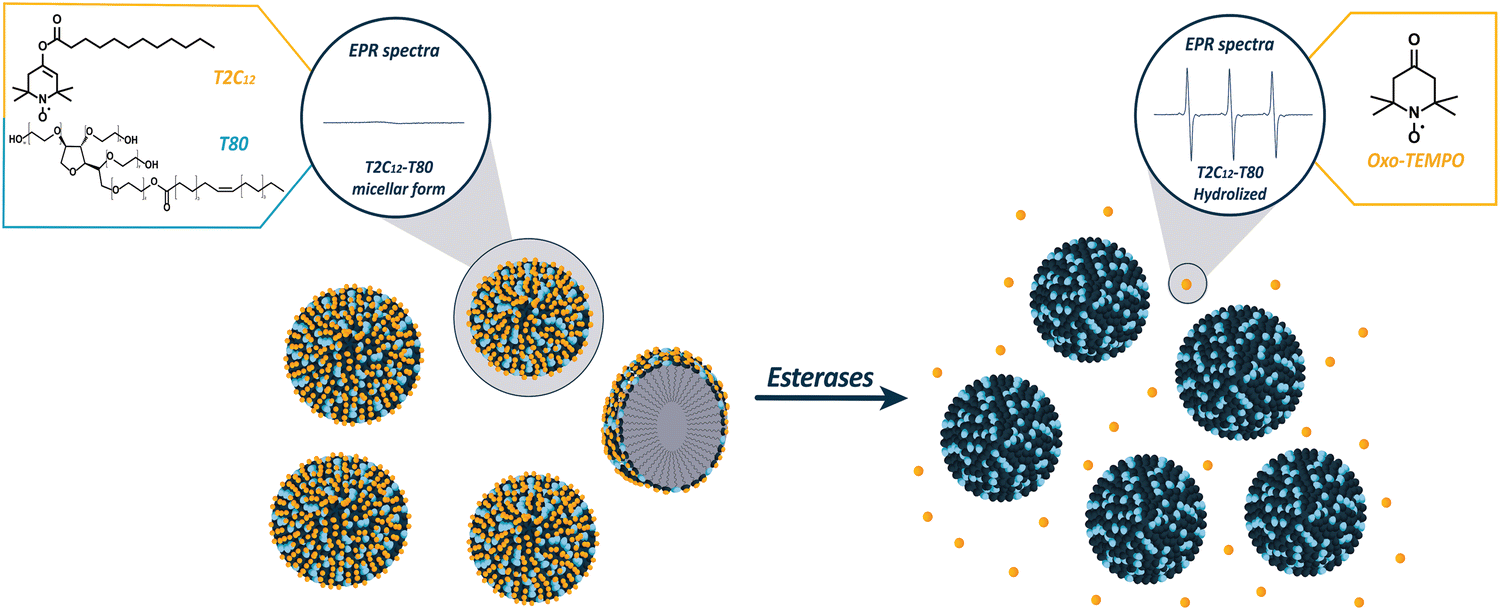
Quantitative detection and monitoring of enzymatic activity play a crucial role in diagnostic medicine by enabling a greater understanding of disease processes, early detection and tailored treatment strategies, leading to the development of novel diagnostic and therapeutic tools.1 Despite its immense diagnostic potential, rapid, sensitive, and high-resolution detection of enzymatic activity in vivo, particularly in relatively deep tissues, remains a significant challenge. Optical imaging modalities that are widely used to assess enzyme activities in cells and animal models are hampered in vivo by well-known issues associated with the penetration and scattering of light by animal tissues.2,3 Analogous PET and SPECT techniques, although endowed with excellent sensitivity, cannot be exploited to monitor chemical transformations in which the radioactive element is the same in the parent and daughter species.4–6 Alternatively, magnetic resonance imaging (MRI), with or without the involvement of exogenous contrast agents,7,8 provides the required properties of high resolution and tissue penetration, but its limited detection sensitivity may represent a drawback. Moreover, it often necessitates the use of complex ratiometric protocols as the detection method frequently entails measuring a change in an NMR parameter (T1, T2, and CEST response) inherently affected by the concentration of the contrast agent. Therefore, it is evident that there is a need to explore innovative methodologies that are capable of addressing sensitivity and tissue penetration as well as are, ideally, accessible at costs, thus permitting widespread access. Low-field and ultra-low-field MRI technologies have recently gained significant interest as an emerging field owing to their unique advantages, such as portability and low cost.9–12 Although low-field MRI has several advantages, it has certain limitations, including a lower signal-to-noise ratio and reduced spatial resolution compared to high-field MRI. One strategy to increase sensitivity is to use dynamic nuclear polarization (DNP), also known as the Overhauser effect. This technique exploits the dipole–dipole interaction between the electron and the nuclear spin, which, upon saturating the Zeeman states of the unpaired electrons, leads to a polarization transfer to the protons in their proximity that become hyperpolarized.13–15 In MRI, this approach has often been deemed Overhauser MRI (OMRI). When the catalytic conversion of a radical containing substrate into the reaction product is accompanied by a change in the EPR signal, OMRI can act as a reporter of the ongoing enzymatic activity. Using this diagnostic strategy, the activity of some enzymes, such as proteases,16,17 elastases,18 and phospholipases,19 have been detected in vitro and in vivo on mice with inflamed lungs20,21 on a low field MRI system operating at 0.19 T. The main drawback of this approach is the EPR irradiation that occurs at microwave wavelengths (GHz or above), thus generating harmful tissue heating and shallow wave penetration. Therefore, the in vivo translation of OMRI to larger animals and humans must imply the use of lower fields (<1 mT). Interestingly, when radical species with hyperfine coupling are employed, it has been demonstrated both theoretically and empirically that the DNP enhancement factor increases as the strength of the static magnetic field decreases.22,23 This effect can be exploited to partially compensate for the loss of the NMR signal at very low fields, paving the way for molecular imaging studies using OMRI. In this context, the University of Bordeaux developed a home-made earth-field double resonance system capable of producing Overhauser-enhanced MR images in living rats by in situ DNP at 206 μT (i.e. very close to the earth's magnetic field) using stable and non-toxic nitroxides.24 Using this system, the protease-driven hydrolysis of a specific nitroxide probe was detected in a preliminary spectroscopy-based approach in which the enzymatic transformation involved a net shift in the EPR frequencies of the parent and daughter species.25 In this context, we deemed it of interest to design an off/on OMRI agent that can report on a specific enzymatic activity. Recently, our group synthesized esters containing TEMPO derivatives, namely TEMPO-C12 (TC12) and TEMPO-2-C12 (T2C12), which formed stable micelles in water, resulting in EPR silence.26 However, narrow and intense EPR signals are produced following the release of the free TEMPO radicals due to the hydrolysis of the ester bond catalysed by esterases, whose activity is directly correlated with the intensity of the observed EPR signals. Esterases, a subclass of the hydrolase enzyme superfamily, are responsible for hydrolysing ester bonds27 in various substrates and are involved in the reprogramming of metabolic pathways, promotion of cancer pathogenesis, drug metabolism and drug toxicity.28,29 Therefore, the detection of esterase activity appears to be a valuable tool for assessing various biological processes, diagnosing and monitoring disease progression and the effect of undertaken therapies.30–32 Our preliminary results showed that the off/on T2C12 probe, upon the hydrolysis of the ester bond, displayed narrow peaks because of the release of small-sized, highly mobile nitroxide radicals in principle suitable for OMRI applications.26 In the present study, a further improvement of the probe sensitivity has been pursued using a new formulation of T2C12 with Tween 80 (T80) that allows the formation of smaller micelles endowed with faster hydrolysis kinetics in the presence of the target enzymes (Fig. 1). By exploiting the DNP effect induced through the radiofrequency saturation of the EPR signal, it was possible to detect, for the first time, a marked enzyme-dependent enhancement in the OMRI image arising from the Overhauser effect at the ultra-low magnetic field strength of 206 μT, i.e. at a value close to earth field (25–65 μT). The enhanced signal acts as an efficient reporter of the enzymatic activity, potentially enabling detection in living subjects. Without DNP, the presence of nitroxide would not have been able to generate any enhancement effect in conventional NMR/MRI experiments because only the application of DNP allows an increase in proton polarization in media containing 4-oxo-TEMPO.
