
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hybrid d0 and d10 electronic configurations promote photocatalytic activity of high-entropy oxides for CO2 conversion and water splitting†
Jacqueline
Hidalgo-Jiménez
 ab,
Taner
Akbay
c,
Xavier
Sauvage
d,
Lambert
van Eijck
e,
Motonori
Watanabe
af,
Jacques
Huot
g,
Tatsumi
Ishihara
af and
Kaveh
Edalati
*af
ab,
Taner
Akbay
c,
Xavier
Sauvage
d,
Lambert
van Eijck
e,
Motonori
Watanabe
af,
Jacques
Huot
g,
Tatsumi
Ishihara
af and
Kaveh
Edalati
*af
aInternational Institute for Carbon-Neutral Energy Research (WPI-I2CNER), Kyushu University, Fukuoka, Japan. E-mail: kaveh.edalati@kyudai.jp; Fax: +81 92 802 6744; Tel: +81 92 802 6744
bDepartment of Automotive Science, Kyushu University, Fukuoka, Japan
cMaterials Science and Nanotechnology Engineering, Yeditepe University, Istanbul, Turkey
dUniv Rouen Normandie, INSA Rouen Normandie, CNRS, Groupe de Physique des Matériaux, UMR6634, 76000 Rouen, France
eDepartment of Radiation Science and Technology, Delft University of Technology, Delft, Netherlands
fMitsui Chemicals, Inc. – Carbon Neutral Research Center (MCI-CNRC), Kyushu University, Fukuoka, Japan
gHydrogen Research Institute, Université du Québec à Trois-Rivières, Trois-Rivières, Canada
First published on 21st October 2024
Abstract
Photocatalysis offers a sustainable solution for essential reactions such as CO2 conversion and water splitting, but constraints in catalyst properties like bandgap and active site availability often limit its efficiency. High-entropy oxides (HEOs), which incorporate five or more different cations, present significant potential for this application due to their elemental diversity. This study explores active HEO development for photocatalytic applications by integrating cations with d0 and d10 electronic configurations. A single-phase HEO with a monoclinic structure was successfully synthesized, comprising elements with d0 (titanium, zirconium, niobium and tantalum) and d10 (zinc) electronic configurations. Comprehensive analyses of its microstructure, chemical composition, optical properties and photocatalytic activity were conducted. The resulting TiZrNbTaZnO10 exhibited superior UV and visible light absorption, a low bandgap of 2.5 eV, minimal radiative electron–hole recombination and high stability under photocatalytic conditions. Remarkably, TiZrNbTaZnO10 outperformed the TiZrHfNbTaO11 photocatalyst which contains solely d0 electronic configuration. This enhanced performance is attributed to the mixed electronic configurations fostering heterogeneous chemical environments, which facilitate efficient charge carrier separation and transfer.
Introduction
The consumption of fossil fuels and uncontrolled use of resources have caused irreversible problems in the environment and living society. Nonetheless, many academic and industrial projects are trying to decrease the consequences of these problems and improve the conditions. Two main strategies are currently under investigation: hydrogen utilization and CO2 conversion. On one hand, hydrogen utilization aims to replace the consumption of fossil fuels as a clean alternative. However, nearly 60% of hydrogen is produced directly from fossil fuels,1 and most of the remaining hydrogen is produced from electricity generated by non-renewable energies. On the other hand, the already emitted CO2 can be trapped, stored and converted into useful products. Both processes can be performed using clean technologies such as photocatalysis,2 which is a light-driven chemical reaction in the presence of a catalyst. In photocatalysis, the light energy can promote the electron excitation from the valence band and transfer them to the conduction band,3 generating positive and negative charges which can take part in redox reactions.Despite the advantages of photocatalytic processes, their practical utilization is still limited. Low light absorbance, the short lifetime of charge carriers and their fast recombination are some limiting factors that photocatalysts face.3–5 Moreover, the limited number of active sites for the adsorption and conversion of water and CO2 is another factor that negatively affects photocatalytic activity.3–5 Aiming to solve this problem, high-entropy materials have been investigated as a new type of photocatalyst. These materials are typically formed by five or more elements, having a high mixing entropy. The increment in the mixing entropy allows high-entropy materials to be energetically stabilized in a single- or dual-phase form by decreasing the Gibbs free energy.6,7 Additionally, the sluggish diffusion effect, cocktail effect and lattice distortion can contribute to the generation of numerous active sites, promote defect formation, produce hybridized orbitals and give them tunable properties for photocatalysis.4,8,9 The combinations of these features make high-entropy materials appropriate catalysts for different reactions such as hydrogen evolution,10–16 CO2 conversion,8,17–20 dye degradation12,13,21 and contaminant removal.22,23
Nonetheless, the inherent nature of high-entropy photocatalysts is not necessarily sufficient to ensure their high performance. Different strategies were employed to enhance the activity of these materials such as increasing the surface area,24 including porosities,8,14 employing the piezo-photocatalysis concept,13 enhancement of lattice distortion25 and increasing light absorbance by nitride26 or oxynitride generation.11,17 Most high-entropy photocatalysts are based on high-entropy oxides (HEOs), in which cations usually have a d0 electronic configuration. TiZrHfNbTaO11 is one of the most popular high-entropy photocatalysts that contains d0 cations. However, it is well-known that the cations with d10 electronic configuration such as zinc and gallium can also show good photocatalytic activity. While the d0 cations are strong electron acceptors, the d10 cations are weaker acceptors and can sometimes act as electron donors. Therefore, the co-presence of d0 and d10 cations can make a heterogenous electronic structure with a chemical environment containing different active site types including electron and hole reaction sites. Despite developing high entropy photocatalysts with d0 electronic configuration, there has been no attempt so far to examine the efficiency of mixing cations with d0 and d10 electronic configurations on the photocatalytic activity of HEOs.
This study reports the synthesis of a new high-entropy photocatalyst, TiZrNbTaZnO10, with mixed d0 (titanium, zirconium, niobium and tantalum) and d10 (zinc) electronic configurations for CO2 conversion and hydrogen production. The sample is synthesized by a high-pressure torsion (HPT) process followed by calcination and examined by different characterization analyses and photocatalytic testing. A comparison of the activity of this HEO with TiZrHfNbTaO11 with only d0 cations confirms the impact of the mixed electronic configuration concept on photocatalysis. This concept can be employed to design new photocatalysts with high activity.
