
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Superior oxygen evolution reaction activities of highly crystalline Ni1−xFex-LDH (0.20 ≤ x ≤ 0.51) synthesized using soft chemistry†
Atsunori
Ikezawa
 *,
Shinji
Koito
and
Hajime
Arai
*,
Shinji
Koito
and
Hajime
Arai
School of Materials and Chemical Technology, Tokyo Institute of Technology, Yokohama 226-8501, Japan. E-mail: ikezawa.a.aa@m.titech.ac.jp
First published on 24th October 2024
Abstract
Developing oxygen evolution reaction (OER) catalysts is an urgent issue for the practical application of electrochemical devices such as water electrolysers and rechargeable batteries. NiFe-based layered double hydroxides (NiFe-LDH) are known as some of the most active OER catalysts. However, LDH is a metastable phase, making it difficult to synthesize it with high crystallinity and controlled composition and morphology. In this study, NiFe-LDHs are synthesized by soft chemistry using NaNi1−xFexO2 as the starting material. The soft chemistry samples have higher crystallinity with more uniform composition and morphology than the conventional coprecipitation samples. In addition, the substitution of Ni for Fe is successfully achieved at 51%, exceeding the conventional solid solution limit. The soft chemistry samples show superior OER specific activity with an overpotential of less than 0.20 V at 1.0 mA cmBET−2 in 1.0 mol dm−3 KOH, exceeding the activity of the conventional NiFe-LDH, indicating that the crystalline site has higher OER activity than the amorphous site. The soft chemistry samples exhibit the lowest Tafel slope (24.7–27.0 mV dec−1) among the reported NiFe-LDH (>27.8 mV dec−1). It is also found that the Fe substitution ratio for maximum OER activity exists in the range of 20–51 at%.
Introduction
Expanding the use of renewable energy is urgently needed to solve energy issues and environmental problems, including global warming. Since most renewable energies are unstable in output, energy storage devices are needed to level the electricity derived from these renewables. Electrochemical devices that utilize the oxygen evolution reaction (OER) in alkaline electrolytes, such as zinc–air secondary batteries1–3 and alkaline water electrolysis,4–6 have attracted attention as such energy storage devices. These devices can satisfy high safety standards because of the aqueous electrolyte solution. In addition, low-cost value is also possible since inexpensive non-precious metal-based catalysts can be used in alkaline electrolyte solutions. However, the wide practical application of these electrochemical devices is hindered by the low energy conversion efficiency due to the high overvoltage of the OER.1–6It is essential to develop highly active OER catalysts as well as create a favourable reaction field7,8 for reducing the OER overvoltage. Precious metal oxide catalysts such as IrO2 and RuO2 have long been known as highly active OER catalysts,9,10 but these catalysts are relatively expensive. Therefore, inexpensive non-precious metal-based catalysts such as perovskite and spinel-type oxides have been actively developed.9,11,12 Among them, NiFe-based layered double hydroxide (NiFe-LDH) is one of the most promising OER catalysts.13–15 NiFe-LDH is known to have higher OER activity than noble metal oxide-based catalysts.13–15 In addition, it can be produced at low cost because it is composed of Ni and Fe, which are relatively inexpensive non-precious metals. NiFe-LDH comprises NixFe1−xO2 layers and interlayer water molecules/ions, also known as the partially Fe-substituted α-Ni(OH)2 phase. Many experimental and theoretical studies have been performed on its OER mechanism.13–15 For example, Dionigi et al. theoretically showed that the Ni–O–Fe site is highly active.16 It was also reported that the partially Fe-substituted γ-NiOOH phase, formed by the oxidation of Ni(II), is the active phase in studies combining operando measurements and theoretical calculations.16,17
The partially Fe-substituted α-Ni(OH)2 and γ-NiOOH phases are metastable and decompose at below 300 °C in air.18,19 Therefore, relatively low-temperature synthesis methods such as coprecipitation, electrodeposition, and hydrothermal method have been used.13,14 However, low-temperature synthesis suffers from low crystallinity and uneven composition/morphology of the product, which makes it difficult to elucidate the factors affecting the OER activity.
Since the crystal growth rate is generally faster at higher temperatures, it is challenging to synthesize highly crystalline samples using low-temperature synthesis. Therefore, the OER activity of NiFe-LDH with high crystallinity has rarely been evaluated so far. In addition, although crystallinity and specific surface area usually have a trade-off relationship, most studies have compared the mass activity,20 which can lead to the overestimation of the specific activity of low-crystallinity samples.
Elemental interdiffusion is generally enhanced at higher temperatures. Therefore, low temperature synthesis can generally lead to non-uniform composition. This causes uncertainty in the optimum amount of Fe substitution for the OER activity, which has been reported to be in the range of 10–50 at%.14 The reported maximum Fe substitution of ca. 25 at% (ref. 17) can also be due to the limited elemental interdiffusion. In addition, it is reported that the amount of Fe substitution affected the material morphology20–22 which can affect the ratio of catalytically active edge planes.17 Since the grain growth rate is generally faster at higher temperatures, low temperature synthesis has limitations in controlling the morphology, which is essential for systematically comparing the specific activities.