 |
| | Fig. 1 Schematic representation of the EPR silent T2C12–T80 probe in the micellar form, followed by hydrolyzation in the presence of esterases and release of oxo-TEMPO, which display an intense EPR signal. | |
Results and discussion
Mixed micelle formulation, characterization, and enzymatic assays
In our previous work,26in vitro experiments with T2C12 micelles and enzymes showed incomplete hydrolysis of the probe. Thus, a novel formulation employing the non-ionic surfactant Tween 80 (T80) was explored to diminish the size of the aggregated nanostructures and boost the performance of the radical containing probe. T2C12 mixed micelles were prepared by simply dissolving T2C12 in DMSO, then diluting the solution in a HEPES-buffered saline medium containing Tween 80 and sonicating the solution using an ultrasonic homogenizer. Two concentrations of T80, 10 mol% and 20 mol%, were considered. In both cases, the mixture of T2C12 and Tween 80 self-assembled into colloidal nanoaggregates characterized by enhanced stability and reduced size.33 The EPR spectra of both formulations exhibited very low and broad EPR signals identical to those previously observed for the T2C12 probe (Fig. S1, ESI†). The micelles made with 20 mol% (T2C12–T80) showed greater stability, with hydrodynamic diameters of about 30–40 nm, which remained below 70 nm when measured after 1 hour from the sonication step. Conversely, when the T80 concentration was brought to 10 mol% (T2C12–T8010), the mixture resulted in reduced micelle stability, as their hydrodynamic diameter increased to 120 nm after 1 hour from sonication. Notably, the diameter of micelles prepared with only T2C12 without T80 was significantly higher (>300 nm) (Fig. 2). Additional characterization with FESEM confirmed the morphology and size of the nanoparticles (Fig. S2, ESI†) and DLS measurements showed neutral zeta potential values with the T2C12–T80 and T2C12–T8010 (Fig. S3, ESI†). The ability of the micellar radical probes to act as a reporter of the enzymatic activity was initially tested with the porcine liver esterases (PLE), and both formulations with 10 mol% and 20 mol% T80 revealed almost complete hydrolysis (>90%), at a significantly faster rate when compared to the micelles without T80 (Fig. 3). Then, a different class of esterases, namely human carboxylesterases (CEs), was explored. The two major isoforms, carboxylesterases 1 (CEs1) and carboxylesterases 2 (CEs2),34 are highly expressed under many pathologic conditions.35,36 As found in our previous study with T2C12,26 the mixed micelles displayed a much higher efficiency with CEs2 than CEs1, demonstrating a nearly complete and faster hydrolysis of T2C12, which confirms the superiority of the new formulation (Fig. 4).
 |
| | Fig. 2 (left) Table with hydrodynamic diameters of T2C12 100 μM, T2C12–T8010 100 μM and T2C12–T80 100 μM measured via DLS 5 min and 1 hour after the sonication step. (right) Number-weighted average size determined for T2C12 100 μM, T2C12–T8010 100 μM and T2C12–T80 100 μM micelles via DLS measurement after 1 hour from the sonication step. | |
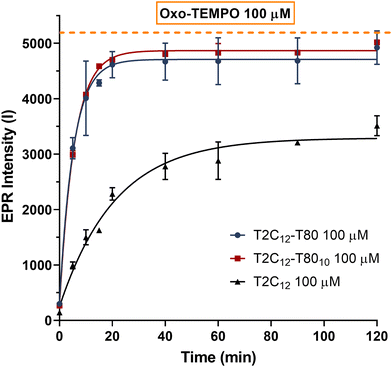 |
| | Fig. 3 EPR enzymatic assays with 0.5 U mL−1 of PLE in the presence of T2C12 100 μM, T2C12–T8010 100 μM or T2C12–T80 100 μM micelles maintained at 37 °C under stirring at 400 rpm. The continuous line is drawn to better show the course of the points. | |
 |
| | Fig. 4 (A) EPR enzymatic assays with 12 U mL−1 of CEs2 in the presence of T2C12 100 μM, T2C12–T8010 100 μM and T2C12–T80 100 μM. (B) Enzymatic assays of T2C12 100 μM, T2C12–T8010 100 μM and T2C12–T80 100 μM with CEs1 12 U mL−1. All kinetics were performed at 37 °C with stirring at 400 rpm, and the continuous line was drawn to better show the course of the points. | |
Micelle stability
The mixed micelles stability was evaluated in human serum (HS) and a buffered solution of human serum albumin (HSA). Fig. 5 shows that a similar hydrolytic behaviour occurred in serum and HSA containing medium, where the protein is at the same concentration found in serum (40 mg mL−1). Thus, the modest hydrolysis observed in HS is likely due to the slight esterolytic activity of HSA as reported by Ascenzi et al.37–39 Even though the physiological significance of HSA esterolytic activity and its natural substrates remain unclear, HSA can hydrolyse various compounds, including40 long- and short-chain fatty acid esters, potentially influencing various pharmacokinetic or toxicokinetic processes.38 The T80-containing mixed micelles showed a significantly faster hydrolysis rate in serum compared to T2C12 alone. Despite the reduced stability of the micelles in serum, due to the addition of T80, the hydrolysis kinetics remained remarkably faster in the presence of specific esterases (Fig. 6). After 1 h of incubation, T2C12–T80 in the presence of PLE and CEs2 produced a much higher EPR signal compared to the ones observed in human serum alone (Fig. 6). Therefore, the formulation with 20 mol% of T80 (T2C12–T80), due to its smaller size and slightly improved stability in HS and HSA-containing solutions, was selected for the following in vitro experiments.
