
Strategic cation exchange induced 2D nickel sulphide nanoplates with enhanced oxygen evolution reaction performance†
Jiayi
Chen
a,
Xiaomin
Xu
 b,
Rundong
Mao
a,
Cuifang
Wang
b,
Hsien-Yi
Hsu
cd,
Zongyou
Yin
e,
Mark A.
Buntine
a,
Alexandra
Suvorova
f,
Martin
Saunders
f,
Zongping
Shao
*b and
Guohua
Jia
*a
b,
Rundong
Mao
a,
Cuifang
Wang
b,
Hsien-Yi
Hsu
cd,
Zongyou
Yin
e,
Mark A.
Buntine
a,
Alexandra
Suvorova
f,
Martin
Saunders
f,
Zongping
Shao
*b and
Guohua
Jia
*a
aSchool of Molecular and Life Sciences, Curtin University, Bentley, WA 6102, Australia. E-mail: guohua.jia@curtin.edu.au
bWA School of Mines: Minerals, Energy and Chemical Engineering (WASM-MECE), Curtin University, Perth, WA 6102, Australia. E-mail: zongping.shao@curtin.edu.au
cSchool of Energy and Environment, City University of Hong Kong, Kowloon Tong, Hong Kong SAR, P. R. China
dShenzhen Research Institute of City University of Hong Kong, Shenzhen, 518057, P. R. China
eResearch School of Chemistry, Australian National University, Canberra, ACT 2600, Australia
fCentre for Microscopy Characterization and Analysis (CMCA), The University of Western Australia, Perth, WA 6009, Australia
First published on 11th September 2024
Abstract
Nickel sulphides stand out as promising, earth-abundant transition metal chalcogenides with significant potential for the electrocatalytic oxygen evolution reaction. However, the realisation of their full potential is hindered by challenges in controlling the size, morphology and phase of nickel sulphide nanocrystals, limiting their broader application. In this study, we introduce a novel method for synthesising two-dimensional Ni9S8 phase-dominated NixS nanoplates via a precisely controlled cation exchange approach. Through meticulous adjustments in surface ligands and reaction temperature, we effectively fine-tune the reaction kinetics, resulting in the production of NixS nanoplates with well-preserved morphology and high crystallinity. Notably, the resulting NixS nanoplates synthesised at 170 °C exhibit exceptional performance in the oxygen evolution reaction, boasting a low overpotential of 329 mV at a current density of 10 mA cm−2 and a Tafel slope of 52 mV dec−1. These findings not only advance our understanding of nickel sulphide nanomaterials but also hold promise for their practical applications in efficient and sustainable electrocatalytic processes.
Introduction
In recent years, the excessive use of traditional fossil fuels has led to severe environmental pollution, making it imperative to gradually replace fossil fuels with clean and sustainable energy. Consequently, the development of clean energy production has become highly demanded.1,2 Electrocatalytic overall water-splitting has emerged as a prominent focus within the scientific community due to its potential as an efficient and sustainable method for generating clean hydrogen energy. However, the widespread implementation of this technology faces significant obstacles. Currently, commercial noble metal oxides like RuO2 and IrO2 exhibit excellent performance in the oxygen evolution reaction (OER), which is a crucial step in the water-splitting process. Nonetheless, the high cost of such materials impedes their widespread use as OER catalysts for efficient electrocatalytic water-splitting.3 Consequently, recent research has placed significant emphasis on exploring earth-abundant metal compounds for the OER. Notably, sulphides based on transition metals like Fe, Co, and Ni exhibit outstanding OER catalytic activities. Given the abundance of these metal elements in the earth's crust, they hold tremendous potential in advancing this field.4,5Previous research has elucidated the polymorphic nature of nickel sulphides, encompassing NiS2, Ni3S4, NiS, Ni9S8, Ni7S6, Ni4S3, and Ni3S2.6–9 These materials exhibit exceptional performance across various fields, underscoring their immense potential. Li et al.10 showcased the synthesis of Ni7S6 hollow spheres and their use in supercapacitors, highlighting their potential in energy storage applications. In contrast, Silva et al.11 synthesised Ni9S8 and NiS microparticles as electrocatalysts for the hydrogen evolution reaction (HER), emphasising the outstanding catalytic performance of nickel sulphides. Furthermore, nickel sulphides have shown promise in the oxygen evolution reaction (OER)12,13 due to surface reconstruction and the formation of oxides/hydroxides, which provide significant stability under highly alkaline conditions. For instance, Zhao et al.14 reported a Ni3S2@Ni9S8 catalyst with high intrinsic conductivity, demonstrating excellent OER performance with a low overpotential of 167 mV at a current density of 10 mA cm−2. Similarly, Rathore et al.15 developed a bi-functional Co0.05Ni8.95S8 electrocatalyst, which requires only −0.151 V to realise a 10 mA cm−2 current density in the HER and 1.557 V to achieve a current density of 30 mA cm−2 in the OER, with a Tafel slope value of 125 and 49.8 mV dec−1, respectively. This catalyst can stably operate for over 40 hours.
The polymorphic nature of nickel sulphide brings many possibilities for applications; however, it also presents significant challenges in synthesising compounds with the desired composition and structure. Controlling the size, morphology, and structure of nickel sulphide nanocrystals has been a formidable challenge. Tailoring these parameters would not only enhance their performance but also broaden their potential applications across various energy conversion and storage technologies. Numerous studies have reported the fabrication of nickel sulphides with diverse morphologies, ranging from spheres and rods to complex structures like flowers, hollow, or cage microparticles.16–19 However, two-dimensional (2D) nickel sulphide nanostructures remain insufficiently explored. 2D materials possess advantages such as better carrier separation, electronic conductivity, larger specific surface area, and abundant surface reactive sites, making them promising catalysts.20,21 Since nickel typically forms non-layered metal chalcogenide structures, the widely used hydrothermal synthesis methods face challenges in precisely controlling the synthesis of 2D nanostructures.22,23 The ion exchange synthesis method has emerged as a potent approach capable of fabricating such nanocrystals with tailored structures and properties.24–28
To the best of our knowledge, the synthesis of 2D nickel sulphide nanoplates through colloidal chemistry, which offer an increased surface area and versatile surface chemistry, remains unexplored in the literature. In this study, we introduce a novel method for the fabrication of 2D NixS nanoplates via a facile and straightforward cation exchange approach, utilising Cu2−xS nanoplates as sacrificial templates. Through optimisation of synthesis conditions, including surface ligands and temperature parameters, we have successfully achieved nickel sulphide nanoplates of a predominant Ni9S8 structure with well-preserved morphology. Importantly, we systematically investigate their potential in the electrocatalytic OER, showcasing their efficacy in energy conversion applications. This work not only contributes to the advancement of nanoplate synthesis methodologies but also underscores the promising prospects of 2D nickel sulphide materials in sustainable energy technologies.