Soft chemistry synthesis is a technique for synthesizing metastable phases with high crystallinity and controlled composition and morphology.23–27 Soft chemistry synthesis applies topochemical reactions, such as insertion/extraction and adsorption/desorption reactions, to high-temperature calcined reactant hosts. In the topochemical reaction, the higher-order structure of the reactant is maintained, and the reactants' crystallinity, composition, and morphology can be transferred to the products.
In this study, we synthesized highly crystalline NiFe-LDHs using soft chemistry. As a control experiment, low-crystallinity NiFe-LDHs were synthesized by the conventional coprecipitation method. The samples synthesized by soft chemistry had higher crystallinity and more uniform composition and morphology than those synthesized by coprecipitation. In addition, we successfully substituted Ni for Fe up to 51 at%, which is above the conventional solid solution limit.17 A comparison of OER activity in 1.0 mol dm−3 KOH revealed that the soft chemistry samples have higher OER activities normalized by Brunauer–Emmett–Teller (BET) surface areas than the coprecipitation samples. It was also found that the Fe substitution ratio for maximum OER activity is 19–51 at%. Furthermore, the soft chemistry samples with Fe substitution rates of 19–51 at% showed mass activity exceeding that of the coprecipitation samples, despite having an order of magnitude smaller BET specific surface areas.
Experimental
Material preparation
Highly crystalline NiFe-LDH was synthesized by soft chemistry with NaNi1−xFexO2 as the reactant.23,24 The reactant NaNi1−xFexO2 was synthesized using the solid phase synthesis.28 NiO was synthesized by calcining basic NiCO3 (Kanto Chemical) with a temperature rise of 2 hours, a holding time of 2 hours, and a holding temperature of 350 °C in an electric furnace (KDF-S70, Denken-Highdental). NiO, Na2O2 (Kanto Chemical), and Fe2O3 (Kanto Chemical) were mixed in an Ar-filled glove box in the ratios shown in Table S1.† The atomic ratio of Fe/Ni + Fe was set in the range of 0.1 to 0.5. It has been reported that phase separation occurred at ratios above 0.5.28 The mixed powder was formed into pellets of φ = 14 mm at 100 MPa. Magnesia crucibles containing the obtained pellets were heated in an electric furnace under an oxygen flow of 1 dm3 min−1 with a temperature rise of 1.5 h, a holding time of 6 h, and a holding temperature of 700 °C. The synthesized NaNi1−xFexO2 pellet was ground in an agate mortar and stirred in 250 cm3 of 1.0 mol dm−3 KOH solution for 1 hour to hydrate and exchange the cation. After centrifugation and removal of the supernatant, the obtained powder was washed with injections of ultrapure water and centrifugation twice. The washed powder was dried at 50 °C for 12 hours in a drying oven (DX-42, Yamato Scientific) to obtain NiFe-LDH.Conventional NiFe-LDHs were synthesized using a coprecipitation method.29–31 100 cm3 of Ni(NO3)2·6H2O (Kanto Chemical) and Fe(NO3)3·9H2O (Kanto Chemical) aqueous solution and 100 cm3 of NaOH (Kanto Chemical) and Na2CO3 (Kanto Chemical) aqueous solution were prepared at the concentrations shown in Table S2.† These solutions were added to 180 cm3 of ultrapure water at around 0.8 cm3 min−1 using syringe pumps (YSP-101, YMC) and stirred with a magnetic stirrer. The pH of the solution was monitored with a pH meter (D-71AL, Horiba), and the addition rates of the solutions were adjusted to maintain pH = 9.5. After adding the entire solutions, the solution was stirred at 25 °C for 2 h and at 50 °C for 48 h. After stirring, the solution was allowed to stand, and the supernatant was removed. Then, the sample was washed by adding ultrapure water to the precipitate, centrifuging, and removing the supernatant. After three times of washing, NiFe-LDH powder was obtained by drying in the oven at 50 °C for 12 h.
Material characterization
The Fe-substitution ratio, Fe/(Ni + Fe), was analyzed by inductively coupled plasma atomic emission spectroscopy (ICP-AES) (ICPS-8100, Shimadzu). The measurement solution was prepared by dissolving 100 mg of sample powder in 10 cm3 of 3.7 wt% HCl aq and then diluting it 100-fold with 3.7 wt% HCl aq.Powder X-ray diffraction (XRD) measurements were performed using an X-ray diffractometer (SmartLab, Rigaku) to identify the crystalline phases of the obtained samples. The scanning range and rate were set to 2θ = 3–50° and 10° min−1, respectively. Mo Kα radiation (60 kV, 150 mA) was used. The crystallite sizes were calculated using Scherrer's equation (eqn (1)) to evaluate the crystallinity of the synthesized samples.
 | (1) |
Field emission scanning electron microscopy (FE-SEM) observations (Regulus 8230, Hitachi High-Tech) and BET specific surface area measurements (Gemini VII 2390p, Micrometrics) were performed to evaluate the morphology of the synthesized samples. Energy dispersive X-ray spectroscopy (EDS) (QUANTAX FlatQUAD 5060F, Bruker) was performed in FE-SEM measurements to evaluate the dispersibility of Fe and Ni. The acceleration voltage, the emission current, and the EDS integration time were set to 20 kV, 15 mA, and 180 s, respectively.
X-ray photoelectron spectroscopy (XPS) (ESCA1700R, ULVAC-PHI) was performed at room temperature using Mg Kα radiation (300 W). The electron take-off angle and pass energy were set at 45° and 117.4 eV.
Electrochemical measurements
Electrochemical measurements were performed using a three-electrode cell with a rotating disk electrode as the working electrode (Fig. S1†). Pt wire, Hg|HgO (RE-61AP, BAS), and 1.0 mol dm−3 KOH solution saturated with O2 were used as the counter electrode, reference electrode, and electrolyte solution, respectively. We also measured the OER activity using graphite as the counter electrode instead of Pt to examine the possible contribution of dissolved Pt species to the OER. As shown in Fig. S2,† almost the same OER activity was obtained in cells with Pt and graphite counter electrodes, verifying that the Pt counter electrode does not affect the OER activity. The working electrode was prepared using the following procedure. Catalyst inks were prepared by adding 10 mg of the synthesized samples and 2.0 mg of the conductive carbon (Vulcan XC-72R) to 1.0 cm3 of 1-hexanol and dispersing ultrasonically. 250 μg cm−2 of the synthesized sample and 50 μg cm−2 of the conductive carbon was coated by dropping 1.77 mm3 of the catalyst ink onto a glassy carbon (GC) disk electrode (φ = 3 mm, BAS). After the electrode was dried at room temperature for 24 hours, 1.72 mm3 of 1-hexanol solution containing 0.2 wt% anion-exchange ionomer (FAA-3, Fumatech) was dropped onto the electrode. Finally, the electrode was dried for 24 h at room temperature.AC impedance measurements, linear sweep voltammetry (LSV), and constant current measurements were performed as electrochemical measurements. All the electrochemical measurements were performed at 25 ± 1 °C using an electrochemical measurement system (Squidstat Plus, Admiral Instruments) and a rotating electrode system (RRDE-3A, BAS). The electrode rotation speed was set at 2000 rpm. AC impedance measurements at open circuit potentials (OCP) were performed for IR correction. The AC amplitude and frequency were set to 10 mV and 1 MHz to 1 Hz, respectively. The real axis intercept of the Nyquist plot was used as the solution resistance. LSV was performed at a scan speed of 10 mV s−1, and a scan range of OCP to 1.70 V vs. RHE. Constant current measurements were performed after the LSV to obtain Tafel plots. The applied current densities were 1.0 mA cmBET−2, 0.5 mA cmBET−2, 0.2 mA cmBET−2, and 0.1 mA cmBET−2 (the current densities per geometric area are shown in Table S3†), and the applied time was 900 s. Long time constant current measurements were also conducted to evaluate durability. The current density was 0.1 mA cmBET−2 and the retention time was 60 h. Differential electrochemical mass spectrometry (DEMS) was performed to measure the partial current of carbon oxidation that occurs in parallel with the OER. A dual thin-layer flow cell combining a microreactor and an ion exchange membrane was used for the DEMS study. The detailed cell configuration is described in our previous paper.32 The working electrode for the DEMS measurements was composed of the LDH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :the conductive carbon:polyvinylidene difluoride = 75:15:10 wt% coated on a graphite sheet (EYG-S121803, Panasonic). Pt wire and Hg|HgO were used as the counter and reference electrodes, respectively. An aqueous solution of 1.0 mol dm−3 KOH saturated with He was allowed to flow using a syringe pump at a flow rate of 0.3 cm3 min−1 as the electrolyte solution. Linear sweep voltammetry (LSV) was performed within the potential range from open circuit potential to 1.63 V vs. RHE at a scan rate of 5 mV s−1. The partial current for the CO2 evolution reaction (carbon oxidation reaction) was obtained from a mass signal of CO2 (m/z = 44) and a calibration constant. The calibration constant was obtained from CO stripping voltammetry of a Pt/C electrode.32
:the conductive carbon:polyvinylidene difluoride = 75:15:10 wt% coated on a graphite sheet (EYG-S121803, Panasonic). Pt wire and Hg|HgO were used as the counter and reference electrodes, respectively. An aqueous solution of 1.0 mol dm−3 KOH saturated with He was allowed to flow using a syringe pump at a flow rate of 0.3 cm3 min−1 as the electrolyte solution. Linear sweep voltammetry (LSV) was performed within the potential range from open circuit potential to 1.63 V vs. RHE at a scan rate of 5 mV s−1. The partial current for the CO2 evolution reaction (carbon oxidation reaction) was obtained from a mass signal of CO2 (m/z = 44) and a calibration constant. The calibration constant was obtained from CO stripping voltammetry of a Pt/C electrode.32
Results and discussion
Characterization of the materials
Inductively coupled plasma atomic emission spectroscopy (ICP-AES) shows that the Fe substitution ratio (Fe/Ni + Fe) of the samples synthesized by soft chemistry and coprecipitation is 11, 20, 32, 41, and 51 at% and 7, 11, and 19 at%, respectively (Table S4†). Hereafter, we denote these samples as soft chemistry 11, 21, 32, 41, and 51% and coprecipitation 7, 11, and 19%, respectively.X-ray diffraction (XRD) patterns of the soft chemistry samples show the desired LDH phases (LDH (α-Ni(OH)2) and γ-NiOOH phases) (Fig. 1(a) and (b)). In both samples, XRD patterns of the γ-NiOOH phase were observed. γ-NiOOH is the oxidized phase of Ni-based LDH (α-Ni(OH)2 phase) and has the same basic crystal structure as the LDH, where Ni is octahedrally coordinated by OH− or O2− to form ABC-stacked slabs.23,24,33 The diffraction patterns of the LDH, which are probably produced from γ-Ni1−xFexOOH by the water oxidation reaction (OER) during the alkaline treatment, are also observed for Fe substitution ratios above 21%. The diffraction intensity of the LDH relative to that of the γ-NiOOH increases with increasing Fe substitution ratio. Considering that the reduction potential of γ-Ni1−xFexOOH increases with increasing Fe substitution ratio,22 it is reasonable that the production of the LDH increases with the Fe substitution ratio. A slight diffraction pattern of β-Ni(OH)2 is also observed in the case of soft chemistry 11%. The Ni-based LDH (α-Ni(OH)2 phase) is known to be stabilized by substituting Ni with trivalent cations.34 We deduce that 11 at% Fe substitution is insufficient to stabilize α-Ni(OH)2 and prevent dehydration to β-Ni(OH)2. Fig. 1(c) shows the XRD patterns of the samples synthesized by the conventional coprecipitation method. Both samples showed diffraction patterns characteristic of LDH. For 7% Fe, the β-Ni(OH)2 peak was observed at a relatively high intensity, probably due to the instability of the hydrous structure at low Fe substitution rates,34 as mentioned above. The presence of the γ-NiOOH phase in the soft chemistry sample also corresponds to the higher binding energy (ca. 856 eV) of the Ni 2p3/2 peak than the coprecipitation samples (ca. 855 eV) (Fig. S3†).35 A comparison of the XRD patterns of the coprecipitation and soft chemistry samples shows that the diffraction peaks of the hydrous phase are sharper in the soft chemistry sample, which indicates that the soft chemistry samples have higher crystallinities of the hydrous phases. To evaluate the crystallinity more quantitatively, Scherrer's equation was applied to the 003 diffraction peak to calculate the crystallite size (Fig. 1(d)). The soft chemistry samples have crystallite sizes about one order of magnitude larger than the coprecipitation samples.
 | ||
| Fig. 1 XRD patterns of the samples synthesized using (a and b) soft chemistry and (c) coprecipitation. (d) Crystallite size of the synthesized samples calculated using the 003 diffraction peak and Scherrer's equation. Lattice constant a of the (e) γ-NiOOH and (f) LDH (α-Ni(OH)2) phases in the soft chemistry samples and (g) the LDH phase in the coprecipitation samples plotted against Fe substitution rate. | ||
To evaluate the uniformity of Fe substitution, we examined whether the lattice constant a follows Vegard's rule. Fig. 1(e) and (f) show the lattice constants a of the γ-NiOOH and LDH (α-Ni(OH)2) phases in the soft chemistry samples obtained by Rietveld refinement (Fig. S4, Table S5†) plotted against Fe substitution rate. The linear relationships following the Vegard's rule indicate that Fe is well soluble in the Ni1−xFexO2 layers in the soft chemistry samples. Notably, Ni in the soft chemistry samples is successfully substituted with Fe even at substitution rates above ca. 25 at%, which is considered the solubility limit of NiFe-LDH.17Fig. 1(g) shows the lattice constant a of the coprecipitation samples calculated from the peak top of the LDH 110 diffraction. Although the lattice constant a decreases with increasing Fe substitution, it is less linear than that of the soft chemistry sample, which indicates that Fe is better solubilized in the soft chemistry samples than in the coprecipitation samples. The increased a in the γ-NiOOH phase and decreased a in the LDH (α-Ni(OH)2 phase) accord with the ionic radius order of Ni2+ > Fe3+ > Ni3+.
Fig. 2 shows FE-SEM/EDS images of the soft chemistry samples. Scale-like particles of about 1–3 μm in diameter are observed in all samples, which indicates that Fe composition does not affect the morphology of the soft chemistry samples. In addition, EDS mappings reveal that Fe and Ni are homogeneously dispersed in the soft chemistry samples. Fig. S5† shows SEM/EDS images of the coprecipitation samples. Unlike the soft chemistry samples, the Fe composition has a relatively large effect on the morphology of the coprecipitation samples. While fine particles of about 100 nm are observed for 7% Fe and 9.5% Fe, relatively coarse particles of 500 nm to 3 μm are observed for 19% Fe. On the other hand, EDS mapping showed that Fe and Ni were homogeneously dispersed in the coprecipitation samples as with the soft chemistry samples.
 | ||
| Fig. 2 FE-SEM/EDS images of the soft chemistry (a and b) 11, (c and d) 20, (e and f) 32, (g and h) 41, and (i and j) 51%. Green and red regions indicate the distributions of Ni and Fe, respectively. | ||
Finally, N2 adsorption isotherms were measured to determine the BET specific surface area of the samples (Fig. 3). The soft chemistry samples show specific surface areas about one order of magnitude smaller than the coprecipitation samples. This is probably because the reactants of the soft chemistry samples were synthesized at a higher temperature, and the particles grew larger. On the other hand, the difference in the specific surface area between the soft chemistry samples is smaller than that between the coprecipitation samples, indicating that the Fe substitution ratio has little effect on the morphology of the soft chemistry samples.
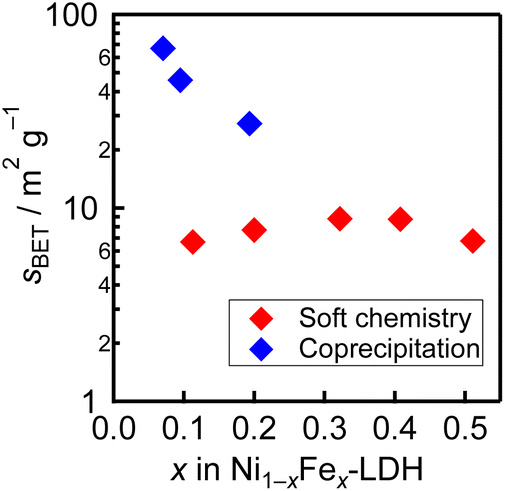 | ||
| Fig. 3 BET specific surface areas of the soft chemistry and the coprecipitation samples plotted against Fe substitution rates. | ||
In summary, using the soft chemistry synthesis, we have successfully obtained NiFe-LDH with higher crystallinity and compositional/morphological uniformity than those of the conventional coprecipitation samples.
Electrochemical measurements
Fig. 4 shows the IR-corrected linear sweep voltammograms (LSVs) of the soft chemistry and coprecipitation samples in O2-saturated 1.0 mol dm−3 KOH aq. Current densities are normalized by geometric surface area as shown in Fig. 4(a), (c) and (e), and by BET surface area as shown in Fig. 4(b), (d) and (f). The coprecipitation sample clearly shows the oxidation peak associated with Ni(II) oxidation, and the onset potential increases with increasing Fe substitution ratio as in previous studies.22 On the other hand, the oxidation current is not observed as a peak but as a shoulder-like shape for the soft chemistry samples, probably due to the already existing Ni(III). Current response attributed to Ni(II) oxidation is hardly observed for soft chemistry 50%. This suggests that the oxidation potential of Ni(II) is higher than the onset potential of OER for soft chemistry 50%, corresponding to the result that more α-Ni(OH)2 phase is observed in 50% Fe after alkaline treatment (Fig. 1(b)). | ||
| Fig. 4 IR-corrected linear sweep voltammograms of the (a and c) soft chemistry and (b and d) coprecipitation samples and (e and f) both samples in O2-saturated 1.0 mol dm−3 KOH aq. Current densities are normalized by (a, b and e) the geometric or (c, d and f) BET surface area. | ||
Fig. 4(e) and (f) compares the LSVs of the soft chemistry and coprecipitation samples. In the high current density region (40–50 mA cmgeo−2), where the effect of Ni(II) oxidation current is relatively small, the soft chemistry sample exhibits the same level of overpotential as the coprecipitation sample (Fig. 4(e)) although their specific surface areas are about one order of magnitude smaller. When compared based on current density normalized by BET surface area, both soft chemistry samples exhibit lower overpotentials than the coprecipitated samples (Fig. 4(f)), suggesting that the soft chemistry samples have higher specific activity for the OER than the coprecipitation samples.
Steady-state polarization curves were obtained by the constant current measurement (Fig. S6†) to analyse the OER activity in more detail. Fig. 5(a) and (b) show a Tafel plot normalized by the BET surface area. Both soft chemistry samples exhibit lower overpotentials than the coprecipitation samples, indicating that the soft chemistry samples have higher OER specific activity. In addition, even though the soft chemistry samples have an order of magnitude smaller specific surface area than the coprecipitation samples, the Tafel plots of soft chemistry 19–51% normalized by mass also show smaller overpotentials than soft chemistry samples (Fig. 5(c) and (d)). To more clearly compare the Tafel plot normalized by mass, a constant current test of 41% Fe was performed at 10 times higher current density, 10 mA cmBET−2, 5.0 mA cmBET−2, 2.0 mA cmBET−2, and 1.0 mA cmBET−2 (Fig. S7 and S8†). Soft chemistry 41% Fe exhibits a lower overpotential than coprecipitation 7% Fe, which exhibits the lowest overvoltage among the coprecipitation samples. From these results, it can be concluded that not only the specific activity but also the mass activity of the soft chemistry sample is higher than that of the conventional coprecipitation sample.
 | ||
| Fig. 5 IR-corrected Tafel plots of the (a and c) soft chemistry and (b and d) coprecipitation samples in O2-saturated 1.0 mol dm−3 KOH aq. Current densities are normalized by the (a and b) BET surface area or (c and d) mass of the catalyst. Overpotentials η at (e) 100 μA cmBET−2 and (f) 1.0 mA cmBET−2. | ||
Table 1 summarizes Tafel slopes and exchange current densities. The lower Tafel slopes of the soft chemistry samples than those of the coprecipitation samples indicate that the active sites in the soft chemistry samples have more enhanced OER kinetics. Since soft chemistry samples have higher crystallinity than coprecipitation samples, the active sites of the soft chemistry and coprecipitation samples are mainly composed of crystalline and amorphous regions, respectively. Previous studies using theoretical calculations have reported that the edge site of the γ-NiOOH phase (crystalline phase) is highly active.16,17 Therefore, it is reasonable from a theoretical point of view that the active sites in the soft chemistry samples have higher OER activity. On the other hand, the soft chemistry samples exhibit lower exchange current densities normalized by BET surface area than the coprecipitation samples. The soft chemistry sample has a higher intensity of 003 diffraction relative to the other diffraction intensities (Fig. 1(a)–(c)), which suggests stronger c-axis orientations. Since edge planes are regarded as active sites,17 the smaller exchange current density of the soft chemistry sample is probably due to lower edge plane ratios.
| Sample | Tafel slope (mV dec−1) | i 0 (nA cmBET−2) |
|---|---|---|
| Soft chemistry 11% Fe | 30.4 ± 0.8 | 1.2 ± 0.6 |
| Soft chemistry 20% Fe | 27.0 ± 0.3 | 0.5 ± 0.2 |
| Soft chemistry 32% Fe | 24.7 ± 0.5 | 0.13 ± 0.06 |
| Soft chemistry 41% Fe | 25.9 ± 0.3 | 0.32 ± 0.07 |
| Soft chemistry 51% Fe | 25.7 ± 0.4 | 0.25 ± 0.08 |
| Coprecipitation 7% Fe | 34.7 ± 0.1 | 1.57 ± 0.08 |
| Coprecipitation 9.5% Fe | 34.7 ± 0.2 | 1.44 ± 0.06 |
| Coprecipitation 19% Fe | 34.7 ± 0.1 | 0.99 ± 0.02 |
Fig. 5(e) and (f) show the superior OER activity of the soft chemistry samples at both 0.1 and 1.0 mA cmBET−2. The overpotential of the soft chemistry sample decreases as the Fe substitution ratio increases from 11 at% to 20 at%, but the overpotential remained almost unchanged from 20 at% to 51 at%. This result indicates that the optimum Fe substitution ratio is at least 20–51 at%. The optimal Fe substitution ratio of 20–51 at% can be a compromise of the number of Fe–O–Ni sites16 increasing with the Fe content and oxidation state of Ni22 decreasing with the Fe content, both of which are considered to be responsible for the high OER activity. The coprecipitation sample shows an increase in overpotential with increasing Fe substitution ratio (7–19 at%), which is a different behaviour from that of the soft chemistry sample. Considering that the coprecipitation samples show the dependence of the crystallinity and morphology on the Fe substitution ratio and the relatively poor Fe solubility, we deduce that the impact of Fe substitution on the OER specific activity of the coprecipitation samples was not strictly evaluated.
Fig. S9† shows the relationship between overpotential and turn over frequency (TOF). TOF was calculated with the applied current in the constant current test, the BET surface area, and the atomic densities of the edge plane ({1 1 0} plane). The atomic density of the edge plane was calculated with the lattice constant of each sample. The soft chemistry samples exhibit larger TOFs at the same overvoltage, which indicates that their OER activity per surface metal atom was also higher than that of the coprecipitation samples. The variation of TOF among the soft chemistry samples was the smallest for Fe, which strongly suggests the presence of Fe in the active sites.
The OER activities of the samples synthesized in this study are compared with other reported samples. First, the overpotentials at 1.0 mA cmBET−2 are compared to those of other OER catalysts. Our soft chemistry (η < 0.22 V for 10% Fe, η < 0.20 V for 20–51% Fe) and coprecipitation (η < 0.25 V) samples show lower overpotentials at 1.0 mA cmBET−2 than other oxide catalysts, including Ba0.5Sr0.5Co0.8Fe0.2O3−δ (η ≈ 0.33 V), La0.5Sr0.5CoO3−δ (η ≈ 0.31 V), and Sr2Co0.8Fe0.2O3Cl (η ≈ 0.28 V),36–38 which indicates that our soft chemistry samples as well as the conventional LDH have significantly high OER specific activity. We then compared the OER activity of our soft chemistry samples with other LDH samples. Since only a few reports normalize current density by the BET specific surface area, OER specific activity was compared using the Tafel slope,13–15 a strength-determining variable. The coprecipitation sample exhibited an average Tafel slope (ca. 35 mV dec−1) for NiFe-LDH. In contrast, the soft chemistry samples with the Fe substitution ratio of 20 –51 at% exhibit Tafel slopes (24.7–27.0) lower than 28 mV dec−1, the lowest value for NiFe-LDH-based materials in 1.0 mol dm−3 KOH aq,39,40 which clearly indicates that highly crystalline Ni1−xFex-LDH (0.20 ≤ x ≤ 0.51) synthesized by soft chemistry has a higher specific activity than conventional NiFe-LDHs. We also compared the mass activity of our soft chemistry samples with other LDHs using an overvoltage with a current flow of 10 mA cmgeo−2 in the LSV measurement (η10). η10 is often used to compare LDH activity but includes the effect of nickel oxidation current. Therefore, it should be noted that the soft chemistry samples with high nickel oxidation states possibly show larger η10 due to the smaller nickel oxidation current. While the soft chemistry samples show η10 of 252–279 mV, other NiFe-LDHs evaluated at similar catalyst loading densities (200–300 μg cm−2) showed η10 of 229–300 mV.39,40 Considering the smaller contribution of the nickel oxidation current in the soft chemistry sample, the soft chemistry sample could have a relatively high mass activity among the NiFe-LDHs. The soft chemical samples synthesized in this study have a lower specific surface area than other NiFe-LDHs. If NiFe-LDH with both high specific surface area and high crystallinity can be developed, it is strongly expected that higher mass activity will be obtained.
Finally, to compare the durability of the soft chemistry and coprecipitation samples, constant current measurements were performed at 0.1 mA cmBET−2 for 60 h. While the coprecipitation sample shows an overvoltage increase of about 45 mV, the soft chemistry sample showed an overvoltage increase of about 20 mV (Fig. 6(a)), indicating that the soft chemistry sample has higher durability than the conventional co-precipitation sample. To investigate the factors that contribute to high durability, DEMS measurements are used to analyze the CO2 produced by carbon oxidation. Fig. 6(b) and (c) show LSVs for the faradaic current and CO2 evolution (carbon oxidation) current of soft chemistry 40% Fe and coprecipitation 9.5% Fe. In both samples, the CO2 evolution current is about one order of magnitude smaller than the faradaic current, indicating that most of the faradaic current is the OER current. While both samples showed a maximum CO2 generation current of about 1 mA, soft chemistry 40% Fe showed about twice the faradaic current compared to the coprecipitation method 9.5% Fe. This result indicates that the carbon oxidation rate is slower for soft chemistry 40% Fe under constant current conditions, suggesting that the lower carbon corrosion activity partially contributes to the higher durability.
 | ||
| Fig. 6 (a) Chronoamperograms of soft chemistry 41% Fe and coprecipitation 9.5% Fe at 100 μA cmBET−2. LSVs for the faradaic current and CO2 evolution current of (b) the soft chemistry 40% Fe (1.7 mg cm−2) and (c) the coprecipitation 9.5% Fe (3.3 mg cm−2). | ||
Conclusions
NiFe-LDH with high crystallinity and uniform composition and morphology were synthesized using soft chemistry, and their OER activities in 1.0 mol dm−3 KOH were evaluated. We achieve 51 at% Fe substitution, which exceeds the conventional solid solution limit. Soft chemistry samples with 20–51 at% Fe substitution showed almost the same and higher OER activity than the 11 at% Fe-substituted samples. The good OER activity at high Fe substitution rates is a favorable result from an elemental strategic point of view. The soft chemistry samples with 20–51 at% Fe substitution exhibit higher specific and mass activities than those synthesized by the conventional coprecipitation method, indicating that the crystalline site has higher OER activity than the amorphous site. In addition, the soft chemistry samples with 20–51 at% Fe substitution show the lowest Tafel slopes among the NiFe-LDH samples. Considering the relatively low specific surface area of the soft chemical samples synthesized in this study, the synthesis of NiFe-LDH with both high crystallinity and high specific surface area should lead to higher OER active catalysts. Furthermore, it is strongly expected from this study that soft chemistry enables the evaluation of the crystalline region's activity and the influence of composition on the activity for metastable phases other than NiFe-LDH.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JRP-LEAD with DFG Grant Number PJSJRP20221602 and JSPS KAKENHI Grant Number JP23K13819. The XPS measurement was performed using research equipment shared in the MEXT Project for promoting public utilization of advanced research infrastructure (Program for supporting introduction of the new sharing system) Grant Number JPMXS0420900521. The authors thank Materials Analysis Division, Open Facility Center, Tokyo Institute of Technology, for ICP-AES analysis. The authors thank Prof. Masaaki Hirayama and Prof. Kenta Watanabe, Tokyo Institute of Technology, for SEM/EDS observations. The authors thank Ms Hisako Murakami and Mr Shugo Shimizu, Tokyo Institute of Technology, for the DEMS measurement.References
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- J. Zhang, Q. Zhou, Y. Tang, L. Zhang and Y. Li, Chem. Sci., 2019, 10, 8924–8929 RSC.
- K. Zeng and D. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS.