 |
| | Fig. 5 (A) Incubation of T2C12 100 μM, T2C12–T8010 100 μM and T2C12–T80 100 μM with human serum. (B) Incubation of T2C12 100 μM, T2C12–T8010 100 μM and T2C12–T80 100 μM with HAS containing medium 40 mg mL−1. The assays were performed at 37 °C under stirring at 400 rpm and measured after 3 min, 1 h and 2 h of incubation. The results are reported as a percentage of hydrolyzation (% hydrolyzation) that was calculated using a calibration curve of 4-oxo-TEMPO (Fig. S4, ESI†). | |
 |
| | Fig. 6 EPR intensity detected upon 1 h incubation of T2C12 100 μM + T80 20 μM with PLE, CEs2 and human serum at 37 °C under stirring at 400 rpm. | |
Because biological systems require a reducing environment to maintain the redox status of living cells, we examined the resistance to the reduction of the nitroxide radicals within the micelles. This evaluation occurred in the presence of ascorbic acid (AC), which is an abundant molecule in the bloodstream.41 As already reported, nitroxide radicals are generally not stable in a reducing environment.42,43 In fact, with AC 1 mM, a complete reduction of the parent 4-oxo-TEMPO to the respective EPR silent hydroxylamine was observed in only few min. (Fig. 7). Interestingly, T2C12–T80 micelles exhibited significantly enhanced radical stability when incubated with AC 1 mM, with only an 11 ± 6% and 60% reduction in the EPR signal after 20 minutes and 2 hours (Fig. S5, ESI†), respectively. The stability was also investigated at higher micelle concentrations (2 mM), which was used in the below OMRI experiments. In the presence of human serum (Fig. 8A), minor hydrolyzation of the probe (12% compared to PLE) was observed after 1 hour of incubation. However, a slower but complete kinetic was detected in the presence of PLE, confirming the ability of the probe to assess the esterase activity despite slight hydrolyzation by the serum. Although in the presence of 0.1 mM and 0.2 mM of AC (Fig. 8B), corresponding to the endogenous concentrations present in plasma and tissue, respectively,44 only a 16% reduction in the free radical released following the hydrolyzation was noted. This observation underscores the remarkable stability of radicals when encapsulated within micelles, even in the presence of reducing agents, underlining a clear advantage for their utilization in in vivo studies.
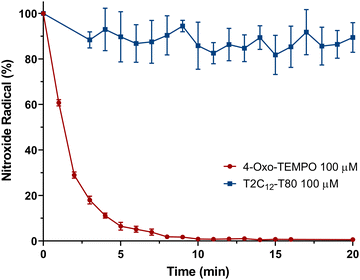 |
| | Fig. 7 Residual nitroxide radical (%) of 4-oxo-TEMPO 100 μM (red) and T2C12–T80 100 μM (blue) in the presence of ascorbic acid 1 mM at room temperature for 20 min. | |
 |
| | Fig. 8 Stability assessment with T2C12–T80 2 mM. (A) Incubation of the micelle probe HS and enzymatic kinetics with PLE 10 U mL−1 in HEPES-buffered saline solution or in human serum. (B) Enzymatic kinetics with PLE 10 U mL−1 in the absence and presence of ascorbic acid of 0.1 and 0.2 mM. All the assays were performed at room temperature. | |
In vitro esterase assessment
To evaluate the expression of esterases under different pathologic conditions, preliminary tests were performed using cytosolic cell extracts of the human pancreatic cancer (Hs766T), human hepatocarcinoma (HepG2) and fibroblast (3T3) cell lines. Hs766T and HepG245–48 were chosen as tumour models due to the significantly higher levels of cytosolic esterases compared to healthy cells, represented in this study by 3T3 fibroblast cells.49,50 To validate the reported higher levels of cytosolic esterases in cancer cells compared to healthy cells, a fluorometric assay based on fluorescein diacetate (FDA) was employed. FDA is an esterified fluorogenic substrate widely used for assessing esterase activity in bacteria.51,52 It permeates cells by undergoing hydrolysis of its diacetate (DA) groups into fluorescent fluorescein by intracellular nonspecific esterases. As depicted in Fig. 9, consistent with the literature findings, both tumour cell lines (Hs766T and HepG2) exhibit significantly elevated esterase concentrations in their cytosolic extracts compared to healthy 3T3 cells. Moreover, FDA hydrolysis exhibits relatively rapid kinetics, allowing for differentiation between different cell lines within an hour. Similarly, the incubation of the T2C12–T80 probe with the cytosolic cell extracts showed a significant increase in the EPR signal with cytosolic extracts of tumour cells (Hs766T and HepG2) (Fig. S6, ESI†), whereas no change was detected with the cytosolic extracts of healthy cells (3T3 fibroblasts) (Fig. 10A). The observed increase corresponds to the hydrolysis of approximately 70% of the radical containing species as the result of the esterase activity in the cytoplasmatic extracts. More insights into the specificity of the T2C12–T80 radical probe were gained by repeating the experiment in the presence of an excess of bis-nitrophenylphosphate (BNPP),34 a specific inhibitor of carboxylesterases (Fig. S7, ESI†). In the case of the two tumour cell lines, a remarkable decrease in the intensity, with residual activity around 30–40%, was observed, while only a 20% reduction was recorded in the case of healthy 3T3 (Fig. 10B). Following the promising results obtained by comparing cell lysates, specifically by Hs766T and 3T3, the response of the T2C12–T80 incubated in the presence of intact living cells was investigated (Fig. 11). The obtained results showed a significant difference (P value ≤0.001) in the EPR intensity between the tumour (Hs766T) and healthy (3T3) cell lines after 30 min of incubation of T2C12–T80 in the cell culture medium. This trend continued even after 1 hour of incubation, with a P-value of ≤0.0001. The normalized EPR intensity with the number of cells present on the dishes still revealed a significant difference (P value ≤0.001) between the two cell lines. Therefore, the high EPR signal observed in the experiment with the pancreatic tumour cells confirmed the upregulation of the hydrolytic enzymes accessible to the probe (T2C12–T80). These observations do not provide information on the enzyme location, specifically whether they are found on the cell membrane or released into the extracellular medium.53–55 Uptake assays were carried out using T2C12–T80 and 4-oxo-TEMPO on Hs766T cells. In both cases, after 24 hours of incubation, no EPR signal was detected in the cell pellets (Fig. S8, ESI†), which indicates a fast reaction with ROS present in the intracellular environment. The toxicity of T2C12–T80 and T2C12 micelles was evaluated against Hs766T and 3T3 cell lines using an MTT assay to ensure that, within the concentration range used to assess esterase activity, there was no observed toxicity, and that the compounds were biosafe within the concentration range investigated (<300 μM) (Fig. S9, ESI†). The excellent sensitivity, biocompatibility and stability exhibited by T2C12–T80 micelles make them a highly suitable option for serving as a specialized sensor for assessing the enzymatic activity in the first OMRI experiment at ultra-low magnetic strengths.