Experimental materials and procedures
Sample synthesis
The synthesis was performed using binary oxides, as starting materials, to fabricate a HEO with a nominal composition of TiZrNbTaZnO10. TiO2 anatase powder (Sigma-Aldrich, 99.8%), ZrO2 (Kojundo, 97 mol%), Nb2O5 (Kojundo, 99%), Ta2O5 (Kojundo, 99.9%) and ZnO (Kanto Chemical Co. Inc., 99.0%) were manually mixed in a mortar using acetone for 30 min. The powder mixture with about 350 mg was pressed to disc shapes with 10 mm diameter and subsequently processed by HPT under a pressure of 6 GPa for 6 turns to obtain a better nano-scale mixture. The HPT process was performed at room temperature while the rotation speed was kept at 1 rpm. The resulting material was a disc with 10 mm diameter and about 0.8 mm thickness. The HPT-processed disc was crushed and calcined at 1373 K for 24 h to synthesize a single-phase material. To have a better homogeneity, the calcined powder was subjected to HPT for another 3 turns and subsequently calcined for another 24 h.Characterization
The crystal structure in every step of the synthesis process was examined by X-ray diffraction (XRD) using Cu Kα radiation. The final sample was additionally examined by neutron diffraction using a wavelength of 1.67 Å (ref. 27) and diffraction patterns were analyzed using Rietveld refinement to obtain accurate lattice parameters and atomic positions. The characteristic crystal vibrational modes were studied using Raman spectroscopy with a 532 nm laser source. The oxidation state of elements was examined by X-ray photoelectron spectroscopy (XPS) using a radiation source of Al Kα.The elemental distribution was examined at different scales. First, at the microscopic scale, scanning electron microscopy (SEM) coupled with energy-dispersive X-ray spectroscopy (EDS) under an acceleration voltage of 15 keV was used. Second, at the nanometric scale, scanning-transmission electron microscopy (STEM) coupled with EDS was employed at an acceleration voltage of 200 keV. The sample for STEM was prepared by crushing the HEO and dispersing it on a copper grid covered with a carbon film. Third, at the atomic scale, atom probe tomography (APT) was used. The sample for APT was prepared using a focused-ion beam technique. APT analysis was conducted using a CAMECA LEAP-5000XS instrument under ultra-high vacuum (10−11 mbar) at a cryogenic temperature of 40 K using laser pulses with a frequency of 200 kHz. Analyzed volumes were reconstructed using IVAS (Integrated Visualization & Analysis Software), and data analysis was carried out using GPM3DSoft (Groupe de Physique des Matériaux 3D-soft) software.
The microstructure of the sample was analyzed by SEM using an acceleration voltage of 15 keV and transmission electron microscopy (TEM) with an acceleration voltage of 200 keV. For TEM, bright-field images, selected area electron diffraction (SAED) and high-resolution images were recorded. For the examination of oxygen vacancies, electron spin resonance (ESR) utilizing a 9.4688GHz microwave was conducted.
The light absorbance of the sample was examined by UV-vis spectroscopy, and subsequently, the bandgap was estimated using the Kubelka–Munk theory. The radiative recombination of photogenerated charge carriers was analyzed by photoluminescence spectroscopy using a 325nm laser source. The lifetime of the charge carriers was investigated using time-resolved photoluminescence decay (PL decay) with a 340 nm laser source and a longpass filter of 385 nm. XPS was used to estimate the valence band top, and to finally obtain the band structure by combining with the data from UV-vis spectroscopy.
Photocatalytic activity
The photocatalytic activity of the sample was evaluated using two reactions: CO2 conversion and water splitting. For CO2 conversion, 100 mg of sample, 4.2 g of NaHCO3 and 500 ml of deionized water were placed in a continuous flow quartz reactor of 858 ml volume. In the inner space of the reactor, a high-pressure mercury light source with a power of 400 W was inserted. The light irradiated on the photocatalyst was equivalent to 14 W cm−2. On the top of the reactor, two pipes were placed: one for inputting a constant CO2 gas flow of 30 ml min−1, and one for outputting the generated gases which was connected to two gas chromatographs. One gas chromatograph was a Shimadzu GF-8A to analyze the hydrogen and oxygen concentrations, and the other one was a GL Science GC-4000 Plus equipped with a methanizer (GL Science MT 221) to evaluate the concentration of CO and CH4. The temperature was controlled to 291–296 K using a water chiller and the dispersion of particles in suspension was homogenized using a magnetic stirrer with a rotation speed of 420 rpm. The CO2 gas was first injected for 1 h without light irradiation to confirm that no air was left in the reactor, and no CO2 conversion occurred.The photocatalytic hydrogen production from water splitting was analyzed in a reactor coupled with a gas chromatograph (Shimadzu GF-8A) under a full arc of 300 W UV xenon (Xe) light. For the solution preparation, 50 mg of sample, 27 ml of deionized water, 8 ml of methanol (as sacrificial agent) and 0.25 ml of H3PtCl6·6H2O (0.01 M) were mixed. The photocatalytic test was performed for 180 min, and sampling was conducted every 30 min using a volume of 3.2 ml. The light irradiated on the photocatalyst was equivalent to 15 W cm−2. Before the photocatalytic measurement, a blank test without light irradiation and with catalyst addition was conducted for 30 min.
Results
Crystal structure and the microstructure
The progress of crystal structure transformation obtained by XRD during the synthesis process is shown in Fig. 1(a). The initial powder mixture shows all the characteristic peaks for the binary oxides utilized. After the first HPT processing with 6 turns, the peaks become broad due to the crystallite size reduction and defect introduction.28 Subsequently, the XRD pattern after the first calcination shows much sharper peaks and a completely different crystal structure than the powder mixture. Nonetheless, the analysis of this pattern shows the presence of minor amounts of binary oxides, thus, additional 3 HPT turns followed by calcination were performed. After the second calcination, a single-phase material with the monoclinic P2/c crystal structure is obtained. The results of Rietveld refinement, as shown in Fig. 1(b), give the lattice parameters included in Table 1. | ||
| Fig. 1 Formation of single-phase high-entropy oxide TiZrNbTaZnO10 with a monoclinic structure. (a) XRD patterns of the material obtained after different steps of synthesis, (b) Rietveld refinement analysis of the XRD pattern after the final step of the synthesis, (c) neutron diffraction and corresponding Rietveld refinement analysis after the final step of the synthesis, and (d) Raman spectra taken at three random positions after the final step of the synthesis. C in (a) indicates calcination at 1373 K. | ||
| Lattice parameters (Å) | |
|---|---|
| a | 4.7405 ± 0.0002 |
| b | 5.6459 ± 0.0002 |
| c | 5.0550 ± 0.0002 |
| α (°) | 90 |
| β (°) | 91.114 ± 0.003 |
| γ (°) | 90 |
The crystal structure was also examined by neutron diffraction. Like XRD, this technique can give additional information about the crystal structure and atomic positions. The main difference is that neutron diffraction analysis is more sensitive to light elements. Fig. 1(c) shows the neutron diffraction and corresponding Rietveld analysis. The Rietveld refinement confirms the monoclinic crystal structure with the P2/c space group. Moreover, it gives some hints about the specific atomic positions. Using Nb2ZrZnO8 as a similar model structure, 3 main Wyckoff positions of 4g, 2f and 2e are refined. It is confirmed that the 4g positions of TiZrNbTaZnO10 correspond to the oxygen atoms, 2f positions correspond to niobium and tantalum atoms, and 2e positions correspond to zirconium and zinc. Regarding the position of titanium, the best fit of the Rietveld refinement was obtained when titanium was distributed in the two positions of 2f and 2e.
