
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fast Mg-ion insertion kinetics in V2Se9†
Matthew A.
Wright
 abc,
Jungwoo
Lim
ab,
Raul A.
Pacheco Muino
a,
Anna E.
Krowitz
a,
Cara J.
Hawkins
a,
Mounib
Bahri
dg,
Luke M.
Daniels
a,
Ruiyong
Chen
a,
Luciana
Gomes Chagas
e,
James
Cookson
e,
Paul
Collier
e,
Alan V.
Chadwick
f,
Nigel D.
Browning
dg,
John B.
Claridge
a,
Laurence J.
Hardwick
*ab and
Matthew J.
Rosseinsky
*a
abc,
Jungwoo
Lim
ab,
Raul A.
Pacheco Muino
a,
Anna E.
Krowitz
a,
Cara J.
Hawkins
a,
Mounib
Bahri
dg,
Luke M.
Daniels
a,
Ruiyong
Chen
a,
Luciana
Gomes Chagas
e,
James
Cookson
e,
Paul
Collier
e,
Alan V.
Chadwick
f,
Nigel D.
Browning
dg,
John B.
Claridge
a,
Laurence J.
Hardwick
*ab and
Matthew J.
Rosseinsky
*a
aDepartment of Chemistry, University of Liverpool, Liverpool, L69 7ZD, UK. E-mail: hardwick@liverpool.ac.uk; M.J.Rosseinsky@liverpool.ac.uk
bStephenson Institute for Renewable Energy, Liverpool L69 7ZF, UK
cMaterials Research Laboratory, Materials Department, University of California, Santa Barbara, California CA 93106, USA
dAlbert Crewe Centre, University of Liverpool, Research Technology Building, Elisabeth Street, Pembroke Place, Liverpool, L69 3GE, UK
eJohnson Matthey Technology Centre, Sonning Common, Reading, RG4 9NH, UK
fSchool of Physical Sciences, University of Kent, Canterbury, CT2 7NH, UK
gSchool of Engineering, Department of Mechanical, Materials and Aerospace Engineering, University of Liverpool, Liverpool, L69 7ZD, UK
First published on 24th October 2024
Abstract
V2Se9 displays facile electrochemical insertion of up to 1.6 Mg2+ per unit formula with fast diffusion (coefficients of 10−10–10−12 cm2 s−1) surpassing best-in-class materials like Mo6S8. Detailed structural characterization of synchrotron X-ray diffraction data with ab initio Maximum Entropy Method analysis reveals Mg2+ insertion into octahedral sites within the large vdW space between [V4Se18]∞ chains. Fast rate performance is attributed to low structural perturbation and low diffusion barriers, calculated by bond valence pathway analysis, resulting from the low charge-per-size of anionic selenium. X-ray photoelectron spectroscopy and X-ray absorption spectroscopy reveal that reversible insertion of Mg2+ is facilitated by V5+/V3+ redox. V2Se9 demonstrates that selenides, despite their larger molecular weight, offer potential as fast-rate positive electrode materials for magnesium batteries over well-explored oxides and sulfides.
Introduction
Performance limitations, safety concerns, and the availability of the necessary raw materials for rechargeable Li-ion batteries have created a demand for innovative energy storage technologies beyond lithium.1,2 The high natural abundance, low cost and high volumetric capacity of magnesium (3833 mA h cm−3) compared to that of lithium (2046 mA h cm−3) and sodium (1125 mA h cm−3) make rechargeable Mg batteries an appealing alternative.3–6The most significant challenge preventing the commercialization of magnesium batteries is a lack of suitable positive electrode materials capable of suitable kinetics for Mg2+ insertion. Despite the comparable ionic radii of Mg2+ and Li+ (0.57 vs. 0.59 Å),7 the high charge density of Mg2+ results in strong electrostatic interaction between the anionic sub-structure of the host electrode and the inserting ion.8,9 As a result, Mg-storage is hindered both thermodynamically, with limited reaction reversibility; and kinetically, with sluggish Mg2+ insertion into the host structure.10
Increasing the polarizability of the anion framework has resulted in improved kinetic performance of insertion electrodes: layered VSe2 and TiSe2 demonstrate faster Mg2+ migration than the isostructural sulfide and oxide materials, albeit at a lower working potential.11–17 Similarly, capacity retention has been improved in well-studied electrode materials like the Chevrel phase Mo6S8−xSex (0 < x ≤ 2)18,19 and CuS1−xSex (0 < x ≤ 0.25)20–22 by partial substitution of S for Se. The enhanced kinetics of Mg2+ storage following the trend of Se > S > O can be attributed to both weakening electrostatic interactions as well as increasing anionic radii resulting in larger channels for Mg2+ diffusion.23
Recently, one-dimensional chain materials like VS4,24–27 V2PS10,28 and TiS3 (ref. 29 and 30) have attracted interest as positive electrode materials for Mg batteries. Their structures are comprised of one-dimensional chains formed from trans-facially connected [MS8] polyhedra bound loosely by weak van der Waals (vdW) forces with large inter-chain spacing (5.38 Å in VS4); through which Mg2+ can diffuse. These materials are noted for the complex insertion mechanisms, emanating from anionic redox on the disulfide (S2)2− site.31 The highly active anion chemistry of these materials provides motivation to explore S/Se substitution.
The structure of V2Se9, like VS4 and V2PS10, consists of infinite one-dimensional chains of [V4Se18]∞ formed by trans-facial stacking of distorted rectangular anti-prismatic [VSe8] polyhedra. These chains extend along the [302] direction and are weakly bound by vdW forces. The large anionic radius of Se results in large inter-chain spacings of 6.366–6.610 Å for V2Se9, significantly longer than those observed in VS4 and V2PS10. The oxidation states in V2Se9 can be considered by the asymmetric unit V25+(Se2)2–4Se2−, where all Se are coordinated to a V5+ metal center and exist in either a (Se2)2− or Se2− oxidation state (a detailed structural description is provided in the ESI, Fig. S1 and Table S1†).
Recently, V2Se9 was investigated as an electrode material in monovalent Li+ and Na+ batteries and was shown to operate by a combination of insertion and conversion mechanisms.32–34 Initially, up to nine Li+ or Na+ are inserted between the [V4Se18]∞ chains by an intercalation-type process. At lower potentials, conversion reactions result in the formation of metallic V and either Li2Se or Na2Se.
Herein, Mg2+ insertion into V2Se9 with fast kinetics is reported. Through detailed structural analysis of synchrotron X-ray diffraction (SXRD) data, combined with maximum entropy method analysis and bond valence diffusion pathway analysis, Mg2+ sites are located within the structure and highlight the inter-connected three-dimensional diffusion pathways and low structural perturbation as the origin of fast Mg2+ insertion. Ex situ X-ray photoelectron (XPS) and X-ray absorption spectroscopic (XAS) characterization and Raman spectroscopy were used to elucidate an insertion mechanism resulting from a reversible V5+/V3+ redox couple, with no evidence of parasitic conversion reactions which were seen for Mg2+ insertion into VS4.