 |
| | Fig. 9 Fluorescence-detected enzymatic assay, achieved after 1 hour of incubation at 37 °C with stirring at 400 rpm with samples containing FDA 30 nM and 3T3, Hs766T and HepG2 cell lysate. Cells per mL indicate the number of cells used to obtain cell lysates and incubated with FDA before dilution for fluorescence measurements. | |
 |
| | Fig. 10 EPR assay of carboxylesterase enzymatic activity. (A) Incubation of T2C12–T80 100 μM with cytosolic extracts of 3T3, Hs766T, and HepG2 cells, each obtained from 250 × 103 cells at 37 °C under stirring at 400 rpm for 3 hours. (B) Residual activity measured after 3 hours of incubation in the presence of the carboxylesterase inhibitor (BNPP 600 μM); 100% correspond to the values obtained in the absence of the inhibitor. Statistical significance was determined using an unpaired student's t-test calculated with GraphPad prism version 8.0.2 (P > 0.05 is ns, P ≤ 0.05 is *, P ≤ 0.01 is **, P ≤ 0.001 is ***, and P ≤ 0.0001 is ****). | |
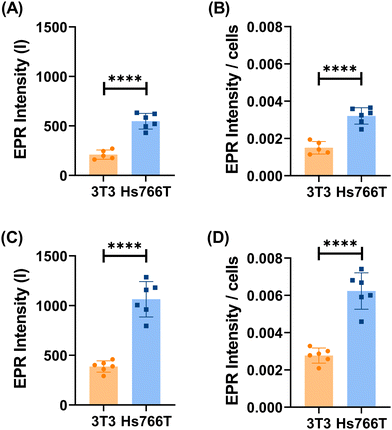 |
| | Fig. 11
In vitro assessment of esterase activity with 3T3 and Hs766T cells with T2C12–T80 100 μM at 37 °C. Absolute EPR intensities (A) and their values normalized to the number of cells (B) after 30 and after 1 hour (C) and (D) of incubation. Statistical significance was determined via an unpaired student's t-test calculated with GraphPad prism version 8.0.2 (P > 0.05 is ns, P ≤ 0.05 is *, P ≤ 0.01 is **, P ≤ 0.001 is ***, and P ≤ 0.0001 is ****). | |
Assessment of the esterase activity by OMRI at ultra-low field
Based on the above-reported results, an OMRI experiment was performed at an ultra-low field of 206 μT to monitor the enzymatic kinetics with T2C12–T80 and PLE. The experiment was executed with a phantom containing four sample tubes made of three controls (HEPES-buffered saline solution, 4-oxo-TEMPO at 0.4 mM and T2C12–T80 2 mM) and one with T2C12–T80 2 mM and PLE 1 U mL−1. The spatial distribution of the samples in the phantom could be clearly delineated in conventional MR spin-echo images after Zeeman pre-polarization at 20 mT, i.e. at a field that is two orders of magnitude higher than that used in the OMRI experiment. This step was mandatory in the absence of DNP because no measurable NMR signal would have been observed with the experimental magnetic spin polarization achievable at 206 μT. The OMRI kinetics of enzymatic hydrolysis were measured at room temperature and followed for 200 min with the acquisition of a new image every 6 minutes. The results reported in Fig. 12 demonstrate that the signals arising from the 4-oxo-TEMPO reference solution and the enzyme-free T2C12–T80 micelles solution remained fairly constant over time, with a complete “quenched” signal in the case of the micelles. Conversely, the OMRI signal increased over time in the enzyme containing solution. The reaction appeared complete after 200 minutes and the concentration of the radical in the OMRI-reference and in the enzyme containing sample, as measured by EPR, yielded values of 0.42 mM and 1.35 mM, respectively. During the OMRI experiment, the sample temperature showed a small increase, from 27 °C to 31 °C, due to RF deposition (15 W EPR continuous wave irradiation; 30 s duration at the frequency of 71.6 MHz). This temperature variation does not significantly affect the reaction kinetics, thus maintaining the validity of the proposed method. The obtained results clearly demonstrate the ability of the ultra-low field OMRI instrument to detect and measure in vitro the contrast built up from the enzyme activity on a properly designed off/on responsive agent. Notably, as depicted in Fig. 12, one can estimate that T2C12 concentration as low as 0.2 mM, corresponding grossly to time-point t0 + 19 minutes, is sufficient for generating enough contrast in 2D-OMRI in half a minute imaging time. An analogous sensitivity was observed in the previously published in vitro experiments.24 This low concentration of a non-toxic nitroxide appears compatible with values encountered in in vivo experiments in rodents with no significant adverse effects.20,21,24
 |
| | Fig. 12 OMRI monitoring of the enzymatic reaction between T2C12–T80 2 mM and PLE 1 U mL−1 for 200 minutes at room temperature. The phantom contained the following 4 sample tubes: (blue) T2C12–T80 2 mM with PLE 1 U mL−1, (orange) HEPES-buffered saline solution, (red) 4-oxo-TEMPO at 0.42 mM in HEPES-buffered saline solution and (light blue) T2C12–T80 2 mM in HEPES-buffered saline solution. Comparison with the MR image obtained for the phantom upon the application of Zeeman pre-polarization at 20 mT is also shown (green border). | |
Conclusions
In our previous paper,26 we reported on the synthesis and characterization of a TEMPO-based radical functionalized with an aliphatic chain (T2C12) through the insertion of an ester bond that can be easily hydrolysed by esterases. The T2C12 forms in water EPR silent micelles that can be activated by the enzymes. Following the release of the free TEMPO radicals, narrow and intense EPR signals are produced due to the hydrolysis of the ester bond catalysed by esterases whose activity is directly correlated with the intensity of the observed EPR signals. The results reported in this study showed that the novel formulation of the nitroxide radical T2C12 using the non-ionic surfactant Tween 80 (T80) exhibited a higher sensitivity and selectivity to detect esterase activity in vitro and in the presence of tumour cells (HepG2 and Hs766T) characterized by esterase overexpression. Interestingly, the resistance to the reduction in the nitroxide radicals forming the micelles was significantly improved with respect to the free oxo-tempo radical, and the “off–on” transition made the methodology semi-quantitative, avoiding the use of complex ratiometric corrections to eliminate the contribution arising from the nonhydrolyzed probes. Moreover, it was shown that OMRI in vitro could serve in the follow-up of an enzymatic conversion at an ultra-low field. As observed from our previous study,24 the bio-distribution of a conventional nitroxide was measured in living rats in 3D with the same OMRI system within a few minutes of imaging time. In both studies, significant signal enhancement due to DNP could be observed with very limited amounts of nitroxides, i.e. for concentrations lower than 0.5 mM. In this context, the indications gained in this work will provide useful insights for the design of further improved responsive agents. Actually, one may envisage additional designs for reporting on the activity of degradation enzymes, such as matrix metalloproteinases (MMPs) and plasminogen activators, in the tumor ECM. Regarding detection sensitivity, ULF-OMRI is beneficial over OMRI at higher fields (e.g. 0.2 T)19,20 because Overhauser enhancements are much lower in the latter case. Moreover, ULF-OMRI is safe in terms of specific absorption rate and allows investigation in large animals. Therefore, a consistent continuation of the present work is the investigation of the fate of the proposed micellar contrast agent in vivo in healthy rodents but also in the context of molecular imaging of disease-associated carboxylesterase activity in rodents.