The crystal nature of TiZrNbTaZnO10 was also examined by Raman spectroscopy, as shown in Fig. 1(d). Raman spectra from three different random positions in Fig. 1(d) show similar features, suggesting good homogeneity in the crystal structure. The vibrational modes in Raman spectra can be determined using Nb2ZrZnO8 as a reference with a similar crystal structure. Table 2 summarizes the vibrational modes assigned to TiZrNbTaZnO10. The first peak observed at 130 cm−1 was identified as a characteristic vibration of the lattice.29 The peaks observed at 185, 355, 405 and 465 cm−1 are related to vibrational modes of Zr–O bonding.30 Ti–O vibrational modes are observed at 632 and 671 cm−1.30 The peak observed at 755 cm−1 can be related to the Ti–O bond; however, it can be associated with Nb/Ta–O asymmetric vibrations as well.29,30 Finally, the modes observed at 280, 843 and 890 cm−1 are associated with Nb/Ta–O vibrational modes.30,31 The differences between TiZrNbTaZnO10 and Nb2ZrZnO8 are due to the differences in the interatomic distances due to the addition of titanium and tantalum to the lattice.30
| Raman shift (cm−1) | Vibrational mode | Reference |
|---|---|---|
| 130 | Lattice vibration | 29 |
| 185 | Zr–O stretching | 30 |
| 249 | Oxygen–cation–oxygen bending | 29 |
| 280 | Nb/Ta–O bonding | 31 |
| 307 | Zn–O symmetric stretching | 31 |
| 355 | O–Zr–O bending | 30 |
| 405 | Zr–O stretching | 30 |
| 465 | O–Zr–O bending | 30 |
| 511 | Zr–O bonding | 30 |
| 593 | Nb/Ta–O asymmetric vibrations | 31 |
| 632 | Zr–O bonding | 30 |
| 671 | Ti–O bonding | 30 |
| 755 | Ti–O or Nb/Ta–O asymmetric vibrations | 29 and 31 |
| 843 | Nb–O asymmetric stretching | 30 and 31 |
| 890 | Nb–O symmetric stretching | 30 |
Fig. 2 shows the XPS results for the sample TiZrNbTaZnO10. The full oxidation state of each element (i.e. Ti4+, Zr4+, Nb5+, Ta5+, Zn2+ and O2−) is confirmed. The peaks in the spectra are asymmetric, but small shoulders to lower energy sites are observed in some elements such as zinc. The shoulders indicate the possible presence of small amounts of oxygen vacancies in the sample.4 The presence of oxygen vacancies is also suggested using ESR spectra in which a double peak with a turning point at a g value of 2.01 is observed (Fig. 3). Lattice defects such as oxygen vacancies can act as active sites for photocatalysis, particularly when they are located on the surface.3
 | ||
| Fig. 2 Fully oxidized states of elements in high-entropy oxide TiZrNbTaZnO10. XPS analysis for (a) titanium, (b) zirconium, (c) niobium, (d) tantalum, (e) zinc, and (f) oxygen. | ||
 | ||
| Fig. 3 Presence of oxygen vacancies in high-entropy oxide TiZrNbTaZnO10 by ESR analysis. | ||
The chemical analysis of TiZrNbTaZnO10 was performed using three different techniques. The SEM-EDS and STEM-EDS mappings are shown in Fig. 4. The SEM-EDS maps (Fig. 4(a)) confirm a homogeneous distribution of the element at the micrometer scale. The general atomic concentrations of elements are 4.6 ± 0.4 at% Ti, 5.2 ± 0.4 at% Zr, 5.2 ± 0.5 at% Nb, 5.5 ± 0.5 at% Ta, 6.8 ± 0.1 at% Zn and 72.3 ± 1.7 at% O, which are close to the nominal composition within the precision limits of SEM-EDS. Fig. 4(b) shows the STEM-EDS mappings, confirming the homogenous distribution of elements at the nanometer scale. Similarly, the three-dimensional APT analysis of a tip of HEO (Fig. 5(a)) shows the chemical homogeneity at the atomic scale. The percentage of each element obtained along the tip is shown in Fig. 5(b). The APT analysis shows an average composition of ∼6 at% of each element being slightly higher for niobium and lower for titanium. Nonetheless, all the results demonstrate that the five cations are well distributed in the sample with concentrations close to nominal composition. The homogenous distribution of elements at the atomic scale confirms that each cation should be surrounded by different elements, making a heterogeneous chemical environment all over the sample, which is a positive feature for heterogeneous photocatalysts.32
 | ||
| Fig. 4 Homogeneous distribution of elements at the micrometer and nanometer scales in high-entropy oxide TiZrNbTaZnO10. (a) SEM-EDS and (b) STEM-EDS mappings. | ||
 | ||
| Fig. 5 Homogeneous distribution of elements at the atomic scale in high-entropy oxide TiZrNbTaZnO10. (a) Three-dimensional chemical composition distribution and (b) percentage of elements along the sample tip. | ||
The microstructural analysis of TiZrNbTaZnO10 is shown in Fig. 6. The SEM micrographs in Fig. 6(a) and (b) show the morphology of the powder, containing micro-sized particles with an average size of 32 μm. This average particle size corresponds to a small surface area of 0.033 m2 g−1. This small surface area should be due to the synthesis method which includes the application of high pressure during the HPT process and high temperature during the calcination.28,33 Examination of microstructure by TEM bright-field analysis (Fig. 6(c)) indicates that the crystal sizes are either at the submicrometer or micrometer sizes. Here, it should be noted that the Rietveld analysis suggests an average crystallite size of 140 nm, confirming the large sizes of powder particles. The SAED analysis (Fig. 6(d)), high-resolution images and corresponding fast Fourier transforms (Fig. 6(d)) confirm that the monoclinic is the only detected phase in the crystal structure.