Experimental section
V2Se9 synthesis
Powdered vanadium (99.5% Alfa Aesar) and selenium (99.5% Sigma Aldrich) were combined in stoichiometric quantities in an agate pestle and mortar in an inert Ar atmosphere in a glovebox (MBraun LabMaster 130; H2O and O2 levels < 0.1 ppm) before being pressed into pellets (13 mm diameter at 0.5 tonnes) and sealed in fused silica ampules under a vacuum of 1 × 10−5 mbar before heating to 623.15 K in a Carbolite RHF 1600 furnace for 21 days using heating and cooling rates of 5 K min−1 (Fig. S2 and Table S2†). After synthesis, the pellets were collected and homogenized by further grinding. Elemental analysis by ICP-OES yielded a composition of V2.00(2)Se9.4(3) (Fig. S3 and Table S3†). TEM-EDX yielded a Se/V ratio of 4.31(18) (Fig. S4 and S5 and Table S4†). Further details for ICP-OES, TEM and TEM-EDX measurements is provided in the ESI.† Se and VSe2 were observed as minor impurity phases by XRD in the as-made V2Se9 and the Mg-intercalated powders. The relative weight ratios of these impurity phases did not change between the pristine and intercalated materials.Electrochemical cell assembly and testing
Stainless steel coin cells (CR2032, Pi-KEM) assembled under an inert Ar atmosphere inside a glovebox (MBraun LabMaster Pro-Eco) with O2 and H2O levels < 0.1 ppm. Positive electrodes consisted of a slurry of V2Se9 powder, sieved through a 20 μm mesh (Impact Test Systems), conductive carbon (Pi-KEM, dried), and binder (PVDF, dried) in proportions of 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:10 w/w combined with in N-methyl-2-pyrrolidone. The slurry was cast onto carbon-coated aluminium foil and cut into disks (16 mm diameter) and dried under vacuum overnight. Active material loadings were typically between 0.25 and 0.35 mg cm−2, with an electrode thickness of 100 μm. Negative electrodes consisted of polished Mg foil discs with a diameter of 18 mm. Electrodes were separated by Whatman GF/D borosilicate glass fibre membranes soaked with 500–750 μL of an electrolyte consisting of a 0.5 M solution of magnesium bis(trifluoromethane)sulfonimide (Mg(TFSI)2, 99.5%, Solvionic) dissolved in a mixture of dimethoxyethane (DME, Sigma Aldrich, anhydrous, inhibitor-free, 99.5%) and 1-methoxypropyl-2-amine (MPA, Sigma Aldrich, 99%) in a 3.84/1 w/w ratio (Fig. S6†).35 The Mg(TFSI)2 had been dried at 220 °C under a dynamic vacuum of ≈5 mbar for 12 h. DME was further dried over activated molecular sieves (Sigma Aldrich, 3 Å, heated to 250 °C under vacuum). MPA was degassed by freeze–pump–thaw with liquid N2 and dried by distillation over CaH2 before storing over activated molecular sieves. A final water content of <10 ppm was recorded by Karl-Fischer titration (Table S5†). Electrolyte stability was confirmed by cyclic voltammetry and Mg plating and stripping (Fig. S7 and S8†).
:20:10 w/w combined with in N-methyl-2-pyrrolidone. The slurry was cast onto carbon-coated aluminium foil and cut into disks (16 mm diameter) and dried under vacuum overnight. Active material loadings were typically between 0.25 and 0.35 mg cm−2, with an electrode thickness of 100 μm. Negative electrodes consisted of polished Mg foil discs with a diameter of 18 mm. Electrodes were separated by Whatman GF/D borosilicate glass fibre membranes soaked with 500–750 μL of an electrolyte consisting of a 0.5 M solution of magnesium bis(trifluoromethane)sulfonimide (Mg(TFSI)2, 99.5%, Solvionic) dissolved in a mixture of dimethoxyethane (DME, Sigma Aldrich, anhydrous, inhibitor-free, 99.5%) and 1-methoxypropyl-2-amine (MPA, Sigma Aldrich, 99%) in a 3.84/1 w/w ratio (Fig. S6†).35 The Mg(TFSI)2 had been dried at 220 °C under a dynamic vacuum of ≈5 mbar for 12 h. DME was further dried over activated molecular sieves (Sigma Aldrich, 3 Å, heated to 250 °C under vacuum). MPA was degassed by freeze–pump–thaw with liquid N2 and dried by distillation over CaH2 before storing over activated molecular sieves. A final water content of <10 ppm was recorded by Karl-Fischer titration (Table S5†). Electrolyte stability was confirmed by cyclic voltammetry and Mg plating and stripping (Fig. S7 and S8†).
Electrochemical measurements were conducted using a BioLogic VSP300 potentiostat at 30 °C. Galvanostatic charge–discharge was performed between potential limits of 0.8–2.2 V vs. Mg2+/Mg at current densities of 20–200 mA g−1, recording the change in potential every 5 mV. Ex situ cell testing was performed on powder-based Swagelok cells. The cathode composition was 70% active material, 30% carbon black, without binder with typical loadings of 15–20 mg. Cells were discharged and charged with a constant current of 20 mA g−1 to the required potentials. Powders were collected, washed in DME, and dried under vacuum at 60 °C for 12 h.
Potentiostatic Intermittent Titration Technique (PITT) utilised chronoamperometric pulses imposed from open circuit potential to 0.8 V vs. Mg2+/Mg with an interval of 20 mV until the current reach 0.75 mA g−1 at each potential step. Diffusion coefficients (D) were calculated by differentiation of the finite-diffusion Cottrell equation for the long-time region:
 485 C mol−1), A surface area (cm2), ΔC for concentration gradient within the particles, t for time (s) and a for particle radius (cm).
485 C mol−1), A surface area (cm2), ΔC for concentration gradient within the particles, t for time (s) and a for particle radius (cm).