Experimental section
Materials and methods
All the compounds were purchased from Sigma-Aldrich, and all the solvents from VWR. EPR spectra were acquired using the Adani EPR spectrometer Spinscan X (9.2–9.55 GHz) with the following parameters: center field = 336.50 mT, sweep width = 8 mT, sweep time = 30 s, modulation amplitude = 150 μT, and attenuation = 20 dB. All the enzymatic incubations were done in Starlab Thermomixer-Mixer HC at 37 °C under stirring at 400 rpm. The ultrasonic homogeniser Sonicator Bandelin Sonoplus HD 2070 with an M72 tip was used for the sonication of the micelles. The hydrodynamic mean diameter was determined using a Malvern dynamic light-scattering spectrophotometer (Malvern Instruments, Malvern, UK). A fluorescence assay was performed using the spectrofluorometer (Fluoro-Max-4, Horiba Jobin Yvon). Cell lines were purchased from ATCC, while cell culture medium and supplements (foetal bovine serum FBS, glutamine, glucose) were purchased from Euroclone. All the measurements were repeated at least three times, and the results are shown as mean ± standard deviation.
Micelle preparation
T2C12 was prepared according to the previously reported procedure, as illustrated by Elkhanoufi et al., 2022.26 T2C12 micelles were obtained by dissolving the nitroxide radical derivative in dimethyl sulfoxide (DMSO) to a final concentration of 33.3 mM. Then, 9 μL of T2C12 33.3 mM was diluted in 991 μL of HEPES-buffered saline solution at pH 7.4 to obtain T2C12 300 μM in micellar form. The solution was then sonicated for 2 min at 15% power using an ultrasonic homogenizer in an ice bath and diluted to 100 μM in HEPES-buffered saline solution (with a final DMSO concentration of 0.3%). The mixed micelles were obtained using the same procedure by diluting 9 μL of T2C12 33.3 mM in 984 μL HEPES-buffered saline and 7.4 μL of Tween 80 8.12 mM to obtain T2C12 300 μM + T80 60 μM (T2C12–T80 300 μM), or 987 μL of HEPES-buffered saline and 3.7 μL of Tween 80 8.12 mM for the T2C12 300 μM + T80 30 μM (T2C12–T8010 300 μM, respectively). Similarly, the samples were sonicated for 2 min at 15% power and then diluted to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 to achieve T2C12 100 μM + T80 20 μM (T2C12–T80 100 μM) or T2C12 100 μM + T80 10 μM (T2C12–T8010 100 μM).
:3 to achieve T2C12 100 μM + T80 20 μM (T2C12–T80 100 μM) or T2C12 100 μM + T80 10 μM (T2C12–T8010 100 μM).
DLS measurements
All samples (T2C12 300 μM, T2C12–T8010 300 μM and T2C12–T80 300 μM) were diluted in the ratio of 1:3 in HEPES-buffered saline solution (pH 7.4) after the sonication, freshly filtered (cut-off = 0.2 μm), and measured via DLS at 25 °C immediately after dilution and after 1 hour.
Enzymatic kinetic assays
Enzyme kinetic assays were performed using final concentrations of T2C12, T2C12–T80 or T2C12–T8010 100 μM and of CEs2 12 U mL−1, CEs1 12 U mL−1 or PLE 0.5 U mL−1, corresponding to concentrations of 23 μg mL−1, 7 μg mL−1 and 5 μg mL−1, respectively. All the experiments were carried out at 37 °C under stirring at 400 rpm, and at different intervals, a small sample was taken from the solutions for EPR measurement.
Stability in albumin and serum
The stability of the radicals was evaluated in the presence of human serum albumin (HSA) by adding 10 mg of the protein to 250 μL of T2C12, T2C12–T80 or T2C12–T8010 100 μM to achieve a final HSA concentration of 40 mg mL−1. The serum stability was evaluated using lyophilised human serum (seronorm human) resuspended with 200 μL of 100 μM micelles solutions (T2C12, T2C12–T80 and T2C12–T8010). All the experiments were carried out at 37 °C under stirring at 400 rpm, and at different intervals, a small volume was taken from the solutions for EPR measurements.
Stability of ascorbic acid
Ascorbic acid solution was prepared by dissolving 26.8 mg of ascorbic acid (AC) in 1.9 mL of HEPES-buffered saline solution and 150 μL of NaOH 1 M to obtain a solution of AC 74 mM at pH 7.4. The stability assessment of the radicals was performed by incubating 50 μL of oxo-TEMPO 200 μM with 47.3 μL of buffered HEPES saline solution and 2.7 μL of AC 37 mM. Moreover, with the micelles, 50 μL of T2C12–T80 300 μM were mixed with 96 μL of HEPES-buffered saline solution and 4 μL of AC 37 mM. At different intervals, a small sample was taken from the solutions for EPR measurement after the addition of AC.