 | ||
| Fig. 6 Formation of micro-sized particles with a monoclinic structure in high-entropy oxide TiZrNbTaZnO10. (a and b) SEM micrographs at different magnifications, (c) TEM bright-field micrograph, (d) SAED pattern, and (e) high-resolution image and corresponding fast Fourier transform. | ||
Optical properties
The results for light absorbance and band structure of the HEO TiZrNbTaZnO10 are illustrated in Fig. 7. Fig. 7(a) shows UV-vis spectroscopy results, indicating good light absorbance in the UV and visible ranges of the light spectrum. The light absorbance is a desirable property to improve the efficiency of the photocatalysts. The analysis of the light absorbance spectrum using the Kubelka–Munk theory to obtain the bandgap is shown in Fig. 7(b). TiZrNbTaZnO10 has a bandgap of 2.5 eV, and a defect state close to 1.8 eV. Compared to the bandgap of each utilized binary oxide, TiZrNbTaZnO10 has a smaller bandgap which coincides with the visible light region. Bandgap reduction to the visible light region is a good finding because there are significant efforts to discover visible-light-active photocatalysts.34,35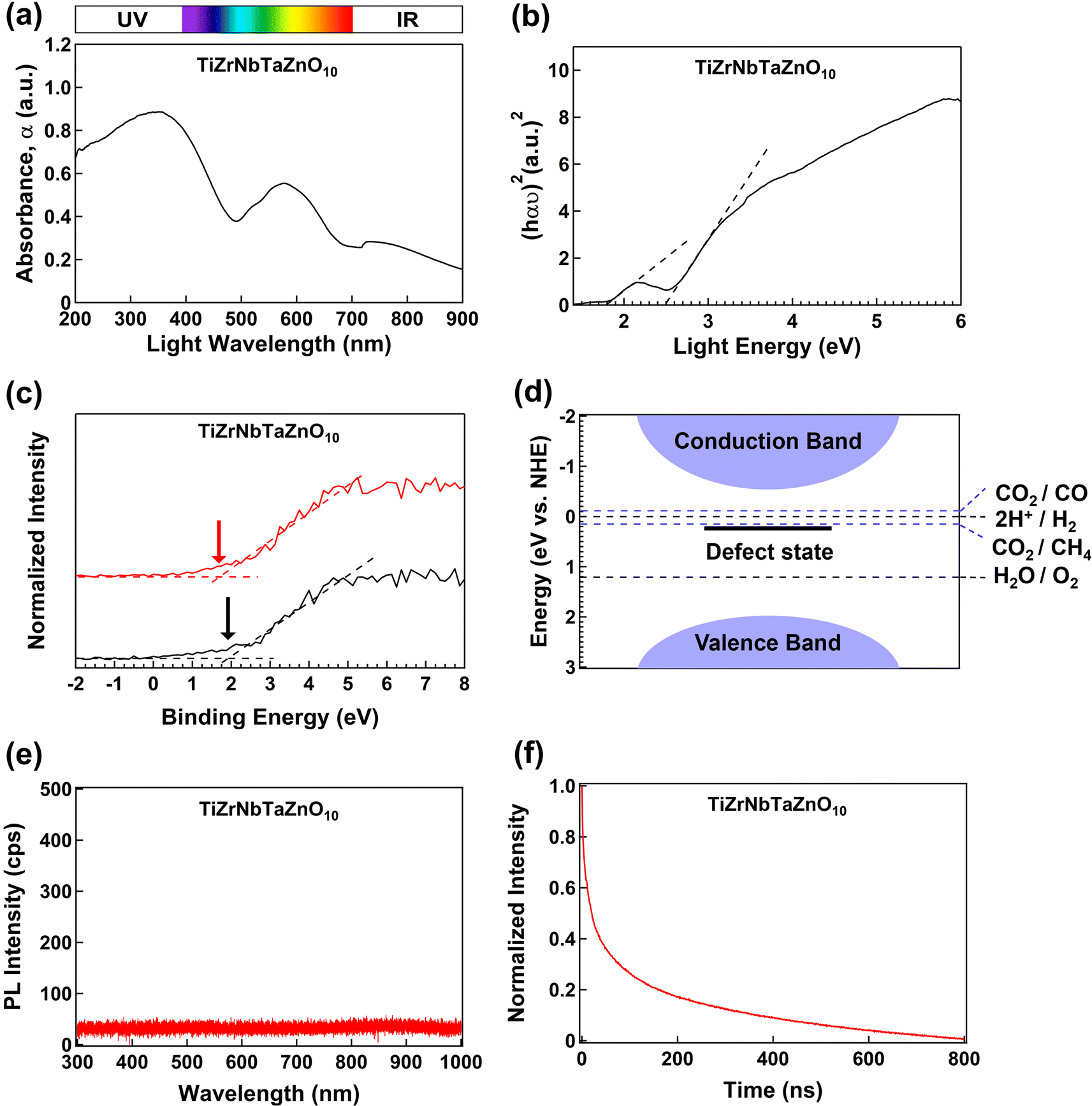 | ||
| Fig. 7 High UV and visible light absorbance and the appropriate band structure of high-entropy oxide TiZrNbTaZnO10 for photocatalytic CO2 conversion and water splitting. (a) UV-vis spectra, (b) Kubelka–Munk analysis for bandgap calculation, (c) XPS spectra for valence band top determination, (d) band structure and potential energies for different reactions, (e) photoluminescence (PL) spectroscopy for radiative recombination evaluation, and (f) PL decay spectroscopy to examine the lifetime of charge carriers. | ||
XPS analysis was performed to calculate the valence band top. Fig. 7(c) shows the result of XPS in two different random positions of the sample. The valence band top is located close to 2.0 ± 0.1 eV for both spectra. Combining the XPS results with the bandgap obtained by UV-vis and Kubelka–Munk theory determines the band structure shown in Fig. 7(d). The valence band is located below the energy required for the oxidation of H2O to O2 (1.23 eV), and the conduction band is located above the energy required for the reduction of H+ to H2 (0 eV).35,36 CO2 to CO and CO2 to CH4 are also located between the bands.4,35 Therefore, the TiZrNbTaZnO10 band structure is appropriate for both CO2 conversion and water-splitting photocatalytic reactions shown below at pH = 0.
| CO2 + 2H+ + 2e− → CO + H2O potential vs. NHE: −0.11 eV (ref. 4 and 35) | (1) |
| CO2 + 8H+ + 8e− → CH4 + 2H2O potential vs. NHE: 0.17 eV (ref. 4 and 35) | (2) |
| H2O + 2h+ → 1/2O2 + 2H+ potential vs. NHE: 1.23 eV (ref. 35 and 36) | (3) |
| 2H+ + 2e− → H2 potential vs. NHE: 0 eV (ref. 35 and 36) | (4) |
Fig. 7(e) shows photoluminescence spectroscopy results used to analyze the radiative recombination of photogenerated charge carriers. There is no evident peak for TiZrNbTaZnO10, indicating low radiative recombination of charge carriers. Low photoluminescence intensity is usually related to disordered atomic arrangements and is beneficial for photocatalytic activity.37,38
PL decay analysis is shown in Fig. 7(f) which can provide evidence for the lifetime of the photogenerated carriers in TiZrNbTaZnO10. An exponential equation can be used to analyze the plot in Fig. 7(f) as given below.
I(t) = A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ) exp(−t/τ) | (5) |
In this equation, A corresponds to the amplitude of the exponential function, t is the time and τ is the lifetime of the photogenerated carriers. The fitting of this function suggests that the value for τ is 11.63 ns. Compared with the lifetime of the carriers reported for TiZrHfNbTaO11 (10.7 ns), TiZrNbTaZnO10 exhibits slower radiative recombination of the carriers. The delay in the recombination of the carriers can be attributed to the mixed electronic configuration obtained by including zinc.
Photocatalytic activity
The photocatalytic activity of TiZrNbTaZnO10 was examined by two methods, CO2 conversion and water splitting. The results for both tests are shown in Fig. 8, while the activity of binary oxides mixed by HPT is also included for comparison. TiZrNbTaZnO10 constantly transforms CO2 to CO (Fig. 8(a)) and CH4 (Fig. 8(b)) during 24 h when it is irradiated with the full arc of a mercury lamp. The CO average production during the first 24 h is close to 25.2 μmol h−1 g−1 while the CH4 average production during the first 24 h stays around 9.9 μmol h−1 g−1. The generation of CH4 is usually difficult due to the involvement of eight electrons and photocatalysis usually ends with CO production which requires only four electrons. However, the amount of CH4 in this study is comparable to CO, indicating good activity of current HEO. For water splitting, TiZrNbTaZnO10 generates up to 0.6 mmol g−1 with the full arc of a xenon lamp, as shown in Fig. 8(c). Despite the small surface area,33,39 TiZrNbTaZnO10 showed promising performance as a photocatalyst. Fig. 8 confirms that the activity of TiZrNbTaZnO10 is higher than that of the powder mixture for CO, CH4 and H2 production, although the powder mixture contains TiO2 and ZnO which are considered good photocatalysts.40,41 | ||
| Fig. 8 Enhanced photocatalytic CH4, CO and H2 production on high-entropy oxide TiZrNbTaZnO10 compared to binary oxides mixed by HPT. (a) CO and (b) CH4 production rates versus time during CO2 conversion using a mercury lamp. (c) H2 concentration versus time during water splitting using a xenon lamp. Stability of TiZrNbTaZnO10 evaluated by (d) water splitting cycle test for 4 cycles and (e) XRD before and after photocatalytic tests. | ||
Another important characteristic of photocatalysts is their stability. Fig. 8(d) and (e) demonstrate the stability of TiZrNbTaZnO10 using two different testing methods. First, the sample was tested during four cycles for photocatalytic water splitting as shown in Fig. 8(d). The hydrogen production rate obtained for the different cycles is reasonably similar demonstrating the high photocatalytic activity and catalyst stability during a prolonged time. Second, Fig. 8(e) shows the XRD patterns for the sample before and after CO2 conversion and water splitting. It is confirmed that the crystal structure does not change due to the interaction with the light or other compounds used. It should be noted that an additional CO2 conversion experiment was conducted using a different measurement system (due to a technical problem in the authors' facility) for 3 cycles and a total period of 75 h. This experiment also confirmed that TiZrNbTaZnO10 remains photocatalytically active for a prolonged time.