Experimental
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and centrifugation for another 3 min at 5000 rpm. The final products were naturally dried in a glovebox. Hereinafter, the washing and centrifugation process will use the same method without the specific explanation.
:1 and centrifugation for another 3 min at 5000 rpm. The final products were naturally dried in a glovebox. Hereinafter, the washing and centrifugation process will use the same method without the specific explanation.
Characterisation
Transmission electron microscopy (TEM), high-resolution transmission electron microscopy (HRTEM), scanning transmission electron microscopy (STEM) and energy dispersive X-ray spectroscopy (EDS) images were taken using a JEOL F200 Electron Microscope or FEI Titan G2 80-200 high-resolution transmission electron microscope. X-ray diffraction (XRD) patterns were acquired on an X'per PRO Panalytical Empyrean X-ray diffractometer with a Cu Kα radiation (λ = 1.54 Å) source at 40 kV/40 mA. Ultraviolet-visible (UV-vis) absorption spectroscopy was conducted on a Cary 4000 UV-Vis spectrophotometer. X-ray photoelectron spectroscopy (XPS) spectra were acquired using a Kratos Axis Ultra DLD spectrometer. The binding energy scale and high-resolution spectrum of all samples were calibrated and fitted with a Gaussian–Lorentzian (70–30%) line shape using a linear background. The analysis was performed on a TA Instruments SDT Q600 simultaneous differential thermal analysis (DTA) and thermal gravimetric analysis (TGA) analyzer.Electrochemical measurement
The as-prepared sample was annealed under an inert N2 atmosphere at 430 °C for 1.5 h to partially remove the surface ligands. 2.5 mg of the annealed catalyst powder was mixed with 0.5 mg of carbon black (Super P®, Alfa Aesar) before being suspended in 225 μL of absolute ethanol and 25 μL of 5 wt% Nafion® 117 solution (Sigma-Aldrich). This mixture was sonicated for 1 h to yield a homogeneous ink. 5 μL of the catalyst ink was drop cast onto a glassy carbon electrode (GCE, 5 mm diameter) to get a catalyst loading of 0.255 mg cm−2.Electrochemical tests were performed under ambient conditions by employing a three-electrode configuration. The catalyst-modified GCE served as the working electrode, Pt wire as the counter electrode and Ag/AgCl as the reference electrode. The electrolyte was a freshly prepared 1.0 M KOH solution, which was saturated with O2 and maintained under this atmosphere throughout the tests to ensure the O2/H2O equilibrium at 1.23 V vs. RHE. The electrochemical data were collected using a potentiostat (CHI650D, CH Instruments). The electrocatalytic activity was assessed by cyclic voltammetry (CV) at 10 mV s−1. The working electrode was rotated at 3000 rpm to get rid of any gaseous oxygen bubbles evolved on the catalyst surface. To avoid any interference from the Ni redox (prior to the OER), the cathodic scan of the CV data was selected and iR-corrected to compare the OER activity. The potential reported was converted to the RHE scale following the equation: ERHE = EAg/AgCl + 0.199 + 0.0591 × pH (V). The Tafel plot was obtained by plotting the logarithm of current density versus the potential using the cathodic part of the CV data. The electrochemical stability was assessed using the i–t curve at a constant potential of 0.65 V vs. Ag/AgCl (corresponding to 1.65 V vs. RHE, without iR correction).
Results and discussion
Morphology and structure study
Direct synthesis of 2D-shaped NixS nanoparticles poses significant challenges. In this study, we address this issue by employing as-grown 2D hexagonal Cu2−xS nanoplates as sacrificial templates for the preparation of NixS nanoplates via a cation exchange method. Detailed synthesis procedures are provided in the Experimental section. The resulting NixS nanoplates exhibit well-maintained morphology and dimensions, with the cation exchange reaction inducing a slight roughening and wrinkling of the basal plane, as depicted in Fig. 1A. This structural preservation underscores the effectiveness of the cation exchange approach in facilitating the synthesis of 2D NixS nanoplates with desired characteristics. | ||
| Fig. 1 (A) Schematic illustration of the cation exchange process in the formation of NixS nanoplates from Cu2−xS sacrificial templates. TEM images of (B) Cu2−xS and (C) NixS nanoplates. The photo of cuvettes shows the Cu2−xS (left) and NixS (right) nanoplates dispersed in hexane. (D) Histogram of the diameter of Cu2−xS nanoplates. (E and F) Histograms of the length and width of NixS nanoplates. (G) UV-Vis spectra of Cu2−xS nanoplates and NixS nanoplates. | ||
Fig. 1B depicts the morphology and crystal structure of Cu2−xS nanoplates obtained from the thermal decomposition of a single-source precursor of CuSCN. The resulting Cu2−xS exhibits a 2D hexagonal shape with an average lateral size of 105 ± 5 nm, determined from measurements of more than 200 nanosheets (Fig. 1D). Notably, the high-contrast rod-like particles observed in the image represent Cu2−xS nanoplates standing on their edges, with a thickness of approximately 13 nm. Fig. 1C shows the TEM image that showcases the as-obtained NixS nanoplates obtained from the cation exchange reaction. These nanoplates exhibit a highly uniform oval-like morphology, featuring noticeable wrinkles and humps on the surface following the cation exchange process. The formation of wrinkles and humps on the NixS nanoplates after cation exchange can be attributed to the strain induced by the lattice volume change and the large later size of the nanoplates during the exchange process. This strain can lead to the reconstruction of the anion sublattice, which in turn causes morphological changes.30 As the system evolves towards a thermodynamically more stable configuration before reaching its final equilibrium state, these morphological changes can manifest as surface irregularities.31 Consequently, the surface of the resulting NixS nanoplates is less smooth compared to the original Cu2−xS template. NixS nanoplates have a length of ∼97 ± 5 nm and a width of ∼84 ± 5 nm, as indicated in the sizing histograms in Fig. 1E and F. Importantly, compared with the original Cu2−xS templates, the width of the obtained NixS nanoplates is slightly narrow, suggesting that structural modifications may occur during the cation exchange reaction. Similarly, the thickness of the NixS nanoplates, measured on rod-like particles standing on their edges, remains consistent at approximately 13 nm. Moreover, upon dispersing both Cu2−xS and NixS nanoplates in hexane, a noticeable difference in colour from the brownish for Cu2−xS to the absolute black for NixS nanoplate is observed. This colour alteration further corroborates the transformation from Cu2−xS to NixS during the cation exchange reaction. The absorption spectrum reveals distinct features of nanoplates before and after the cation exchange reaction. Prior to the exchange, Cu2−xS nanoplates (illustrated by the orange curve in Fig. 1G) exhibit a broad absorption peak around 468 nm. Compared to bulk Cu2S, a blue shift in the band edge is observed, indicating that the Cu2−xS structure exhibits quantum size effects.32–34 This shift was caused by the smaller particle size, which enhances quantum confinement effects and leads to the observed changes in optical properties. Following the exchange, NixS nanoplates exhibit higher absorption across both the UV and visible ranges. This increased absorption can be attributed to the metallic nature of NixS, which enhances its ability to interact with a broader spectrum of light. The absorption characteristics of NixS nanoplates align well with values reported in the literature,35–37 confirming the successful transformation and the distinct optical properties of the resulting nanoplates.
A further structural analysis of individual Cu2−xS and NixS nanoplates was performed using HRTEM. Fig. 2A–C present HRTEM images and corresponding FFT patterns of Cu2−xS nanoplates, providing detailed insights into the structural characteristics of Cu2−xS nanoplates. The observed interplanar distances of d = 1.9805 and d = 1.8759 are consistent with the (110) and (103) planes of the hexagonal Cu2S structure (PDF 26-1116), respectively. Notably, the presence of (211) and (103) planes aligns with the orthorhombic anilite-type Cu1.75S crystal structure (PDF 33-0489). The observed crystalline phases and lattice plane spacings of the as-prepared Cu2−xS nanoplates are in line with the previously reported coexistence of the Cu2S phase with the Cu1.75S structure.29 The HRTEM images and FFT results depicted in Fig. 2D–F provide detailed insights into the lattice structure of NixS nanoplates. The FFT analysis unveils two distinct patterns. The measured interplanar distance of 0.332 nm corresponds to the (220) plane, along with planes (440) and (116), all of which can be attributed to the Ni9S8 phase (ICCD 96-901-3881). Additionally, the lattice spacings of 0.344 nm and 0.193 nm correspond to the (022) and (242) planes of the Ni3S4 phase (ICCD 96-900-9864), respectively. Furthermore, the HRTEM images of the NixS nanoplates standing on their edges, as depicted in Fig. 2G and H, reveal lattice spacings of 0.288 nm and 0.281 nm, corresponding to the (303) and (008) planes of the Ni9S8 phase (ICCD 96-901-3881), respectively. Analysis of the crystal lattices indicates that the top and bottom basal plane of the NixS nanoplates is (008) and (00![[8 with combining macron]](https://www.rsc.org/images/entities/char_0038_0304.gif) ), as illustrated in Fig. 2I.
), as illustrated in Fig. 2I.
 | ||
| Fig. 2 (A) TEM, (B) HRTEM images of the areas marked by green rectangles in (A) and (C) corresponding fast Fourier transform (FFT) pattern of Cu2−xS nanoplates. (D) TEM and (E) HRTEM images of the areas marked by green rectangles in (D) and (F) corresponding FFT pattern of NixS nanoplates. (G) HRTEM image and (H) corresponding FFT pattern of the side view of NixS nanoplates, and (I) schematic illustration of the (008) plane of NixS nanoplates. (J) HAADF-STEM image of NixS nanoplates and corresponding STEM-EDS elemental mapping of (K) Ni and (L) S. | ||
Both Ni9S8 and Ni3S4 phases are present in NixS nanoplates, which can be explained by the tendency of nickel sulfides to form nonstoichiometric polymorphs. This intrinsic property leads to the coexistence of different phases within the material. The cation exchange process is the substitution of cations within the crystal lattice while preserving the original morphology. Notably, this low-temperature, rapid ion-exchange process can yield metastable phases and compositions that are otherwise inaccessible through conventional synthesis techniques.38,39 In this reaction, Ni cations are adsorbed onto the precursor compound and react with the Cu cations, leading to their replacement. Concurrently, diffusion drives the redistribution of cations throughout the structure, resulting in a homogeneous composition.40,41 This method enables the synthesised NixS nanoplates to retain the morphology of the original Cu2−xS nanoplates while allowing the conversion of their chemical composition.
Following this, the composition of the as-prepared NixS nanoplates was examined using high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) coupled with EDS elemental mapping (depicted in Fig. 2J–L), which was used to further affirm the presence of both Ni and S within these nanoplates. The areas with higher contrast in the elemental mapping are attributed to the overlapping of nanoplates, consistent with the HAADF image, suggesting a homogeneous distribution of Ni and S elements across all nanoplates. The overlapping signals of both Ni and S elements in Fig. S1† confirm the formation of NixS nanoplates. As the TEM grids used for electron microscope measurements were composed of copper, to mitigate the error arising from the detection of Cu, the EDS spectra from both sample areas and the background were extracted to compare the Cu content (Fig. S2A†). As illustrated in Fig. S2B,† the presence of the Cu signal was detected in both the sample area and the background, as expected. The comparable intensities indicate only trace amounts of Cu residue present in the obtained NixS nanoplates, which is anticipated to stabilise the NixS structure following the cation exchange reaction.42 To validate this assumption, an inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis was performed, and the findings are detailed in Table S1.† The analysis reveals the presence of only a tiny quantity of copper residue within the sample, which is approximately 0.227 at%. Considering the extremely low concentration of Cu atoms present within the NixS nanoplates after the cation exchange process, their impact on phase formation during the cation exchange reaction is anticipated to be insignificant.
The prepared NixS nanoplates were further characterised via XRD and XPS testing. Fig. 3A illustrates the XRD pattern of Cu2−xS templates (depicted in orange), alongside exchanged NixS nanoplates (shown in blue). The significant difference between the two patterns confirms the complete cation exchange from Cu2−xS to NixS. In the diffraction pattern of Cu2−xS nanoplates, the strong peak at 28° indicates the dominant Cu2S structure, with additional peaks matching the pattern of bulk anilite-type Cu1.75S, being consistent with previous TEM results. Furthermore, the diffraction patterns of NixS reveal a combination of Ni9S8 and Ni3S4 phases. Peaks labelled with solid blue rhombuses correspond to the diffraction patterns of bulk Ni9S8 (ICCD 96-901-3881), as displayed at the bottom of Fig. 3A in green. Conversely, only a few peaks with relatively low intensity can be indexed to the Ni3S4 phase (ICCD 96-900-9864), as marked by hollow blue rhombuses. This suggests that a Ni9S8-dominated Ni9S8/Ni3S4 mixed phase is formed in the cation-exchanged NixS nanoplates. Notably, the significantly intense diffraction peak at 31.3° suggests a large number of crystal lattice repetition along the (008) plane of Ni9S8, which is consistent with the morphology structure configuration of NixS nanocrystals revealed by TEM measurements (Fig. 2G–I). XRD results indicate that the thickness of the Ni9S8 layer corresponding to the (008) crystal plane is 14.885 nm. This value was calculated using the Scherrer–Debye formula,43
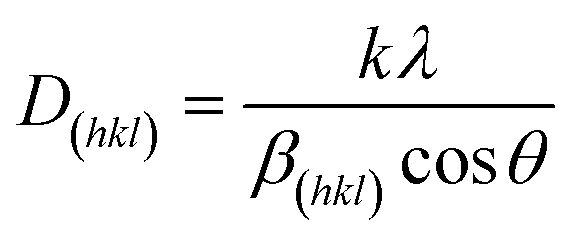
 | ||
| Fig. 3 (A) XRD patterns of the initial Cu2−xS sacrificial templates and NixS nanoplates obtained from the cation exchange reaction. The blue and green lines on the top of (A) are diffraction patterns of bulk Cu2S (PDF 26-1116) and anilite-type Cu1.75S (PDF 33-0489), respectively; the green and orange lines on the bottom of (A) correspond to bulk Ni9S8 (ICCD 96-901-3881) and Ni3S4 (ICCD 96-900-9864), respectively. (B–D) XPS spectra of NixS nanoplates and (B) Ni 2p spectrum. The blue, orange and green curves indicate the Ni3+ and Ni2+ species as well as the satellite peaks, respectively; (C) S 2p spectrum, S 2p3/2, S 2p3/2 and satellite peaks are depicted by blue, orange and green curves, respectively; and (D) Cu 2p spectrum, the Cu 2p3/2 and Cu 2p1/2 are depicted by blue and pink curves, respectively. | ||
The XPS test was conducted to gain insight into the chemical state and bonding of the final NixS product. As depicted in Fig. S3,† the XPS survey spectra reveal the presence of Ni, S, C, and O, with no apparent peak of Cu. The peaks corresponding to C and O may be attributed to oxidation reactions and ligand residues, which are very common occurrences in wet-chemical syntheses.46 The high-resolution XPS spectrum in Fig. 3B shows the spin–orbit doublets of Ni 2p. The Ni 2p3/2 peak at 852.79 eV and Ni 2p1/2 peak at 869.89 eV belong to Ni3+ species, while the Ni 2p3/2 peak at 855.56 eV and Ni 2p1/2 peak at 873.54 eV are indexed to Ni2+ species. The strong shake-up satellite peaks (denoted as Sat.) at 859.74 eV from Ni 2p3/2 and 878.69 eV further confirm the Ni2+-dominated co-existence of divalent and trivalent nickel sulphides, which is consistent with the literature results.47 The core-level S 2p deconvolution peaks are shown in Fig. 3C, and the binding energy values at 161.85 eV and 164.01 eV can be assigned to S 2p3/2 and S 2p1/2, respectively, while the satellite peak is located at 165.96 eV.37 The low signal-to-noise ratio combined with the relatively low intensity of peaks in Cu 2p spectra (Fig. 3D) indicates the trace amount of Cu existing in this sample which is in accordance with both EDS and ICP-AES results. It is expected that a trace amount of Cu in these samples may be beneficial in the stabilisation of the NixS structure. All XPS results match these from previous reports of Ni9S8,11,15 which indicates that Ni9S8 is the dominant phase of the as-prepared NixS nanoplates.