- C. Hu, L. Zhang and J. Gong, Energy Environ. Sci., 2019, 12, 2620–2645 RSC.
- D. Zhou, P. Li, W. Xu, S. Jawaid, J. Mohammed-Ibrahim, W. Liu, Y. Kuang and X. Sun, ChemNanoMat, 2020, 6, 336–355 CrossRef CAS.
- A. Ikezawa, K. Miyazaki, T. Fukutsuka and T. Abe, Electrochem. Commun., 2017, 84, 53–56 CrossRef CAS.
- A. Ikezawa, K. Seki and H. Arai, Electrochim. Acta, 2021, 394, 139128 CrossRef CAS.
- M. Plevová, J. Hnát and K. Bouzek, J. Power Sources, 2021, 507, 230072 CrossRef.
- C. Wang, L. Jin, H. Shang, H. Xu, Y. Shiraishi and Y. Du, Chin. Chem. Lett., 2021, 32, 2108–2116 CrossRef CAS.
- N. Yuan, Q. Jiang, J. Li and J. Tang, Arabian J. Chem., 2020, 13, 4294–4309 CrossRef CAS.
- X. Xie, L. Du, L. Yan, S. Park, Y. Qiu, J. Sokolowski, W. Wang and Y. Shao, Adv. Funct. Mater., 2022, 32, 2110036 CrossRef CAS.
- Z. Cai, X. Bu, P. Wang, J. C. Ho, J. Yang and X. Wang, J. Mater. Chem. A, 2019, 7, 5069–5089 RSC.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
- S. Anantharaj, K. Karthick and S. Kundu, Mater. Today Energy, 2017, 6, 1–26 CrossRef.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik and D. Fan, Nat. Commun., 2020, 11, 2522 CrossRef CAS PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- Y. Tian, G. Wang, F. Li and D. G. Evans, Mater. Lett., 2007, 61, 1662–1666 CrossRef CAS.
- Lakhanlal, O. Rimon, W. Moschkowitsch, G. C. Taguri, L. Elbaz and M. Caspary Toroker, Mol. Catal., 2024, 561, 1141364 Search PubMed.
- M. Wilhelm, A. Bastos, C. Neves, R. Martins and J. Tedim, Mater. Des., 2021, 212, 110188 CrossRef CAS.
- Y. Han, Z.-H. Liu, Z. Yang, Z. Wang, X. Tang, T. Wang, L. Fan and K. Ooi, Chem. Mater., 2008, 20, 360–363 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- C. Delmas, Y. Borthomieu, C. Faure, A. Delahaye and M. Figlarz, Solid State Ionics, 1989, 32, 104–111 CrossRef.
- C. Delmas, J. Braconnier, Y. Borthomieu and M. Figlarz, Solid State Ionics, 1988, 28, 1132–1137 CrossRef.
- H. Arai, M. Tsuda, M. Hayashi, H. Ohtsuka and Y. Sakurai, Electrochim. Acta, 2005, 50, 1821–1828 CrossRef CAS.
- R. Uppuluri, A. S. Gupta, A. S. Rosas and T. E. Mallouk, Chem. Soc. Rev., 2018, 47, 2401–2430 RSC.
- X. Wang, G. Liu, T. Iwao, M. Okubo and A. Yamada, J. Phys. Chem. C, 2014, 118, 2970–2976 CrossRef CAS.
- K. Imai, A. Ikezawa, S. Sato and H. Arai, Chem. Commun., 2024, 60, 10192–10195 RSC.
- Y. Zhao, F. Li, R. Zhang, D. G. Evans and X. Duan, Chem. Mater., 2002, 14, 4286–4291 CrossRef CAS.
- M. A. Oliver-Tolentino, J. Vázquez-Samperio, A. Manzo-Robledo, R. d. G. González-Huerta, J. L. Flores-Moreno, D. Ramírez-Rosales and A. Guzmán-Vargas, J. Phys. Chem. C, 2014, 118, 22432–22438 CrossRef CAS.
- K. Miyazaki, Y. Asada, T. Fukutsuka, T. Abe and L. A. Bendersky, J. Mater. Chem. A, 2013, 1, 14569–14576 RSC.
- A. Ikezawa, J. Kida, K. Miyazaki and H. Arai, Electrochem. Commun., 2024, 159, 107647 CrossRef CAS.
- P. Oliva, J. Leonardi, J. F. Laurent, C. Delmas, J. J. Braconnier, M. Figlarz, F. Fievet and A. d. Guibert, J. Power Sources, 1982, 8, 229–255 CrossRef CAS.
- G. Mishra, B. Dash and S. Pandey, Appl. Clay Sci., 2018, 153, 172–186 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. S. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771–1779 CrossRef CAS.
- Y. Miyahara, T. Fukutsuka, T. Abe and K. Miyazaki, Chem. Mater., 2020, 32, 8195–8202 CrossRef CAS.
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, 1806296 CrossRef PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS PubMed.
- L. Yu, H. Zhou, J. Sun, F. Qin, F. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2017, 10, 1820–1827 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05347h |
| This journal is © The Royal Society of Chemistry 2024 |