Two issues should be considered regarding Fig. 8. First, the authors examined the photocatalytic activity of TiZrNbTaZnO10 under visible light conditions for water splitting for 24 h (due to its low bandgap), however, the reaction products could not be detected within the measurement limits of the gas chromatograph. Second, the optical and electronic properties of TiO2,39,42 ZrO2,43,44 Nb2O5,45,46 Ta2O5 (ref. 47) and ZnO40,41,48 have been widely studied for photocatalytic applications. These materials often show bandgap in the range of 3.2 eV (for TiO2 (ref. 39)) to 5.3 eV (for Ta2O5 (ref. 47)) and good light absorbance in the UV region. The commercial powders of these binary oxides contain cations with fully oxidized states and negligible vacancy concentration, but there have been attempts to introduce oxygen vacancies in them by different methods such as HPT for enhancing light absorbance and bandgap narrowing.39,44,48 Nonetheless, the bandgap of all these binary oxides is still larger than that of TiZrNbTaZnO10 which is 2.5 eV. The optical and electronic properties of TiZrHfNbTaO11 photocatalyst were also previously reported.4,10 TiZrHfNbTaO11 shows mainly UV light absorbance with a bandgap of 2.9 eV, but it can also exhibit some visible light absorbance due to the presence of defects such as oxygen vacancies and dislocations in its structure4,10 Nonetheless, the light absorbance and bandgap of TiZrHfNbTaO11 are not as good as TiZrNbTaZnO10 for photocatalysis.
Discussion
A newly synthesized HEO TiZrNbTaZnO10 is introduced as an active photocatalyst. The lattice strain, vacancy-type defects and the presence of different chemical environments due to the presence of elements with different atomic sizes and electronic structures make HEOs attractive for photocatalysis. Nonetheless, these characteristics appear in various HEOs, but additional concepts need to be employed to further enhance their activity. This study attempts to demonstrate that the co-presence of cations with d0 and d10 electronic configurations is an additional strategy to develop efficient high-entropy photocatalysts. Two important questions should be discussed in detail: (1) how significant is the difference between the activity of HEOs with d0 and d0 + d10 electronic configurations? and (2) what is the impact of combining d0 and d10 electronic configuration elements?To answer the first question, zinc with the d10 electronic configuration was added to titanium, zirconium, niobium and tantalum with the d0 electronic configuration in this study. The comparison of TiZrNbTaZnO10 having mixed d0 and d10 cations with TiZrHfNbTaO11 containing only d0 cations is shown in Fig. 9. Fig. 9(a) and (b) show CO2 to CO conversion rates per unit mass and surface area of the catalysts, respectively, and Fig. 9(c) and (d) illustrate hydrogen production from water splitting per catalyst mass and surface area. TiZrNbTaZnO10 has better performances for both CO2 conversion and water splitting, and the difference is more noticeable when comparing per surface area. Since surface area and morphology have a stronger impact on the photocatalytic activity compared to the catalyst mass,8,14 comparing the results per surface area is more reasonable. The difference observed for TiZrNbTaZnO10 and TiZrHfNbTaO11 in Fig. 9 can be related to the contribution of d10 cations. Table 3 compares the photocatalytic activity of TiZrNbTaZnO10 with some famous binary catalysts such as TiO2 and ZnO, some active composites containing heterojunctions and some high-entropy catalysts.48–54 The comparison suggests that high-entropy materials, particularly those containing elements with the d10 electronic configuration like gallium or zinc, show better performance for photocatalysis compared to binary oxides and composites. Particularly, TiZrNbTaZnO10 shows one of the best performances for both CO2 conversion and water splitting when comparing the activity per catalyst surface area.
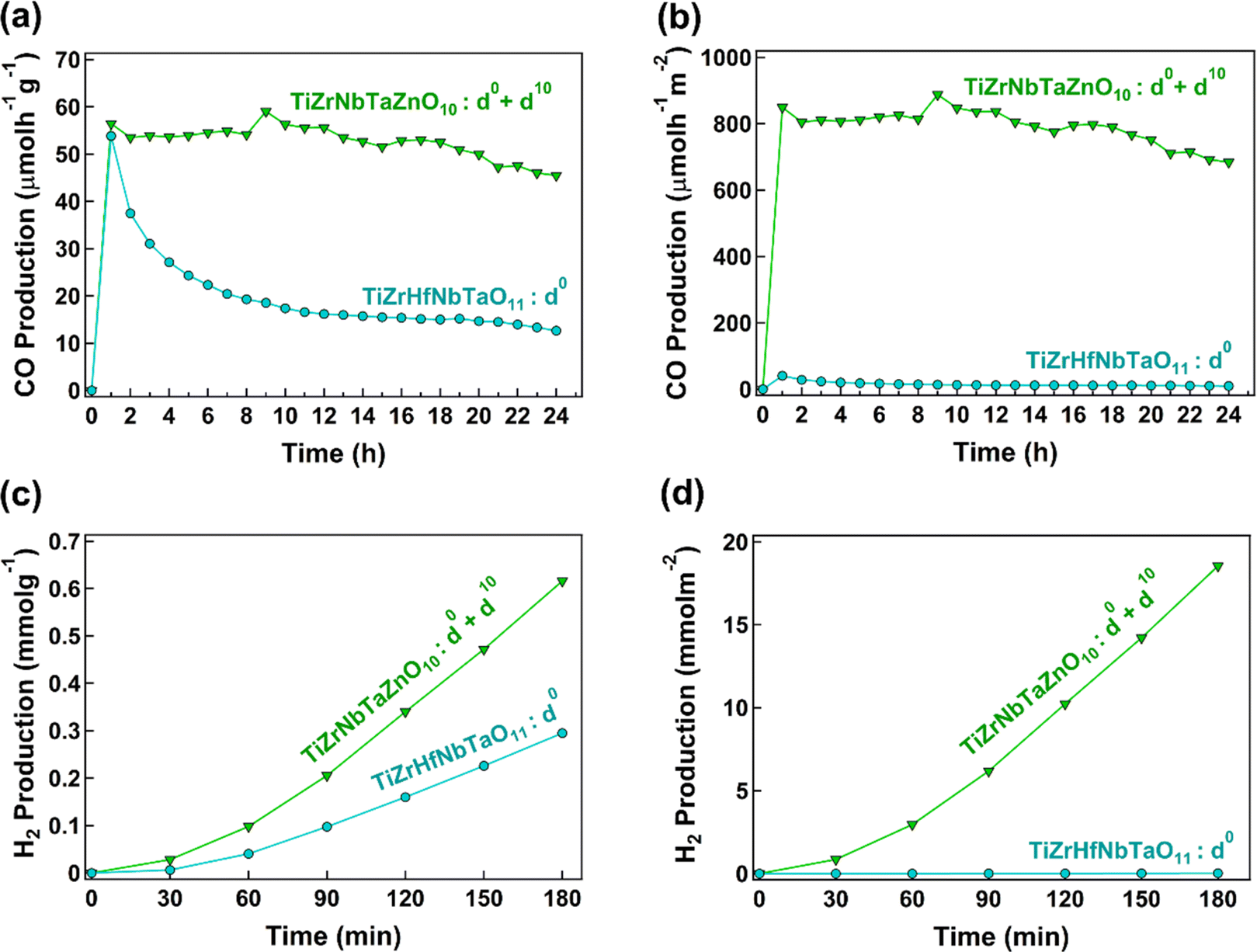 | ||
| Fig. 9 Enhancement of the photocatalytic activity of high-entropy oxide TiZrNbTaZnO10 compared to TiZrHfNbTaO11 due to the mixed d0 and d10 electronic configuration. CO2 to CO conversion rate per (a) unit mass and (b) surface area of the two catalysts. H2 production from water splitting per (c) unit mass and (d) surface area of the two catalysts. | ||
| Photocatalyst | Mass (mg) | Surface area (m2 g−1) | Light source | CO2 conversion (μmol h−1 g−1) | CO2 conversion (μmol h−1 m−2) | Water splitting (mmol h−1 g−1) | Water splitting (mmol h−1 m−2) | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|---|
| CO | CH4 | CO | CH4 | H2 | H2 | |||||
| TiO2 anatase | 50 | 13.5 | 300 W Xe | 0.3189 | 0.47 | 39 | ||||