Synthesis optimisation
To comprehensively understand the intricate formation process of these NixS nanosheets, a detailed investigation involving a partial exchange was undertaken to yield Cu2−xS/NixS heterostructures by adjusting the quantity of reactants. As depicted in Fig. 4A, the distinct presence of NixS and Cu2−xS domains within a single particle provides visual confirmation of their distinct crystal structures, as supported by their respective FFT patterns. This observation elucidates the evolutionary nature of their formation. The observed uneven exchange process suggests that the disparate growth rates and crystal structures of Ni9S8 and Ni3S4 may play a pivotal role. Consolidating all the aforementioned findings, it can be conclusively affirmed that NixS nanoplates emerge from sacrificial Cu2−xS templates via cation exchange reactions, as shown by the process depicted in Fig. 4B. | ||
| Fig. 4 (A) HRTEM image and FFT patterns showing NixS and Cu2−xS domains within a partially exchanged single Cu2−xS/NixS nanoplate. (B) Schematic illustration depicting the cation exchange process of NixS nanoplates. (C) TEM and HRTEM images of products obtained from Cu2−xS cation exchange reactions with different surfactants and at different temperatures. Structural diagrams of the respective surfactants are shown in the bottom right corner. Blue, red, and orange atoms indicate N, O and P, respectively. | ||
The reaction conditions were carefully examined to optimise the synthesis process. It is well established that surface ligands play a crucial role in morphological and size control in wet-chemical methods. In our reaction setup, the Ni precursor was combined with OA and OAm surfactants before being injected into a dispersion of Cu2−xS nanoplates in OAm. A series of cation exchange reactions were then conducted using either the Ni–OA–OAm complex or OAm as the sole surfactant, alongside the manipulation of other variables, to elucidate the influence of surface ligands on the formation of NixS nanoplates.
With the particular attention given to the presence of both OA and OLA, and the use of TOP to extract copper ions, at a reaction temperature of 170 °C, as depicted in Fig. 4C (left side), nickel sulfide nanosheets with a smoother surface and more uniform morphology, alongside enhanced crystallinity, were achieved in the presence of both OA and OLA. Conversely, in the absence of OA, particles displaying numerous wrinkles and humps were observed. The deprotonated OA, owing to its high electro-donating capacity, exhibits the ability to coordinate with the Ni precursor, thereby forming a stable Ni–OA–OAm complex. This complex, characterised by a strong bond between the Ni precursor and OA, is believed to mitigate their reactivity and modulate the growth kinetics during the reaction process. Consequently, this controlled modulation results in a more uniform distribution of Ni ions upon hot injection, thereby slowing down the incorporation of the Ni precursor into the Cu2−xS lattice. Such meticulous control facilitates the fabrication of NixS nanoplates with well-retained morphology and enhanced crystallinity.48 In addition, the role of TOP was also investigated. It was primarily employed as a weak Lewis base to reduce the lattice binding energy of the sacrificial templates for constitutional transformation.49 In the absence of TOP, cation exchange was completely hindered, with all particles retaining the Cu2−xS structure even after a reaction duration of 12 hours, underscoring the pivotal role of TOP in facilitating the extraction of Cu+ ions to enable the cation exchange reaction.
Furthermore, given the substantial evidence highlighting the criticality of temperature control in nickel sulphide growth and phase control,50 a comprehensive exploration of the impact of temperature on nickel sulphide formation was undertaken in this study. In addition to the optimised temperature of 170 °C, the cation exchange reactions were conducted at temperatures of both 100 °C and 220 °C. The nanoparticles synthesised at these varying temperatures exhibited notable differences in morphology and structure compared to those obtained at 170 °C, as evidenced by the presence of wrinkles, corrugations, and even cracks, as depicted in the right panel of Fig. 4C. The observed variations suggest that deviations from the optimal temperature can lead to an uncontrolled exchange process, resulting in the formation of undesired polyphase structures.
Electrocatalytic OER measurement
Before conducting electrochemical measurements, all prepared samples underwent calcination in a nitrogen atmosphere at 430 °C. This process aimed to partially remove surface ligands and thereby enhance conductivity, a common practice for nanoparticles synthesised via wet-chemical methods prior to electrocatalysis.51–53 OLA, a frequently used ligand, can be effectively removed through annealing, typically without altering the morphology of the prepared nanocrystals, although their dispersibility in organic solvents may decrease.54 As shown in Fig. S4A,† the morphology of these nanoplates remained unchanged after thermal treatment. The HRTEM images in Fig. S4B and C† clearly demonstrate a d-spacing of 0.241 nm, corresponding to the (208) plane of the Ni9S8 phase (ICCD 96-901-3881), consistent with the structure observed before calcination. Thus, the NixS nanoplates retained their morphology and crystallinity post-calcination. Additionally, optical UV-Vis spectroscopy was conducted on the annealed NixS nanoplates and compared with those subjected to electrocatalysis (Fig. S5†). The absorption spectra of the annealed and catalysed NixS nanoplates closely resemble those prior to annealing, indicating the preservation of their optical properties. TGA was conducted on the NixS nanoplates under a nitrogen atmosphere over a temperature range of 0–500 °C, with the results shown in Fig. S6.† The initial weight loss of approximately 1% is likely due to the evaporation of residual organic solvents remaining from the purification. A significant weight loss starts at 183.6 °C, reaching around 7%, which can be attributed to the removal of surface ligands. Beyond this point, the weight loss rate slows down and continues gradually up to 500 °C. This observation is corroborated by the trends seen in the differential thermal analysis (DTA) curve. Additionally, a small exothermic peak observed in the DTA curve can be attributed to changes in the crystal structure with increasing temperature.55 These TGA and DTA results are consistent with reference data,56,57 confirming that thermal annealing effectively removes surface ligands from the NixS nanoplates.The capability of nickel sulphides in the OER has been extensively reported with high electrical conductivity and low price as promising electrocatalysts in the substitution of commercial noble-metal catalysts. Therefore, as a proof-of-concept study, the as-prepared NixS nanoplates were evaluated for their efficacy as an electrocatalyst toward the OER under alkaline conditions. Fig. 5 shows the electrochemical OER performance of various NixS catalysts prepared at different temperatures in a 1.0 M KOH electrolyte. Interestingly, a redox peak was observed prior to the onset of the OER for all samples (Fig. 5A), which could be assigned to the redox of the Ni cation. Of note, the position and intensity of such peaks were found to correlate with the OER activity. The more negative the peak position and the stronger the peak intensity, the better the OER activity. It was found that the NixS sample synthesised at 170 °C gave the best OER activity. This is more obviously the case when one compares the overpotential at a given current density like 10 mA cm−2 (a metric associated with solar fuel production)58 or 100 mA cm−2 (a metric more relevant for practical applications);59 the smaller the overpotential, the better the OER activity. To avoid the possible interference of the oxidation peak on the assessment of the OER activity, the cathodic branch of the CV data was selected. As depicted in Fig. 5B, NixS synthesised at 170 °C only required an overpotential of 329 mV to reach 10 mA cm−2, which is lower than that synthesised at 100 °C (395 mV) and 220 °C (374 mV) and also superior to a noble metal benchmark tested under identical conditions (i.e., RuO2 with an overpotential of 368 mV).54 A similar activity trend was observed for the overpotential to reach 100 mA cm−2, following the order of 411 mV (for 170 °C sample) < 486 mV (for 220 °C sample) < 503 mV (for 100 °C sample). Tafel analysis can also be utilised to evaluate the kinetics information of the electrocatalysts.60–62 As illustrated in Fig. 5C, the Tafel plot revealed that NixS obtained at 170 °C displayed a Tafel slope of 52 mV dec−1, smaller than that of the counterparts obtained at 220 °C (62 mV dec−1) and 100 °C (66 mV dec−1). This Tafel slope is also much lower than that of the RuO2 standard (with the Tafel slope of 200.1 mV dec−1).47 These results suggest that the NixS sample synthesised at 170 °C shows the optimum OER activity, which could be related to its unique and advantageous morphological features and improved crystallinity. The electrochemical stability of this sample was further analysed under constant potential conditions (Fig. 5D). In general, the sample was able to maintain a current density of around 50 mA cm−2 during a 10000 second test. This further suggests the promise of NixS as an efficient OER catalyst. A close look at the stability data revealed that the current density first experienced an increase to some extent until reaching a more stable state. The initial increase of current density could be associated with the surface reconstruction of the nickel sulphide.63
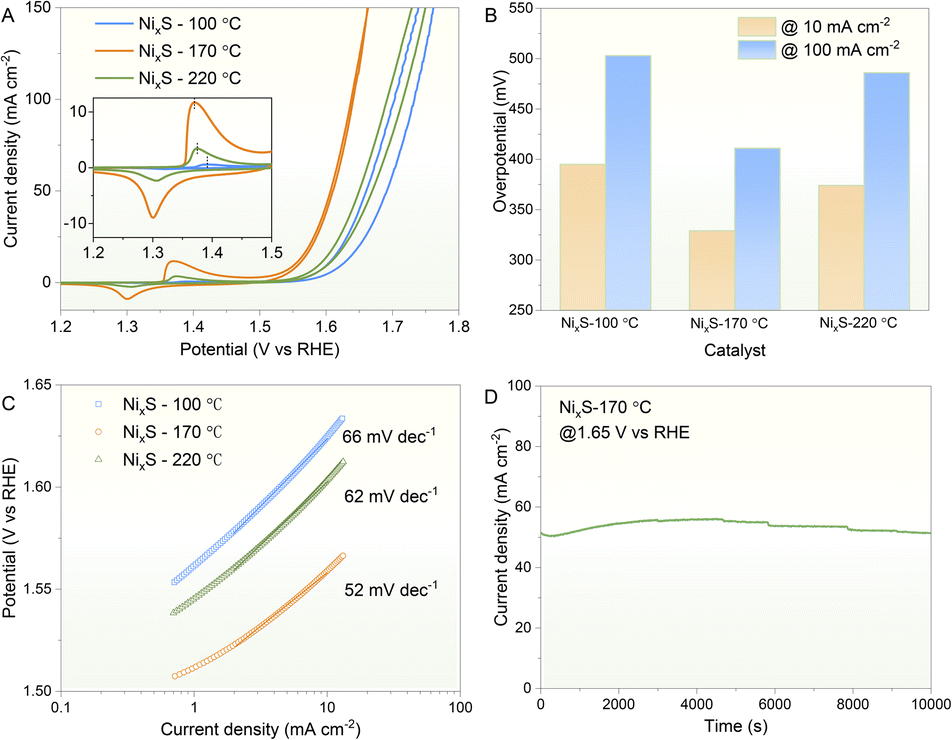 | ||
| Fig. 5 (A) CV curves of various NixS samples synthesised at different temperatures. (B) Comparison of the overpotentials needed for reaching current densities of 10 and 100 mA cm−2 (C) Tafel plot. (D) Electrochemical stability assessed using the i–t curve at a constant potential of 1.65 V vs. RHE. | ||
Conclusions
In conclusion, we successfully synthesised NixS with distinct 2D nanoplate morphology via a cation exchange reaction using as-prepared Cu2−xS as sacrificial templates. The constitution and structure of these NixS nanoplates were thoroughly confirmed through a series of advanced characterisation methods, including TEM, XRD, XPS and ICP analyses. Our investigation into the reaction conditions revealed that OA, TOP, and an appropriate reaction temperature are critical factors for the formation of NixS nanoplates with well-preserved 2D morphology and high-purity crystallinity. These factors play a significant role in modifying the reaction kinetics, thereby ensuring a controlled and efficient cation exchange process. The electrochemical performance demonstrated a low overpotential of 329 mV at a current density of 10 mA cm−2, along with a Tafel slope value of 52 mV dec−1. These results indicate that the NixS nanoplates exhibit promising catalytic activity and stability, making them suitable candidates for efficient OER catalysis. The successful synthesis and characterisation of NixS nanoplates highlight the importance of precise control over reaction conditions to achieve desired structural and morphological control, and their promising electrochemical performance indicates their potential for practical applications in energy conversion and storage technologies.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
J. Chen: synthesis of reported materials, characterisation, writing – original draft; X. Xu: conducted OER experiments, writing and editing of the draft; R. Mao: synthesis of reported materials, experimental data analysis, writing – review & editing; C. Wang, H.-Y. Hsu, Z. Yin, M. A. Buntine, A. Suvorova, and M. Saunders: experimental data analysis, writing – review & editing; Zongping Shao and Guohua Jia: conceived the idea, funding acquisition, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Australian Research Council (ARC) Future Fellowship Scheme (FT210100509), ARC Discovery Project (DP220101959), the Hebrew University of Jerusalem – Zelman Cowen Academic Initiatives (ZCAI) Joint Projects 2021 and the Innovation and Technology Commission (Grant no. MHP/104/21). The authors acknowledge the facilities, and the scientific and technical assistance of Microscopy Australia at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments.Notes and references
- J. Su, R. Ge, Y. Dong, F. Hao and L. Chen, J. Mater. Chem. A, 2018, 6, 14025–14042 Search PubMed.
- C. Y. Lin, L. Zhang, Z. Zhao and Z. Xia, Adv. Mater., 2017, 29, 1606635 Search PubMed.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 Search PubMed.
- K. Zhu, X. Zhu and W. Yang, Angew. Chem., Int. Ed., 2019, 58, 1252–1265 CrossRef.
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2021, 80, 105514 CrossRef.
- X. Long, G. Li, Z. Wang, H. Zhu, T. Zhang, S. Xiao, W. Guo and S. Yang, J. Am. Chem. Soc., 2015, 137, 11900–11903 CrossRef PubMed.
- S. Anantharaj, S. Kundu and S. Noda, J. Mater. Chem. A, 2020, 8, 4174–4192 RSC.
- B. Tang, Y. Lv, J. Du, Y. Dai, S. Pan, Y. Xie and J. Zou, ACS Sustainable Chem. Eng., 2019, 7, 11101–11109 CrossRef.
- A. Ghezelbash, M. B. Sigman and B. A. Korgel, Nano Lett., 2004, 4, 537–542 CrossRef.
- Z. Li, J. Han, L. Fan and R. Guo, CrystEngComm, 2015, 17, 1952–1958 RSC.
- M. G. S. da Silva, C. M. Leite, M. A. L. Cordeiro, V. R. Mastelaro and E. R. Leite, ACS Appl. Energy Mater., 2020, 3, 9498–9503 CrossRef.
- C. Z. Yuan, Z. T. Sun, Y. F. Jiang, Z. K. Yang, N. Jiang, Z. W. Zhao, U. Y. Qazi, W. H. Zhang and A. W. Xu, Small, 2017, 13, 1604161 CrossRef PubMed.
- O. Mabayoje, A. Shoola, B. R. Wygant and C. B. Mullins, ACS Energy Lett., 2016, 1, 195–201 CrossRef.
- G. Zhao, Y. Xing, Y. Liu, X. Wang, B. Zhang, L. Mu, W. Liao and X. Xu, Mater. Today Chem., 2023, 34, 101758 CrossRef.
- D. Rathore, S. Ghosh, J. Chowdhury and S. Pande, ACS Appl. Nano Mater., 2022, 5, 11823–11838 CrossRef.
- J. Zhang, Y. Li, X. Liang, Q. Liu, Q. Chen and M. Chen, Small, 2022, 18, e2106074 CrossRef.
- S. Kim, S. H. Lee, M. Cho and Y. Lee, Biosens. Bioelectron., 2016, 85, 587–595 CrossRef PubMed.
- X. Y. Yu, L. Yu, H. B. Wu and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 5331–5335 CrossRef.
- H. Q. Fu, L. Zhang, C. W. Wang, L. R. Zheng, P. F. Liu and H. G. Yang, ACS Energy Lett., 2018, 3, 2021–2029 CrossRef.
- S. A. Thomas, J. Cherusseri and D. N. Rajendran, Energy Technol., 2024, 12, 2301641 CrossRef.
- Y. Luo, L. Shi, H. He, G. Yang, G. Cong, C. Zhu and J. Xu, Carbon, 2021, 182, 194–202 CrossRef.
- Y. P. Xie, Y. Zheng, Y. Yang, R. Jiang, G. Wang, Y. Zhang, E. Zhang, L. Zhao and C.-Y. Duan, J. Colloid Interface Sci., 2018, 514, 634–641 CrossRef.
- N. Uddin, H. Zhang, Y. Du, G. Jia, S. Wang and Z. Yin, Adv. Mater., 2020, 32, 1905739 CrossRef.
- C. Zhang, J. Chen, L. Turyanska, J. Wang, W. Wang, L. Wang, L. Kong, K. Wu, J. Yao, H. Yao, Z. Yang, W. Li, Y. Bekenstein, Y. Wang, G. Jia and X. Yang, Adv. Funct. Mater., 2023, 33, 2211466 CrossRef.
- C. Zhang, J. Chen, S. Wang, L. Kong, S. W. Lewis, X. Yang, A. L. Rogach and G. Jia, Adv. Mater., 2020, 32, e2002736 CrossRef PubMed.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef PubMed.
- J. Chen, D. Hao, W. Chen, Y. Liu, Z. Yin, H. Y. Hsu, B. J. Ni, A. Wang, S. W. Lewis and G. Jia, Chin. J. Chem., 2023, 41, 3050–3062 CrossRef.
- D. H. Son, S. M. Hughes, Y. Yin and A. Paul Alivisatos, Science, 2004, 306, 1009–1012 CrossRef.
- A. Wang, W. Wang, J. Chen, R. Mao, Y. Pang, Y. Li, W. Chen, D. Chen, D. Hao, B. J. Ni, M. Saunders and G. Jia, J. Phys. Chem. Lett., 2020, 11, 4990–4997 CrossRef PubMed.
- Z. Li, M. Saruyama, T. Asaka, Y. Tatetsu and T. Teranishi, Science, 2021, 373, 332–337 CrossRef PubMed.
- O. Ramirez, P. Ramasamy, Y. Choi and J. Lee, Chem. Mater., 2018, 31, 268–276 CrossRef.
- O. Elimelech, J. Liu, A. M. Plonka, A. I. Frenkel and U. Banin, Angew. Chem., Int. Ed., 2017, 56, 10335–10340 CrossRef PubMed.
- Y. Zhao, H. Pan, Y. Lou, X. Qiu, J. Zhu and C. Burda, J. Am. Chem. Soc., 2009, 131, 4253–4261 CrossRef PubMed.
- N. Li, X. Zhang, S. Chen, W. Yang, H. Kang and W. Tan, CrystEngComm, 2011, 13, 6549–6554 RSC.
- X. Liu and J. Li, Inorg. Chem., 2020, 59, 17184–17190 CrossRef.
- G. S. Varalakshmi, C. Pawar, R. Selvam, W. G. Pearl, V. Manikantan, A. S. Pillai, A. Alexander, N. R. Prasad, I. V. Enoch and P. Dhanaraj, Int. J. Pharm., 2023, 643, 123282 CrossRef.
- A. Li, Z. Peng and X. Fu, J. Mater. Sci.: Mater. Electron., 2021, 32, 21643–21657 CrossRef CAS.
- J. Rivest and P. Jain, Chem. Soc. Rev., 2013, 42, 89–96 RSC.
- L. Liu, B. Bai, X. Yang, Z. Du and G. Jia, Chem. Rev., 2023, 123, 3625–3692 CrossRef CAS PubMed.
- H. Hou, C. Bowen, D. Yang and W. Yang, Chem, 2024, 10, 800–831 CAS.
- B. Bai, C. Zhang, Y. Dou, L. Kong, L. Wang, S. Wang, J. Li, Y. Zhou, L. Liu, B. Liu, X. Zhang, I. Hadar, Y. Bekenstein, A. Wang, Z. Yin, L. Turyanska, J. Feldmann, X. Yang and G. Jia, Chem. Soc. Rev., 2023, 52, 318–360 RSC.
- A. E. Powell, J. M. Hodges and R. E. Schaak, J. Am. Chem. Soc., 2016, 138, 471–474 CrossRef CAS PubMed.
- A. Patterson, Phys. Rev., 1939, 56, 978 CrossRef CAS.
- U. De Silva, J. See, W. P. Liyanage, J. Masud, J. Wu, W. Yang, W.-T. Chen, D. Prendergast and M. Nath, Energy Fuels, 2021, 35, 4387–4403 CrossRef CAS.
- A. Molla, M. Sahu and S. Hussain, Sci. Rep., 2016, 6, 26034 CrossRef CAS.
- N. Jiang, Q. Tang, M. Sheng, B. You, D.-e. Jiang and Y. Sun, Catal. Sci. Technol., 2016, 6, 1077–1084 RSC.
- Q. Gao, W. Luo, X. Ma, Z. Ma, S. Li, F. Gou, W. Shen, Y. Jiang, R. He and M. Li, Appl. Catal., B, 2022, 310, 121356 CrossRef CAS.
- S. Mourdikoudis, M. Menelaou, N. Fiuza-Maneiro, G. Zheng, S. Wei, J. Perez-Juste, L. Polavarapu and Z. Sofer, Nanoscale Horiz., 2022, 7, 941–1015 RSC.
- B. J. Beberwyck and A. P. Alivisatos, J. Am. Chem. Soc., 2012, 134, 19977–19980 CrossRef CAS.
- G. Barim, S. R. Smock, P. D. Antunez, D. Glaser and R. L. Brutchey, Nanoscale, 2018, 10, 16298–16306 RSC.
- M. Ledendecker, J. S. Mondschein, O. Kasian, S. Geiger, D. Göhl, M. Schalenbach, A. Zeradjanin, S. Cherevko, R. E. Schaak and K. Mayrhofer, Angew. Chem., Int. Ed., 2017, 56, 9767–9771 CrossRef CAS PubMed.
- Q. Lu, G. S. Hutchings, W. Yu, Y. Zhou, R. V. Forest, R. Tao, J. Rosen, B. T. Yonemoto, Z. Cao and H. Zheng, Nat. Commun., 2015, 6, 6567 CrossRef CAS PubMed.
- T. Zhang, X. Liu, X. Cui, M. Chen, S. Liu and B. Geng, Adv. Mater. Interfaces, 2018, 5, 1800359 CrossRef.
- S. Javaid, X. Xu, W. Chen, J. Chen, H.-Y. Hsu, S. Wang, X. Yang, Y. Li, Z. Shao, F. Jones and G. Jia, Nano Energy, 2021, 89, 106463 CrossRef CAS.
- O. O. Balayeva, A. A. Azizov, M. B. Muradov, A. M. Maharramov, G. M. Eyvazova, R. J. Gasimov and Z. X. Dadashov, Mater. Sci. Semicond. Process., 2017, 64, 130–136 CrossRef CAS.
- M. Seyhan, W. Kucharczyk, U. E. Yarar, K. Rickard, D. Rende, N. Baysal, S. Bucak and R. Ozisik, Nanotechnol., Sci. Appl., 2017, 69–77 CrossRef.
- S. Nagaveena and C. Mahadevan, J. Alloys Compd., 2014, 582, 447–456 CrossRef.
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612–13614 CrossRef PubMed.
- H. Sun, X. Xu, H. Kim, W. Jung, W. Zhou and Z. Shao, Energy Environ. Mater., 2023, 6, e12441 CrossRef.
- A. Grimaud, K. May, C. Carlton, Y. Lee, M. Risch, W. Hong, J. Zhou and S. Yang, Nat. Commun., 2013, 4, 2439 CrossRef PubMed.
- B. Zhao, L. Zhang, D. Zhen, S. Yoo, Y. Ding, D. Chen, Y. Chen, Q. Zhang, B. Doyle, X. Xiong and M. Liu, Nat. Commun., 2017, 8, 14586 CrossRef CAS PubMed.
- F. Abdelghafar, X. Xu, D. Guan, Z. Lin, Z. Hu, M. Ni, H. Huang, T. Bhatelia, S. Jiang and Z. Shao, ACS Mater. Lett., 2024, 6, 2985–2994 CrossRef CAS.
- M. Lee, H.-S. Oh, M. K. Cho, J.-P. Ahn, Y. J. Hwang and B. K. Min, Appl. Catal., B, 2018, 233, 130–135 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta05191b |
| This journal is © The Royal Society of Chemistry 2024 |