
Solvent-free synthesis of biostable segmented polyurethane shape memory polymers for biomedical applications
Maryam
Ramezani
a,
Dariya
Getya
bc,
Ivan
Gitsov
 abc and
Mary Beth Browning
Monroe
*a
abc and
Mary Beth Browning
Monroe
*a
aDepartment of Biomedical and Chemical Engineering, BioInspired Syracuse, Syracuse University, Syracuse, NY, USA. E-mail: mbmonroe@syr.edu
bDepartment of Chemistry, State University of New York ESF, Syracuse, NY, USA
cThe Michael M. Szwarc Polymer Research Institute, Syracuse, NY, USA
First published on 19th December 2023
Abstract
Biostable shape memory polymers that remain stable in physiological conditions are beneficial for user-defined shape recovery in response to a specific stimulus. For potential commercialization and biocompatibility considerations, biomaterial synthesis must be simple and scalable. Hence, a library of biostable and cytocompatible shape memory polymers with tunable thermomechanical properties based on hard segment content was synthesized using a solvent-free method. Polymer surface chemistry, thermomechanical and shape memory properties, and biostability were assessed. We also investigated the effects of processing methods on thermomechanical and shape memory properties. All polymers showed high glass transition temperatures (>50 °C), which indicates that their temporary shape could be preserved after implantation. Polymers also demonstrate high shape fixity (73–80%) and shape recovery (93–95%). Minimal mass loss (<5%) was observed in accelerated oxidative (20% H2O2) and hydrolytic (0.1 M NaOH) media. Additionally, minimal shape recovery (∼0%) occurred in programmed samples with higher hard segment content that were stored in degradation media. After 40 days of storage in media, programmed samples recovered their primary shapes upon heating to temperatures above their transition temperature. Annealing to above the polymer melting point and solvent casting of polymers improved shape memory and thermal properties. To enable their potential use as biomaterial scaffolds, fiber formation of synthesized polyurethanes was compared with those of samples synthesized using a previously reported solvent-based method. The new method provided polymers that can form fibrous scaffolds with improved mechanical and shape memory properties, which is attributed to the higher molecular weight and crystalline content of polymers synthesized using the new, solvent-free approach. These biostable segmented polyurethanes could be coupled with a range of components that respond to specific stimuli, such as enzymes, magnetic field, pH, or light, to enable a specific shape change response, which could be coupled with drug and/or bioactive material delivery in future work.
1. Introduction
Shape memory polymers (SMPs) are smart polymers that can remember their primary shape. If an external stimulus, commonly heat, is applied to the polymer, the chains become more flexible to enable programming (e.g., straining, compressing, or folding) into a temporary shape. Upon re-exposure to a second stimulus, polymer chains release the tension and recover their primary shape.1–6 These smart materials have been employed in an extensive range of applications in tissue regeneration and repair,7,8 drug and antimicrobial delivery,9,10 wound care,11 sutures,7 and biofilm/infection assessment/treatment.12 Different applications of SMPs require varying degrees of stability, depending upon the goals and functions.13,14 Some applications, such as hemostasis and sutures, need rapid, non-specific shape change upon implantation15,16 while others require a slower rate of shape change and/or degradation, such as polymers for tissue regeneration or infection treatment.12,13,17SMPs can be designed to respond to user-defined stimulation or unique signals from the environment, which requires materials that do not undergo unwanted degradation or actuation from exposure to physiological conditions.3,14 For example, in our previous work, we designed SMPs that only respond to magnetic field1,9 and bacterial proteases12 by altering responsive moieties within a biostable SMP system. A wide range of cues could be coupled with this stable SMP that respond to changes in pH, environmental proteases, oxidative species, light, or magnetic field to provide a specific response to the healing environment and/or clinician-applied external stimuli. The shape/dimension alteration could be coupled with drug or bioactive factor delivery,9 it could be used to control cell or bacteria interactions,12 or it could be used as tool for diagnosis.12,18
Segmented thermoplastic polyurethanes (STPUs) are copolymers containing hard and soft alternate blocks chemically bonded together (Fig. 1). To synthesize STPUs, long chain macromonomers with hydroxyl groups (soft segments), small molecule chain extenders (hard segments), and coupling agents are required. Macromonomers confer flexibility to system, while chain extenders act as rigid segments when reacted with coupling agents to form hydrogen bonding sites. The secondary interactions between chains contribute to the crystal structure and enable maintenance of programmed SMP structures.19,20 Microphase separation due to incompatibility between hard and soft segments is crucial for STPU performance and can define polymer properties, including shape memory abilities.21–23 The transition temperature (typically the glass transition temperature, Tg) of STPU-based SMPs is the temperature at which soft segments become flexible to enable the SMP to be deformed/stretched. If the Tg of a SMP is well above body temperature, programmed polymers will not be passively actuated by heating after implantation in the body. To further ensure that SMPs do not undergo significant shape changes corresponding with alterations in their thermal and mechanical properties over time, they need to be biostable in physiological conditions. Thus, SMPs with high transition temperatures and high biostability can be designed to be responsive to specific user-defined stimuli.24,25
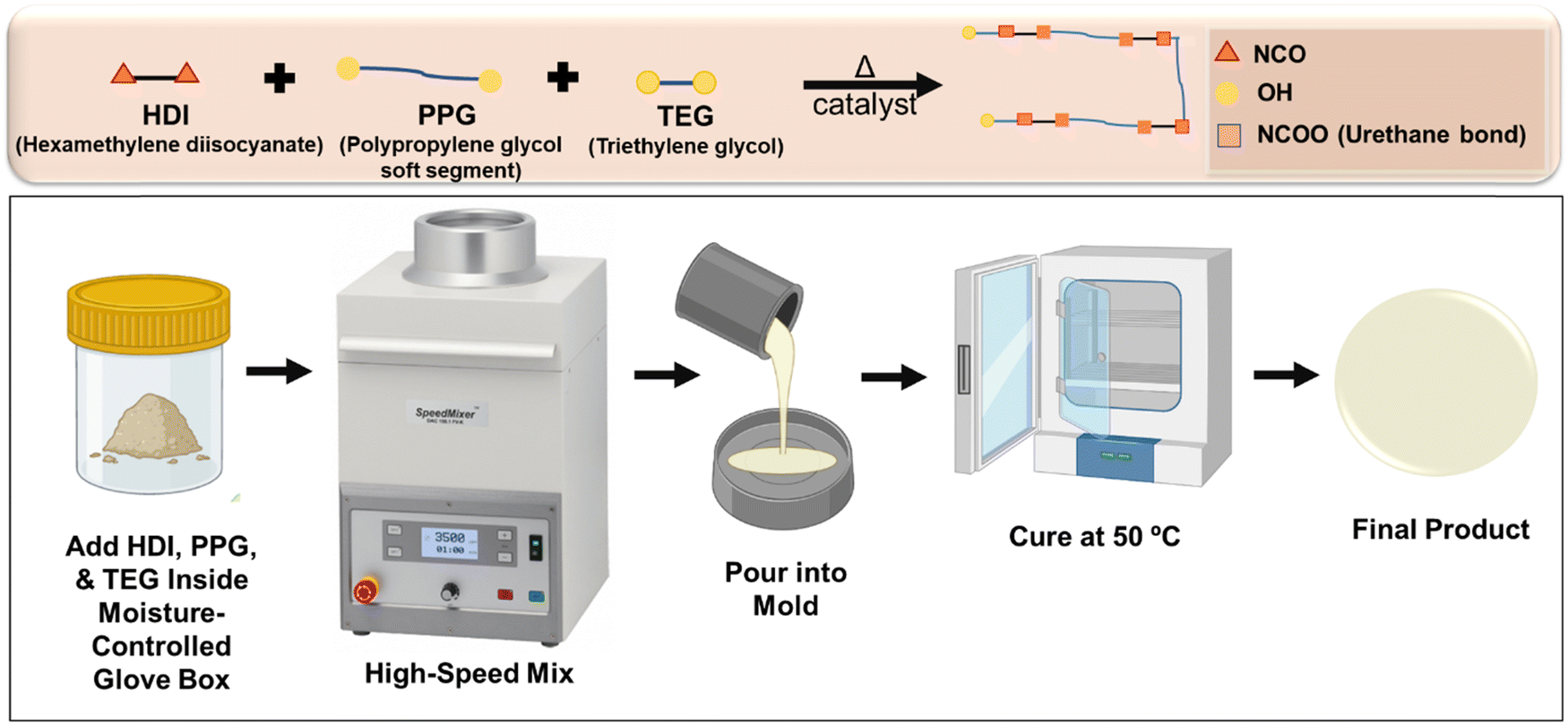 | ||
| Fig. 1 Schematic representation of solvent-free method for segmented thermoplastic polyurethane synthesis. | ||
In our previous work, we proved that a STPU based on hexamethylene diisocyanate (HDI), polypropylene glycol (PPG), and triethylene glycol (TEG) provides a promising platform for user-defined responsive materials due to their high tunability and biostability.1 However, the previously reported synthesis process employed a solvent (tetrahydrofuran) and was time- and labor-intensive (∼3 days). In this study, we aimed to improve the efficiency of the synthesis protocol and make it more environmentally friendly by removing the solvent from the system. This solvent-free method is very straightforward and user-friendly, since it does not require extra titration or stoichiometric balance steps, the synthesis can be performed in one pot, and the hands-on preparation time is reduced to ∼15 min.
We characterized the synthesized polymers in terms of surface chemistry, molecular mass, and thermomechanical and shape memory properties. We also characterized the biostability of the synthesized STPUs in accelerated oxidative (20% H2O2) and hydrolytic (0.1 M NaOH) media in terms of mass loss, surface chemistry, changes in thermal properties, and their ability to maintain programmed temporary shapes over time.1,26–28 Then, we assessed the influence of different processing methods on microphase separation and shape memory/thermal properties of synthesized STPUs. To extend the potential utility of this system, we fabricated fibrous, porous scaffolds from the synthesized materials. We employed electrospinning, a commonly used processing method in biomedical applications, to characterize the fiber formation ability of synthesized STPUs compared with polymers synthesized by the previously reported solvent-based method. Tensile and shape memory testing was utilized to characterize properties of resulting fiber mats. Overall, this study can open new windows for synthesizing flexible, cytocompatible, and biostable SMPs for biomedical application using an easy, environmentally-friendly, and efficient method. This technique could also be extended to other STPU synthesis platforms to expand their utility and enhance scale-up efforts.