Cell lines
Hs766T, human pancreatic cancer cell line, Hep-G2, human hepatocellular carcinoma cell line and NIH/3T3 (3T3), mouse embryo fibroblast, were obtained from ATCC. The Hs766T were cultured in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) Fetal bovine serum (FBS), and 2.5 mM of L-glutamine (Lonza). The Hep-G2 cells were cultured in Eagle's minimum essential medium containing 2.5 mM glutamine and EBSS and supplemented with 10% (v/v) FBS and 1 mM sodium pyruvate. The 3T3 cells were cultured in Dulbecco's modified Eagle's medium containing 2.5 mM glutamine and supplemented with 10% (v/v) FBS. All media contained 100 U mL−1 penicillin and 100 U mL−1 streptomycin. All the cell lines were maintained at 37 °C in a 5% CO2 incubator.
Cell lysate experiments
Hs766T, HepG2, and 3T3 cells were seeded in 175 cm diameter flasks at the following densities: 1 × 106, 5 × 105, and 3 × 105 cells, respectively. Following an incubation period of nearly 72 hours at 37 °C with 5% CO2 in a cell incubator, when the confluence reached approximately 70–80%, the cells were washed three times with 10 mL of ice-cold PBS and subsequently detached using a solution containing 0.05% trypsin and 0.02% EDTA. After detachment, all the cell samples were transferred to Falcon tubes and resuspended in 600 μL of PBS, and the cell samples in each condition were counted using trypan blue (Sigma). The cells were then sonicated at 30% power for 1 minute on ice. Subsequently, aliquots of cells with concentrations of 0.5 × 103, 1 × 103, 2 × 103, and 3 × 103 cells per mL were prepared for the fluorimetry experiment, and aliquots containing 250 × 103 cells were prepared for the EPR experiment.
Fluorimetry with cell lysate
Fluorescein diacetate (FDA) solution was dissolved in DMSO to a final concentration of 333 mM. Then, 3 μL of the solution was diluted with 997 μL of HEPES-buffered saline solution to obtain FDA 1 μM. Afterwards, 100 μL of FDA 1 μM was incubated for 60 min at 37 °C and stirred at 400 rpm with a small aliquot of cell lysate (3T3, Hs766T, HepG2), corresponding to 0.5 × 103, 1 × 103, 2 × 103, and 3 × 103 cells per mL. After the incubation, 90 μL was taken from the samples, diluted with 2.91 mL of HEPES-buffered saline solution (FDA/fluorescein final concentration of 30 nM) and measured using the fluorescence spectrometer. The excitation and emission parameters were set at 490 nm and 510 nm, respectively.
EPR with cell lysate
The experiment was carried out by adding 100 μL of T2C12–T80 300 μM to a volume of cell lysate corresponding to 250 × 103 cells and diluted with HEPES-buffered saline solution to a final volume of 300 μL. The samples were then incubated at 37 °C and 400 rpm, and after 1, 2 and 3 hours, a small aliquot was taken for the EPR measurement. The inhibition experiment was performed using bis(4-nitrophenyl)phosphate (BNPP). After solubilizing the inhibitor in HEPES-buffered saline solution to a final concentration of 5.4 mM, the pH was corrected to 7.4 and sonicated in an ultrasonication bath at 30 °C for 30 minutes. To inhibit the enzyme activity, preincubation was done by adding 16 μL of BNPP 5.4 mM to the cell lysate solution and HEPES-buffered saline solution at 37 °C and 400 rpm for 30 min. Then, 100 μL of micelle solution was added to obtain a solution with T2C12–T80 100 μM with BNPP 600 μM and 250 × 103 cell lysate (final volume of 300 μL).
EPR with living cells
Hs766T and 3T3 cells were seeded in 3 cm dishes at 10 × 104 and 20 × 104 densities for 3T3 and Hs766T, respectively. After 24 h of incubation, the medium was replaced with a serum-free medium, and after 30 min, the cells were incubated with 100 μM T2C12–T80 in a final volume of 1 mL at 37 °C for 30 and 60 min, respectively. At these time intervals, 60 μL of the medium was collected and analysed using EPR. The cells were then washed once with 0.5 mL of ice-cold PBS and detached using a solution of 0.05% trypsin and 0.02% EDTA. The cell samples in each condition were counted using trypan blue.
MTT
In a 96-well microtiter plate, 11 × 104 and 6.5 × 103 cells per well were seeded for the Hs766T and NIH/3T3 cell lines, respectively. Following a 24-hour incubation period at 37 °C and 5% CO2, the cells were subjected to escalating concentrations of T2C12–T80 and T2C12 (ranging from 0 to 300 μM) and further incubated for an additional 24 hours under normoxic conditions (at 37 °C and 5% CO2). The concentration of dimethyl sulfoxide (DMSO) in the cell medium did not surpass 0.3% (v/v) under all tested conditions. After the incubation period, the medium was aspirated, and each well was treated with thiazolyl blue tetrazolium bromide (Sigma Aldrich) dissolved in the medium at a concentration of 0.45 mg mL−1. The cells were then incubated for 4 hours at 37 °C and 5% CO2. Subsequently, the medium was removed, and 150 μL of DMSO was added to each well to dissolve the formazan salt crystals produced due to live cell metabolism. The microplate was incubated at room temperature for 30 minutes, after which the absorbance was measured at 565 nm using the GloMax® Discover Microplate Reader (Promega Corporation, Milano, Italy). Cell viability was assessed as the percentage of dead cells observed in the treated samples relative to the non-treated control cells.
OMRI experiments
Ultra-low field Overhauser-enhanced magnetic resonance imaging was performed using a system already described (Boudries et al., 2023).24 Briefly, the system is composed of concentric cylinder-shaped elements: a gradient coil (280 mm diameter), a Zeeman prepolarization coil allowing 20 mT B0-field (150 mm diameter), an NMR coil operating at 8.77 kHz (second-order solenoid gradiometer, 80 mm diameter) and an EPR coil operating at 72 MHz (8-leg birdcage, 62 mm diameter, 15 W power). The entire system was inserted in a B0-cage capable of local earth's field cancelation replaced by a vertical B0-field of 206 μT. Details on the hardware were demonstrated by Boudries et al., 2023.24 The system could operate in two distinct modes: either with a Zeeman pre-polarization step of 2 seconds at 20 mT prior to conventional MRI acquisition at 206 μT (8.77 MHz proton frequency), or with Overhauser enhancement with a constant EPR irradiation at 71.6 MHz throughout proton MRI acquisition at 206 μT.
The MRI sequences used are as follows:
– Zeeman pre-polarization: 2D spin echo (TE/TR: 85/2230 ms), no slice selection, field-of-view: 72 × 72 mm, resolution = 1.1 × 1.1 mm, acquisition time: 270 seconds, and 4 averages.
– OMRI: 2D balanced steady-state free-precession (TE/TR: 67/155 ms), no slice selection, field-of-view: 72 × 72 mm, resolution = 1.1 × 1.1 mm, acquisition time: 30 seconds, and 4 averages. In kinetics experiments, this sequence was repeated every 6 minutes.
Author contributions
Conceptualization: G. C. S., T. E.; funding acquisition: G. C. S., T. E., F. J. M., Ma. P., Me. P., P. E.; resources: G. C. S., T. E., F. J. M., M. P., M. P., P. E., S. R., A. D.; methodology: E. S., R. S., N. M. J., B. D., P. T. J; sample preparation and data acquisition: E. S., R. S., N. M. J., B. D., P. T. J.; data analysis: E. S., R. S., B. D.; data interpretation: E. S., R. S., B. D., Me. P.; visualization: G. C. S., T. E.; supervision: G. C. S., T. E.; writing – original draft: E. S., R. S., G. C. S., T. E. Ma. P.; writing – review and editing: all authors.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 86309, Primogaia project and FSE REACT-EU (dotazione PON 2014-2020, Azione IV.5). We acknowledge prof. Silvio Aime for the fruitful discussion and valuable input to this paper.
References
- R. H. Wijdeven, J. Neefjes and H. Ovaa, Trends Cell Biol., 2014, 24, 751–760 CrossRef CAS PubMed.
- C. Yang, Q. Wang and W. Ding, RSC Adv., 2019, 9, 25285–25302 RSC.
- R. Yan and D. Ye, Sci. Bull., 2016, 61, 1672–1679 CrossRef CAS.
- Q. Zhang, T. Liu, J. Ding, N. Zhou, Z. Yu, Y. Ren, X. Qin, P. Du, Z. Yang and H. Zhu, Mol. Pharm., 2022, 19, 4149–4156 CrossRef CAS PubMed.
- W. Lee, G. Il An, H. Park, S. Sarkar, Y. S. Ha, P. T. Huynh, A. Bhise, N. Bhatt, H. Ahn, D. N. Pandya, J. Y. Kim, S. Kim, E. Jun, S. C. Kim, K. C. Lee and J. Yoo, ACS Nano, 2021, 15, 17348–17360 CrossRef CAS PubMed.
- T. Lindner, A. Altmann, S. Krämer, C. Kleist, A. Loktev, C. Kratochwil, F. Giesel, W. Mier, F. Marme, J. Debus and U. Haberkorn, J. Nucl. Med., 2020, 61, 1507–1513 CrossRef CAS PubMed.
- I. Daryaei and M. D. Pagel, Res. Rep. Nucl. Med., 2015, 5, 19–32 Search PubMed.
- D. V. Hingorani, B. Yoo, A. S. Bernstein and M. D. Pagel, Chem. – Eur. J., 2014, 20, 9840–9850 CrossRef CAS PubMed.
- M. Sarracanie and N. Salameh, Front. Phys., 2020, 8 DOI:10.3389/fphy.2020.00172.
- M. E. Halse, A. Coy, R. Dykstra, C. Eccles, M. Hunter, R. Ward and P. T. Callaghan, J. Magn. Reson., 2006, 182, 75–83 CrossRef CAS PubMed.
- L. L. Wald, P. C. McDaniel, T. Witzel, J. P. Stockmann and C. Z. Cooley, J. Magn. Reson. Imaging, 2020, 52, 686–696 CrossRef PubMed.
- A. Mohoric, G. Planinsic, M. Kos, A. Duh and J. Stepisnik, Instrum. Sci. Technol., 2004, 32, 655–667 CrossRef CAS.
- A. Abragam and M. Goldman, Rep. Prog. Phys., 1978, 41, 395 CrossRef CAS.
- D. J. Lurie, D. M. Bussell, L. H. Bell and J. R. Mallard, J. Magn. Reson. (1969-1992), 1988, 76, 366–370 CrossRef CAS.
- D. Grucker, Magn. Reson. Med., 1990, 14, 140–147 CrossRef CAS PubMed.
- P. Mellet, P. Massot, G. Madelin, S. R. A. Marque, E. Harte, J.-M. Franconi and E. Thiaudière, PLoS One, 2009, 4, e5244 CrossRef PubMed.
- G. Audran, L. Bosco, P. Brémond, J.-M. Franconi, N. Koonjoo, S. R. A. Marque, P. Massot, P. Mellet, E. Parzy and E. Thiaudière, Angew. Chem., Int. Ed., 2015, 54, 13379–13384 CrossRef CAS PubMed.
- N. Jugniot, I. Duttagupta, A. Rivot, P. Massot, C. Cardiet, A. Pizzoccaro, M. Jean, N. Vanthuyne, J.-M. Franconi, P. Voisin, G. Devouassoux, E. Parzy, E. Thiaudiere, S. R. A. Marque, A. Bentaher, G. Audran and P. Mellet, Free Radical Biol. Med., 2018, 126, 101–112 CrossRef CAS PubMed.
- D. Alberti, E. Thiaudiere, E. Parzy, S. Elkhanoufi, S. Rakhshan, R. Stefania, P. Massot, P. Mellet, S. Aime and S. Geninatti Crich, Sci. Rep., 2023, 13, 13725 CrossRef CAS PubMed.
- N. Koonjoo, E. Parzy, P. Massot, M. Lepetit-Coiffé, S. R. A. Marque, J.-M. Franconi, E. Thiaudiere and P. Mellet, Contrast Media Mol. Imaging, 2014, 9, 363–371 CrossRef CAS PubMed.
- A. Rivot, N. Jugniot, S. Jacoutot, N. Vanthuyne, P. Massot, P. Mellet, S. R. A. Marque, G. Audran, P. Voisin, M. Delles, G. Devouassoux, E. Thiaudiere, A. Bentaher and E. Parzy, ACS Omega, 2021, 6, 15012–15016 CrossRef CAS PubMed.
- T. Guiberteau and D. Grucker, Phys. Med. Biol., 1998, 43, 1887 CrossRef CAS PubMed.
- M. E. Halse and P. T. Callaghan, J. Magn. Reson., 2008, 195, 162–168 CrossRef CAS PubMed.
- D. Boudries, P. Massot, E. Parzy, S. Seren, P. Mellet, J.-M. Franconi, S. Miraux, E. Bezançon, S. R. A. Marque, G. Audran, M. Muetzel, S. Wintzheimer, F. Fidler and E. Thiaudiere, J. Magn. Reson., 2023, 348, 107383 CrossRef CAS PubMed.
- E. Parzy, D. Boudries, S. Jacoutot, M. Albalat, N. Vanthuyne, J.-M. Franconi, P. Mellet, E. Thiaudiere, G. Audran, S. R. A. Marque and P. Massot, J. Magn. Reson., 2021, 333, 107095 CrossRef CAS PubMed.
- S. Elkhanoufi, R. Stefania, D. Alberti, S. Baroni, S. Aime and S. Geninatti Crich, Chem. – Eur. J., 2022, 28, e202104563 CrossRef CAS PubMed.
- R. Kumari, M. M. Majumder, J. Lievonen, R. Silvennoinen, P. Anttila, N. N. Nupponen, F. Lehmann and C. A. Heckman, Br. J. Cancer, 2021, 124, 1428–1436 CrossRef CAS PubMed.
- T. Fukami and T. Yokoi, Drug Metab. Pharmacokinet., 2012, 27, 466–477 CrossRef CAS PubMed.
- R. A. Kohnz, M. M. Mulvihill, J. W. Chang, K.-L. Hsu, A. Sorrentino, B. F. Cravatt, S. Bandyopadhyay, A. Goga and D. K. Nomura, ACS Chem. Biol., 2015, 10, 1624–1630 CrossRef CAS PubMed.
- K. Na, M. Kim, C.-Y. Kim, J.-S. Lim, J.-Y. Cho, H. Shin, H. J. Lee, B. J. Kang, D. H. Han, H. Kim, J.-H. Baik, M. Swiatek-de Lange, J. Karl and Y.-K. Paik, J. Proteome Res., 2020, 19, 4867–4883 CrossRef CAS PubMed.
- J. Liu, B. Yao, L. Gao, Y. Zhang, S. Huang and X. Wang, Biochem. Pharmacol., 2022, 205, 115250 CrossRef CAS PubMed.
- G. Xu, W. Zhang, M. K. Ma and H. L. McLeod, Clin. Cancer Res., 2002, 8, 2605–2611 CAS.
- I. K. Mkam Tsengam, M. Omarova, E. G. Kelley, A. McCormick, G. D. Bothun, S. R. Raghavan and V. T. John, J. Phys. Chem. B, 2022, 126, 2208–2216 CrossRef CAS PubMed.
- D. Wang, L. Zou, Q. Jin, J. Hou, G. Ge and L. Yang, Acta Pharm. Sin. B, 2018, 8, 699–712 CrossRef PubMed.
- S. J. Park, H. W. Lee, H.-R. Kim, C. Kang and H. M. Kim, Chem. Sci., 2016, 7, 3703–3709 RSC.
- H. J. Braddick, W. J. Tipping, L. T. Wilson, H. S. Jaconelli, E. K. Grant, K. Faulds, D. Graham and N. C. O. Tomkinson, Anal. Chem., 2023, 95, 5369–5376 CrossRef CAS PubMed.
- G. De Simone, A. di Masi and P. Ascenzi, Int. J. Mol. Sci., 2021, 22, 10086 CrossRef CAS PubMed.
- P. Ascenzi, L. Leboffe, A. di Masi, V. Trezza, G. Fanali, M. Gioia, M. Coletta and M. Fasano, PLoS One, 2015, 10, e0120603 CrossRef PubMed.
- P. Ascenzi, M. Gioia, G. Fanali, M. Coletta and M. Fasano, Biochem. Biophys. Res. Commun., 2012, 424, 451–455 CrossRef CAS PubMed.
- S. B. Tove, Biochim. Biophys. Acta, 1962, 57, 230–235 CrossRef CAS PubMed.
- C. S. Johnston, C. G. Meyer and J. C. Srilakshmi, Am. J. Clin. Nutr., 1993, 58, 103–105 CrossRef CAS PubMed.
- X. Zhuang, C. Xiao, K. Oyaizu, N. Chikushi, X. Chen and H. Nishide, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5404–5410 CrossRef CAS.
- J. Fuchs, N. Groth, T. Herrling and G. Zimmer, Free Radical Biol. Med., 1997, 22, 967–976 CrossRef CAS PubMed.
- J. Du, J. J. Cullen and G. R. Buettner, Biochim. Biophys. Acta, Rev. Cancer, 2012, 1826, 443–457 CrossRef CAS PubMed.
- M. Capello, M. Lee, H. Wang, I. Babel, M. H. Katz, J. B. Fleming, A. Maitra, H. Wang, W. Tian, A. Taguchi and S. M. Hanash, JNCI, J. Natl. Cancer Inst., 2015, 107, djv132 Search PubMed.
- K. Kailass, O. Sadovski, M. Capello, Y. Kang, J. B. Fleming, S. M. Hanash and A. A. Beharry, Chem. Sci., 2019, 10, 8428–8437 RSC.
- Y. Wen, N. Jing, M. Zhang, F. Huo, Z. Li and C. Yin, Adv. Sci., 2023, 10, 2206681 CrossRef CAS PubMed.
- K. Na, S.-K. Jeong, M. J. Lee, S. Y. Cho, S. A. Kim, M.-J. Lee, S. Y. Song, H. Kim, K. S. Kim, H. W. Lee and Y.-K. Paik, Int. J. Cancer, 2013, 133, 408–415 CrossRef CAS PubMed.
- N. Qiu, X. Liu, Y. Zhong, Z. Zhou, Y. Piao, L. Miao, Q. Zhang, J. Tang, L. Huang and Y. Shen, Adv. Mater., 2016, 28, 10613–10622 CrossRef CAS PubMed.
- W. Duan, S. Ji, Y. Guan, X. Mu, S. Fang, Y. Lu, X. Zhou, J. Sun and Z. Li, Biomacromolecules, 2020, 21, 5093–5103 CrossRef CAS PubMed.
- P. Breeuwer, J. L. Drocourt, N. Bunschoten, M. H. Zwietering, F. M. Rombouts and T. Abee, Appl. Environ. Microbiol., 1995, 61, 1614–1619 CrossRef CAS PubMed.
- D. Hoefel, W. L. Grooby, P. T. Monis, S. Andrews and C. P. Saint, J. Microbiol. Methods, 2003, 52, 379–388 CrossRef CAS PubMed.
- J. Lamego, P. Ferreira, M. Alves, A. Matias and A. L. Simplício, Mol. Cell. Probes, 2015, 29, 215–222 CrossRef CAS PubMed.
- S. Wang, M. Xu, X. Li, X. Su, X. Xiao, A. Keating and R. C. Zhao, J. Hematol. Oncol., 2018, 11, 82 CrossRef PubMed.
- J. Conde-Vancells, E. Gonzalez, S. C. Lu, J. M. Mato and J. M. Falcon-Perez, Expert Opin. Drug Metab. Toxicol., 2010, 6, 543–554 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Rachele
Stefania
b,
Rachele
Stefania