| TiO2 (HPT, ∼70% columbite) | 50 | 0.1 | 300 W Xe | 0.005 | 1.08 | 39 | ||||
| TiO2 (HPT and annealing) | 100 mg | 6.8 | 300 W Hg | 1.39 | 0.15 | 49 | ||||
| ZnO (HPT) | 50 | 0.585 | 0.45 | 48 | ||||||
| CeO2−x | 50 | 20.5 | 300 W Xe | 1.68 | 0.081 | 50 | ||||
| Nb2O5/TiO2 | 100 | 57.3 | 200 W Xe | 0.18 | 10.31 | 46 | ||||
| S-doped Ta2O5–CdS | 5 | 0.017 | 300 W Xe | 0.2725 | 16.03 | 51 | ||||
| MnCo/CN | 300 W Xe | 47 | 52 | |||||||
| Cd1−xZnxS | 45 | 119 | 100 W LED | 2.9 | 0.22 | 0.096 | 0.01 | 53 | ||
| TiZrHfNbTaO11 | 100/50 | 0.089 | 400 W Hg/300 W Xe | 4.6 | — | 5.16 | — | 0.0361 | 0.2 | 4 and 10 |
| TiZrHfNbTaO11 (mechano-thermal synthesis) | 50 | 3 | 300 W Xe | 0.027 | 134.76 | 54 | ||||
| TiZrHfNbTaO11 (laser crushing) | 100 | 2.69 | 400 W Hg | 50 | 200 | 1.66 | 6.66 | 24 | ||
| TiZrHfNbTaO6N3 | 100/50 | 2.3 | 400 W Hg, 300 W Xe | 11.6 | — | 0.5 | — | 0.0319 | 0.006 | 11 and 17 |
| Ce0.2Zr0.2La0.2Pr0.2Y0.2O2 | 2 | 61.4 | 11 W UV | 9.2 | 0.0002 | 12 | ||||
| ZrYCeCrO2-based + 38 at% Ca | — | 150 W Xe | 0.415 | 13 | ||||||
| Li(NbVTaCrMoWCo)O3 | 50 | 11.19 | 300 W Xe, 420 nm cutoff filter | 6.61 | 0.29 | 15 | ||||
| (Co, Mn, Ni, Zn)O-metal organic framework | 10 | 300 W Xe | 13.24 | 16 | ||||||
| Ce0.2Zr0.2La0.2Pr0.2Y0.2O2 | 2 | 61.4 | 11 W UV | 9.2 | 0.0002 | 12 | ||||
| (Ga0.2Cr0.2Mn0.2Ni0.2Zn0.2)3O4 | 20 | 16.71 | 300 W Xe | 23.01 | 2.89 | 0.03 | 0.65 | 8 | ||
| Cu–(Ga0.2Cr0.2Mn0.2Ni0.2Zn0.2)3O4 | 20 | 42.08 | 300 W Xe | 5.66 | 33.84 | 0.002 | 0.02 | 19 | ||
| (NiCuMnCoZnFe)3O4 | 30 | 66.48 | — | 15.89 | 8.03 | 0.007 | 0 | 20 | ||
| TiZrNbTaZnO10 | 100/50 | 0.03 | 400 W Hg/300 W Xe | 25.2 | 9.9 | 761.3 | 301 | 12 | 2.95 | This study |
Concerning the second question, it should be noted that a combination of elements with different electronic configurations leads to (i) geometrical and (ii) electronic effects in crystal structure. First, the lattice distortion is caused by different cations with diverse atomic sizes. These highly distorted lattices produce distortion in octahedral and tetrahedral coordination which can influence the ionic properties, electron transfer and photocatalytic activity of the material by producing dipole moments.5,55,56 Moreover, vacancies are formed to compensate for such distortion in long-range order. The presence of such vacancies, confirmed by the ESR analysis (Fig. 3), is usually beneficial as active sites for photocatalysis. Second, the electronic configuration plays an important role in photocatalytic activity. The elements with d0 electronic configuration (titanium, zirconium, niobium and tantalum) work as strong electron donor elements, while d10 cations (zinc) can also function as acceptors. The elements with d0 have usually a lower electronegativity, making a stronger binding with the absorbed molecules of CO2 and H2O.57 It should be noted that the charge transfer between CO2 and catalyst or H2O and catalyst is a critical aspect that determines the reactant absorbance on the surface by controlling binding energy, as discussed earlier by first-principles calculations.57 First-principles calculations showed that in TiZrHfNbTaO11, the elements with lower electronegativity such as hafnium or zirconium have stronger binding energy for water molecules, while niobium and tantalum with a higher electronegativity have a good electron transfer performance especially when they are placed in the surrounding of the low-electronegativity cations.57 Moreover, in elements with d10 electronic configurations, the contribution of sp orbitals to band structure improves the mobility of photoexcited carriers and promotes charge separation.5,56 Therefore, the presence of cations with mixed d0 and d10 electronic configurations can provide a heterogeneous chemical environment containing sites for both giving and accepting charge carriers leading to better charge transfer as well as stronger CO2 and H2O absorbance. More specifically for zinc (d10 electronic configuration element), it was reported that adding Zn2+ can enhance the activity when utilized as a dopant.58 Zn2+ can weakly trap and release the carriers due to its stronger redox ability. This property can enhance the carrier mobility on the surface and suppress the recombination of the photogenerated carriers.58–60 These features of Zn2+ in the presence of d0 cations, which are strong electron acceptors, can promote charge separation and transfer. Evidence of this interaction can be observed in the reduction of bandgap to the visible light region (Fig. 7) and the weak irradiative recombination, as evident from negligible photoluminescence intensity (Fig. 7(e)). Considering all these accepts, combining cations with d0 and d10 electronic structures can be considered as a new strategy to develop active high-entropy photocatalysts.
Conclusions
A new high-entropy oxide with mixed electronic configurations was successfully synthesized using titanium, zirconium, niobium and tantalum (d0 configuration) mixed with zinc (d10 configuration). TiZrNbTaZnO10 demonstrated promising optical properties such as high light absorbance in the UV and visible light regions as well as low bandgap and low irradiative recombination of the photogenerated carriers. The combination of d0 and d10 improved the photocatalytic activity for hydrogen production and CO2 conversion compared to a high-entropy oxide TiZrNbTaZnO10 having only the d0 electronic configuration. This study confirms the high potential of high-entropy oxides with mixed d0 + d10 electronic configuration cations as a new type of efficient photocatalyst for both CO2 conversion and water splitting.Data availability
The data will be made available upon request from the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Mr Fabien Cuvilly of the University of Rouen Normandy for APT sample preparation and analyses. The author JHJ acknowledges a scholarship from the Q-Energy Innovator Fellowship of Kyushu University. This study is supported partly by Mitsui Chemicals, Inc., Japan, partly through Grants-in-Aid from the Japan Society for the Promotion of Science (JP22K18737), partly by Japan Science and Technology Agency (JST), the Establishment of University Fellowships Towards the Creation of Science Technology Innovation (JPMJFS2132), partly by the University of Rouen Normandy through project BQRI MaP-StHy202, partly by the CNRS Federation IRMA-FR 3095, and partly by the ASPIRE project of the Japan Science and Technology Agency (JST) (JPMJAP2332).Notes and references
- G. H. Patel, J. Havukainen, M. Horttanainen, R. Soukka and M. Tuomaala, Green Chem., 2024, 26, 992–1006 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- B. Liu, X. Zhao, C. Terashima, A. Fujishima and K. Nakata, Phys. Chem. Chem. Phys., 2014, 16, 8751–8760 RSC.
- S. Akrami, Y. Murakami, M. Watanabe, T. Ishihara, M. Arita, M. Fuji and K. Edalati, Appl. Catal., B, 2022, 303, 120896 CrossRef CAS.
- Y. Inoue, Energy Environ. Sci., 2009, 2, 364–386 RSC.
- C. Oses, C. Toher and S. Curtarolo, Nat. Rev. Mater., 2020, 5, 295–309 CrossRef CAS.
- C. M. Rost, E. Sachet, T. Borman, A. Moballegh, E. C. Dickey, D. Hou, J. L. Jones, S. Curtarolo and J. P. Maria, Nat. Commun., 2015, 6, 8485 CrossRef CAS PubMed.
- Z. Jiang, R. Zhang, H. Zhao, J. Wang, L. Jia, Y. Hu, K. Wang and X. Wang, Appl. Surf. Sci., 2023, 612, 155809 CrossRef CAS.
- A. Haeussler, S. Abanades, J. Jouannaux and A. Julbe, J. Membr. Sci., 2021, 634, 119387 CrossRef CAS.
- P. Edalati, Q. Wang, H. Razavi-Khosroshahi, M. Fuji, T. Ishihara and K. Edalati, J. Mater. Chem. A, 2020, 8, 3814–3821 RSC.
- P. Edalati, X. F. Shen, M. Watanabe, T. Ishihara, M. Arita, M. Fuji and K. Edalati, J. Mater. Chem. A, 2021, 9, 15076–15086 RSC.
- S. Nundy, D. Tatar, J. Kojčinović, H. Ullah, A. Ghosh, T. K. Mallick, R. Meinusch, B. M. Smarsly, A. A. Tahir and I. Djerdj, Adv. Sustain. Syst., 2022, 6, 1–20 Search PubMed.
- S. C. Chang, H. Y. Chen, P. H. Chen, J. T. Lee and J. M. Wu, Appl. Catal., B, 2023, 324, 122204 CrossRef CAS.
- M. Einert, A. Waheed, S. Lauterbach, M. Mellin, M. Rohnke, L. Q. Wagner, J. Gallenberger, C. Tian, B. M. Smarsly, W. Jaegermann, F. Hess, H. Schlaad and J. P. Hofmann, Small, 2023, 19, 2205412 CrossRef CAS PubMed.
- H. Ling, M. Sun, H. Han, L. Lu, L. Cai, Y. Lan, R. Li, P. Chen, X. Tian, X. Bai and W. Wang, J. Phys. Chem. Lett., 2024, 15, 5103–5111 CrossRef CAS PubMed.
- S. Qi, K. Zhu, T. Xu, H. Zhang, X. Guo, J. Wang, F. Zhang and X. Zong, Adv. Mater., 2024, 36, 1–9 Search PubMed.
- S. Akrami, P. Edalati, Y. Shundo, M. Watanabe, T. Ishihara, M. Fuji and K. Edalati, Chem. Eng. J., 2022, 449, 137800 CrossRef CAS.
- Y. Li, X. Bai, D. Yuan, C. Yu, X. San, Y. Guo, L. Zhang and J. Ye, Nat. Commun., 2023, 14, 1–10 Search PubMed.
- Y. Zhang, Z. Jiang, R. Zhang, K. Wang and X. Wang, Appl. Surf. Sci., 2024, 651, 159226 CrossRef CAS.
- L. Zhang, S. Xia, X. Zhang, Y. Yao, Y. Zhang, S. Chen, Y. Chen and J. Yan, ACS Nano, 2024, 18, 5322–5334 CAS.
- H. Fu, S. Li, Y. Lin, X. Wu, T. Lin, C. Zhao, M. Gao and C. Lin, Ceram. Int., 2024, 50, 9159–9168 CrossRef CAS.
- Y. Xu, L. Wang, Z. Shi, N. Su, C. Li, Y. Huang, N. Huang, Y. Deng, H. Li, T. Ma, X. Y. Kong, W. Lin, Y. Zhou and L. Ye, Energy Environ. Sci., 2023, 16, 1531–1539 RSC.
- Y. Yu, S. Liu, H. Wang, S. Zhang, N. Wang, W. Jiang, C. Liu, W. Ding, Z. Zhang and C. Dong, J. Solid State Chem., 2023, 317, 123694 CrossRef CAS.
- Z. Pourmand Tehrani, T. Fromme, S. Reichenberger, B. Gökce, T. Ishihara, T. Lippert and K. Edalati, Adv. Powder Technol., 2024, 35, 104448 CrossRef CAS.
- A. Gautam, S. Das and M. I. Ahmad, Surf. Interfaces, 2024, 46, 104054 CrossRef CAS.
- W. Li, Y. Sun, L. Ye, W. Han, F. Chen, J. Zhang and T. Zhao, J. Am. Ceram. Soc., 2022, 105, 3729–3734 CrossRef CAS.
- L. Van Eijck, L. D. Cussen, G. J. Sykora, E. M. Schooneveld, N. J. Rhodes, A. A. Van Well and C. Pappas, J. Appl. Crystallogr., 2016, 49, 1398–1401 CrossRef CAS.
- K. Edalati, A. Bachmaier, V. A. Beloshenko, Y. Beygelzimer, V. D. Blank, W. J. Botta, K. Bryła, J. Čížek, S. Divinski, N. A. Enikeev, Y. Estrin, G. Faraji, R. B. Figueiredo, M. Fuji, T. Furuta, T. Grosdidier, J. Gubicza, A. Hohenwarter, Z. Horita, J. Huot, Y. Ikoma, M. Janeček, M. Kawasaki, P. Král, S. Kuramoto, T. G. Langdon, D. R. Leiva, V. I. Levitas, A. Mazilkin, M. Mito, H. Miyamoto, T. Nishizaki, R. Pippan, V. V. Popov, E. N. Popova, G. Purcek, O. Renk, Á. Révész, X. Sauvage, V. Sklenicka, W. Skrotzki, B. B. Straumal, S. Suwas, L. S. Toth, N. Tsuji, R. Z. Valiev, G. Wilde, M. J. Zehetbauer and X. Zhu, Mater. Res. Lett., 2022, 10, 163–256 CrossRef CAS.
- Z. Huang, J. Qiao and L. Li, J. Am. Ceram. Soc., 2023, 106, 5855–5867 CrossRef CAS.
- R. Xiang, H. Su, Q. Zhang, Y. Li and X. Tang, J. Mater. Sci.: Mater. Electron., 2020, 31, 4769–4779 CrossRef CAS.
- K. Liu, H. Zhang, C. Liu, L. Shi, X. Wang, J. Li, Y. Liao and D. Zhang, J. Am. Ceram. Soc., 2023, 106, 1089–1101 CrossRef CAS.
- A. O. Ibhadon and P. Fitzpatrick, Catalysts, 2013, 3, 189–218 CrossRef CAS.
- K. Edalati, Adv. Eng. Mater., 2019, 21, 1–10 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- E. Kalamaras, M. M. Maroto-Valer, M. Shao, J. Xuan and H. Wang, Catal. Today, 2018, 317, 56–75 CrossRef CAS.
- K. Shimura and H. Yoshida, Energy Environ. Sci., 2011, 4, 2467–2481 RSC.
- X. Wang, B. Liu, S. Ma, Y. Zhang, L. Wang, G. Zhu, W. Huang and S. Wang, Nat. Commun., 2024, 15, 1–11 Search PubMed.
- H. Yin, J. Chen, P. Guan, D. Zheng, Q. Kong, S. Yang, P. Zhou, B. Yang, T. Pullerits and K. Han, Angew. Chem., Int. Ed., 2021, 60, 22693–22699 CrossRef CAS PubMed.
- J. Hidalgo-Jiménez, T. Akbay, T. Ishihara and K. Edalati, J. Mater. Chem. A, 2023, 11, 23523–23535 RSC.
- C. B. Ong, L. Y. Ng and A. W. Mohammad, Renewable Sustainable Energy Rev., 2018, 81, 536–551 CrossRef CAS.
- S. Patial, R. Kumar, P. Raizada, P. Singh, Q. Van Le, E. Lichtfouse, D. Le Tri Nguyen and V. H. Nguyen, Environ. Res., 2021, 197, 111134 CrossRef CAS PubMed.
- K. Nakata and A. Fujishima, J. Photochem. Photobiol., C, 2012, 13, 169–189 CrossRef CAS.
- N. C. S. Selvam, A. Manikandan, L. J. Kennedy and J. J. Vijaya, J. Colloid Interface Sci., 2013, 389, 91–98 CrossRef CAS PubMed.
- Q. Wang, K. Edalati, Y. Koganemaru, S. Nakamura, M. Watanabe, T. Ishihara and Z. Horita, J. Mater. Chem. A, 2020, 8, 3643–3650 RSC.
- I. Ahmad, A. Al-Qattan, M. Z. Iqbal, A. Anas, M. A. Khasawneh, A. J. Obaidullah, A. Mahal, M. Duan, W. Al Zoubi, Y. Y. Ghadi, N. Al-Zaqri and C. Xia, Adv. Colloid Interface Sci., 2024, 324, 103093 CrossRef CAS PubMed.
- J. Yan, G. Wu, N. Guan and L. Li, Appl. Catal., B, 2014, 152–153, 280–288 CrossRef CAS.
- V. Gurylev, Mater. Today Sustain., 2022, 18, 100131 Search PubMed.
- Y. Shundo, T. Tam Nguyen, S. Akrami, P. Edalati, Y. Itagoe, T. Ishihara, M. Arita, Q. Guo, M. Fuji and K. Edalati, J. Colloid Interface Sci., 2024, 666, 22–34 CrossRef CAS PubMed.
- S. Akrami, M. Watanabe, T. H. Ling, T. Ishihara, M. Arita, M. Fuji and K. Edalati, Appl. Catal., B, 2021, 298, 120566 CrossRef CAS.
- T. Ye, W. Huang, L. Zeng, M. Li and J. Shi, Appl. Catal., B, 2017, 210, 141–148 CrossRef CAS.
- H. Liu, L. Zhao, J. Yu, G. Xiong, Z. Liu, X. Zhang, B. Chu, X. Liu, H. Liu and W. Zhou, Chem. Eng. J., 2022, 436, 1–8 Search PubMed.
- H. Ou, S. Ning, P. Zhu, S. Chen, A. Han, Q. Kang, Z. Hu, J. Ye, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202206579 CrossRef CAS PubMed.
- E. A. Kozlova, M. N. Lyulyukin, D. V. Markovskaya, D. S. Selishchev, S. V. Cherepanova and D. V. Kozlov, Photochem. Photobiol. Sci., 2019, 18, 871–877 CrossRef CAS PubMed.
- Ö. Güler, M. Boyrazlı, M. G. Albayrak, S. H. Güler, T. Ishihara and K. Edalati, Materials, 2024, 17, 853 CrossRef PubMed.
- K. Su, C. Zhang, Y. Wang, J. Zhang, Q. Guo, Z. Gao and F. Wang, Chin. J. Catal., 2022, 43, 1894–1905 CrossRef CAS.
- H. Nishiyama, H. Kobayashi and Y. Inoue, ChemSusChem, 2011, 4, 208–215 CrossRef CAS PubMed.
- J. Hidalgo-Jiménez, T. Akbay, T. Ishihara and K. Edalati, Scr. Mater., 2024, 250, 116205 CrossRef.
- L. G. Devi, B. N. Murthy and S. G. Kumar, Mater. Sci. Eng. B, 2010, 166, 1–6 CrossRef CAS.
- J. Guo, X. Liao, M.-H. Lee, G. Hyett, C.-C. Huang, D. W. Hewak, S. Mailis, W. Zhou and Z. Jiang, Appl. Catal., B, 2019, 243, 502–512 CrossRef CAS.
- W. Wang, M. O. Tadé and Z. Shao, Chem. Soc. Rev., 2015, 44, 5371–5408 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04689g |
| This journal is © The Royal Society of Chemistry 2024 |