X-ray diffraction (XRD) techniques
Synchrotron X-ray diffraction (SXRD) was performed at Diamond Light Source, U.K., on high-resolution beamline I11.36,37 The patterns were recorded in the transmission mode [0° < 2θ < 90°] using a position sensitive detector (PSD, λ = 0.824399 Å) at room temperature on samples introduced into 0.3 mm diameter borosilicate glass capillaries. Rietveld refinements were carried out using TOPAS Academic Version 7.38 The background was fitted manually using a 36-part linear profile. Fundamental parameters of the beamline were modelled, including Lorentz-polarization, la, lo and lh Voight coefficients, source-to-sample radius & slit widths. Peak profiles were fitted using the Stephens model in the monoclinic setting (b-axis unique). Simple axial model was initially constrained to 5.0 before being refined after initial peak profile fitting.Maximum Entropy Method (MEM) was used to calculate electron density distributions. The observed structure factor was extracted from data using a Rietveld refinement performed in Jana 2006.39 The refinement models were taken from Topas refinements and atomic positions were kept fixed. Peak profiles were fit empirically using a pseudo-Voight function and the Stephens model for anisotropic strain broadening in the monoclinic setting, as was done in Topas. The background was fit manually using a 60-point linear interpolation to ensure that peak intensities were extracted to the highest degree of accuracy. The electron densities were generated with MEM using the Sakata–Sato algorithm in BayMEM40 on a grid of 300 × 300 × 300 voxels and run till the combined FG constraint met its convergence criteria of 1.0. Electron densities were visualized using VESTA.41
Mg2+ diffusion pathways in V2Se9 were analyzed by bond valence pathway analysis (BVPA) using the software SoftBV GUI (Version 1.3.1).42 Three-dimensional energy landscape maps were generated for Mg2+ ion diffusion using soft bond valence parameters calculated using the CIF generated from SXRD data for V2Se9. Maximum voxel resolution of 0.1 Å was applied. The resulting volumetric data was visualized using an isosurface level of 5.0 α0−3 (α0: Bohr radius) using the VESTA software.41
Elemental analysis
Scanning Transmission Electron Microscopy (STEM) imaging, and EDX analysis were performed using a Cs-corrected TEM/STEM Jeol 2100FCs microscope operating at 200 kV. For EDX mapping, a JEOL silicon drift detector (SDD) was used with a detection area of 100 mm2.Further details regarding Transmission Electron Microscopy (TEM) and Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) can be found in the ESI.†
Raman spectroscopy
Ex situ Raman measurements were collected using a Raman microscope (Renishaw, in via reflex coupled with an inverted Leica microscope), with a 532 nm laser as the excitation source (power <300 μW), focused onto the sample using a ×50 objective (Olympus). The collected Raman spectra were baseline-corrected. The spectral measurement time was 60 seconds with 5 accumulations to ensure a good signal-to-noise ratio and well-resolved peaks. To check for potential laser damage, V2Se9 was measured with varying laser intensities and exposure times. No changes in the spectra were observed up to 10% power for 60 seconds.X-ray photoelectron and X-ray absorption analysis
X-ray Photoelectron Spectroscopy (XPS) analysis was performed using a Thermo NEXSA XPS fitted with a monochromatic Al Kα X-ray source (1486.7 eV), a spherical sector analyser and multichannel resistive plate, 128 channel delay line detectors. All data were recorded at 19.2 W with an X-ray beam size of 400 × 200 μm2. Survey scans were recorded at a pass energy of 200 eV, and high-resolution scans were recorded at a pass energy of 40 eV. Electronic charge neutralization was achieved using a dual-beam low-energy electron/ion source (Thermo Scientific FG-03). The ion gun was operated at a current of 150 μA and a voltage of 45 V. All sample data were recorded at a pressure below 10−8 Torr and at 294 K. Data was processed using CasaXPS (v2.3.19PR1.0) using a Shirley background and peak fitting with Gaussian–Lorentzian functions. The Fermi edge of Ni foil measured using the same equipment was used to calibrate the data and eliminate any effects of surface charging.X-ray Absorption Spectroscopy (XAS) was measured at beamline B18 (ref. 43) at Diamond Light Source, U.K. Pellets of sample diluted with cellulose were prepared with an optimized density for X-ray absorption measurements in transmission mode at the V K-edge (5465 eV) and the Se K-edge (12658 eV) at ambient temperature. The samples were hermetically sealed in Al pouches under argon. The spectra were calibrated by fixing the maximum of the derivative of V and Se foil references placed after the samples for the corresponding edges, respectively, and normalized with the Athena software.44
Results and discussion
Cyclic voltammetry was used to probe the operational potential window and redox activity of V2Se9 towards electrochemical (de)insertion of Mg2+ in 0.5 M magnesium bis(trifluoromethane)sulfonimide (Mg(TFSI)2) in dimethoxyethane (DME) and 1-methoxy-2-propylamine (3.84:1 w/w) electrolyte (Fig. S6–S8 and Table S5†). From an open circuit potential (OCP) of 1.8 V vs. Mg2+/Mg the cell potential was swept at a rate of 100 μV s−1 while the reductive potential was lowered, from 0.9 V to 0.2 V (Fig. 1a). When cycled between 0.8 V and 2.4 V, a sharp reductive peak is observed at 1.72 V, with additional weaker peaks observed at 1.36 V, 1.25 V and 1.13 V. Strong oxidative peaks are observed above 1.6 V, centered at 1.72 V and 1.95 V, though several smaller less resolved peaks are also present. The inflection at 2.4 V was shown to be oxidation of the electrolyte solution, which continued as the potential was further increased. This observation is similar to other reports which use Mg(TFSI)2 electrolyte solutions in the literature and is in-line with the potential at which [TFSI]− has been shown to corrode Al in Li-ion based systems.45,46 When the potential is swept below 0.6 V there is an almost immediate loss of redox activity, suggesting the system undergoes irreversible changes below ca. 0.7 V (Fig. S9†). Low-potential conversion reactions are observed in V2Se9 in Li+ and Na+ cells when cycled below 1.5 V vs. Li+/Li (0.83 V vs. Mg2+/Mg) and 1.0 V vs. Na+/Na (0.66 V vs. Mg2+/Mg).32–34 Similar conversion reactions are likely occurring with Mg2+ at potentials below 0.7 V. This is indeed the case for structurally related one-dimensional chain materials containing sulfur, such as VS4 and TiS3, which display low-potential conversion reactions in which MgS and metallic V are formed.47–49 While the formation of Li2Se and Na2Se is shown to be reversible, the formation of MgS is not, and results in significant capacity decay over the first few cycles. It is therefore reasonable to expect that the formation of MgSe is also irreversible. The large charge-per-size of Mg2+ compared to Li+ and Na+ will result in stronger Mg–Se bonding in MgSe than Li–Se and Na–Se bonds in Li2Se and Na2Se, respectively. As such, a suitable potential window of 0.8 V to 2.2 V was selected for galvanostatic cycling.
 | ||
| Fig. 1 (a) Cyclic voltammogram recorded for MgxV2Se9 at a sweep rate of 100 μV s−1 between 0.8 V and 2.4 V. (b) Voltage profiles of cycles 1–12 for V2Se9 in 0.5 M Mg(TFSI)2 in dimethoxyethane/1-methoxypropyl-2-amine (3.84/1, w/w) at 200–600 mA g−1. (c) Potentiostatic intermittent titration technique of the discharge of V2Se9 to calculate the Mg2+ diffusion coefficients as a function of potential, from 1.3 V to 0.8 V. (d) Second cycle discharge potential profile for V2Se9/carbon loose powder electrode in a Swagelok cell at a current density of 50 mA g−1 for ex situ analysis. Spheres represent the potentials at which cells were stopped and the materials collected: cyan for discharged (magnesiated) MgxV2Se9 and orange for charged (de-magnesiated) MgxV2Se9. Typically, a second discharge capacity of 90 mA h g−1 (i.e., 1.35 Mg2+/V2Se9) is achieved by 0.8 V. Samples of MgxV2Se9 were collected at 1.6 V (x = 0.15); 1.3 V (x = 0.47); and 0.8 V (x = 1.35) during discharge; and 1.65 V (x = 0.97) and 2.2 V (x = 0.00) during charging. | ||
During the first cycle, an initial discharge capacity of 107 mA h g−1 is observed at a current density of 200 mA g−1. The first cycle shows a different initial potential profile to subsequent cycles. The potential drops rapidly to 1 V, suggesting a large overpotential activation for Mg2+ insertion whereas upon further cycling, Mg begins to be inserted at a much higher potential of 1.6 V. Subsequent cycles achieve a decreasing amount of capacity from 106 to 88 mA h g−1 (i.e., 1.6–1.3 Mg2+/V2Se9) at 200 mA h g−1 with potential profiles remaining similar; with plateaus at 1.75 V, 1.50 V and 1.35 V (Fig. 1b), consistent with the peak positions observed in cyclic voltammetry. The capacity decreases at higher current densities, with 50 mA h g−1 (0.76 Mg2+/V2Se9) at 400 mA g−1 and 31 mA h g−1 (0.47 Mg2+/V2Se9) at 600 mA g−1 being maintained for multiple cycles. This is lower than the capacity achieved by the insertion of Li+ or Na+ into V2Se9, which can be estimated to be 300 mA h g−1 from insertion reactions alone.32,33 This can likely be attributed to the associated thermodynamic and kinetic difficulties with the insertion of high charge-per-size Mg2+ relative to Li+ and Na+.
Mg2+ de-insertion appears to be a more complex process with voltage hysteresis observed during charging. Plateaux are observed at 1.65 V, 1.72 V, and 1.85–1.90 V, again in agreement with peaks observed during cyclic voltammetry. An extended plateau is observed above 2.1 V that originates from the oxidation of the electrolyte as the potential approaches the upper limit of the electrochemical stability window (Fig. S8†), accounting for the larger capacity observed on charging.50 As a result, this difference in capacity is observed for cycles at slower rates, with higher current densities achieving higher coulombic efficiencies (Fig. S10†).
Magnesium insertion diffusion was studied using Potentiostatic Intermittent Titration Technique (PITT); the calculated diffusion coefficients from the selected points are illustrated in Fig. 1c. The Mg2+ diffusion coefficients in MgxV2Se9 are ≈10−10–10−12 cm2 s−1, and change as a function of potential indicating Mg content-dependent insertion kinetics. The kinetic performance of V2Se9 towards Mg2+ insertion is faster than V2PS10 (10−14–10−11 cm2 s−1 between 1.5 and 0.1 V)28 and Mo6S8 (10−14–10−11 cm2 s−1 between 1.4–1.0 V);18,51 and is comparable to monovalent charge carrier (Li+, Na+) insertion into layered oxide materials.52,53 The observed rapid kinetics of V2Se9 can be attributed to the low-charge-density selenide framework, helping to overcome the sluggish kinetics of Mg batteries.
To understand the storage mechanism of V2Se9, Swagelok-type cells with cathodes consisting of approximately 20 mg of 70/30 w/w V2Se9/carbon black powder (no binder) were used to prepare samples of MgxV2Se9 for ex situ diffraction, XPS, and XAS measurements. Typically, a second discharge capacity of 90 mA h g−1 (i.e., 1.35 Mg2+/V2Se9 giving Mg1.35V2Se9) is achieved by 0.8 V (Fig. 1d). The lower capacity is the result of reduced conductivity between the loose powder electrode in Swagelok cells compared to cast electrodes in coin cells. The long plateau above 2.1 V is attributed to electrolyte oxidation, as previously discussed.
STEM-EDX false color micrographs shown in Fig. 2 confirm that Mg is homogeneously distributed throughout the bulk of V2Se9 crystallites after insertion. ICP-OES analysis gives a composition of Mg1.20(3)V2.00(8)Se9.4(5), in good agreement with the expected Mg content from the electrochemical discharge capacity (Table S6†). Ex situ SXRD analysis was performed on MgxV2Se9 (0 ≤ x ≤ 1.35) prepared at various potentials during discharge: 1.6 V (x = 0.15); 1.3 V (x = 0.47); 0.8 V (x = 1.35); and recharge: 1.65 V (x = 0.97) and 2.2 V (x = 0.00).
 | ||
| Fig. 2 STEM dark field and false color EDX micrographs of (a) V2Se9 (b) and Mg1.35V2Se9 (after discharge to 0.8 V; shown in for separate areas) using an acceleration voltage of 200 kV with a probe current of 7.475 nA. V is shown in blue, Se in green and Mg in orange. The Mg content cannot be resolved due to overlap between the Mg Ka (1.253 keV) and Se La (1.379 keV) peaks. Mg signal is within the limits of noise in V2Se9. For Mg1.35V2Se9 Mg is homogeneously inserted throughout the crystallite. | ||
Analysis of SXRD data for Mg1.35V2Se9 (discharged to 0.8 V vs. Mg2+/Mg) indicates that the structural symmetry (C2/c) and framework of V2Se9 is retained for cycled MgxV2Se9 (Fig. 3a). Fourier difference analysis reveals two separate regions of electron density as suitable positions for Mg2+ within the unit cell (Fig. 3b and c and S11†). Magnesium was placed on each of these sites with an initial occupancy of 1.0 (equivalent to Mg2V2Se9) before refinement. The proximity of magnesium positions to V and Se sites was restrained using a penalty function during final refinement, and the Biso values for both Mg sites were constrained to be equal. The final structural model (shown in Fig. 3a) achieved fit statistics of Rwp = 0.521%, improved from 0.624% for a model without any Mg present (Fig. S12 and Table S7†).
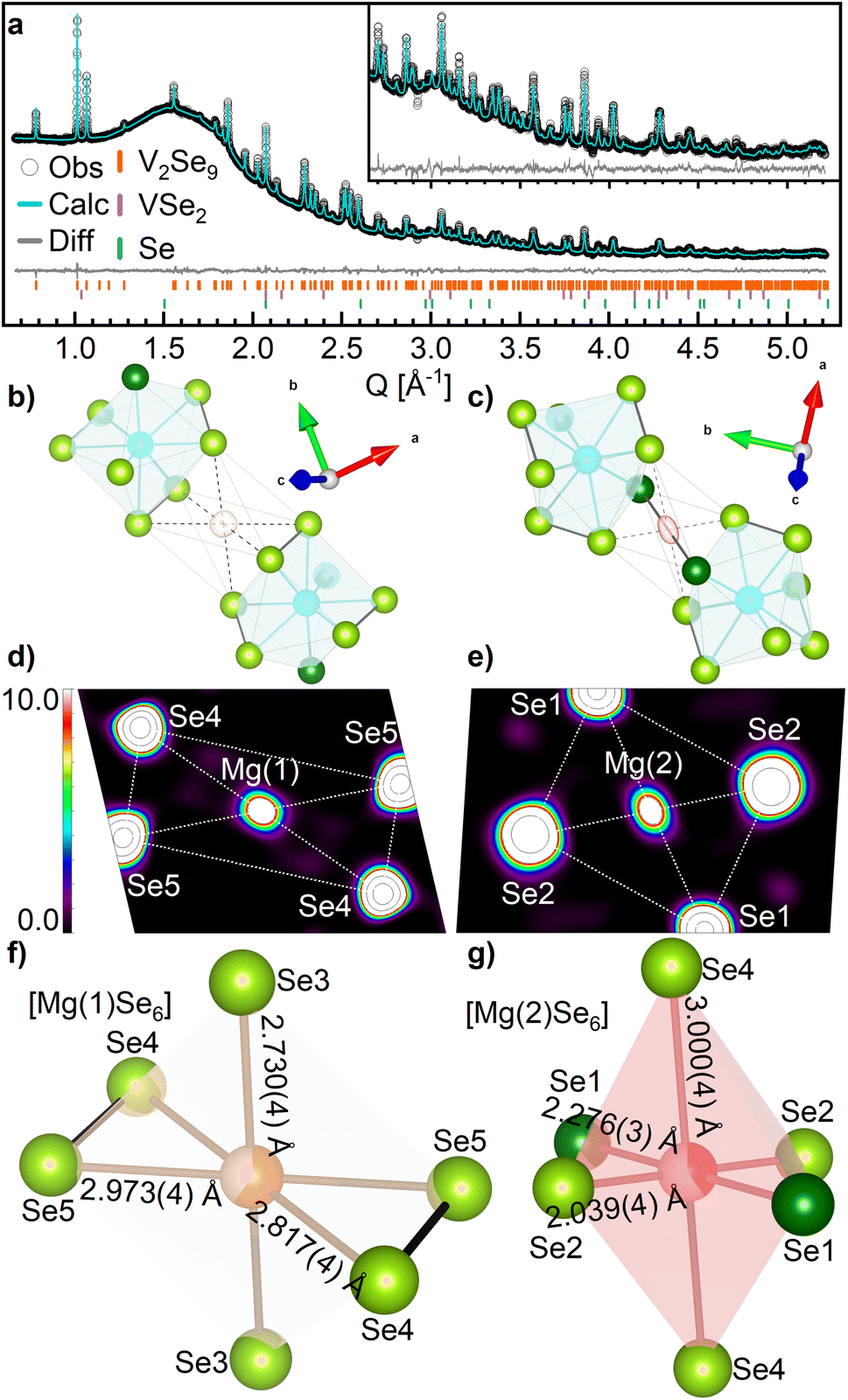 | ||
| Fig. 3 (a) Rietveld refinement of Mg1.35V2Se9 against SXRD data collected at I11 beamline. Observed data are shown by white circles. The calculated fit is shown by the cyan line. The difference between the calculated and observed data are shown by the grey line. The Bragg reflection positions of V2Se9, VSe2 and Se are given by the orange, purple and green ticks, respectively. Inset is an enlarged region of the fit between 2.5–5.2 Å−1. Refined parameters are summarized in Table S7.† (b–g) Coordination of Mg sites in Mg1.35V2Se9. V is shown by blue spheres, Mg(1) by orange spheres, Mg(2) by red spheres, Se2− by dark green spheres and Se1− by light green spheres. Se–Se bonds are shown by bold black lines and V–Se and Mg–Se bonds are shown by grey lines. Electron density isosurfaces of the Mg(1) and Mg(2) sites in Mg1.35V2Se9 generated by (b and c) Fourier difference analysis and (d and e) maximum entropy method. 2D planes bisect the MgSe6 octahedrons in the equatorial plane (see Fig. S13†). (f and g) Local coordination environments of Mg(1) and Mg(2). | ||
The location of the two refined Mg positions is further confirmed through analysis of the SXRD data via maximum entropy method (MEM) calculations by generating the electron density distribution ab initio from the observed diffraction data.40 Analysis was performed on both pristine V2Se9 and Mg1.35V2Se9 to visualize the change in charge density upon reduction of the host structure. Fig. 3d and e shows electron density observed between neighboring [V4Se18]∞ chains of Mg1.35V2Se9 which is not present in V2Se9, further confirming Mg is present on these sites (Fig. S13†). Notably, the electron density observed on the Mg sites is more localized than that of the larger, and therefore less densely charged, neighboring Se sites.
The refined occupancies of the two Mg sites were 0.596(19) and 0.402(16), providing a refined composition of Mg0.99(3)V2Se9, compared to Mg1.35V2Se9 calculated from electrochemical capacity. Identification of discrete Mg sites suggests a homogeneous distribution of Mg2+ within bulk V2Se9, indicative of an insertion-type electrochemical process. Additionally, no metallic V or MgSe is observed via ex situ diffraction measurements, suggesting that no conversion processes take place when cycling between 0.8–2.2 V.
Mg is octahedrally coordinated to Se in Mg1.35VSe9, with MgSe6 octahedra located in the vdW space between neighboring [V4Se18]∞ chains. Selenide materials containing octahedral MgSe6 units involving Se2− only, such as spinel MgM2Se4 (M = In, Al, Sc),54 olivine Mg2MSe4 (M = Ge, Si, Sn)55 and Mg2P2Se6 (ref. 56) have average Mg–Se distances of 2.74(9) Å with a range of 2.3–3.3 Å (Fig. S14†), and average polyhedral volumes of 27.2(4) Å3. In Mg1.35V2Se9, Mg(1) is coordinated entirely to Se1− anions with an average Mg–Se distance of 2.84(12) Å and an octahedral volume of 21.51 Å3 (Fig. 3f). Mg(2) is coordinated to four Se1− and two Se2− anions with an average Mg–Se distance of 2.4(5) Å and an octahedral volume of 17.85 Å3 (Fig. 3g). The smaller Mg(2) octahedra is attributed to a lower Mg occupancy and the smaller anionic radius of Se2−. Additionally, the mixed coordination of Mg(2) to Se1− and Se2− results in a larger octahedral distortion index than that of Mg(1) (0.154 compared to 0.031) due to different anionic radii and charge densities.57
Each VSe8 polyhedron shares faces with two MgSe6 octahedra, with one Mg(1) and one Mg(2); likewise, each MgSe6 octahedron shares two opposite, and identical, faces with two separate VSe8 polyhedra from neighboring [V4Se18]∞ chains. Every (Se2)2− group is coordinated to at least one Mg2+ site. Mg(1) shares two triangular faces with VSe8 formed between Se(1)2−, Se(2)1− and Se(4)2−. Mg(2) shares two triangular faces formed between Se(3)1−, Se(4)1−, and Se(5)2−, such that two edges of Mg(2) Se8 octahedron is formed by the (Se2)2− bond between Se(4) and Se(5) (Fig. 4a). In this way, chains of [V4Se18]∞ that are separated by vdW gaps in V2Se9 are connected in Mg1.35V2Se9 through Se–Mg–Se bonds (Fig. 4b).
 | ||
| Fig. 4 The structure of Mg1.35V2Se9. V is shown by blue spheres, Mg(1) by orange spheres, Mg(2) by red spheres, Se2− by dark green spheres and Se1− by light green spheres. Se–Se bonds are shown by bold black lines and V–Se and Mg–Se bonds are shown by grey lines. (a) The coordination of Mg sites around V2Se9. (b and c) Chains of [V4Se18]∞ that are separated by vdW space in V2Se9 are connected through Se–Mg–Se bonding in Mg1.35V2Se9 in the (011) plane. V–V interchain distances across Mg sites are provided. | ||
The structural model for Mg1.35V2Se9 was used as a starting model for refinement against SXRD data for other ex situ samples of MgxV2Se9 after discharging (0 ≤ x ≤ 1.35): 1.6 V (x = 0.15); 1.3 V (x = 0.47); 0.8 V (x = 1.35); and after charging: 1.65 V (x = 0.97) and 2.2 V (x = 0.00). Refined Rietveld parameters and fits are given in Fig. S15 and Tables S8–S11.†
Refinement of Mg site occupancies shows Mg content in MgxV2Se9 increases as potential is decreased (Fig. S16a†). After discharging from the OCV (1.8 V) to 1.6 V the occupancy of Mg(1) increases to 0.227(17), while Mg(2) is only 0.061(17). Mg(1) occupancy continues to increase rapidly to 0.553(14) by 1.3 V and more slowly to 0.596(11) by 0.8 V; suggesting that Mg(1) is filled more easily in the earlier stages of electrochemical discharge, as would be expected given the larger octahedral volume and low charge density Se1− coordination of this site. The occupancy of Mg(2) increases to 0.263(14) by 1.3 V and 0.402(12) by 0.8 and is therefore filled more easily in the later stages of discharge, but to a lesser degree than Mg(1). This supports the measured PITT data (Fig. 1c), which shows faster kinetics in the early stages of Mg insertion before decreasing later in the discharge process. The preferential occupation of Mg(1) over Mg(2) is likely attributed to favorable kinetics associated with the interaction of high-charge density Mg2+ with the lower charge density Se1− (to which Mg(1) has a higher coordination) compared to Se2−. None of the Mg sites observed through ex situ structural analysis are fully occupied and, likely, the higher capacities recorded for non-ex situ cells (Fig. 1b and d) are the result of greater occupancy of these sites with increased Mg insertion due to the difference in cell preparation. Upon charging from 0.8 to 2.2 V, refinement shows that magnesium is de-intercalated from Mg1.35V2Se9, reforming V2Se9.
To better understand the facile Mg2+ insertion kinetics observed by PITT, Mg2+ ion conduction pathway energy landscapes were calculated from soft bond valence parameters by bond valence pathway analysis (BVPA) (further details are given in the ESI†).58 2D and 2D Mg2+ diffusion pathways are observed in V2Se9, with energy barriers ranging from 0.140 to 0.742 eV (Fig. S17 and S18†).44–46
Locally, this results in minimal strain induced on the individual [VSe8] polyhedra, which is evidenced by minimal change to average V–Se and Se–Se distances between V2Se9 and Mg1.35V2Se9 (2.57(5) Å to 2.55(4) Å for V–Se; and 2.716(12) Å to 2.728(18) Å for Se–Se, respectively). The relatively small changes in V–Se distances are in line with other vanadium chalcogenide materials after electrochemical insertion of Mg2+: V–O distances in MgV2O5 increase by only 0.03 Å compared to V2O5 (1.49 Å to 1.52 Å)59 and V–S distances in Mg1.5VS4 increase by only 0.04 Å compared to VS4 (2.40 Å to 2.44 Å).25 The lower charge density of Se, compared to O and S, as well as the lower Mg/V ratio for Mg1.35V2Se9 result in little to no change in the average V–Se bond length between V2Se9 and Mg1.35V2Se9. The minimal change in Se–Se distances between V2Se9 and Mg1.35V2Se9 indicates that (Se2)2− bonds are maintained after Mg2+ insertion, unlike VS4 (ref. 25) and V2PS10 (ref. 28) where S–S bonds break upon discharge (increasing in length by up to 15%). A full comparison of bond lengths for V2Se9 and MgxV2Se9 (0 ≤ x ≤ 1.35) is provided in Table S12.†
More significant structural perturbation is observed in the relative positions of the [V4Se18]∞ chains across the vdW space, which are influenced by the location of the Mg sites. The V–V interchain distances between vanadium sites adjacent to Mg(1) increase by 0.1% (from 7.394(9) Å in V2Se9 Å to 7.401(9) Å) in Mg1.35V2Se9 (Fig. 4b and c). This is different to the V–V interchain distances between vanadium adjacent to the lesser occupied Mg(2) site contract by 2% (from 6.625(10) Å to 6.495(9) Å). This suggests that at low levels of Mg2+ occupancy, the selenium anionic framework of the [V4Se18]∞ chains are attracted towards the high charge density intercalated ion. For x(Mg) = 1.35 in Mg1.35V2Se9, this attractive force dominates, and the distance between [V4Se18]∞ chains decreases in all directions (Fig. S16b†).25,28,59 The shortest inter-chain Se–Se distances decrease from 3.443(6) to 3.419(7) Å for Se1–Se2 and from 3.521(4) Å to 3.419 Å for Se2–Se5. This leads to contraction of the a, b, and c lattice parameters, each with an approximately equal induced strain of 0.1%, resulting in a 0.3% decrease in unit cell volume from 1063.225(19) Å3 in V2Se9 to 1059.511(18) Å3 in Mg1.35V2Se9 (Fig. S16c†). After charging, the unit cell returns to 1062.988(13) Å3, with lattice parameters close to that of V2Se9.
Spectroscopic methods were used to further examine the changes in the electronic structure of V2Se9 upon Mg insertion. Both V2Se9 and Mg1.35V2Se9 crystalize in C2/c symmetry (crystal class 2/m). Group-theory analysis identifies 33 Raman active modes for V2Se9. V and 4 of the Se sites that make up the (Se2)2− groups occupy 8f Wyckoff positions, each with 3 Ag and 3 Bg active modes, while Se2− occupies a 4e site with 1 Ag and 2 Bg active modes (Tables S13–S15†).60,61 Low structural symmetry results in overlapping modes and produces the convoluted spectra observed for V2Se9 in Fig. 5a. Similarly to Raman spectra of isostructural Nb2Se9 (ref. 62) and related VS4,47 the spectrum can be divided into two regions; with phonons in the low-wavenumber range, between 105 and 175 cm−1, assigned to V–V and V–Se bending and stretching modes, and a high-wavenumber range, between 268 and 346 cm−1 (relating to shorter chemical bonds), assigned to Se–Se stretching and twisting as Se cage-breathing (Table S16†).
 | ||
| Fig. 5 (a) Ex situ Raman spectroscopy of V2Se9 (black), Mg1.35VSe9 (orange) after discharging to 0.8 V and V2Se9 after charging (Ch) to 2.2 V (cyan). (b and c) XPS spectra of MgxV2Se9 collected at various stages of discharge/charge. (b) Mg 2p collected at a pass energy of 200 eV and scaled using the Se 3d signal. (c) Se 3d XPS collected at a pass energy of 40 eV. Both Se 3d5/2 and Se 3d3/2 contributions are shown for each Se species: (Se2)2− (light blue) and Se2− (orange). The sum of orbital contributions is shown as the purple envelope relative to the observed data (black circles). The Se 3d5/2 and 3d3/2 spin–orbit components of each oxidation state were constrained based on relative position, Δ = 0.86 eV, and intensity ratio = 0.735. The relative areas of Se2−/(Se2)2− for V2Se9 were constrained at 1/8, to reflect the nominal formula V25+(Se2)2–4Se2−. Normalized (d) Se K-edge and (e) V K-edge XANES data measured in transmission for V2Se9 (black), Mg1.35V2Se9 (discharged 0.8 V, orange), and V2Se9 (charged 2.2 V, cyan). Inset panels show enlarged profile of the absorption edges at 12658 eV and 5465 eV, respectively. | ||
Ex situ Raman spectra collected for Mg1.35V2Se9 (Fig. 5a) contain similar 33 Raman active Ag and Bg modes as V2Se9 as the octahedrally coordinated Mg2+ occupies 4a and 4b Wyckoff positions and possess no Raman active modes (Table S15†). The decrease in the intensity of signals observed between 268 and 346 cm−1 is the result of electron delocalization away from (Se2)2− towards the Raman inactive Mg site, to which all (Se2)2− are coordinated, resulting in reduced polarizability, and dampening of the Raman signal.63 Additionally, coordination of (Se2)2− to Mg2+, as well as the presence of Mg sites between [V4Se18]∞ chains, will limit the movement of Se sites to stretching, twisting, and breathing, further decreasing signal intensity. After charging to 2.2 V intense peaks in the high-wavenumber range are recovered supporting Mg de-insertion from the bulk.
X-ray photoelectron spectroscopy (XPS) measurements show the formation of peaks at 1303.2 eV, 89.0 eV, and 49.8 eV in Mg1.35V2Se9 after discharge that are not present in V2Se9. These signals relate to Mg 1s, Mg 2s, and Mg 2p core levels, respectively (Fig. S22†), and relate to Mg insertion after discharging to 0.8 V. The low energy of the Mg 2p signal corresponds to higher kinetic energy electrons and is capable of probing deeper than Mg 1s and Mg 2s, and as such suggests that Mg is present beyond the immediate surface in Mg1.35V2Se9.64 The intensities of the Mg peaks is much lower after charging to 2.2 V, supporting Mg de-insertion (Fig. 5b), with the slight Mg signal that remains likely being due to the presence of surface species, which would explain why this feature is more prominent for the more surface sensitive Mg 1s and Mg 2s cores than Mg 2p.65
The Se 3d XPS signals contain contributions from both Se2− and (Se2)2− oxidation states (Fig. 5c and S23†). In V2Se9, the binding energy of the Se2− 3d5/2 signal is at 53.4 eV; comparable to other vanadium selenide materials containing Se2−.66–68 The higher oxidation state Se1− 3d5/2 signal is at a higher binding energy of 54.4 eV due to the increased interaction between the photo-emitted electron and the ion core.69 The (Se2)2− 3d5/2 signal is maintained at 54.4 eV in both V2Se9 and Mg1.35V2S9. The Se2− 3d5/2 shows a slight shift in Mg1.35V2Se9 relative to V2Se9 (from 54.7 eV to 53.7 eV) though its position is maintained after charging to 2.2 V. This is indicative of slight irreversible structural change occurring upon initial discharging supported by the signal broadening observed for both 0.8 V and 2.2 V samples. The relative areas of the Se2−/(Se2)2− signals is maintained at close to 1/8 (as in V2Se9); suggesting that Se is redox inactive between 0.8-2.2 V for x(Mg) 0 ≤ x ≤ 1.35, in agreement with the structural analysis via SXRD.
X-ray Absorption Near-Edge Spectroscopy (XANES) measurements of Se and V K-edges were used to monitor changes in the oxidation state of bulk MgxV2Se9 at the various stages of electrochemical cycling. Similarly to the Se 3d XPS measurements, there is no change in Se edge position observed by XANES upon discharge or charge (Fig. 5d). An average Se oxidation state between −1.08 and −1.11 is maintained between V2Se9, Mg1.35V2Se9, and after charging to V2Se9 indicating the redox inactivity of Se (Fig. S24†). This differs from VS4 (ref. 25) and V2PS10 (ref. 28) which show changing contributions from signals relating to S2− and (S2)2− as S–S bonds are broken and reformed. Se has a lower electronegativity compared to S, meaning Se2− is less readily formed than S2− or O2− which may explain why V2Se9 is anionic redox-inactive. While reversible cleavage of (Se2)2− has been observed in molecular systems,70 in solid materials Se–Se bonding is almost certainly less prone to redox activity than S–S or O–O. Cleavage of (Se2)2− bonding in MgxV2Se9 may be the cause of irreversible electrochemistry below 0.8 V. The V K-edge XANES edge position decreases from 5471.0 eV for V2Se9 to 5470.5 eV for Mg1.35V2Se9 after discharge, indicating a reduction from V5+ (Fig. 5e). Insertion of 0.675 Mg2+ per V requires the reduction of V5+ to V3.65+. Upon charging, the vanadium edge energy and absorption profile return to that of V2Se9 (5471.0 eV), suggesting reversible redox processes take place on the V site and are responsible for Mg intercalation.
Conclusions
V2Se9 is capable of storing up to 1.6 Mg2+ (106 mA h g−1) within the vdW spaces between its constituent one-dimensional chains. Detailed structural characterization by SXRD and MEM supports homogeneous distribution of Mg2+ ions within V2Se9, supporting an insertion-type electrochemical process without the formation of metallic V or MgSe that would be associated with a conversion-type reaction. Insertion of Mg2+ is facilitated by reversible redox on the V site, while (Se2)2− is not redox active over the achieved levels of discharge. The octahedral coordination of Mg2+ results in minimal strain on the [VSe8] polyhedra, allowing for small changes in bond distances during Mg insertion, resulting in V2Se9 displaying exceptional Mg2+ diffusion kinetics, with diffusion coefficients surpassing state-of-the-art cathode materials such as Mg2Mo6S8. Calculated bond valence parameters support diffusion pathways with low energy barriers to ion mobility are available to inserting Mg2+. The one-dimensional structure of V2Se9 combined with the low charge density of coordinating (Se2)2− and Se2− anions enables rapid access to Mg2+ sites which enhances kinetic performance. Despite the larger mass of Se relative to S, V2Se9 can achieve comparable capacity to high-performance sulfide materials like Mo6S8, whilst operating with faster diffusion kinetics, helping to overcome the kinetic limitations that have perpetually hindered Mg2+ electrodes. This work motivates further investigation of tuning the anion chemistry of the wide range of known low-dimensional transition metal chalcogenides to improve kinetic performance of multivalent electrodes.Data availability
The data supporting this article have been included as part of the ESI.† Underlying data collected as part of this work is available via the University of Liverpool data repository at https://datacat.liverpool.ac.uk/id/eprint/2676. The CIF files for the structures of V2Se9 and Mg1.35V2Se9 are deposited with CSD accession codes 2357643 and 2357642, respectively.Author contributions
Matthew A. Wright: conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing – original draft, writing – review & editing; Jungwoo Lim, Raul A. Pacheco Muino: formal analysis, investigation, visualization, data curation; Anna E. Krowitz, Cara J. Hawkins, Mounib Bahri: investigation; Luke M. Daniels, Ruiyong Chen, Luciana Gomes Chagas: conceptualization, supervision, writing – review & editing; James Cookson, Paul Collier, Alan V. Chadwick, Nigel D Browning: funding acquisition, resources; John B. Claridge: conceptualization, supervision; Laurence J. Hardwick, Matthew J. Rosseinsky: conceptualization, funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded under the Engineering and Physical Sciences Research Council (EPSRC; EP/V026887). M. A. W. thanks Johnson Matthey for studentship funding. We acknowledge the ICSF Faraday Institution project “CATMAT – Next Generation Li-ion Cathode Materials” [grant number FIRG016] for funding J. L. We acknowledge NSG and EPSRC for funding A. E. K. We acknowledge Diamond Light Source for access to beamline I11 under CY31578 and for access to B18 under the Energy Materials BAG. The X-Ray Photoelectron (XPS) data collection was performed by Ms Cara J. Hawkins at the EPSRC National Facility for XPS (“HarwellXPS”), operated by Cardiff University and UCL, under Contract No. PR16195.References
- M. Armand and J. M. Tarascon, Nature, 2001, 414, 359–367 CrossRef PubMed.
- M. Mao, T. Gao, S. Hou and C. Wang, Chem. Soc. Rev., 2018, 47, 8804–8841 RSC.
- J. Heelan, E. Gratz, Z. Zheng, Q. Wang, M. Chen, D. Apelian and Y. Wang, JOM, 2016, 68, 2632–2638 CrossRef CAS.
- R. Davidson, A. Verma, D. Santos, F. Hao, C. Fincher, S. Xiang, J. Van Buskirk, K. Xie, M. Pharr, P. P. Mukherjee and S. Banerjee, ACS Energy Lett., 2019, 4, 375–376 CrossRef CAS.
- E. Levi, Y. Gofer and D. Aurbach, Chem. Mater., 2010, 22, 860–868 CrossRef CAS.
- C. Ling, D. Banerjee and M. Matsui, Electrochim. Acta, 2012, 76, 270–274 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., 1976, A32, 751–767 CrossRef CAS.
- Y. Liang, H. Dong, D. Aurbach and Y. Yao, Nat. Energy, 2020, 5, 646–656 CrossRef CAS.
- Z. Guo, S. Zhao, T. Li, D. Su, S. Guo and G. Wang, Adv. Energy Mater., 2020, 10, 1903591 CrossRef CAS.
- M. Mao, T. Gao, S. Hou and C. Wang, Chem. Soc. Rev., 2018, 47, 8804–8841 RSC.
- Y. Liang, R. Feng, S. Yang, H. Ma, J. Liang and J. Chen, Adv. Mater., 2011, 23, 640–643 CrossRef CAS PubMed.
- X. Sun, P. Bonnick, V. Duffort, M. Liu, Z. Rong, K. A. Persson, G. Ceder and L. F. Nazar, Energy Environ. Sci., 2016, 9, 2273–2277 RSC.
- M. Liu, A. Jain, Z. Rong, X. Qu, P. Canepa, R. Malik, G. Ceder and K. A. Persson, Energy Environ. Sci., 2016, 9, 3201–3209 RSC.
- H. D. Yoo, Y. Liang, H. Dong, J. Lin, H. Wang, Y. Liu, L. Ma, T. Wu, Y. Li, Q. Ru, Y. Jing, Q. An, W. Zhou, J. Guo, J. Lu, S. T. Pantelides, X. Qian and Y. Yao, Nat. Commun., 2017, 8, 339 CrossRef PubMed.
- X. Sun, P. Bonnick and L. F. Nazar, ACS Energy Lett., 2016, 1, 297–301 CrossRef CAS.
- Y. Gu, Y. Katsura, T. Yoshino, H. Takagi and K. Taniguchi, Sci. Rep., 2015, 5, 12486 CrossRef CAS PubMed.
- D. Tao and F. Xu, Mater. Lett., 2021, 300, 130221 CrossRef CAS.
- M. D. Levi, E. Lancri, E. Levi, H. Gizbar, Y. Gofer and D. Aurbach, Solid State Ionics, 2005, 176, 1695–1699 CrossRef CAS.
- G. S. Suresh, M. D. Levi and D. Aurbach, Electrochim. Acta, 2008, 53, 3889–3896 CrossRef CAS.
- C. Du, Z. Han, H. Peng, J. Tian, X. Yang, T. Xia, X. Ma, Y. Zhu and C. Cao, J. Power Sources, 2022, 546, 231673 CrossRef CAS.
- D. Chen, Y. Zhang, X. Li, J. Shen, Z. Chen, S. Cao, T. Li and F. Xu, Chem. Eng. J., 2020, 384, 123235 CrossRef CAS.
- Y. Cao, Y. Zhu, C. Du, X. Yang, T. Xia, X. Ma and C. Cao, ACS Nano, 2022, 16, 1578–1588 CrossRef CAS PubMed.
- M. Mao, X. Ji, S. Hou, T. Gao, F. Wang, L. Chen, X. Fan, J. Chen, J. Ma and C. Wang, Chem. Mater., 2019, 31, 3183–3191 CrossRef CAS.
- Z. Li, B. P. Vinayan, P. Jankowski, C. Njel, A. Roy, T. Vegge, J. Maibach, J. M. G. Lastra, M. Fichtner and Z. Zhao-Karger, Angew. Chem., Int. Ed., 2020, 59, 11483–11490 CrossRef CAS PubMed.
- S. Dey, J. Lee, S. Britto, J. M. Stratford, E. N. Keyzer, M. T. Dunstan, G. Cibin, S. J. Cassidy, M. Elgaml and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 19588–19601 CrossRef CAS PubMed.
- E. D. Grayfer, E. M. Pazhetnov, M. N. Kozlova, S. B. Artemkina and V. E. Fedorov, ChemSusChem, 2017, 10, 4805–4811 CrossRef CAS PubMed.
- Y. Wang, Z. Liu, C. Wang, X. Yi, R. Chen, L. Ma, Y. Hu, G. Zhu, T. Chen, Z. Tie, J. Ma, J. Liu and Z. Jin, Adv. Mater., 2018, 30, 1802563 CrossRef PubMed.
- M. A. Wright, T. W. Surta, J. A. Evans, J. Lim, H. Jo, C. J. Hawkins, M. Bahri, L. M. Daniels, R. Chen, M. Zanella, L. G. Chagas, J. Cookson, P. Collier, G. Cibin, A. V. Chadwick, M. S. Dyer, N. D. Browning, J. B. Claridge, L. J. Hardwick and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2024, 63, e202400837 CrossRef CAS PubMed.
- M. Arsentev, A. Missyul, A. V Petrov and M. Hammouri, J. Phys. Chem. C, 2017, 121, 15509–15515 CrossRef CAS.
- K. Taniguchi, Y. Gu, Y. Katsura, T. Yoshino and H. Takagi, Appl. Phys. Express, 2016, 9, 11801 CrossRef.
- A. M. Abakumov, S. S. Fedotov, E. V. Antipov and J.-M. Tarascon, Nat. Commun., 2020, 11, 4976 CrossRef CAS PubMed.
- S. Oh, C. Woo, J. Ahn, T. Y. Kim, X. Dong, Y. Kim, K. H. Choi, S. Chae, X. Zhang, H.-S. Bang, J. Kang, J. Jeon, H.-S. Oh, W.-S. Yoon, H. K. Yu and J.-Y. Choi, ACS Appl. Mater. Interfaces, 2023, 15, 55745–55752 CrossRef CAS PubMed.
- S. Hwang, S. Oh, W. Choi, S. Chae, J. Ahn, W. Lee, H.-S. Oh, J.-Y. Choi and W.-S. Yoon, J. Alloys Compd., 2024, 980, 173541 CrossRef CAS.
- L. Liu, S. Xu, F. Tang, M. Wu, W. Yang, C. Xu and X. Rui, Chem. Commun., 2023, 59, 11365–11368 RSC.
- S. Hou, X. Ji, K. Gaskell, P.-F. Wang, L. Wang, J. Xu, R. Sun, O. Borodin and C. Wang, Science, 2021, 374, 172–178 CrossRef CAS PubMed.
- N. Tartoni, S. P. Thompson, C. C. Tang, B. L. Willis, G. E. Derbyshire, A. G. Wright, S. C. Jaye, J. M. Homer, J. D. Pizzey and A. M. T. Bell, J. Synchrotron Radiat., 2007, 15, 43–49 CrossRef PubMed.
- S. P. Thompson, J. E. Parker, J. Potter, T. P. Hill, A. Birt, T. M. Cobb, F. Yuan and C. C. Tang, Rev. Sci. Instrum., 2009, 80, 75107 CrossRef CAS PubMed.
- A. A. Coelho, J. Appl. Crystallogr., 2000, 33, 899–908 CrossRef CAS.
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallogr.–Cryst. Mater., 2014, 229, 345–352 CrossRef.
- S. van Smaalen, L. Palatinus and M. Schneider, Acta Crystallogr., Sect. A: Found. Crystallogr., 2003, 59, 459–469 CrossRef PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- L. L. Wong, K. C. Phuah, R. Dai, H. Chen, W. S. Chew and S. Adams, Chem. Mater., 2021, 33, 625–641 CrossRef CAS.
- A. J. Dent, G. Cibin, S. Ramos, A. D. Smith, S. M. Scott, L. Varandas, M. R. Pearson, N. A. Krumpa, C. P. Jones and P. E. Robbins, J. Phys.: Conf. Ser., 2009, 190, 12039 CrossRef.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. Abouimrane, J. Ding and I. J. Davidson, J. Power Sources, 2009, 189, 693–696 CrossRef CAS.
- I. Shterenberg, M. Salama, H. D. Yoo, Y. Gofer, J.-B. Park, Y.-K. Sun and D. Aurbach, J. Electrochem. Soc., 2015, 162, A7118 CrossRef CAS.
- Y. Wang, Z. Liu, C. Wang, X. Yi, R. Chen, L. Ma, Y. Hu, G. Zhu, T. Chen, Z. Tie, J. Ma, J. Liu and Z. Jin, Adv. Mater., 2018, 30, 1802563 CrossRef PubMed.
- S. