
Surface-segregating zwitterionic copolymers to control poly(dimethylsiloxane) surface chemistry†
A. Aslihan
Gokaltun
 abcd,
Luca
Mazzaferro
c,
Martin L.
Yarmush
abe,
O. Berk
Usta
*ab and
Ayse
Asatekin
*c
abcd,
Luca
Mazzaferro
c,
Martin L.
Yarmush
abe,
O. Berk
Usta
*ab and
Ayse
Asatekin
*c
aCenter for Engineering in Medicine and Surgery, Massachusetts General Hospital, Harvard Medical School, 55 Fruit St., Boston, MA 02114, USA. E-mail: ousta@mgh.harvard.edu
bShriners Hospitals for Children, 51 Blossom St., Boston, MA 02114, USA
cDepartment of Chemical and Biological Engineering, Tufts University, 4 Colby St., Medford, MA 02155, USA. E-mail: Ayse.Asatekin@tufts.edu
dDepartment of Chemical Engineering, Hacettepe University, 06532, Beytepe, Ankara, Turkey
eDepartment of Biomedical Engineering, Rutgers University, 599 Taylor Rd., Piscataway, NJ 08854, USA
First published on 27th November 2023
Abstract
The use of microfluidic devices in biomedicine is growing rapidly in applications such as organs-on-chip and separations. Polydimethylsiloxane (PDMS) is the most popular material for microfluidics due to its ability to replicate features down to the nanoscale, flexibility, gas permeability, and low cost. However, the inherent hydrophobicity of PDMS leads to the adsorption of macromolecules and small molecules on device surfaces. This curtails its use in “organs-on-chip” and other applications. Current technologies to improve PDMS surface hydrophilicity and fouling resistance involve added processing steps or do not create surfaces that remain hydrophilic for long periods. This work describes a novel, simple, fast, and scalable method for improving surface hydrophilicity and preventing the nonspecific adsorption of proteins and small molecules on PDMS through the use of a surface-segregating zwitterionic copolymer as an additive that is blended in during manufacture. These highly branched copolymers spontaneously segregate to surfaces and rearrange in contact with aqueous solutions to resist nonspecific adsorption. We report that mixing a minute amount (0.025 wt%) of the zwitterionic copolymer in PDMS considerably reduces hydrophobicity and nonspecific adsorption of proteins (albumin and lysozyme) and small molecules (vitamin B12 and reactive red). PDMS blended with these zwitterionic copolymers retains its mechanical and physical properties for at least six months. Moreover, this approach is fully compatible with existing PDMS device manufacture protocols without additional processing steps and thus provides a low-cost and user-friendly approach to fabricating reliable biomicrofluidics.
Introduction
The microfluidics market grossed $20.7 billion in 2019 and is estimated to reach $59 billion by 2026.1 This growth is propelled by point-of-care diagnostics, pharmaceutical and life science research (i.e., tissue culture/organs on chips and separations), and therapeutics.1 Poly(dimethyl siloxane) (PDMS) is one of the most widely used materials for microfluidics due to its chemical inertness, high gas permeability, flexibility, optical clarity over a wide range of wavelengths (240–1100 nm), low cost, biocompatibility, and feature reproduction quality.2,3 PDMS allows rapid prototyping and, upon plasma oxidation, can adhere to itself or other materials without adhesives. However, the hydrophobicity of PDMS, evidenced by its high water contact angle (WCA) around ∼108°,4–6 often limits its biomedical applications due to the nonspecific adsorption of proteins7–9 and absorption of small hydrophobic molecules. This leads to the loss of active compounds, influences analyte transport, and affects separation performance and detection sensitivity.10 These limitations complicate quantitative analysis in proteomics, genomics, and cell-based assays.8,11Surface segregating copolymers can be effectively used for creating hydrophilic, fouling-resistant surfaces without added manufacturing steps.12–17 These copolymers are mixed with the bulk commodity polymer material. Devices are then fabricated from this mixture following the standard protocol. During the manufacturing process, these smart copolymers spontaneously segregate to surfaces and create a <1 nm layer when in contact with aqueous solutions that prevent nonspecific adsorption and absorption of organic molecules. In our previous work, we demonstrated that block copolymer comprised of poly(ethylene glycol) (PEG) and PDMS segments (PDMS–PEG) led to a fully wettable (WCA ≈ 0) surface and drastically suppressed protein adsorption (≈99.5%), and created stable hydrophilicity for at least 20 months.12 A more recent study has shown that the use of this approach can also decrease the absorption of small-molecule drugs to some extent,18 though for most drugs, the samples had to be pretreated and saturated with higher dosages of drugs for a week before use to sufficiently reduce drug absorption. This shows the limitation of PEG in some applications. Although PEG has been the gold standard for preventing non-specific protein adsorption in many fields, its polyether backbone is comparatively non-polar. Most PEG-based coatings also include a relatively hydrophobic –O–CH3 terminal group,19 needed to ensure stability and prevent depolymerization. As a result, PEG interacts with many compounds via hydrogen bonding and hydrophobic interactions.20 These interactions limit the fouling resistance of PEG in complex mixtures.20
The limitations of PEG led researchers to seek other chemistries to create more stable and fouling resistant surfaces for PDMS while retaining its key positive attributes. Zwitterionic (ZI) groups, defined by equal numbers of anionic and cationic moieties, are extremely fouling resistant and offer crucial advantages over PEG.21–23 The super-hydrophilicity of ZI groups24 prevents ZI–protein interactions,20 leading to better performance than PEG in complex environments such as blood-contacting microfluidic devices and in vivo studies.19,25–27 ZI polymers grafted onto PDMS provide a hydrophilic surface for longer (∼3 months).28–30 Unlike PEG, ZI groups are incompatible with most organic solvents and small organic molecules. This indicates their preference to interact with water over small organic molecules, reducing their adsorption on PDMS surfaces and curtailing their absorption.
Several groups have focused on modifying PDMS with ZI groups to improve hydrophilicity and decrease non-specific adsorption, utilizing post-processing and surface coating strategies.28–38 Many of these treatments have led to improved surface hydrophilicity. However, the critical bottleneck to these approaches has been their laborious multiple post-processing steps that require special equipment. This complicates their large-scale fabrication and their adoption by a broad user base. These modifications typically use toxic chemicals that may limit biocompatibility in cell-based microfluidics and often affect the optical and mechanical properties of PDMS. Further, while some studies have shown ZI polymers grafted onto PDMS provide a hydrophilic surface for longer than PEO and similar hydrophilic polymers (∼3 months),28–30 most of the treatments did not show long-term stability,32–34 reverting to hydrophobic behavior in hours.31
To circumvent complex post-processing steps for modifying PDMS and provide long term stability, it would be preferable to incorporate ZI groups into the PDMS-based materials during routine device manufacture, simply blending a ZI component into the prepolymer. So far, there is only one study that mixed a zwitterionic monomer with PDMS prepolymer during device manufacture to reduce platelet deposition.39 In this study, the ZI monomer, diallyl terminated sulfobetaine (SB-diallyl), was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) and mixed into PDMS. While the SB-diallyl blended PDMS samples exhibited slightly reduced platelet and blood adsorption, they had water contact angles (105.4°) similar to the PDMS (106.4°). Notably, these samples had lower gas permeability and transparency and higher modulus than PDMS. The optical clarity of SB-diallyl blended PDMS decreased with increased additive concentration, and thicker samples were opaque due to phase separation. The use of HFIP also generates a risk of toxicity for long-term cell cultures. Thus, it is important to design a ZI-containing component that stays well-integrated in PDMS to avoid issues associated with phase separation (e.g., loss of transparency and transport properties).
In this work, we designed and synthesized zwitterionic surface-segregating copolymers (CPs) to create hydrophilic, non-fouling PDMS surfaces by a simple and scalable method, with no added manufacturing/post-processing steps. Notably, only a minute amount of this smart self-segregating additive was sufficient for enhanced performance. This zwitterionic copolymer (CP), poly(poly(dimethylsiloxane) methacrylate-random-2-methacryloyloxyethyl phosphorylcholine) (PDMSMA-r-MPC), features a branched architecture to enhance the surface segregation of the CP during device manufacture via the entropic driving force for chain ends to occupy interfaces.13,14,40 We prepared blends of PDMS with this CP at concentrations between 0.025–0.25 wt%. Upon manufacture, the short PDMS side chains are designed to drive the whole copolymer to the surface. Upon water immersion, local rearrangement of the CP exposes ZI groups to the surface (Fig. 1). We first evaluated the effect of PDMS chain length and composition on surface segregation via changing PDMS monomer molecular weight and PDMSMA/MPC monomer ratios. Then, we tested the surface wettability and stability of CP-blended PDMS samples prepared with the optimal CP composition. PDMSMA-r-MPC CP significantly reduced the water contact angle from 102° to 55.8° using as little as 0.025 wt% copolymer. This surface hydrophilicity was stable for at least six months. More importantly, using only 0.025 wt% CP in PDMS suppressed protein adsorption and small molecule absorption by over 90%. CP-blended PDMS has similar physical and mechanical properties to PDMS.
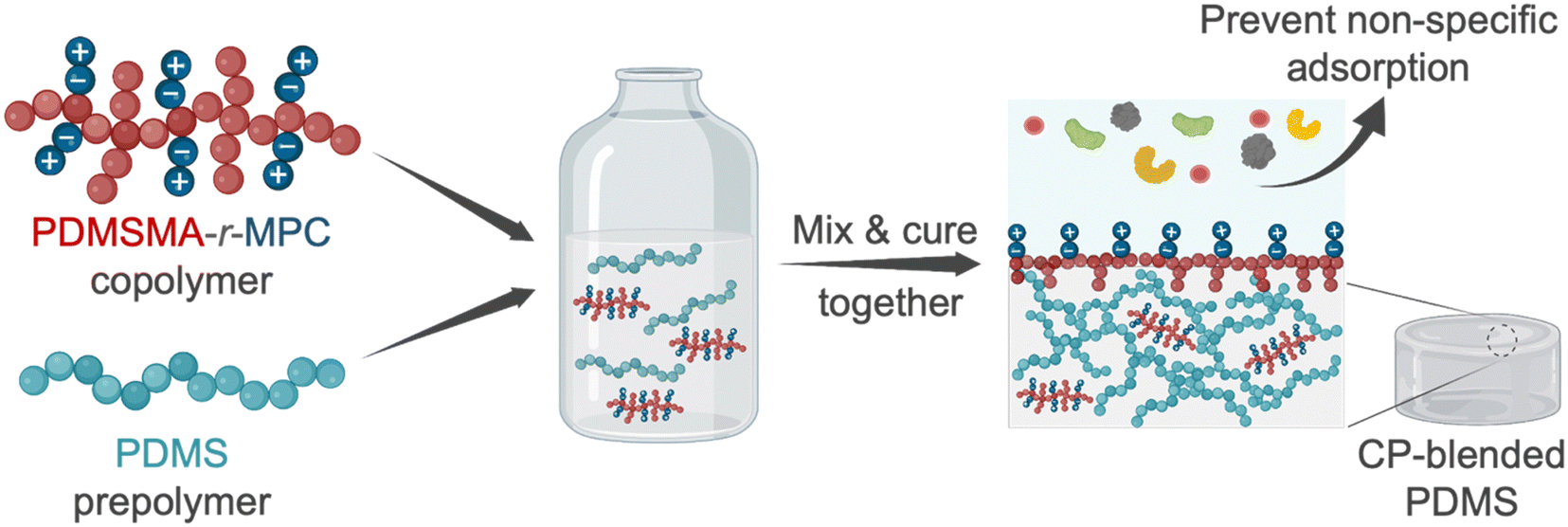 | ||
| Fig. 1 The blending of zwitterionic copolymer in PDMS to create hydrophilic, non-fouling surfaces. We blend a highly branched zwitterionic copolymer (CP), PDMSMA-r-MPC, and PDMS prepolymer and fabricate the samples using standard procedures without additional steps. The CP is expected to segregate to the surface, and ZI groups cover the surface after surface rearrangement occurs in the presence of aqueous solutions. This rearrangement renders the surface more hydrophilic and results in a surface that is highly resistant to nonspecific molecule adsorption and absorption. Figure was created with BioRender.com. | ||
Compared to previous approaches,28–34,41 our manufacturing method differentiates itself by its simplicity and scalability during both manufacture and use, requiring extremely small concentrations of an easily synthesized additive and no added manufacturing steps. Thus, this method promises a straightforward, rapid, and cost-effective approach to manufacturing hydrophilic and fouling resistant surfaces.
2. Materials and methods
2.1. Chemicals
Sylgard 184 silicone elastomer kit was purchased from Dow Corning (Tewksbury, MA). Monomethacryloxypropyl terminated polydimethylsiloxane (PDMSMA), (molecular weight: 5000 g mol−1 (70–80 cSt), 800–1000 g mol−1 (7–9 cst), 600–800 g mol−1 (6–9 cSt)) was obtained from Gelest (Morrisville, PA). 2-Methacryloyloxyethyl phosphorylcholine (MPC), azobisisobutyronitrile (AIBN), deuterated methanol (Methanol-d4), reactive red, vitamin B12, and 2-propanol (IPA) were all received from Sigma Aldrich (St. Louis, MO). Fluorescently labeled protein, bovine serum albumin (BSA) (Alexa Fluor 594-labeled, BSA), was obtained from Thermo Fisher Scientific. Lysozyme (FITC-labeled) was purchased from Nanocs (New York). Rhodamine 6G, BSA (from the chicken egg), lysozyme (from the chicken egg), vitamin B12, and reactive red were purchased from Sigma Aldrich (Burlington, MA). Acetone (ACN), dimethylsulfoxide (DMSO), tetrahydrofuran (THF), methoxyphenol (MEHQ), and reagent alcohol (anhydrous ethyl alcohol 90% ± 1% v/v; methyl alcohol approx. 5% v/v; 2-propanol approx. 5% v/v; labeled as ethanol, Et-OH) were sourced from Fisher Scientific (Hampton, NH). All chemicals and solvents were of reagent grade and used as received.2.2. Synthesis and characterization of PDMSMA-r-MPC copolymer
The random copolymers (CPs) poly(monomethacryloxypropyl terminated dimethylsiloxane-random-2-methacryloyloxyethyl phosphorylcholine) (PDMSMA-r-MPC) were synthesized using free radical polymerization (Fig. 2). First, monomethacryloxypropyl terminated polydimethylsiloxane (PDMSMA) was purified using a basic activated alumina column. MPC, calculated by considering the desired monomer ratios to achieve a total monomer mass of 5 g (for example for PDMSMA: MPC monomer mass ratio of 70/30, 1.5 gr MPC and 3.5 gr PDMSMA were used in the synthesis), was dissolved in 100 mL EtOH in a 250 mL round bottom flask at room temperature. PDMSMA was added to the reaction flask. After dissolution, 1 g of the initiator AIBN was added to the flask. Next, the reaction mixture was purged with nitrogen for 30 min. Then, the reaction mixture was stirred at 250 rpm for 20 hours in an oil bath at 65 °C. The flask was removed from the oil bath, and the reaction was terminated by adding 0.25 g MEHQ. A rotary evaporator (Rotavap) was used to concentrate the reaction mixture into a 10 mL solution. Specifically, PDMSMA: MPC monomer mass ratio of 70/30, the reaction mixture was poured into a 90/10 (v/v) ACN/water to precipitate out the copolymer, followed by three successive ACN washes to eliminate any remaining unreacted monomer. The attained solid copolymer was dried for two days under a fume hood and two more days in a vacuum oven at 50 °C. The product yield was calculated based on the ratio of the mass of the product copolymer to the mass of the monomers used. The chemical composition of the copolymer was measured by 1H NMR (Bruker Avance III 500 MHz spectrometer). Samples were dissolved in methanol-d4 and scanned 32 times using a 10 s relaxation delay.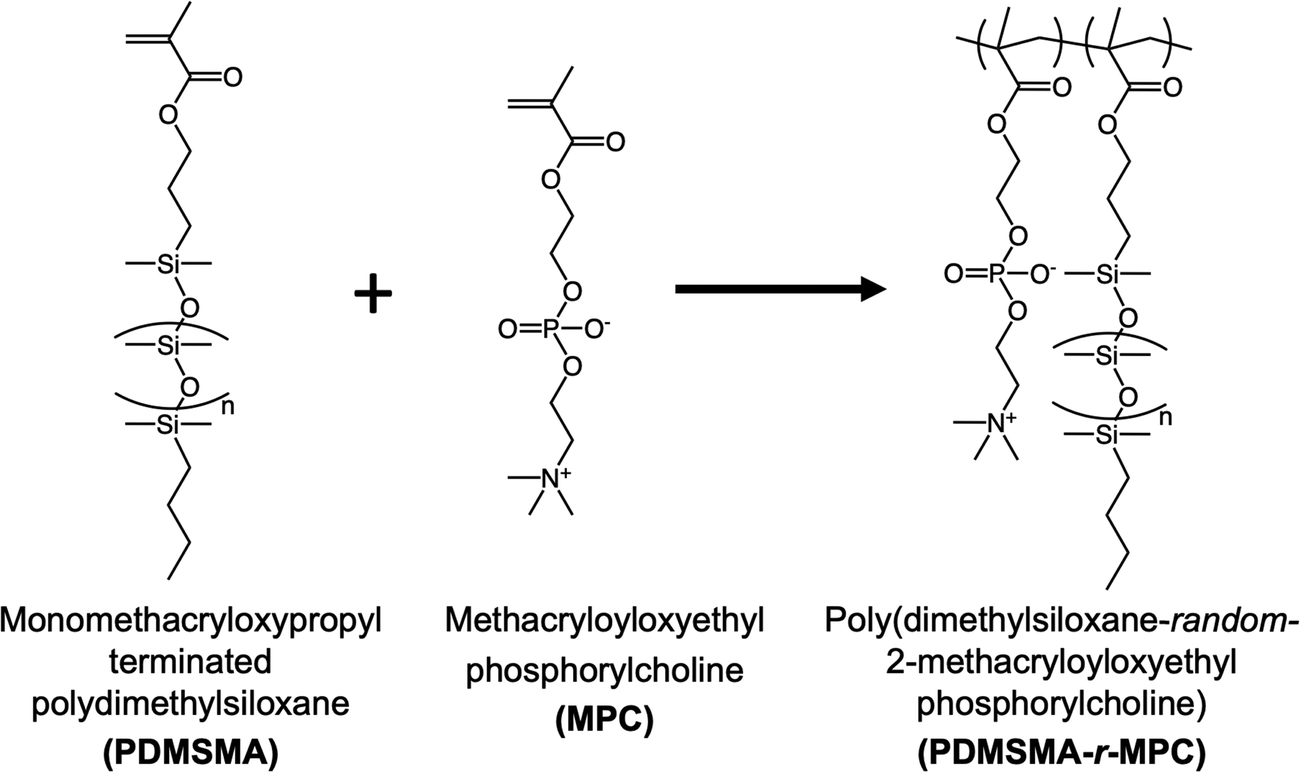 | ||
| Fig. 2 Synthesis scheme for the random zwitterionic PDMSMA-r-MPC copolymer. | ||
2.3. Preparation of CP-blended PDMS
“The zwitterionic PDMSMA-r-MPC CP was dissolved in EtOH (0.2 g mL−1) and used as an additive to modify PDMS. The silicone prepolymer and curing agent were mixed in a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 wt% ratio. We added the desired amount of PDMSMA-r-MPC CP, dissolved in EtOH, to the PDMS polymer base-curing agent mix to achieve final additive concentrations of 0.025%, 0.050%, 0.125%, and 0.250% wt% in the mixtures. The mixtures containing the PDMSMA-r-MPC CP additives were thoroughly blended by glass stirring rods to ensure even distribution within the PDMS base-curing agent mix. The blended mixtures were then poured into 5 cm diameter Petri dishes to manufacture slabs for further experiments. The Petri dishes were kept at 4 °C for 30 minutes to remove trapped air bubbles within the samples, and the mixture was cured at 70 °C for 18–24 hours. The resulting samples had a thickness of 4 mm measured by vernier caliper.”
:1 wt% ratio. We added the desired amount of PDMSMA-r-MPC CP, dissolved in EtOH, to the PDMS polymer base-curing agent mix to achieve final additive concentrations of 0.025%, 0.050%, 0.125%, and 0.250% wt% in the mixtures. The mixtures containing the PDMSMA-r-MPC CP additives were thoroughly blended by glass stirring rods to ensure even distribution within the PDMS base-curing agent mix. The blended mixtures were then poured into 5 cm diameter Petri dishes to manufacture slabs for further experiments. The Petri dishes were kept at 4 °C for 30 minutes to remove trapped air bubbles within the samples, and the mixture was cured at 70 °C for 18–24 hours. The resulting samples had a thickness of 4 mm measured by vernier caliper.”
2.4. Characterization of CP-blended PDMS
To characterize the rearrangement of the copolymer at the surface of PDMS, samples (1cm × 1cm and thickness: 4 mm) were analyzed using X-ray photoelectron spectroscopy (XPS) (K-Alpha + XPS system (Thermo Scientific), Harvard University Center for Nanoscale Systems). An Aluminum k-X-ray line with an energy of 1.4866 keV and an X-ray spot size of 400 μm with a 90-degree take-off angle (sampling depth was roughly 10 nm from the surface) was used as the probe for the measurement. A flood gun, which provides low-energy electrons and ions, was employed throughout the experiment for sample surface charge compensation. At each sample, survey spectra and high-resolution scan data were acquired. The scan was accomplished by averaging five scans in 1 eV increments with passing energy at 200 eV from −10 eV to 1350 eV binding energy for survey spectra. The data for high-resolution scans were collected by averaging 10 scans in 0.1 eV increments with passing energy at 50 eV for the Si 2p, O 1s, and C 1s photoelectron lines.
mm in length, and 4 mm in thickness. Experiments were performed 1 day after plasma treatment and subsequently after 24 h IPA soaking. Each sample was immersed in the rhodamine 6G solution (0.02% wt/v in distilled water) for 30 seconds and then removed. These samples were rinsed twice for 30 seconds in distilled water and dried with Kimwipes. Surfaces were cleaned with scotch tape to remove excess intensity and highlight the MPC interaction. Subsequently, images of the stained samples were captured using a fluorescence microscope (Evos FL Imaging System, ThermoFisher Scientific, RFP filter). The fluorescence image intensity of samples was quantified using ImageJ by adjusting the threshold and then measuring the mean gray value.
2.5. Adsorption and absorption characteristics of CP-blended PDMS
For adsorption and absorption experiments, PDMS and CP-blended PDMS samples (4 mm dia. × 4mm height) were prepared using a dermal punch (Ted Pella Inc.). Samples were soaked in phosphate-buffered saline (PBS, pH 7.4) for twohours to equilibrate. Fluorescently labeled proteins, bovine serum albumin (BSA), and lysozyme were dissolved separately in PBS to have a final concentration of 0.5 mg mL−1. To investigate protein adsorption, 50 μL of fluorescently labeled protein solution was placed on each sample and incubated in the dark at 37 °C for 1.5 h. After incubation, samples were washed off with PBS (500 μL) and dried with Kimwipes. Images were captured using a fluorescence microscope (Evos FL Imaging System, ThermoFisher Scientific). Protein adsorption was then quantified using ImageJ by adjusting the threshold and then measuring the mean gray value.
Quantitative small molecule absorption experiments were also performed with PDMS and CP-blended PDMS (4 mm dia. × 4 mm height). 5 μM aqueous solutions of vitamin B12 and reactive red were prepared separately. 200 μL of each solution was added into the wells of 96-well plates. Samples were immersed into the wells containing small molecule solutions and incubated for 2 hours at 37 °C. The samples were then removed. The amounts of absorbed vitamin B12 and reactive red were calculated by measuring the initial and final concentration of each solute by a UV-vis spectrophotometer (Bio-rad spectrophotometer), using absorbances at 363 nm and 515 nm, respectively. In adsorption and absorption experiments, three samples (N = 3) were used in each group.
2.6. Statistical analysis
Origin Pro 2021 Graphing & Analysis Software v.9.0.8.200 (Origin Lab, Northampton, Massachusetts) was used to analyze data. All quantitative data are presented as the mean ± standard error of the mean (SEM) from at least three samples from different batches. The statistical significance of the results was assessed using one-way ANOVA with Tukey multiple comparisons. Statistical significance is defined as p ≤ 0.05 for all experiments.3. Results and discussion
3.1. Synthesis, design, and formulations of CP-blended PDMS
Most approaches to fabricating PDMS with improved fouling resistance involve modification of PDMS with different strategies such as surface activation, physisorption, and chemical modification.2,3 Here, we developed a radically different approach to create hydrophilic, adsorption/absorption resistant PDMS surfaces by blending a zwitterionic branched copolymer (CP) and following the standard protocol for preparation without any added steps. Branched and brush polymers are known to segregate to the surface of a polymeric object because locating chain ends on the surface leads to enhanced entropy3–6. The presence of a surface or interface forces polymer chains that approach the surface to loop or reflect back into the bulk, resulting in decreased entropy. Chain ends, however, are not constrained by this; locating chain ends at the surface results in higher entropy for the system. As a result, studies have shown that short polymer chains or branched polymers segregate to interfaces in the absence of significant enthalpic effects. Highly branched copolymer chains are enriched near the surface and depleted towards the bulk of an object.6. The highly branched copolymer used in this study is, therefore, expected to be enriched near the surface of the PDMS sample due to entropic contributions, even in the absence of a hydrophilic/aqueous solution. In the air, we expect these highly branched, short chains to be enriched near the surface, with PDMS chain ends at the interface. Upon exposure to water, the copolymer conformationally rearranges to minimize surface energy, exposing zwitterionic MPC units to the aqueous solution. The high mobility of the PDMS chains and the fact that the polymer was already near the surface makes this possible. While the preparation of CP-blended PDMS is simple, its design for sufficient and efficient surface segregation without negative effects on PDMS properties requires thorough consideration and testing to ensure its success. PDMS devices are manufactured in the air, but PDMS surfaces are exposed to aqueous solutions during operation. Thus, CPs are expected segregate to surfaces during manufacture in air and have sufficient mobility to rearrange upon exposure to aqueous solutions, creating a hydrophilic surface that resists nonspecific adsorption/absorption. The polarity mismatch between PDMS and zwitterionic groups is a significant design challenge during the PDMS fabrication. Thus, copolymer architecture can distinctly influence surface segregation and fouling resistance.With these in mind, we designed and synthesized random copolymers of a hydrophobic PDMS macromonomer, monomethacryloxypropyl terminated polydimethylsiloxane (PDMSMA), and the zwitterionic monomer, 2-methacryloyloxyethyl phosphorylcholine (MPC) by free radical polymerization, resulting in a structure with ZI groups along the backbone, with PDMS branches incorporated through the macromonomer (Fig. 2). We first screened several CPs to explore the effect of the PDMS chain length and the PDMSMA/MPC segment ratio on surface segregation, surface properties, and nonspecific adsorption. PDMS side-chain length was varied by using PDMSMA monomers with different molecular weights (5000, 800–1000, 600–800 g mol−1 according to manufacturer). We also used varying PDMSMA/MPC monomer ratios (90/10, 80/20, 70/30, 60/40 wt%) (Table 1). A comparatively high initiator concentration was used in the synthesis to keep the total polymer molar mass low so that the polymer has high enough mobility to surface-segregate, as determined in our previous studies.45
| CP number | PDMSMA monomer MW (g mol−1) | PDMSMA/MPC monomer mass ratio | Reaction solvent |
|---|---|---|---|
| DMSO: dimethyl sulfoxide, THF: tetrahydrofuran, IPA: isopropyl alcohol, Et-OH: ethanol | |||
| CP-1 | 5000 | 80/20 | DMSO |
| CP-2 | 800–1000 | 90/10 | THF + IPA |
| CP-3 | 800–1000 | 80/20 | Et-OH |
| CP-4 | 800–1000 | 60/40 | Et-OH |
| CP-5 | 600–800 | 90/10 | Et-OH |
| CP-6 | 600–800 | 80/20 | Et-OH |
| CP-7 | 600–800 | 70/30 | Et-OH |
| CP-8 | 600–800 | 60/40 | Et-OH |
A sample 1H NMR spectrum of the PDMSMA-r-MPC CP, specifically CP-7 (Table 1), is given along with peak assignments in Fig. 3. The synthesized copolymer contained 46 (w/w%) MPC. The determination of the weight fraction of the CP-7 via NMR is detailed in ESI.† Each MPC unit was associated with nine protons around 3.3 ppm′ (g′). The peaks around 3.7 ppm′ (c′, d′, e′) and 4.3 ppm′ (f′) were attributed to the CH2 protons from MPC. The peak at 2.1 ppm (b) was assigned to the CH2 protons from the PDMS polymer backbone, whereas the peak at 1.9 ppm′ (b′) was assigned to the CH2 protons from the MPC polymer. The molecular weight of the CP-7 and CP-8 were estimated by dynamic light scattering (DLS) measurements in ethanol. The hydrodynamic radii of copolymer CP-7 and CP-8 were determined to be 27.5 and 23.08 nm in ethanol. Using the Mark–Houwink equation based on PDMS in toluene, the relative molecular weight of the copolymer was calculated to be 5.3 × 103 g mol−1 and 4.8 × 103 g mol−1 for CP-7 and CP-8, respectively.46 While this is only a relative molar mass value indicative of coil size (somewhat similar to values reported for gel permeation chromatography when other calibrations are used), it suggests that this particular copolymer chain is quite short, as desired for high copolymer mobility and surface segregation.
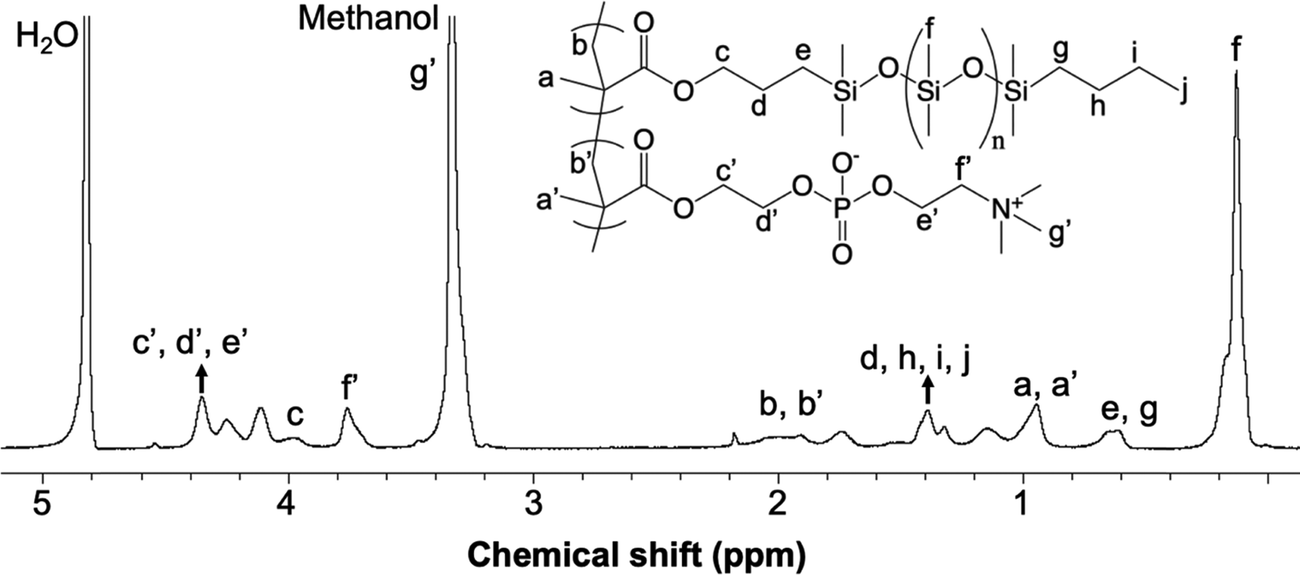 | ||
| Fig. 3 1H-NMR spectrum of the random PDMSMA-r-MPC copolymer. PDMSMA: MPC (wt%): 70/30. | ||
3.2. Preparation of CP-blended PDMS
To prepare CP-blended PDMS, we dissolved the PDMSMA-r-MPC CP in an appropriate blending solvent and mixed it with PDMS prepolymer and curing agent. We then polymerized the mixture at 68 °C for 24 hours.CPs synthesized with high MWs of PDMSMA monomer (5000, 800–1000 g mol−1) resulted in low polymerization yield (∼10–15%) (CP-1, CP-2), low solubility in blending solvent (CP-1, CP-3), poor miscibility with PDMS and curing agent (CP-1, CP-3, CP-4) and/or low transparency (CP-2). Polymerization yields obtained with PDMSMA-r-MPC CP synthesized with the PDMSMA monomer with shorter side chains (600–800 g mol−1) were higher (58%). Furthermore, these CPs had better miscibility with the PDMS prepolymer without problems such as bubble formation and low transparency. Thus, the side-chain length of the PDMSMA monomer considerably influences CP solubility in blending solvent as well as its miscibility in the PDMS prepolymer. We continued our characterization with PDMS blended with the best-performing CP in terms of solubility in blending solvent and miscibility in PDMS prepolymer, CP-5, CP-6, CP-7, and CP-8.
3.3. Surface hydrophilicity and stability of CP-blended PDMS
We performed sessile drop water contact angle (WCA) measurements to characterize the hydrophilicity of CP-blended PDMS. We expected that the surface of these samples would be mostly PDMS, and thus hydrophobic, upon initial manufacture in air. However, when exposed to water, the surface segregation and rearrangement of the CP would decrease the hydrophobicity with time and create a stable surface. Furthermore, to fabricate microfluidic devices for biomedical applications, specifically cell-based research, PDMS has to be sterilized with alcohol (i.e., IPA). In our previous study, we had also established that 24 h IPA soaking was enough to sterilize and remove all low molecular weight copolymers that might likely behave as cytotoxic surfactants, leaching out of the PDMS during experiments, which could negatively affect cell viability.45 Then, the PDMS device parts are treated with O2 plasma and bonded to a glass or another piece of PDMS. This is a required step for assembling microfluidic devices and a common method for generating reactive species that create silanol groups on the PDMS surface and decrease hydrophobicity. However, the hydrophilicity of plasma-treated PDMS is not long-lasting. The surface returns to its initial hydrophobic state within a few days due to the reorientation of low molecular weight species from the bulk to the surface, as observed in our previous study.45 These manufacturing steps may affect surface hydrophilicity.Thus, in our experiments, we performed the same steps that would be used for device fabrication and followed surface property changes. We measured the WCA of samples as manufactured (labeled BS before soak). We soaked the samples in IPA for 24 hours, based on the guidance from our previous study (samples labeled AS). We then treated them with O2 plasma and stored the samples for one week (samples labeled AS + PT 1 week). We measured the WCA in each step (BS, AS, and AS + PT 1 week). We then, investigated whether these treatments result in differences in the surface segregation of PDMSMA-r-MPC CP and, ultimately, in water contact angle and long-term stability.
As an initial screen of these copolymer concentrations, we measured the final WCA (t = 45 min) of CP-5, CP-6, CP-7, and CP-8 with varying concentrations (0.025–0.25%) before IPA soaking (BS), after IPA soaking (AS) and after IPA soaking and one week storage after O2 plasma treatment (AS + PT 1 week) (Fig. S1, ESI†). CP-5, CP-6, CP-7, and CP-8 in PDMS bulk polymer did not significantly decrease in WCA over time before and after IPA soaking compared to PDMS (Fig. S1, ESI†). After IPA soaking and one week of storage after O2 plasma treatment, CP-5 and CP-6 had higher WCA compared to CP-7 and CP-8 (Fig. S1, ESI†). This may be due to the low mass fraction of MPC in CP-5 and CP-6 compared to CP-7 and CP-8, which was not enough to create hydrophilic surfaces. CP-7 was selected for further studies described below.
Fig. 4a shows the time course for WCA of CP-blended PDMS prepared with different concentrations of the selected PDMSMA-r-MPC CP, CP-7, after IPA soaking and one week storage after O2 plasma treatment (AS+ PT 1wk). The initial contact angle of the PDMS-only control was around 102°, whereas that of all CP-blended PDMS were between 70–80°. This indicates that the sample surface is partially decorated with MPC segments even at the beginning. The WCA of PDMS showed a minimal decrease with time, staying above 90°. In contrast, the WCA of CP-blended PDMS decreased in time throughout the 45 minutes. Surfaces became considerably more hydrophilic with CP-blended PDMS compared to PDMS. It should be noted that blending as little as 0.025% PDMSMA-r-MPC CP led to a final contact angle of 55.8° (Fig. 4a). These findings reveal that upon contact with water, the PDMSMA-r-MPC CP self-assembles into PDMS and rearranges at the interface to form a hydrophilic zwitterionic layer even with a minute CP concentration of 0.025 wt%. Interestingly, higher CP concentrations do not lead to significantly lower final contact angles. Compared with other reports in the literature, this method can result in WCA values lower than previous studies for additive-blended PDMS materials (84°–63°) with much lower additive concentrations.47,48 The improved surface hydrophilicity of IPA-soaked & plasma-treated (AS + PT) CP-blended PDMS was stable throughout at least six months of storage under ambient conditions (Fig. 4b).
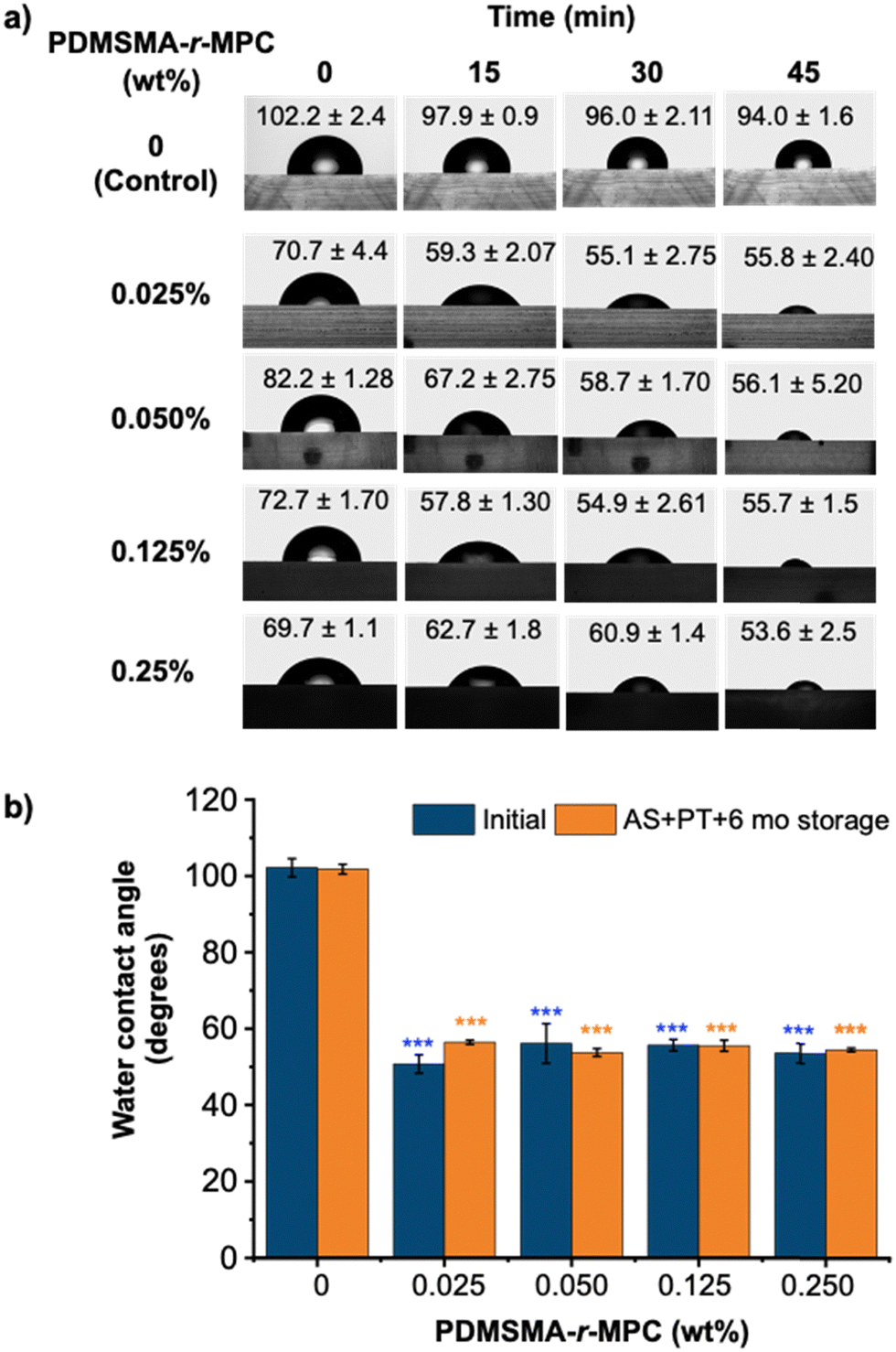 | ||
| Fig. 4 The hydrophilicity of CP-blended PDMS samples, indicated by water contact angle (WCA) measurements after IPA soak and plasma treatment. (a) Water contact angle (WCA) of CP-blended PDMS at concentrations between 0.025–0.25 wt% at different time intervals. Before measurement, samples were soaked in IPA for 24hours and treated with O2 plasma. Measurements were taken a week after O2 treatment. (b) Stability of final water contact angle through 6 months of ambient storage. “Initial” indicates after IPA soak, plasma treatment, and 1 week of storage. PDMSMA/MPC wt%: 70/30 (CP-7). The data are reported as mean ± SE (N = 3). We use ***: p ≤ 0.001 by Tukey-test for significance comparisons between PDMS and CP-blended PDMS groups (*** = statistically lower than PDMS). | ||
3.4. Physical characterization of CP-blended PDMS
| PDMSMA-r-MPC (wt%) | Treatments | O 1s (at%) | C 1s (at%) | Si 2p (at%) |
|---|---|---|---|---|
| 0 | BS | 27.80 | 45.16 | 27.04 |
| AS | 27.85 | 44.99 | 26.16 | |
| AS + PT-1 d | 40.59 | 32.37 | 27.04 | |
| AS + PT-1 week | 27.79 | 45.34 | 26.87 | |
| 0.05 | BS | 28.02 | 44.92 | 27.06 |
| AS | 28.00 | 45.14 | 26.85 | |
| AS + PT-1 d | 46.32 | 26.77 | 26.92 | |
| AS + PT-1 week | 41.61 | 30.82 | 27.56 |
 | ||
| Fig. 5 Rhodamine 6G staining confirmed the presence of MPC units on the surface of CP-blended PDMS. Representative staining images of (a) PDMS and (b) CP-blended PDMS (0.25 wt%). The images were captured using a fluorescence microscope (RFP filter) from three batches (N = 3). PDMSMA/MPC wt%: 70/30 (CP-7). Image scale bar: 400 μm. | ||
nm before and after IPA soaking (Fig. 6). Transparency for the center wavelengths of blue light (480nm) and green light (540nm) of PDMS and CP-blended PMDS are also shown separately in Table S1 (ESI†).
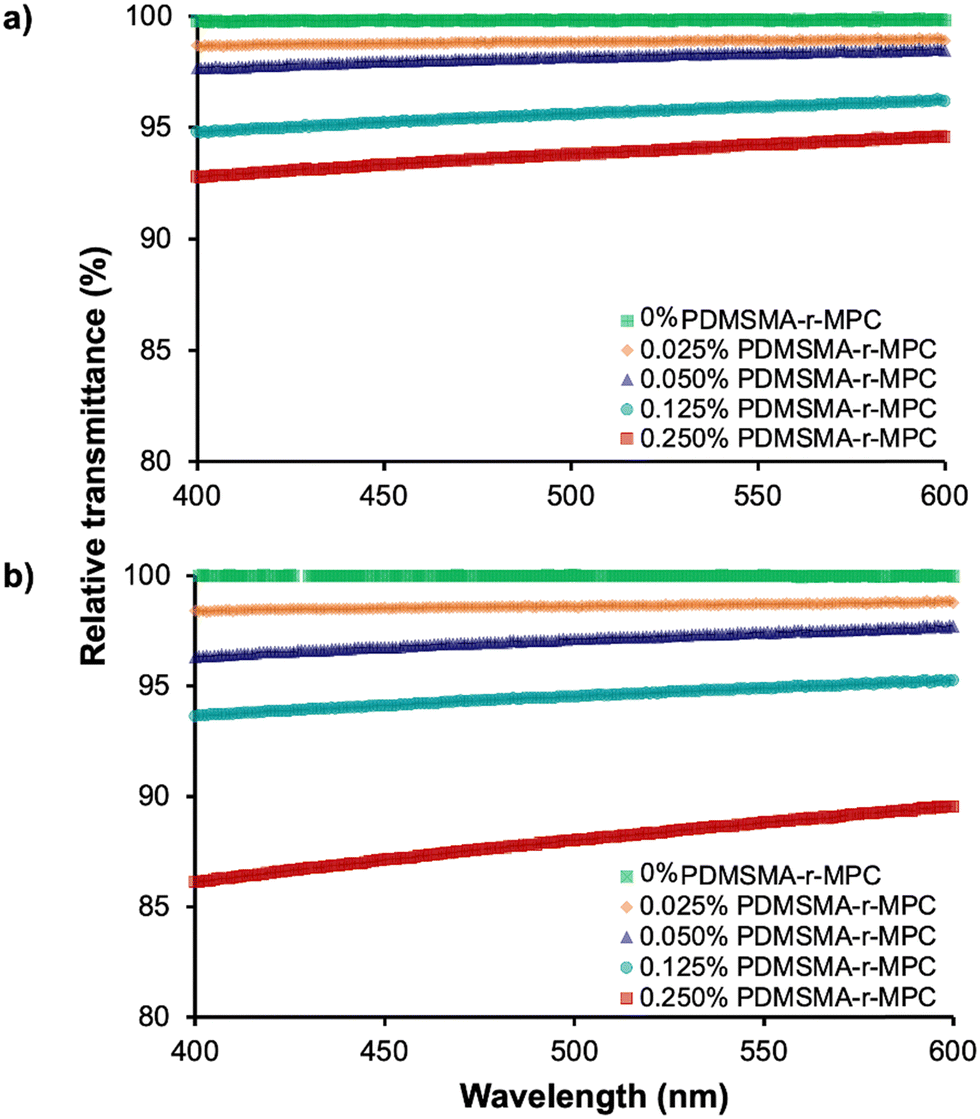 | ||
| Fig. 6 Relative transmittance of CP-blended PDMS (a) before and (b) after IPA soaking between 400–600 nm. All transmittance values are reported relative to the transmittance of “pure” PDMS. Before and after IPA soaking, the transmittance of CP-blended PDMS at 0.25 wt% concentration slightly decreased. PDMSMA/MPC wt%: 70/30 (CP-7). The data are reported as mean ± SE (N = 3). Standard error bars are in between (0.001–0.005) and are smaller than the size of markers. | ||
Before IPA soaking (Fig. 6a), CP-blended PDMS samples with up to 0.25% CP had transmittance above 90% at all wavelengths tested, comparable to PDMS. Transmittance values for the sample containing 0.025 wt% CP were indistinguishable from pure PDMS between 400–600 nm. After IPA soaking (Fig. 6b), the optical clarity of CP-blended PDMS samples slightly decreased but was still higher than 90% for 0.025 wt% to 0.125 wt% CP. Even with the highest CP concentration (0.25 wt%), the average transmittance of CP-blended PDMS was around 88%. This slight decrease is possibly due to the micellization of PDMSMA-r-MPC CP within the bulk PDMS at a higher concentration. These results indicated that, after IPA soaking, if used below a concentration of 0.25 wt%, optically clear devices can be manufactured from PDMSMA-r-MPC CP blends while still achieving increased surface hydrophilicity.
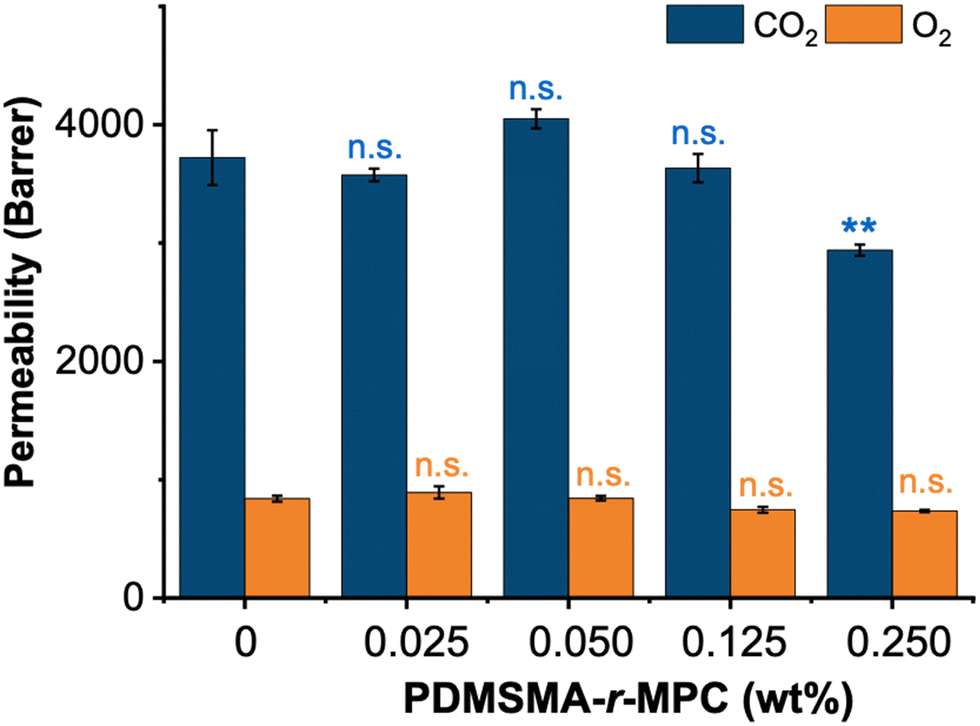 | ||
| Fig. 7 CP-blended PDMS demonstrates similar CO2 and O2 permeability compared to PDMS. Permeability tests were performed without any treatment to samples (no IPA soaking and plasma treatment). PDMSMA/MPC wt%: 70/30 (CP-7). The data are presented as the mean ± SE (n = 3). We use n.s.: non-significant, **: p ≤ 0.01 by Tukey-test were conducted for significance comparisons between the controls and CP-blended PDMS groups (n.s. = statistically similar with PDMS, ** = statistically lower than then PDMS). | ||
3.5. Adsorption and absorption characteristics of CP-blended PDMS
The main focus of this work was to effectively reduce the adsorption and absorption of proteins and small molecules in PDMS-based devices. To test the effectiveness of our approach, we measured the adsorption of fluorescently labeled proteins (albumin and lysozyme) and absorption of small molecules (vitamin B12 and reactive red) on PDMS and CP-blended PDMS and right after manufacturing (without any treatment) and after treatments that imitate biomicrofluidic device fabrication for biomedical applications (24 h IPA soak and one week storage after O2 plasma treatment).The fluorescence intensity of albumin and lysozyme on the CP-blended PDMS was significantly lower than that of pure PDMS, both before soaking and after IPA soaking and plasma treatment (Fig. 8a and b). The adsorption of proteins was suppressed at CP concentrations as low as 0.025 wt%. Pure PDMS samples exhibited considerably higher protein adsorption compared to CP-blended PDMS samples at all concentrations, which was confirmed by the normalized intensity of albumin and lysozyme. (Fig. 8c and d). The addition of CP lowered the adsorption of albumin and lysozyme by >85%, both as prepared and after treatments that are used in microfluidic device manufacture. In addition, CP-blended PDMS exhibited statistically much lower vitamin B12 and reactive red absorption compared to PDMS at all concentrations, both as prepared and after treatments mimicking device manufacture. The addition of only 0.025 wt% PDMSMA-r-MPC CP to PDMS lowered the absorption of vitamin B12 and reactive red by over 90% compared to PDMS (Fig. 9a and b).
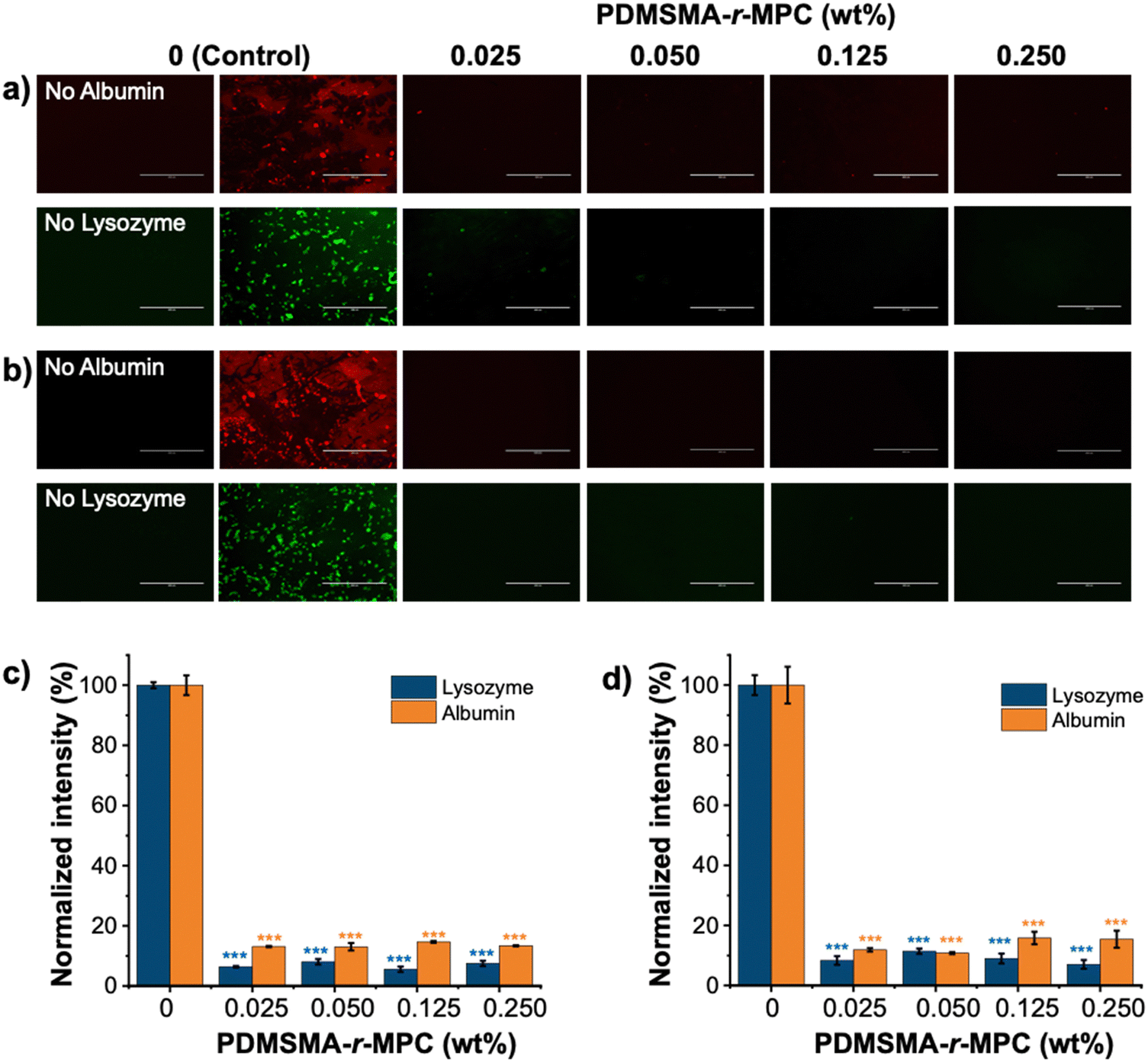 | ||
| Fig. 8 CP-blended PDMS drastically reduces protein adsorption. Protein adsorption of PDMS and CP-blended PDMS (0.025–0.25 wt%). Fluorescently tagged albumin and lysozyme (0.5 mg mL−1) adsorption onto CP-blended PDMS (a) before soaking, and (b) after IPA soaking and one week storage after O2 plasma treatment. Samples were covered with fluorescently tagged albumin and lysozyme for 90 minutes. Image scale bar: 400 μm. Normalized fluorescence intensity of albumin and lysozyme (c) before soaking, and (d) after IPA soaking, and one week storage after O2 plasma treatment. PDMSMA/MPC wt%: 70/30 (CP-7). We use ***: p ≤ 0.001 by Tukey-test for significance comparisons between the controls and CP-blended PDMS groups (*** = statistically lower than PDMS). | ||
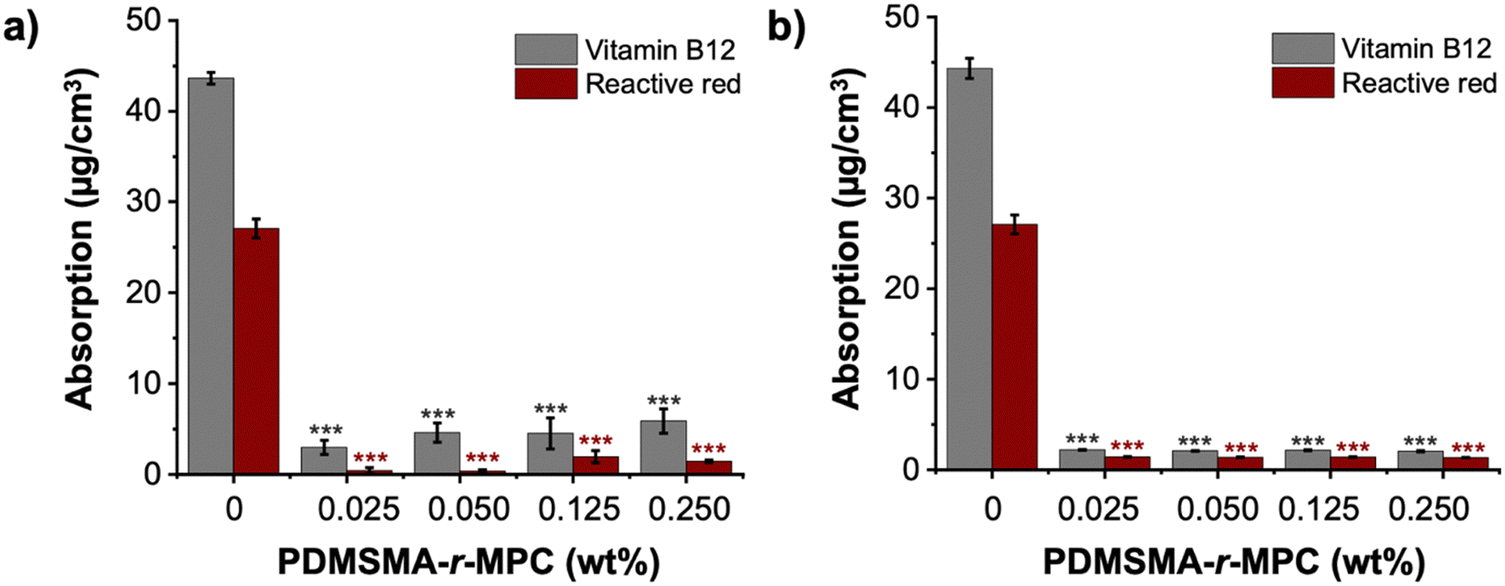 | ||
| Fig. 9 CP-blended PDMS drastically reduces small molecule absorption. Vitamin B12 and reactive red absorption of PDMS and CP-blended PDMS (a) as prepared/before soaking, (b) after IPA soak, and one week storage after O2 plasma treatment. 5 μM aqueous solutions of vitamin B12 and reactive red were prepared separately. 200 μL of each solution was added into the wells of 96-well plates. Samples were immersed into the wells containing small molecule solutions and incubated for 2 hours at 37 °C. PDMSMA/MPC wt%: 70/30 (CP-7). The data are the mean ± SE (N = 3). We use ***: p ≤ 0.001 by Tukey-test for significance comparisons between the controls and CP-blended PDMS groups (*** = statistically lower than then PDMS). | ||
Conclusion
We introduced and demonstrated a simple method to improve the hydrophilicity of PDMS-based materials and decrease non-specific adsorption and absorption of solutes by simply adding a rationally-designed, highly branched zwitterionic copolymer, PDMSMA-r-MPC, into bulk PDMS during manufacture, with no additional post-processing steps. This copolymer segregated to the material/device surface through thermodynamic driving forces, creating a more hydrophilic surface featuring zwitterionic groups upon exposure to aqueous media. This created hydrophilic PDMS surfaces resistant to the adsorption of proteins and absorption of small molecules without adverse effects on mechanical properties, optical transparency, or gas permeability. Interestingly, extremely small concentrations of this additive were sufficient to achieve the desired changes in surface properties. As little as 0.025 wt% CP additive led to a decrease in the final contact angle from ∼94° to ∼55°. This hydrophilicity was stable for at least 6 months, even after conventional manufacturing processes (e.g., soaking in IPA and plasma treatment), surpassing previous reports.39,47,48,55 Impressively blending only 0.025 wt%, PDMSMA-r-MPC CP decreased protein adsorption and small molecule absorption by ∼93% and ∼95% respectively, comparable to or better than the highest reductions in the previous reports.39,55,56 This degree of fouling resistance has typically been observed in samples with much lower contact angles than measured in this study,29–31 implying the presence of zwitterions in a hydrophobic matrix may lead to mechanisms of fouling resistance beyond hydrophilicity. This observation is consistent with other studies in different contexts,57,58 motivating a deeper future study on adsorption and absorption phenomena on amphiphilic zwitterionic surfaces.Unlike previous PDMS modification methods (coating/grafting), our method does not change PDMS microfabrication protocols by designing the zwitterionic copolymers. Thus, this method is easy to scale up and compatible with large-scale manufacturing. We believe this method will positively alter the microfluidics research and industry landscape and improve the accessibility of micro-devices to end users (patients, researchers, industry) by providing a low-cost and user-friendly approach to fabricating reliable biomicrofluidics. Aside from microfluidic applications, we anticipate that our discovery will remove constraints that presently hinder the use of PDMS in crucial commercial applications such as those in the pharmaceutical and biomedical industries.
Author contributions
A. Aslihan Gokaltun: conceptualization, investigation, methodology, validation, visualization, formal analysis, writing – original draft, Luca Mazzaferro: investigation, writing – review & editing, Martin L. Yarmush: funding acquisition, writing – review & editing, resources, Ayse Asatekin: conceptualization, resources, supervision, funding acquisition, writing – review & editing, project administration, O. Berk Usta: conceptualization, resources, supervision, funding acquisition, writing – review & editing, project administration.Conflicts of interest
The authors declare no competing financial or nonfinancial interests.Acknowledgements
Funding from the National Institutes of Health (NIH 1R21GM141683, NIH 1R21GM136002, and NIH 5R01HL145031, NIH R01AR081529, NIH 5R21GM141683), National Science Foundation (NSF Grant CHE-1904465, EEC-1941543), a Massachusetts General Hospital (MGH) Executive Committee on Research (ECOR) Interim Support Fund, and a Shriners Hospital Research Grant (SHC 85125-BOS-19, 85128) are acknowledged. We also gratefully acknowledge the support and use of facilities at the Morphology and Imaging Shared Facility and Regenerative Medicine Shared Facility provided at the Shriners Hospital for Children – Boston. The authors thank Dr David Wilbur for his assistance with NMR data collection and analysis.References
- MarketsandMarkets. Microfluidic Market Size, Growth by Product (Devices, Components (Chip, Sensor, Pump, Valve)) Application (IVD (POC, Clinical, Veterinary), Research, Manufacturing, Therapeutics), End User (Hospital, Diagnostic Center, Academic Institutes) & Region - Global Forecasts to 2026. Report Code: AST 7541 ( 2021) Search PubMed.
- M. P. Wolf, G. B. Salieb-Beugelaar and P. Hunziker, PDMS with designer functionalities—Properties, modifications strategies, and applications, Prog. Polym. Sci., 2018, 83, 97–134 CrossRef CAS.
- A. Gokaltun, M. L. Yarmush, A. Asatekin and O. B. Usta, Recent advances in nonbiofouling PDMS surface modification strategies applicable to microfluidic technology, Technology, 2017, 5, 1–12, DOI:10.1142/S2339547817300013.
- S. H. Tan, N. T. Nguyen, Y. C. Chua and T. G. Kang, Oxygen plasma treatment for reducing hydrophobicity of a sealed polydimethylsiloxane microchannel, Biomicrofluidics, 2010, 4, 32204, DOI:10.1063/1.3466882.
- Y. J. Chuah, S. Kuddannaya, M. H. Lee, Y. Zhang and Y. Kang, The effects of poly(dimethylsiloxane) surface silanization on the mesenchymal stem cell fate, Biomater. Sci., 2015, 3, 383–390, 10.1039/c4bm00268g.
- M. H. Wu, J. P. Urban, Z. Cui and Z. F. Cui, Development of PDMS microbioreactor with well-defined and homogenous culture environment for chondrocyte 3-D culture, Biomed. Microdevices, 2006, 8, 331–340, DOI:10.1007/s10544-006-9597-y.
- R. Mukhopadhyay, When PDMS isn’t the best. What are its weaknesses, and which other polymers can researchers add to their toolboxes?, Anal. Chem., 2007, 79, 3248–3253 CrossRef CAS PubMed.
- M. W. Toepke and D. J. Beebe, PDMS absorption of small molecules and consequences in microfluidic applications, Lab Chip, 2006, 6, 1484–1486, 10.1039/b612140c.
- X. Su, et al., Microfluidic cell culture and its application in high-throughput drug screening: cardiotoxicity assay for hERG channels, J. Biomol. Screen, 2011, 16, 101–111 CrossRef CAS.
- D. Belder and M. Ludwig, Surface modification in microchip electrophoresis, Electrophoresis, 2003, 24, 3595–3606, DOI:10.1002/elps.200305648.
- J. N. Lee, C. Park and G. M. Whitesides, Solvent compatibility of poly(dimethylsiloxane)-based microfluidic devices, Anal. Chem., 2003, 75, 6544–6554, DOI:10.1021/ac0346712.
- A. Gökaltun, Y. B. Kang, M. L. Yarmush, O. B. Usta and A. Asatekin, Simple Surface Modification of Poly(dimethylsiloxane) via Surface Segregating Smart Polymers for Biomicrofluidics, Sci. Rep., 2019, 9, 7377, DOI:10.1038/s41598-019-43625-5.
- D. Walton and A. Mayes, Entropically driven segregation in blends of branched and linear polymers, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1996, 54, 2811 CrossRef CAS.
- D. Walton, et al., Creation of stable poly (ethylene oxide) surfaces on poly (methyl methacrylate) using blends of branched and linear polymers, Macromolecules, 1997, 30, 6947–6956 CrossRef CAS.
- J. Hester, P. Banerjee and A. Mayes, Preparation of protein-resistant surfaces on poly (vinylidene fluoride) membranes via surface segregation, Macromolecules, 1999, 32, 1643–1650 CrossRef CAS.
- P. Kaner, A. V. Dudchenko, M. S. Mauter and A. Asatekin, Zwitterionic copolymer additive architecture affects membrane performance: fouling resistance and surface rearrangement in saline solutions, J. Mater. Chem. A, 2019, 7, 4829–4846 RSC.
- P. Kaner, E. Rubakh and A. Asatekin, Zwitterion-containing polymer additives for fouling resistant ultrafiltration membranes, J. Membr. Sci., 2017, 533, 141–159 CrossRef CAS.
- D. B. Mair, et al., PDMS–PEG block copolymer and pretreatment for arresting drug absorption in microphysiological devices, ACS Appl. Mater. Interfaces, 2022, 14, 38541–38549 CrossRef CAS.
- B. Li, et al., Trimethylamine N-oxide–derived zwitterionic polymers: A new class of ultralow fouling bioinspired materials, Sci. Adv., 2019, 5, eaaw9562 CrossRef CAS PubMed.
- C. Leng, et al., Probing the Surface Hydration of Nonfouling Zwitterionic and PEG Materials in Contact with Proteins, ACS Appl. Mater. Interfaces, 2015, 7, 16881–16888, DOI:10.1021/acsami.5b05627.
- S. F. Chen and S. Y. Jiang, A new avenue to nonfouling materials, Adv. Mater., 2008, 20, 335 CrossRef CAS.
- G. S. Georgiev, et al., Self-assembly, antipolyelectrolyte effect, and nonbiofouling properties of polyzwitterions, Biomacromolecules, 2006, 7, 1329–1334, DOI:10.1021/bm050938q.
- Q. Shao, Y. He and S. Jiang, Molecular Dynamics Simulation Study of Ion Interactions with Zwitterions, J. Phys. Chem. B, 2011, 115, 8358–8363, DOI:10.1021/jp204046f.
- Q. Shao and S. Jiang, Molecular Understanding and Design of Zwitterionic Materials, Adv. Mater., 2015, 27, 15–26, DOI:10.1002/adma.201404059.
- K. Welsher, S. A. McManus, C.-H. Hsia, S. Yin and H. Yang, Discovery of Protein- and DNA-Imperceptible Nanoparticle Hard Coating Using Gel-Based Reaction Tuning, J. Am. Chem. Soc., 2015, 137, 580–583, DOI:10.1021/ja511297d.
- L. Zhang, et al., Zwitterionic hydrogels implanted in mice resist the foreign-body reaction, Nat. Biotechnol., 2013, 31, 553, DOI:10.1038/nbt.2580.
- B. McVerry, et al., A readily scalable, clinically demonstrated, antibiofouling zwitterionic surface treatment for implantable medical devices, Adv. Mater., 2022, 34, 2200254 CrossRef CAS.
- A. J. Keefe, N. D. Brault and S. Jiang, Suppressing Surface Reconstruction of Superhydrophobic PDMS Using a Superhydrophilic Zwitterionic Polymer, Biomacromolecules, 2012, 13, 1683–1687, DOI:10.1021/bm300399s.
- A. Zhang, L. Cheng, S. Hong, C. Yang and Y. Lin, Preparation of anti-fouling silicone elastomers by covalent immobilization of carboxybetaine, RSC Adv., 2015, 5, 88456–88463, 10.1039/C5RA17206C.
- T. J. Plegue, K. M. Kovach, A. J. Thompson and J. A. Potkay, Stability of Polyethylene Glycol and Zwitterionic Surface Modifications in PDMS Microfluidic Flow Chambers, Langmuir, 2018, 34, 492–502, DOI:10.1021/acs.langmuir.7b03095.
- P. Shivapooja, et al., Modification of Silicone Elastomer Surfaces with Zwitterionic Polymers: Short-Term Fouling Resistance and Triggered Biofouling Release, ACS Appl. Mater. Interfaces, 2015, 7, 25586–25591, DOI:10.1021/acsami.5b09199.
- H. Wang, C. Zhang, J. Wang, X. Feng and C. He, Dual-Mode Antifouling Ability of Thiol–Ene Amphiphilic Conetworks: Minimally Adhesive Coatings via the Surface Zwitterionization, ACS Sustainable Chem. Eng., 2016, 4, 3803–3811, DOI:10.1021/acssuschemeng.6b00525.
- A. Vaterrodt, et al., Antifouling and antibacterial multifunctional polyzwitterion/enzyme coating on silicone catheter material prepared by electrostatic layer-by-layer assembly, Langmuir, 2016, 32, 1347–1359 CrossRef CAS.
- S.-B. Yeh, C.-S. Chen, W.-Y. Chen and C.-J. Huang, Modification of Silicone Elastomer with Zwitterionic Silane for Durable Antifouling Properties, Langmuir, 2014, 30, 11386–11393, DOI:10.1021/la502486e.
- T. Goda, T. Konno, M. Takai, T. Moro and K. Ishihara, Biomimetic phosphorylcholine polymer grafting from polydimethylsiloxane surface using photo-induced polymerization, Biomaterials, 2006, 27, 5151–5160 CrossRef CAS.
- J.-H. Seo, R. Matsuno, T. Konno, M. Takai and K. Ishihara, Surface tethering of phosphorylcholine groups onto poly (dimethylsiloxane) through swelling–deswelling methods with phospholipids moiety containing ABA-type block copolymers, Biomaterials, 2008, 29, 1367–1376 CrossRef CAS PubMed.
- K. Nagahashi, Y. Teramura and M. Takai, Stable surface coating of silicone elastomer with phosphorylcholine and organosilane copolymer with cross-linking for repelling proteins, Colloids Surf., B, 2015, 134, 384–391 CrossRef CAS.
- H. Nakano, S. Kakinoki and Y. Iwasaki, Long-lasting hydrophilic surface generated on poly (dimethyl siloxane) with photoreactive zwitterionic polymers, Colloids Surf., B, 2021, 205, 111900 CrossRef CAS PubMed.
- A. Mercader, et al., PDMS-Zwitterionic Hybrid for Facile, Antifouling Microfluidic Device Fabrication, Langmuir, 2022, 38, 3775–3784 CrossRef CAS PubMed.
- A. Asatekin, S. Kang, M. Elimelech and A. M. Mayes, Anti-fouling ultrafiltration membranes containing polyacrylonitrile-graft-poly (ethylene oxide) comb copolymer additives, J. Membr. Sci., 2007, 298, 136–146 CrossRef CAS.
- Q. Cheng, et al., Antifouling and antibacterial polymer-coated surfaces based on the combined effect of zwitterions and the natural borneol, ACS Appl. Mater. Interfaces, 2021, 13, 9006–9014 CrossRef CAS.
- V. A. Haug and G. Meyerhoff, Hydrodynamisches verhalten gelöster polydimethylsiloxane, Macromol. Chem. Phys., 1962, 53, 91–102 CrossRef.
- T. Merkel, V. Bondar, K. Nagai, B. Freeman and I. Pinnau, Gas sorption, diffusion, and permeation in poly (dimethylsiloxane), J. Polym. Sci., Part B: Polym. Phys., 2000, 38, 415–434 CrossRef CAS.
- J. Xu, A. Asatekin and K. K. Gleason, The design and synthesis of hard and impermeable, yet flexible, conformal organic coatings, Adv. Mater., 2012, 24, 3692–3696 CrossRef CAS PubMed.
- A. Gökaltun, Y. B. Kang, M. L. Yarmush, O. B. Usta and A. Asatekin, Simple surface modification of poly (dimethylsiloxane) via surface segregating smart polymers for biomicrofluidics, Sci. Rep., 2019, 9, 7377 CrossRef.
- H. Takimoto, C. Forbes and R. Laudenslager, A viscosity–molecular weight relationship for polydimethylsiloxanes, J. Appl. Polym. Sci., 1961, 5, 153–156 CrossRef CAS.
- Y. Xiao, X. D. Yu, J. J. Xu and H. Y. Chen, Bulk modification of PDMS microchips by an amphiphilic copolymer, Electrophoresis, 2007, 28, 3302–3307 CrossRef CAS PubMed.
- Z. Wu and K. Hjort, Surface modification of PDMS by gradient-induced migration of embedded Pluronic, Lab Chip, 2009, 9, 1500–1503 RSC.
- Y. Yang, K. Kulangara, R. T. Lam, R. Dharmawan and K. W. Leong, Effects of topographical and mechanical property alterations induced by oxygen plasma modification on stem cell behavior, ACS Nano, 2012, 6, 8591–8598 CrossRef CAS.
- R. Katayama, M. Ikeda, K. Shiraishi, A. Matsumoto and C. Kojima, Formation of hydrophobic domains on the poly (MPC-co-dodecyl methacrylate)-coated surface recognized by macrophage-like cells, Langmuir, 2019, 35, 12229–12235 CrossRef CAS.
- J. H. Wang, et al., The use of rhodamine 6G and fluorescence microscopy in the evaluation of phospholipid-based polymeric biomaterials, J. Microsc., 2005, 217, 216–224 CrossRef CAS PubMed.
- J. Ge, et al., Standard fluorescent imaging of live cells is highly genotoxic, Cytometry, Part A, 2013, 83, 552–560 CrossRef PubMed.
- E. Berthier, E. W. Young and D. Beebe, Engineers are from PDMS-land, Biologists are from Polystyrenia, Lab Chip, 2012, 12, 1224–1237 RSC.
- H. J. Eash, H. M. Jones, B. G. Hattler and W. J. Federspiel, Evaluation of plasma resistant hollow fiber membranes for artificial lungs, ASAIO J., 2004, 50, 491–497 CrossRef PubMed.
- J. Zhou, H. Yan, K. Ren, W. Dai and H. Wu, Convenient method for modifying poly (dimethylsiloxane) with poly (ethylene glycol) in microfluidics, Anal. Chem., 2009, 81, 6627–6632 CrossRef CAS PubMed.
- Y. Li, A. J. Keefe, M. Giarmarco, N. D. Brault and S. Jiang, Simple and robust approach for passivating and functionalizing surfaces for use in complex media, Langmuir, 2012, 28, 9707–9713 CrossRef CAS.
- S. Ozcan, P. Kaner, D. Thomas, P. Cebe and A. Asatekin, Hydrophobic antifouling electrospun mats from zwitterionic amphiphilic copolymers, ACS Appl. Mater. Interfaces, 2018, 10, 18300–18309 CrossRef CAS PubMed.
- P. Bengani-Lutz, E. Converse, P. Cebe and A. Asatekin, Self-assembling zwitterionic copolymers as membrane selective layers with excellent fouling resistance: effect of zwitterion chemistry, ACS Appl. Mater. Interfaces, 2017, 9, 20859–20872 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb02164e |
| This journal is © The Royal Society of Chemistry 2024 |