2. Materials and methods
2.1 Materials
Hexamethylene diisocyanate (HDI, Thermo Fisher Scientific, Waltham, MA), polypropylene glycol (PPG, M.W. 2000 g mol−1, Fisher), triethylene glycol (TEG, Fisher), diethyl ether (anhydrous, Fisher), NaOH (Sigma-Aldrich, Burlington, MA), dibutyltin dilaurate (DBTDL, Sigma), hydrogen peroxide (H2O2, Certified ACS, 30%, Fisher), phosphate buffered saline (PBS, Fisher), penicillin–streptomycin (P/S, Fisher), fetal bovine serum (FBS, Fisher), Dulbecco's modified Eagle's medium (DMEM, Fisher), Live/Dead® assay (Fisher), and ethanol (reagent grade, Fisher) were purchased as indicated above and used as received unless otherwise specified.2.2 Segmented thermoplastic polyurethane synthesis
A library of polymers (Table 1) was synthesized using a one-step process (Fig. 1). A mixture of reagents containing PPG as the soft segment and TEG as a chain extender was prepared in a speed mixer cup. Then, HDI was added at a 20% excess to the hydroxyl mixture (1.2 isocyanates: 1 hydroxyl). Dibutyltin dilaurate (1 wt%) was added as a catalyst. All additions were performed in a glove box (Labconco, Kansas City, MO, USA) to maintain a dry inert atmosphere using nitrogen passed through a drying train (Labconco, Kansas City, MO, USA) to have a relative humidity below 200 ppm. The cup then was placed in a speed mixer (Flacktek, Landrum, SC, USA) at 3500 rpm for 30 seconds. Afterwards, the mixture was poured into a Petri dish with a Teflon liner and then placed in an oven at 50 °C for 48 hours. Fourier transform infrared (FTIR) spectroscopy was carried out on the final product to track the depletion of hydroxyl groups of PPG and TEG and of isocyanate groups of HDI while assessing formation of urethane groups.| Sample ID | Formulation | HDI (mol%) | PPG (mol%) | TEG (mol%) |
|---|---|---|---|---|
| HDI-3 | 3 HDI: 1 PPG: 2 TEG | 50 | 16 | 34 |
| HDI-4 | 4 HDI: 1 PPG: 3 TEG | 50 | 12 | 38 |
| HDI-5 | 5 HDI: 1 PPG: 4 TEG | 50 | 10 | 40 |
| HDI-6 | 6 HDI: 1 PPG: 5 TEG | 50 | 8 | 42 |
Films were prepared for further characterization using a hot press (Carver 3851-0, Carver Inc. Wabash, IN, USA). Polymer samples were placed between hot press plates at a temperature of 100 °C for 20 minutes with an applied pressure of 1 bar. Then, the films were cooled to room temperature. To compare the polymer properties with those of the previously reported solvent-based method, the synthesis method from our previous work was utilized to prepare controls as previously described.1
2.3 Spectroscopic analysis
FTIR spectroscopy (Nicolet iS5, Fisher Scientific, Waltham, MA, USA) was employed to characterize the surface chemistry of synthesized polymers on a thin layer of polymer at 0.8 cm−1 resolution to analyze formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and NH bonds of the urethane, and OH and NCO depletion of TEG/PPG and HDI, respectively.
O and NH bonds of the urethane, and OH and NCO depletion of TEG/PPG and HDI, respectively.
2.4 Molecular mass measurement
Size-exclusion chromatography (SEC) was utilized to compare molecular mass of polymers synthesized using solvent with those made with the solvent-free method. Each sample (5–7 mg) was dissolved in 1 mL of distilled THF and kept overnight. The next day, samples were analyzed on a system with a M510 pump, U6K universal injector, and 486 tunable absorbance detector (all from Waters Corporation, Milford, MA, USA) and 250 dual refractometer/viscometer (Malvern Panalytical Inc. Westborough, MA, USA). The separation was achieved over a set of three 5 μm Styragel columns (HR 2, 3 and 5, Waters) and calibrated with 17 narrow dispersity PSt standards with molecular masses between 0.162 kDa and 956 kDa. Number average molecular mass (Mn) and dispersity index (Đ) were determined using OmniSEC 5.0 software (Malvern Panalytical).2.5 Cytocompatibility
NIH Swiss mouse 3T3 fibroblasts were employed to assess cytocompatibility of a representative STPU formulation (HDI-5). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (P/S, Gibco) at 37 °C/5% CO2. Cells were seeded in 24 well plates at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well and allowed to adhere for 24 hours while incubating at 37 °C/5% CO2. Cylindrical film samples (6 mm diameter, 1 mm thickness, n = 3) were cleaned with DI water and PBS, exposed to UV light for 10 minutes per side, and then added to Transwell® inserts above pre-seeded wells to incubate with cells. After 24 hours, the inserts/samples were removed, and a Live/Dead® assay (live: Calcein AM, green; dead: BOBO-3 Iodide, red, Thermo Fisher Scientific) was employed to stain cells. Wells with empty inserts were employed as positive, cytocompatible controls, and cells exposed to 200 μl of 70% methanol were taken as negative, cytotoxic controls. Each well was then imaged (3 field views/sample) using an inverted microscope (Leica, DMI6000). In a second set of samples, a resazurin assay was used to quantify relative cell numbers after exposure to HDI-5 samples under the same conditions. For this assay, inserts were removed after 24 hours, and cells were washed with PBS. Then, each well was incubated with 600 μl of 10% resazurin stain for two hours. Solutions (200 μl) were transferred to a fresh 96 well plate, and florescence was measured (530 excitation, 590 emission) using a plate reader (FLx800, Bio-Tek Instruments, Inc.). Relative cell viability was calculated as:
000 cells per well and allowed to adhere for 24 hours while incubating at 37 °C/5% CO2. Cylindrical film samples (6 mm diameter, 1 mm thickness, n = 3) were cleaned with DI water and PBS, exposed to UV light for 10 minutes per side, and then added to Transwell® inserts above pre-seeded wells to incubate with cells. After 24 hours, the inserts/samples were removed, and a Live/Dead® assay (live: Calcein AM, green; dead: BOBO-3 Iodide, red, Thermo Fisher Scientific) was employed to stain cells. Wells with empty inserts were employed as positive, cytocompatible controls, and cells exposed to 200 μl of 70% methanol were taken as negative, cytotoxic controls. Each well was then imaged (3 field views/sample) using an inverted microscope (Leica, DMI6000). In a second set of samples, a resazurin assay was used to quantify relative cell numbers after exposure to HDI-5 samples under the same conditions. For this assay, inserts were removed after 24 hours, and cells were washed with PBS. Then, each well was incubated with 600 μl of 10% resazurin stain for two hours. Solutions (200 μl) were transferred to a fresh 96 well plate, and florescence was measured (530 excitation, 590 emission) using a plate reader (FLx800, Bio-Tek Instruments, Inc.). Relative cell viability was calculated as: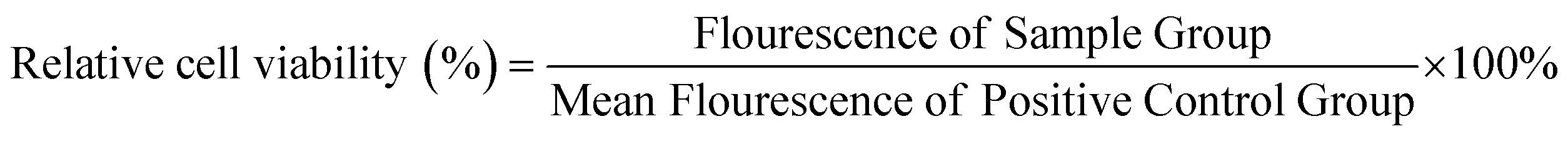 | (1) |
2.6 Thermal analysis
Thermal gravimetric analysis (TGA) was carried out on a TA-Q500 instrument (TA Instruments, Waters) to examine the thermal degradation of the polymers. Samples (n = 3, 5–10 mg) were placed in platinum pans and heated to 450 °C at 10 °C min−1. The temperature associated with 3% mass loss was identified as the thermal degradation temperature for all samples. Further thermal processing and analysis steps were conducted below this temperature.A differential scanning calorimeter TA-Q200 (TA Instruments, Waters) was used to track glass transition temperatures (Tg's) and melting transitions (Tm's) of the polymers. Samples (n = 3, 3–5 mg) were placed in t-zero aluminum pans and equilibrated at −40 °C, heated to 150 °C at 10 °C min−1, held isothermally at 150 °C for 2 min, cooled to 50 °C at 5 °C min−1, held isothermally for 20 min, cooled to 140 °C, held isothermally for 2 min, and re-heated to 150 °C at a rate of 10 °C min−1. Transition temperatures were evaluated using the second heating cycle according to the mid-point of the endothermic inflection (Tg) and the minimum of the endothermic peak (Tm). The enthalpy of melting (ΔH) was characterized by quantifying the area under the endothermic crystal melting curve. If an exothermic cold crystallization peak was observed before the Tm, the area under that peak was subtracted from the measured area under the melting curve.
2.7 Shape memory behavior
To evaluate shape fixity and recovery of the synthesized polymers, a dynamic mechanical analyzer (DMA, TA-Q800, TA Instruments, Waters) in a controlled-force mode was employed. Samples were cut into dog bones (ASTM D638 type IV, scaled down by a factor of 4) with a gauge length of 6.25 mm and width of 1.5 mm. The samples were heated to 60 °C (above their Tg), and a controlled force was applied to stretch them to 40% strain at 0.03 N min−1, followed by cooling to −5 °C at 2 °C min−1. Samples were unloaded at 0.1 N min−1 and re-heated to 60 °C at 2 °C min−1 to assess the shape recovery ratio. This cycle was completed three times and the second cycle was used for analysis. Shape recovery (Rr) and shape fixity (Rf) were measured using eqn (2) and (3) for cycle number N, in which εu is the remaining strain after unloading (fixed strain), εm is maximum strain at loading, and εp is the remaining strain after recovery (permanent strain).1,29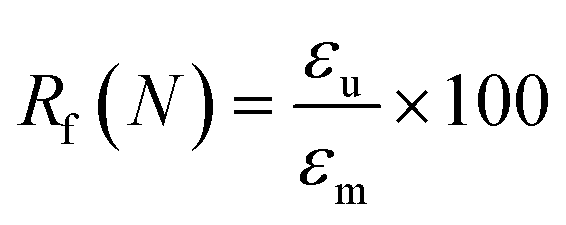 | (2) |
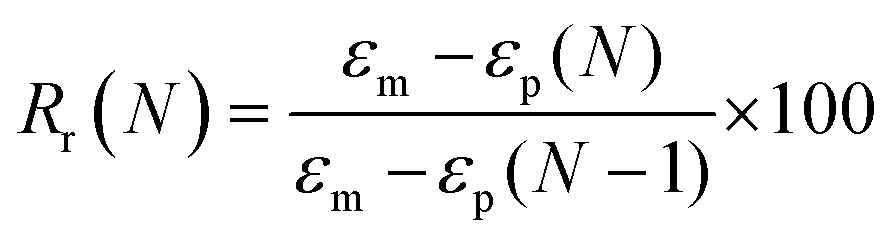 | (3) |
2.8 Mechanical properties
The dog bone die described in Section 2.7 was used to cut samples for mechanical testing. Samples (n = 3) were placed into a tensile tester (100P Universal Testing Machine, Test Resources, Inc., Shakopee, MN) with a 24 N load cell and strained at a rate of 2 mm min−1 until rupture to measure tensile strength, Young's modulus, and elongation at break from resulting stress/strain curves.2.9 Degradation analysis
Samples were cut from polymer films using an 8 mm diameter punch, rinsed with DI water, and dried under vacuum. Then, a gravimetric scale was used to obtain initial masses. The samples were placed in 20% H2O2 (accelerated oxidative media) or 0.1 M NaOH (accelerated hydrolytic media) at 37 °C. Samples were washed with DI water every 4 days and dried under vacuum for 48 hours at room temperature. Masses were recorded after each drying process, and then samples were returned to media for up to 40 days. Thermal properties (DSC) and surface chemistry (FTIR) were assessed on sacrificial samples at each time point.302.10 Shape stability
Dog bones (n = 3) were cut from each polymer film, heated above their transition temperature using an oven, folded in half lengthwise, and cooled to room temperature while folded to lock in the temporary shape. A digital caliper was used to measure the initial distance between two folded edges. The folded samples were placed in oxidative (3% H2O2) or hydrolytic (0.1 M NaOH) degradation media at 37 °C. Every day (up to 40 days), the distance between folded edges was measured using a caliper and used to determine the angle of unfolding (θ, degrees) to assess shape recovery of samples. After 40 days, the samples were placed in a water bath at 75 °C to characterize shape recovery after degradation. This temperature was selected to ensure that samples were well above their transition temperatures (determined using DSC as described in Section 2.5) and that full shape recovery could be characterized. The samples were imaged using a camera, and the amount of unfolding was used to quantify shape recovery.2.11 Film processing effects
To evaluate the influence of processing methods on polymer properties and microphase separation, three different methods were used: (1) a polymer film was formed directly from the polymerization step, (2) a polymer film was made using a hot press described in Sections 2.2, and (3) a polymer film was made by solvent casting (i.e., dissolved 2 g of polymer in 10 mL chloroform, casted into a Teflon mold, and dried under vacuum). After processing films, samples were cut using the dog bone die described in Section 2.6. Then DSC (only for HDI-6 as a representative sample) and DMA (for all formulations) were used to evaluate thermal and shape memory properties, respectively.2.12 Annealing effects
To investigate the effect of annealing on thermal behaviors of the polymers and their microphase separation, polymers that were formed directly from polymerization were used and compared with polymer films formed using solvent casting described in Section 2.11. Samples (3–5 mg) were placed in DSC aluminum pans and incubated at varied temperatures for 30 minutes: below their Tg (40 °C), slightly above their Tg (60 °C), slightly below their Tm (80 °C), and above their Tm (120 °C). A control sample was employed that did not undergo thermal treatment. The pans were removed from the oven and cooled to room temperature. Then, the pans were placed in the DSC and characterized using the cycle described in Section 2.5.2.13 Electrospinning
To evaluate the ability of synthesized polymers to form fibrous mats, a custom electrospinner was utilized. Each polymer (1.5 g) was dissolved in 4 mL chloroform/1 mL DMF and placed into a Spraybase® electrospinning syringe pump set to dispense the solution at 2 mL h−1. The distance between the needle and the collecting drum (covered with a sheet of aluminum foil) was set to 10 cm, and a positive charge of 12.6 kV was applied to the needle tip with 50% humidity at room temperature. The collecting drum (95.6 mm diameter, 300 mm width) rotation rate was set at 2000 rpm.31 After electrospinning for 2.5–3 hours, the mats were removed from the drum and dried under vacuum to remove solvent residue. A Denton Vacuum sputter coater was utilized to sputter coat the samples. Then, they were imaged with a scanning electron microscope (SEM, Jeol JSM 5600) at 300× magnification and 20 kV high vacuum to assess the morphology and thickness of the fibers formed from polymers synthesized with and without solvent. Shape memory and mechanical properties of the fiber mats were evaluated and compared using the methods described in Sections 2.6–2.7.2.14 Statistical analysis
Data are presented as mean ± standard deviation. Sample sizes are provided with data presented in each figure. ANOVA with Tukey's post hoc test was performed on data using Microsoft Excel. Statistical significance was taken as p < 0.05.3. Results and discusion
3.1 Polymer synthesis
Fig. 2(a) shows the FTIR spectra of synthesized STPUs, which confirm the successful synthesis of polyurethanes with similar chemical structures as those reported in our previous work.32 Namely, the appearance of peaks corresponding to urethane bonds N–H (∼3320 cm−1) and CO (∼1677 cm−1) was observed. Additionally, the disappearance of the vibration band at ∼2270 cm−1 proves the complete consumption of isocyanate groups during synthesis, which is also complemented by a reduction in the OH peak at ∼3400 cm−1.12,32,33 SEC results (Fig. 2(b)) showed higher Mn for all formulations synthesized using the solvent-free method as compared with polymers of the same chemistry synthesized using the previously reported solvent-based method. Additionally, HDI-3, 4, and 6 synthesized using the solvent-free method had lower Đ values. This result is attributed to a higher concentration of monomers during reaction in solvent-free conditions compared to diluted monomers in the solvent-based method, which may reduce the occurrence of side reactions by increasing the speed of reaction and/or increasing monomer reaction efficiency.
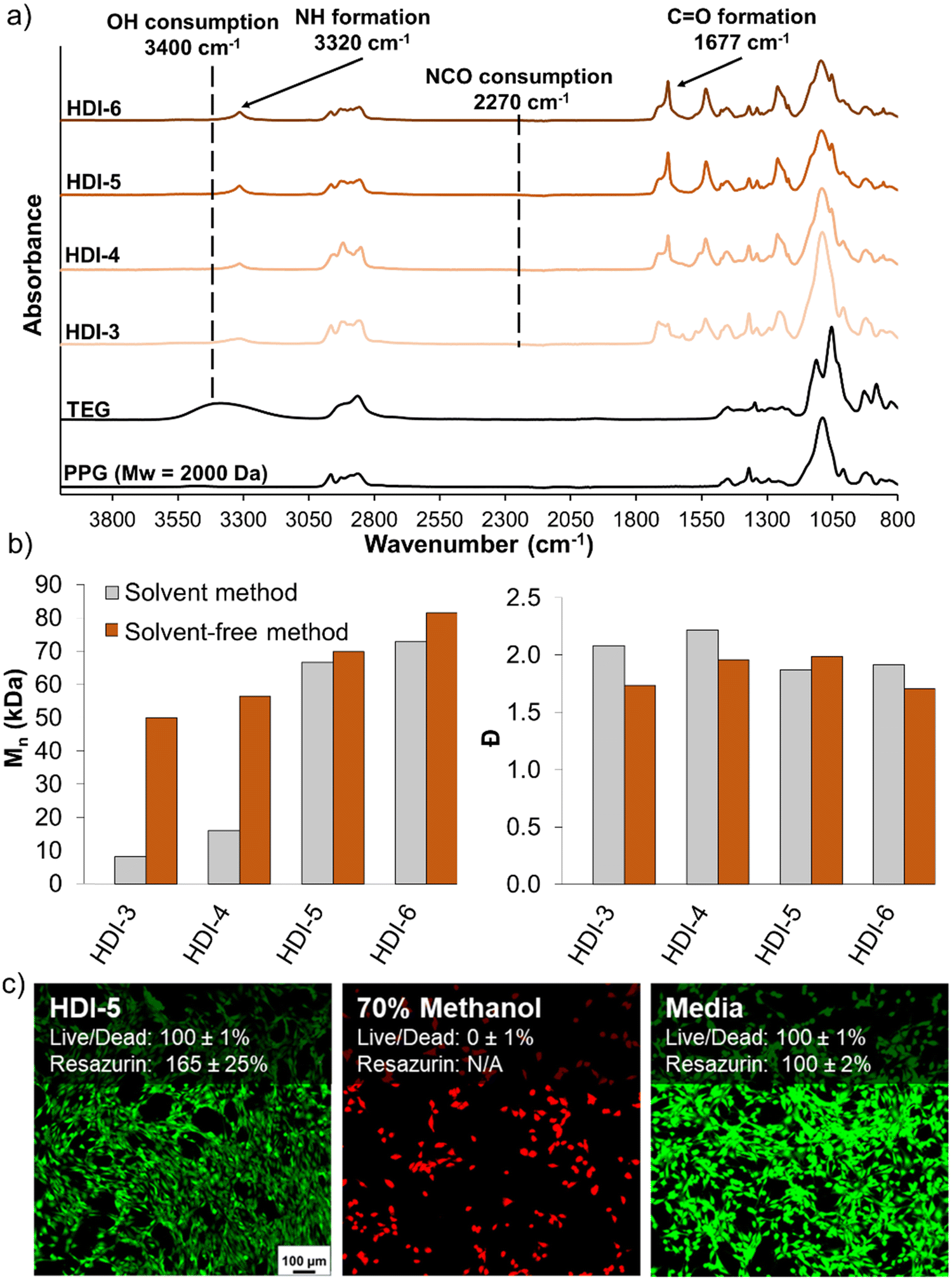 | ||
| Fig. 2 (a) Characterization of synthesized segmented thermoplastic polyurethanes using Fourier transform infrared (FTIR) spectroscopy. (b) Number average molecular mass (Mn, left) and polydispersity index (Đ, right) of polymers synthesized with and without solvent (n = 1). (c) Cytocompatibility of representative segmented thermoplastic polyurethane (HDI-5) measured using live/dead and resazurin assays (n = 3, mean ± standard deviation displayed, scale bar applies to all images). | ||
After synthesis of polymers, HDI-5 was taken as a representative STPU formulation on which to assess cytocompatibility, as all polymers were synthesized from the same components, Fig. 2(c). Using both live/dead and resazurin assays, the STPU shows high cytocompatibility, with higher relative cell numbers (∼165%) relative to the positive control measured using the resazurin assay. While further characterization of cell/material interactions would be required before employing these materials in biomedical applications, these studies provide an initial indication of the high cytocompatibility of STPU's synthesized using the solvent-free method.
3.2 Effects of synthesis route on thermomechanical properties
In this segmented polyurethane system, we observed an initial glass transition temperature (Tg) at approximately 0 °C, which is attributed to the Tg of the PPG soft segment. When a polyol is reacted into a polyurethane, its Tg may or may not be fully apparent in the DSC results. If it does appear, the actual value of the soft segment Tg typically differs from that of the individual polyol material. In this case, the low Tg at 0 °C is higher than that of the individual PPG component (approximately −70 °C34). However, if this temperature is observable in DSC, its measurement serves as an indicator of successful phase separation within segmented polyurethanes.35 Additionally, as a result of this low Tg, the polyurethanes exhibit flexibility at room temperature. This study primarily focuses on the second observed Tg (∼55 °C) that is attributed to the hard segment, as this is the temperature at which shape memory programming can occur.Fig. 3(a) shows hard segment glass transition temperatures (Tg), melting points (Tm), and enthalpy of melting (ΔH) of polymers synthesized using the previously reported solvent-based method and the new solvent-free method. The values for the solvent-based method samples were previously reported by us.1 As shown, thermal properties of polymers synthesized using the two methods are very similar. There was a slight increase in Tm of HDI-3 and HDI-4 using the solvent-free method, which may be due to better microphase separation of polymers synthesized without solvents and/or higher molecular weight.36 Based on the measured enthalpies of melting, as we increase the hard segment content in our library, the polymers have higher contents of crystalline lamellae, which is expected based on more interaction sites for hydrogen bonds between hard segments on chains. There were no observable differences in crystallinity based on the two synthesis methods. However, once polymers achieve a Mn of at least 50 kDa (all polymers except for HDI-3 and HDI-4 synthesized using the solvent method) there is a direct correlation (R2 = 0.94) between Mn and enthalpy of melting, regardless of synthesis method, indicating that as polymer chain molecular weight is increased, the crystallinity is also increased.
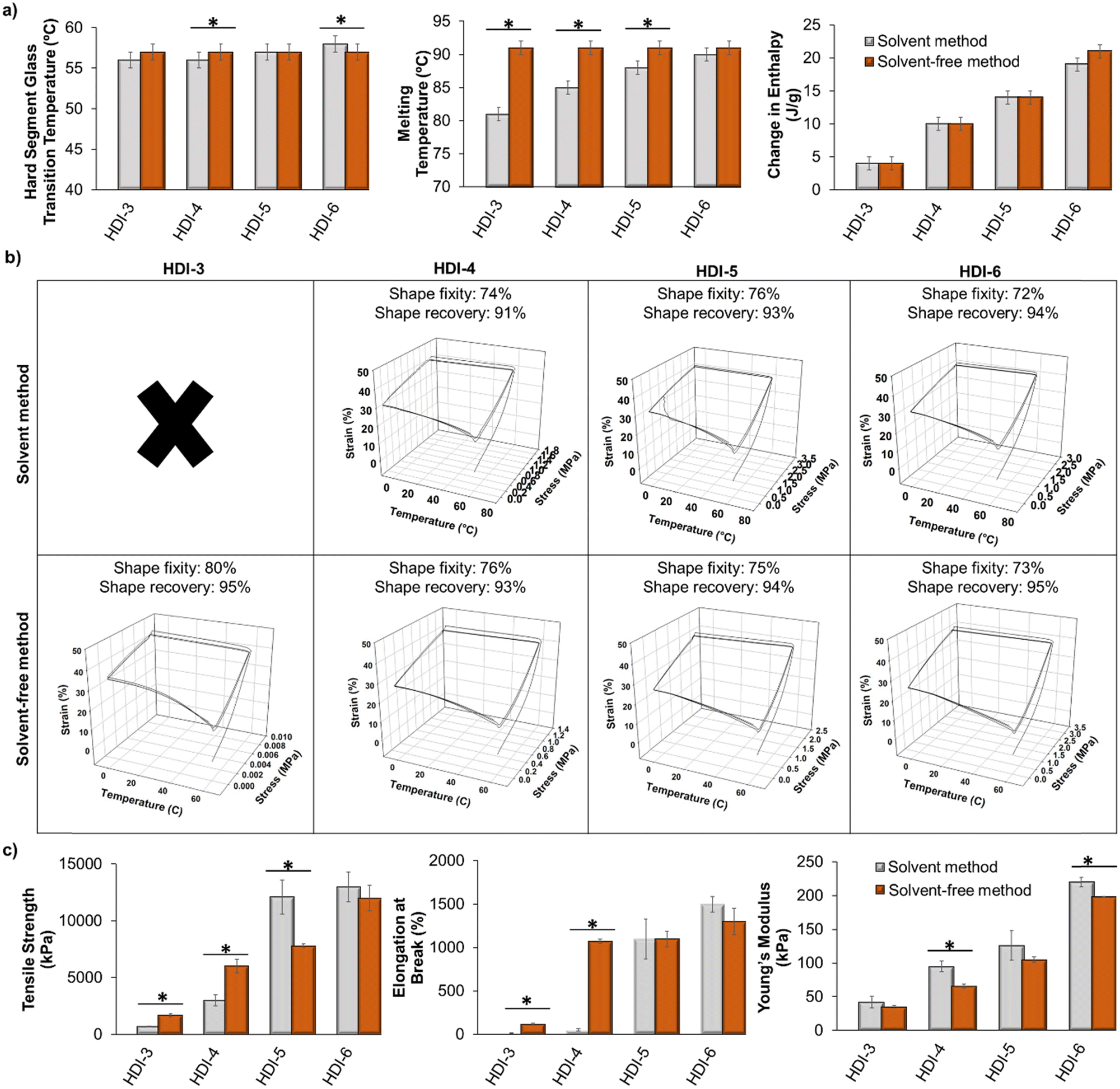 | ||
| Fig. 3 Comparison of (a) thermal (n = 3), (b) shape memory (n = 1, ×: no shape memory properties were observed), and (c) tensile (n = 3) properties of polymers synthesized using solvent-based (data are from our previous work32) and solvent-free methods. Mean ± standard deviation displayed. *p < 0.05 relative to corollary sample synthesized using solvent-based method. | ||
Overall, all transition temperatures (hard segment Tg) are well above body temperature. This property could ensure stable temporary shape fixation after implantation while in physiological conditions, as body temperature heating would not be a trigger for shape recovery. Therefore, these polymers could provide a platform for incorporation of specific stimuli-responsive components to induce shape changes only in response to the desired stimulus. For example, our prior work employed the solvent-based polymers with high transition temperature to incorporate magnetic nanoparticles that enable user-defined shape recovery upon application of a magnetic field.9 The magnetic field induces localized heating within the polymer samples to drive shape change while maintaining safe (∼40 °C) temperatures in surrounding media. We also incorporated peptides that are degraded by bacterial proteases to provide environmentally-responsive SMPs that change shape in the presence of bacteria without the need for external input of a heat stimulus.12 We envision that other groups could use similar approaches to provide biomaterials the respond to light, electric fields, and/or other enzymes.
Fig. 3(b) displays the shape memory properties of polymers synthesized by the two methods. HDI-3 synthesized by the previously reported solvent method does not show shape memory properties, but by using the solvent-free method, these polymers showed high shape fixity and recovery, which is likely due to the increased molecular weight. In general, all polymers with Mn above 15 kDa demonstrate shape memory, with no clear trends between shape memory properties and thermal properties (i.e., enthalpies of melting, Tg, or Tm). For other formulations, shape fixity and recovery were approximately the same for the two methods. Fig. 3(c) shows tensile properties of polymers synthesized using the solvent-based and solvent-free methods. The solvent-free method resulted in significantly increased tensile strength and elongation at break of HDI-3 and 4. The modulus of HDI-3 was not impacted, while the modulus of HDI-4 decreased with the new synthesis approach. Additionally, HDI-5 synthesized using the solvent-based method had higher tensile strength than the new solvent-free HDI-5, but elongation at break and modulus were unaffected by the synthesis method. HDI-6 synthesized by the two methods showed similar tensile strength and elongation at break, but the modulus was slightly reduced in the HDI-6 synthesized using the solvent-free method. Overall, this data proves that tensile strength, modulus, and elongation at break of the polymers are tunable with the new synthesis method. Namely, as we increase the hard segment content, these properties increase.
We can attribute the tunable mechanical properties to stronger interactions between chains, higher crystallinity, and increased phase separation that is achieved with higher hard segment content and increased molecular weight. When comparing mechanical properties with molecular weight, there is a general correlation between higher molecular weight and increased tensile strength and ultimate elongation (R2 = 0.70 and 0.79, respectively). Modulus does not correlate well when evaluating the entire set of polymers (R2 = 0.44); however, when only higher molecular weight polymers are assessed (Mn > 50 kDa), a correlation is apparent between increased molecular weight and increased stiffness (R2 = 0.80). Clear correlations occurred with increased enthalpy of melting and increased tensile strength, ultimate elongation, and modulus (R2 = 0.86, 0.75, and 0.91, respectively). Thus, the enthalpy of melting or crystalline content of these STPUs provides a better indication of increased mechanical properties than molecular weight.
In general, the solvent-free approach provides a mechanism for increasing reaction efficiency to achieve higher molecular weight polymers, particularly in lower hard segment (HDI-3 and HDI-4) formulations. This increased molecular weight drastically affects Tm (Fig. 3(a), middle), change in enthalpy of melting (Fig. 3(a), right), and mechanical properties (Fig. 3(c)), while providing SMPs with similar transition temperatures (hard segment Tg, Fig. 3(a), left) and similar shape fixity and recovery (Fig. 3(b)). Thus, this method enables broad tuning of molecular weight and crystallinity to alter mechanical properties and melting behavior independently of shape memory properties.
3.3 Degradation and secondary shape stability
Fig. 4(a) displays mass loss of synthesized STPUs in oxidative (left) and hydrolytic (right) degradation media over 40 days. As shown HDI-4, 5, and 6 experienced less than 5% mass loss in accelerated oxidative media, while HDI-3 showed 9% mass loss. The increased stability of higher hard segment polymers is in concurrence with our previous work; however, HDI-3 has higher oxidative stability than the previously reported polymer synthesized using the solvent-based method, which underwent ∼28% mass loss over 40 days.32 The improved oxidation resistance may be due to better microphase separation and higher molecular weight of synthesized STPU's using the solvent-free method. In accelerated hydrolytic media, all formulations showed very low levels of mass loss (∼3%), which was expected based on the lack of hydrolyzable groups in the polymer backbones.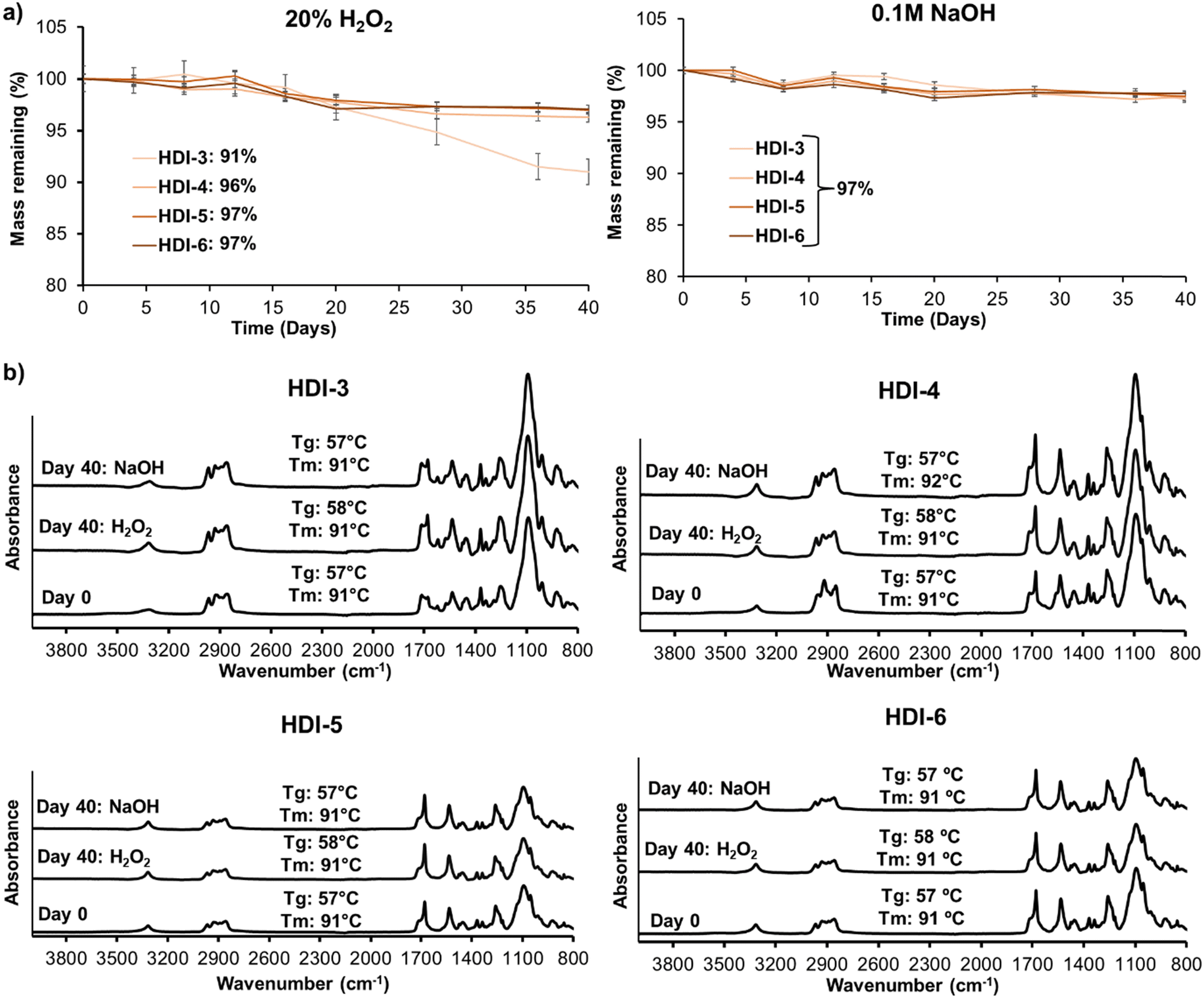 | ||
| Fig. 4 (a) Mass remaining of segmented thermoplastic polyurethanes synthesized by solvent free method in (left) 20% H2O2 and (right) 0.1 M NaOH over 40 days. p > 0.05 between day 0 and 40 for each formulation and between all formulations at day 40. (b) FTIR spectra and corresponding Tg and Tm (n = 1) at 0 and 40 days of incubation in hydrolytic and oxidative media for HDI-3, HDI-4, HDI-5, and HDI-6. | ||
Overall, no significant differences were observed in mass losses over time, suggesting that these materials exhibit good resistance to degradation. It should be noted that these experiments were carried out in accelerated oxidation and hydrolysis conditions and that biological conditions contain much lower concentrations of reactive oxygen species and a more neutral pH. Thus, we expect that these materials would be highly stable in physiological conditions and would experience negligible mass loss in more physiologically relevant degradation media (e.g., 3% H2O2 or phosphate buffered saline). This result is encouraging, especially in the context of biomedical applications where long-term stability and durability are desired. These results also indicate that these materials do not undergo substantial erosion, which is crucial for maintaining their structural integrity and functional properties over extended periods. High stability may be important for applications such as wound dressings, implants, or drug delivery systems, where the materials need to maintain their effectiveness and structural integrity during the intended use. However, it is important to note that the lack of significant mass loss alone may not provide a complete picture of the material performance.
To extend our understanding of the polymer degradation profiles, Fig. 4(b) shows surface chemistry and thermal properties of STPUs after 0 and 40 days of incubation in accelerated oxidative (20% H2O2) and hydrolytic (0.1 M NaOH) media. FTIR spectra did not show observable changes in peaks; and thermal properties were unchanged and remained well above body temperature throughout the experiments. These results reveal that accelerated degradation media did not affect the polymer backbone in synthesized STPUs, which confirms high biostability. Thus, oxidative degradation (from reactive oxygen species produced during healing) and hydrolytic degradation (from water in the body) would be negligible in these polymers after implantation.12,24,32,37,38 Additional analyses, such as mechanical testing over time, should be considered in future work to comprehensively evaluate the material performance and suitability for any intended application.
As Fig. 5(a) and (b) show, programmed HDI-5 and 6 samples (folded into a secondary shape) experienced minimal shape recovery over 40 days in both oxidative and hydrolytic degradation media. Shape recovery of HDI-4 was also low, with ≤10% in hydrolytic and oxidative media. HDI-3 samples unfolded by ∼65%, which was expected due to their weaker interchain interactions and lower crystallinity. The HDI-3 synthesized using the previously reported solvent-based method did not have shape memory properties;32 thus, the solvent-free method confers improved mechanical and shape memory properties to HDI-3. After incubation, retention of shape recovery capabilities was confirmed by heating samples to above their Tg's and macroscopically analyzing unfolding to their primary shapes, Fig. 5(c) and (d). Synthesized STPUs maintained their shape memory properties even after long-term exposure to degradation media, with full recovery to their primary shape upon input of a heat stimulus. Overall, these results demonstrate that HDI-5 and 6 have exceptional ability to maintain their secondary shape in physiological conditions.12,32 This property could enable the use of these polymers for applications that require specific responses to external cues, such as enzymes and magnetic fields, after insertion of desired stimuli-responsive components.
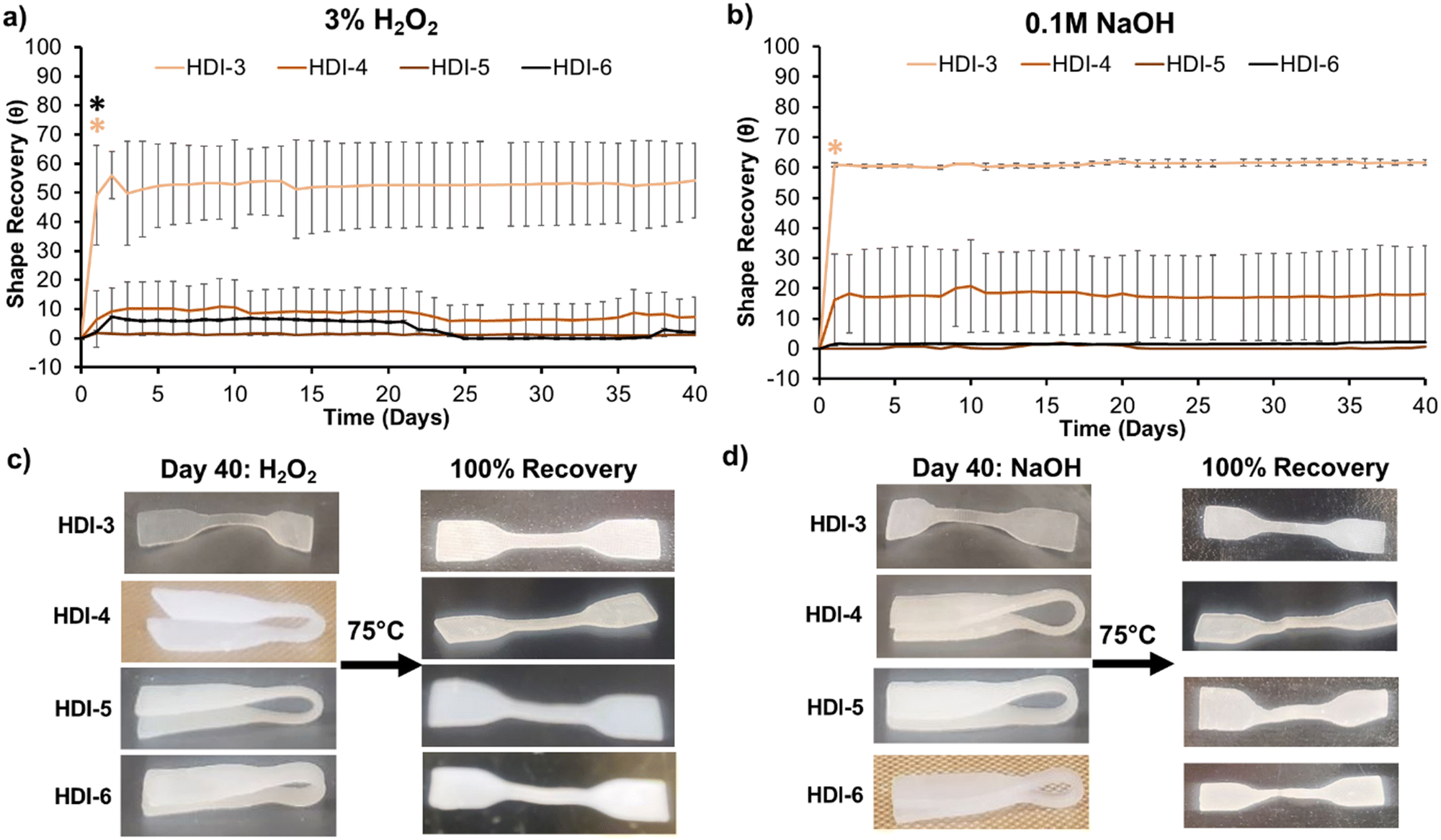 | ||
| Fig. 5 Shape recovery of programmed (folded) segmented thermoplastic polyurethanes measured as the angle of folded samples (θ, degrees), determined based on distance between folded sample ends in (a) 3% H2O2 and (b) 0.1 M NaOH over 40 days of incubation at 37 °C. n = 3, mean ± standard deviation displayed. *p < 0.05 relative to day 0 for all remaining time points, with font color matching corollary sample line color. †p < 0.05 relative to day 0 at given time point, with font color matching corollary sample line color. Shape recovery of samples upon external heating to 75 °C after 40 days of incubation in (c) 3% H2O2 and (d) 0.1 M NaOH. | ||
3.4 Processing effects
Microphase separation of hard and soft segments plays an important role in STPU shape memory and thermomechanical properties, and polymer processing can be used to control microphase separation.39 In general, during heating and/or dissolution, hard and soft segments become more mobile. Upon cooling or drying, the segments begin to form new intermolecular interactions, such as hydrogen bonds, to induce phase separation.21,39 When the material is cooled or dried, these new intermolecular interactions are stabilized within their respective domains to separate the hard and soft phases, leading to the formation of physically crosslinked networks. By controlling polymer processing conditions, such as heating and cooling, it is possible to influence the microphase separation and morphology of STPUs.40 This, in turn, affects the final material properties, including its shape memory behavior and thermomechanical performance.39 Optimizing the microphase separation through appropriate processing techniques enables customization of STPU materials with desired mechanical properties, shape memory characteristics, and processing capabilities. An understanding of microphase separation and its control is essential for designing and tailoring STPUs. Processing can be employed during or after initial film formation. To understand the effects of film fabrication processes, films formed directly from the solvent-free synthesis were compared with hot pressed and solvent cast films in terms of thermal and shape memory properties.Fig. 6(a) shows DSC thermograms of materials made with the three film formation methods. In amorphous materials, the glass transition has a noticeable impact on thermal conductivity, resulting in a more distinctive peak in DSC. This phenomenon is significantly less pronounced in semi-crystalline polymers, primarily because only the amorphous phase contributes to this behavior, while the crystalline phase exhibits a more dominant influence. Thus, Tg does not change the heat capacity of semi-crystalline polymers to the same degree as that of amorphous polymers.41 Polymers with hard and soft domains have even further complicated thermal transitions. Within the STPU literature, the classification of the multiple endothermic peaks associated with hard segments is somewhat controversial, and different authors have attributed these peaks to various phenomenon in the polymer strcuture.42–45 Commonly, the first endothermic event is reported as the Tg of the hard segment.42 Seymour et al. reported that these endothermic peaks rely on they thermal history of the polymer, and they observed that the annealing temperature and hard segment length have significant effects on the endotherms.45 Yuan et al. reported the endothermic peaks as melting points of portions of the hard segments with different degrees of order.46
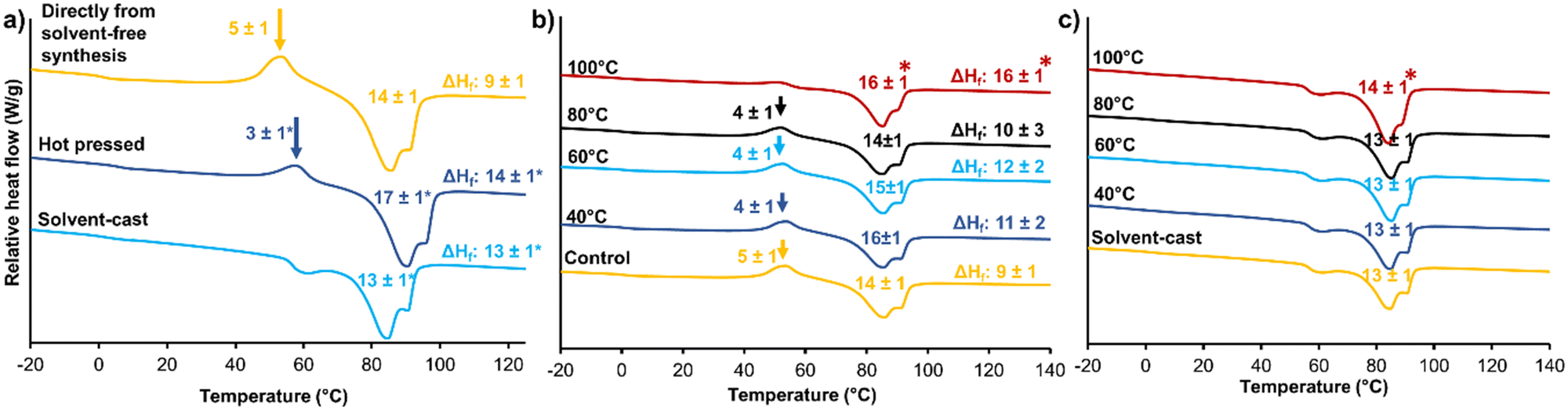 | ||
| Fig. 6 (a) The effect of processing methods on cold crystallization of representative segmented thermoplastic polyurethane (HDI-5) films formed from the solvent-free synthesis method. DSC thermograms of HDI-5 films after annealing at different temperatures for (b) control films formed directly from solvent free method and (c) films formed via solvent casting. All thermograms show the second heating cycle of DSC runs, and overlaid numbers are mean ± standard deviation of crystallization enthalpies, melting enthalpies. ΔHf = ΔHmelt − ΔHcrystallization (J g−1) for n = 3 samples. p < 0.05 relative to control solvent-free film. Arrows point to crystallization peak in heating cycle. | ||
Here, we attribute the first endotherm in this region (around 60 °C) to the Tg of the hard segment due to change in heat capacity before and after the transitions. The other two endotherms at higher temperatures are associated with melting of hard segment domains with varying degrees of order that arise due to differences in the arrangement and alignment of the polymer chains within the material. These domains can vary based on (1) processing conditions, such as temperature and solvent, that can influence the arrangement of polymer chains; (2) degree of crystallinity, based on annealing time/temperature and other processing that might promote a different degree of crystallinity in hard segments with different chain lengths; and (3) cooling rate, which can affect the order of crystalline lamellae (i.e., rapid cooling may result in a less ordered structure compared to slower cooling).47,48
Hot pressed films had reduced exothermic crystallization peaks between the hard segment Tg and Tm endothermic transitions relative to that of the directly made film, while solvent casting completely eliminated the crystallization peak from the heating cycle of DSC. This result indicates that the crystalline phase was able to completely form during solvent casting, eliminating thermal reorganization during heating. The ΔHf (the difference between ΔH melt and ΔH crystallization) values show that the crystalline content in hot pressed and solvent-cast films were similar and that both processing techniques increased crystallinity as compared with films directly formed via solvent-free method. The Tg of the soft segment is still observable at around 0 °C after all three processes (directly from solvent-free synthesis: 1 ± 1 °C, hot pressed 1 ± 1 °C, solvent-cast: 2 ± 1 °C).
Shape recovery was generally unaffected by these film processing methods, Table 2; however, shape fixity was consistently increased in hot pressed films and further increased in solvent cast films. These methods provide more time for chains to form crystalline regions and therefore can improve phase separation to increase crystallinity and enhance shape memory properties.22 This data provides an initial indication of how shape memory properties can be tuned with a single polymer chemistry by altering the film formation process.
| Direct from synthesis Rf & Rr | Hot pressed film Rf & Rr | Solvent-cast film Rf & Rr | |
|---|---|---|---|
| HDI-3 | 77 & 93 | 80 & 95 | 86 & 98 |
| HDI-4 | 66 & 93 | 76 & 93 | 84 & 91 |
| HDI-5 | 61 & 95 | 75 & 94 | 82 & 92 |
| HDI-6 | 61 & 97 | 73 & 95 | 84 & 92 |
3.5 Annealing effects
After polymer films are formed, they can be subjected to additional thermal processing, such as annealing. As shown in Fig. 6(b), heating polymer films formed directly from the solvent-free method to above their Tm's (120 °C) eliminates the crystallization peak in the heating cycle of DSC. During this annealing step, the polymer melts to provide more time for phase separation and crystallization.39 Wei et al. reported a similar trend, where the melt enthalpy increased with increased annealing time, resulting in the formation of more crystals.42 Annealing at lower temperatures did not affect crystallinity (enthalpy of melting), because polymer chains were not mobile enough at these temperatures to allow rearrangement.39Fig. 6(c) shows the effects of annealing on films formed by solvent casting. The solvent-cast film does not have a crystallization peak in its heating cycle. Thus, annealing does not affect the thermal properties of these samples in terms of formation/removal crystallization peak. Overall, these results show that due to the ability to rapidly form polymer films using the solvent-free method, polymer chains do not have enough time to form crystalline lamellae during synthesis. However, film fabrication processes (e.g., solvent casting) and/or post-fabrication annealing can be used to maximize crystallinity after synthesis.3.6 Fiber formation
Beyond bulk film fabrication, porous and fibrous scaffolds can enhance utility of polymeric biomaterials. For example, electrospun fibers can enhance control over drug delivery, provide a scaffold for desired cell/material interactions, or serve as wound dressings or protective coatings.49–51 To expand the potential use of these STPUs, we explored the effects of synthesis route and chemistry on the ability to form electrospun fibers, Fig. 7(a). During the electrospinning process, a charge is applied to a polymer solution inside of a needle to deform the solution droplet into a Taylor cone as it exits the needle. As the electric field increases, the intensity of attractive force between the polymer solution and the collector can overcome the surface tension of the droplet and make it to deform into fibers as it travels toward the rotating collector. During this time, solvent evaporates and dry fibers form.52 Fiber diameter and morphology depend on solution properties, such as viscosity and polymer molecular weight, because fibers must be strong enough to bear the tension from stress and movement until they reach the collector to prevent fiber breakage. Therefore, increasing molecular weight and chain entanglements can be used to provide sufficient force and interactions for fiber jets to withstand the electrospinning process, and variables in polymer synthesis and chemistry can affect fiber formation.52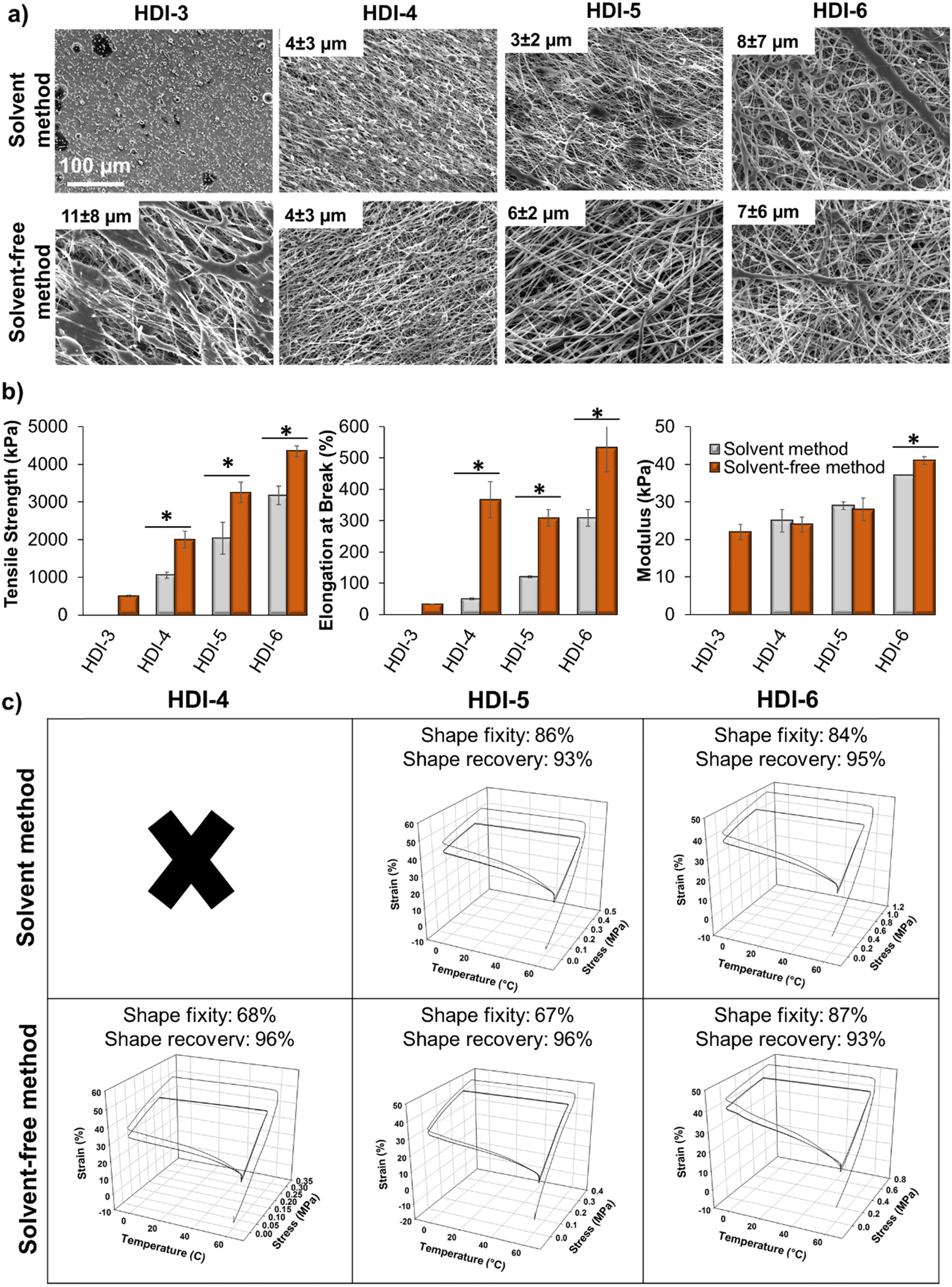 | ||
| Fig. 7 (a) Scanning electron micrographs of electrospun fibers with overlaid fiber diameter (n = 10, quantified using Imagej). Scale bar applies to all images. (b) Tensile properties of fibers. n = 3, mean ± standard deviation displayed. *p < 0.05 between each group. (c) Dynamic mechanical analysis of fibers with corresponding shape fixity and shape recovery values (n = 1; ×: no shape memory properties were observed). | ||
HDI-3 synthesized using the previously reported solvent-based method was not able to form fibers (only evidence of electrospraying was observed in SEM images), while the solvent-free method successfully enabled formation of fiber mats from HDI-3. Molecular weight and chain entanglements of polymers play important role in fiber formation and transitioning from electrospraying to electrospinning capabilities52 to enable application in wound dressings,53,54 drug delivery,55–57 dye removal,58 and scaffolds for tissue engineering.59–61 The solvent-free method sufficiently increases molecular weight of HDI-3 to enable fiber formation. As seen in Fig. 7(a), HDI-4 synthesized using the solvent-based method forms beads, which may be due to low viscosity of the polymer solution52 as a result of lower polymer molecular weight, while the higher molecular weight solvent-free HDI-4 formed stable fibers. As molecular weight and polymer chain interactions increase, less beading/fusion is observed in electrospun scaffolds.
While no significant differences were observed in fiber diameters, Fig. 7(a), HDI-3 (solvent-free) and HDI-6 (both methods) fibers were less uniform with larger standard deviations. This result is attributed to viscosity effects, i.e., HDI-3 solutions were lower viscosity and HDI-6 solutions were higher viscosity, both of which can destabilize fibers.52 In the case of HDI-6, reducing polymer concentration and/or using larger needle gauges may be used to compensate for these negative effects in future work.
Overall, these results show that when a polymer is below a certain molecular weight, only droplets form (HDI-3 synthesized by solvent-based method). As viscosity or molecular weight increases, a mixture of beads and non-uniform fibers form (HDI-3 synthesized by solvent-based method and HDI-4 synthesized by both methods). At appropriate molecular weight and viscosity, uniform fibers form (HDI-5 synthesized by both methods). Further increases in polymer molecular weight (HDI-6) decreases fiber uniformity due to increased solution viscosity.52,62 To understand the effects of these fiber variables on scaffold properties, electrospun scaffolds were characterized in terms of mechanical, Fig. 7(b), and shape memory, Fig. 7(c), properties. Fibers made from HDI-4, 5, and 6 synthesized using the solvent-based method had lower tensile strength and elongation at break than those made from polymers synthesized using the solvent-free method, which may be attributed to higher molecular weight and entanglements that result in higher interactions between polymer chains to provide stronger fibers.52 Specifically, when comparing Mn and fiber tensile strength and modulus of the higher molecular weight samples (Mn > 50 kDa), strong correlations are apparent (R2 = 0.92 and 0.88, respectively). As expected, there are also clear correlations between enthalpy of melting and fiber tensile strength and modulus within this group of higher molecular weight polymers (R2 = 0.89 and 0.90, respectively).
Electrospun HDI-3 scaffolds (both methods) and HDI-4 fibers (solvent-based method) did not exhibit shape memory properties. All other fibers exhibit high shape fixity and recovery, demonstrating that shape memory properties are retained after employing complex scaffold fabrication methods. In future work, these STPU fibers could be adapted for use in wound healing, tissue engineering, or drug delivery. By using this highly stable platform, we could also incorporate additional stimuli-responsive components to enable shape changes in response to controlled environmental cues, including applied magnetic fields,9 enzymes,12 or light.63 These scaffolds could enable controlled biological interactions throughout dynamic shape change processes to expand our understanding of cell/material interactions.
4. Conclusions
In this study, a library of biostable segmented polyurethanes with varying hard segment contents was synthesized using polypropylene glycol, hexamethylene diisocyanate, and triethylene glycol using a one-pot solvent-free method. This new synthesis approach is very quick, easy to do, and environmentally-friendly as compared to the previously reported solvent-based method that was employed with this system. All polymers showed high glass transition temperatures (∼57 °C) which can help to maintain the temporary shape after exposure to physiological conditions. Polymers also show high shape fixity (73–80%) and recovery (93–95%) and minimal mass loss in accelerated oxidative (20% H2O2) and hydrolytic (0.1 M NaOH) media. The polymers can remain stable in their secondary shapes in degradation media, which could be beneficial for designing SMPs with user-defined shape recovery in response to specific stimuli. By post-synthesis processing, we show that annealing to above the melting point and solvent casting can enhance microphase separation to improve thermal and shape memory properties. STPUs synthesized using the solvent-free method showed better ability for fiber formation with higher shape memory and mechanical properties as compared with polymers synthesized using the solvent-based method, which is attributed to higher molecular weight, enhanced crystallinity, and better phase separation. In future work, these biostable STPUs could be coupled with a range of other stimuli-responsive components to enable specific responses for architecture changes and/or controlled bioactive factor delivery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge seed funding from BioInspired Syracuse to support this work.References
- M. Ramezani and M. B. B. Monroe, ACS Appl. Polym. Mater., 2022, 4, 1956–1965 CrossRef CAS.
- M. Sabzi, M. Ranjbar-Mohammadi, Q. Zhang, S. Kargozar, J. Leng, T. Akhtari and R. Abbasi, J. Appl. Polym. Sci., 2019, 136, 1–10 Search PubMed.
- H. T. Beaman, E. Shepherd, J. Satalin, S. Blair, H. Ramcharran, S. Serinelli, L. Gitto, K. S. Dong, D. Fikhman, G. Nieman, S. G. Schauer and M. B. B. Monroe, Acta Biomater., 2022, 137, 112–123 CrossRef CAS PubMed.
- N. Christmas, A. U. Vakil, C. J. Hatch, S. Dong, D. Fikhman, H. T. Beaman and M. B. B. Monroe, J. Biomed. Mater. Res., Part B, 2021, 109, 681–692 CrossRef CAS.
- S. Witold, M. Annick, H. Shunichi, Y. L’Hocine and R. Jean, Biomed. Mater., 2007, 2, S23 CrossRef.
- N. M. Petryk, G. Haas, A. U. Vakil and M. B. B. Monroe, J. Biomed. Mater. Res., Part A, 2022, 110(8), 1422–1434 CrossRef CAS.
- M. Sobczak, Polym.-Plast. Technol. Eng., 2015, 54, 155–172 CrossRef CAS.
- C. Shuai, Z. Wang, S. Peng, Y. Shuai, Y. Chen, D. Zeng and P. Feng, Mater. Chem. Front., 2022, 6, 1456–1469 RSC.
- A. U. Vakil, M. Ramezani and M. B. B. Monroe, Materials, 2022, 15, 7279 CrossRef CAS.
- A. U. Vakil, M. Ramezani and M. B. B. Monroe, ACS Appl. Bio Mater., 2022, 5, 5199–5209 CrossRef CAS.
- M. B. B. Monroe, A. D. Easley, K. Grant, G. K. Fletcher, C. Boyer and D. J. Maitland, ChemPhysChem, 2018, 19, 1999–2008 CrossRef CAS.
- M. Ramezani and M. B. B. Monroe, J. Biomed. Mater. Res., Part A, 2023, 111, 1–17 CrossRef.
- M. B. Browning and E. Cosgriff-Hernandez, Biomacromolecules, 2012, 13, 779–786 CrossRef CAS.
- A. U. Vakil, N. M. Petryk, E. Shepherd, H. T. Beaman, P. S. Ganesh, K. S. Dong and M. B. B. Monroe, ACS Appl. Bio Mater., 2021, 4, 6769–6779 CrossRef CAS.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS.
- A. Utpal Vakil, N. Marie Petryk, E. Shepherd, H. T. Beaman, P. S. Ganesh, K. S. Dong, M. Beth, B. Monroe, A. U. Vakil, N. M. Petryk, E. Shepherd, H. T. Beaman, P. S. Ganesh, K. S. Dong and M. B. B. Monroe, ACS Appl. bio Mater., 2021, 4, 6769–6779 CrossRef.
- L. B. Jiang, D. H. Su, P. Liu, Y. Q. Ma, Z. Z. Shao and J. Dong, Osteoarthr. Cartil., 2018, 26, 1389–1399 CrossRef PubMed.
- X. Xiao, W. Zhao, J. Liang, K. Sauer and M. Libera, Colloids Surf., B, 2020, 192, 110989 CrossRef CAS.
- A. Loredo-Treviño, G. Gutiérrez-Sánchez, R. Rodríguez-Herrera and C. N. Aguilar, J. Polym. Environ., 2012, 20, 258–265 CrossRef.
- I. Yilgör, E. Yilgör and G. L. Wilkes, Polymer, 2015, 58, A1–A36 CrossRef.
- E. G. Bajsic, V. Rek, A. Sendijarevic, V. Sendijarevic and K. C. Frisch, J. Elastomers Plast., 2000, 32, 162–182 CrossRef CAS.
- B.-X. Cheng, W.-C. Gao, X.-M. Ren, X.-Y. Ouyang, Y. Zhao, H. Zhao, W. Wu, C.-X. Huang, Y. Liu, X.-Y. Liu, H.-N. Li and R. K. Y. Li, Polym. Test., 2022, 107, 107489 CrossRef CAS.
- M. V. Mokeev, S. A. Ostanin, N. N. Saprykina and V. V. Zuev, Polymer, 2018, 150, 72–83 CrossRef CAS.
- C. C. Song, F. S. Du and Z. C. Li, J. Mater. Chem. B, 2014, 2, 3413–3426 RSC.
- C. Dunnill, T. Patton, J. Brennan, J. Barrett, M. Dryden, J. Cooke, D. Leaper and N. T. Georgopoulos, Int. Wound J., 2017, 14, 89–96 CrossRef PubMed.
- P. Singhal, W. Small, E. Cosgriff-Hernandez, D. J. Maitland and T. S. Wilson, Acta Biomater., 2014, 10, 67–76 CrossRef CAS.
- M. Nishikawa, M. Hashida and Y. Takakura, Adv. Drug Delivery Rev., 2009, 61, 319–326 CrossRef CAS.
- N. Bryan, H. Ahswin, N. Smart, Y. Bayon, S. Wohlert and J. A. Hunt, Eur. Cells Mater., 2012, 24, 249–265 CrossRef CAS PubMed.
- S. L. Buffington, J. E. Paul, M. M. Ali, M. M. Macios, P. T. Mather and J. H. Henderson, Acta Biomater., 2019, 84, 88–97 CrossRef CAS PubMed.
- A. U. Vakil, N. M. Petryk, E. Shepherd and M. B. B. Monroe, Polymers, 2021, 13, 4084 CrossRef CAS PubMed.
- Y. Zamani, A. Zareein, L. Bazli, R. NasrAzadani, B. P. Mahammod, S. Nasibi and A. M. Chahardehi, J. Compos. Compd., 2020, 2, 215–227 Search PubMed.
- M. Ramezani and M. B. B. Monroe, ACS Appl. Polym. Mater., 2022, 4, 1956–1965 CrossRef CAS.
- M. Sahraro, H. Yeganeh and M. Sorayya, Mater. Sci. Eng., C, 2016, 59, 1025–1037 CrossRef CAS.
- E. Piorkowska, Z. Kulinski, A. Galeski and R. Masirek, Polymer, 2006, 47, 7178–7188 CrossRef CAS.
- A. Padsalgikar, Applications of Polyurethanes in Medical Devices, William Andrew, 2022 Search PubMed.
- E. Yilgör, E. Yurtsever and I. Yilgör, Polymer, 2002, 43, 6561–6568 CrossRef.
- C. Dunnill, T. Patton, J. Brennan, J. Barrett, M. Dryden, J. Cooke, D. Leaper and N. T. Georgopoulos, Int. Wound J., 2017, 14, 89–96 CrossRef.
- S. Deshayes and A. M. Kasko, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3531–3566 CrossRef CAS.
- J. Balko, B. Fernández-d’Arlas, E. Poselt, R. Dabbous, A. J. Muller and T. Thurn-Albrecht, Macromolecules, 2017, 50, 7672–7680 CrossRef CAS.
- H. Wang, L. Zhang, K. W. E. Peh, Q. Yu, Y. Lu, W. Hua and Y. Men, Macromolecules, 2022, 55, 8566–8576 CrossRef CAS.
- W. N. dos Santos, J. A. de Sousa and R. Gregorio, Polym. Test., 2013, 32, 987–994 CrossRef CAS.
- Z. Wei, Y. Yu, C. Zhou, L. Zheng, X. Leng and Y. Li, J. Therm. Anal. Calorim., 2017, 129, 777–787 CrossRef CAS.
- A. Saiani, A. Novak, L. Rodier, G. Eeckhaut, J.-W. Leenslag and J. S. Higgins, Macromolecules, 2007, 40, 7252–7262 CrossRef CAS.
- L. M. Leung and J. T. Koberstein, Macromolecules, 1986, 19, 706–713 CrossRef CAS.
- R. W. Seymour and S. L. Cooper, Macromolecules, 1973, 6, 48–53 CrossRef CAS.
- R. Yuan, S. Fan, D. Wu, X. Wang, J. Yu, L. Chen and F. Li, Polym. Chem., 2018, 9, 1327–1336 RSC.
- J. T. Koberstein and A. F. Galambos, Macromolecules, 1992, 25, 5618–5624 CrossRef CAS.
- Y. Li, T. Gao, J. Liu, K. Linliu, C. R. Desper and B. Chu, Macromolecules, 1992, 25, 7365–7372 CrossRef CAS.
- R. S. Bhattarai, R. D. Bachu, S. H. S. Boddu and S. Bhaduri, Pharmaceutics, 2019, 11(1), 5 CrossRef CAS.
- S. Agarwal, J. H. Wendorff and A. Greiner, Polymer, 2008, 49, 5603–5621 CrossRef CAS.
- B. Yan, Y. Zhang, Z. Li, P. Zhou and Y. Mao, SN Appl. Sci., 2022, 4, 172 CrossRef CAS.
- S. L. Shenoy, W. D. Bates, H. L. Frisch and G. E. Wnek, Polymer, 2005, 46, 3372–3384 CrossRef CAS.
- J. M. Layman, E. R. Kenawy, J. R. Watkins, M. E. Carr, G. L. Bowlin and G. Wnek, Abstracts of Papers of The American Chemical Society, AMER CHEMICAL SOC, 1155 16TH ST, NW, WASHINGTON, DC 20036 USA, 2003, vol. 226, pp.U436–U436 Search PubMed.
- H. Gharib Khajeh, M. Sabzi, S. Ramezani, A. A. Jalili and M. Ghorbani, Colloids Surf., A, 2022, 633, 127891 CrossRef CAS.
- D. Gutschmidt, R. S. Hazra, X. Zhou, X. Xu, M. Sabzi and L. Jiang, Int. J. Pharm., 2021, 594, 120172 CrossRef CAS PubMed.
- E.-R. Kenawy, G. L. Bowlin, K. Mansfield, J. Layman, D. G. Simpson, E. H. Sanders and G. E. Wnek, J. Controlled Release, 2002, 81, 57–64 CrossRef CAS.
- F. Davani, M. Alishahi, M. Sabzi, M. Khorram, A. Arastehfar and K. Zomorodian, Mater. Sci. Eng., C, 2021, 123, 111975 CrossRef CAS.
- S. Aziz, M. Sabzi, A. Fattahi and E. Arkan, J. Polym. Res., 2017, 24, 140 CrossRef.
- E. D. Boland, Front. Biosci., 2004, 9, 1422 CrossRef CAS PubMed.
- J. A. Matthews, G. E. Wnek, D. G. Simpson and G. L. Bowlin, Biomacromolecules, 2002, 3, 232–238 CrossRef CAS.
- W.-J. Li, C. T. Laurencin, E. J. Caterson, R. S. Tuan and F. K. Ko, J. Biomed. Mater. Res., 2002, 60, 613–621 CrossRef CAS.
- P. Gupta and G. L. Wilkes, Polymer, 2003, 44, 6353–6359 CrossRef CAS.
- A. Lendlein, H. Jiang, O. Jünger and R. Langer, Nature, 2005, 434, 879–882 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |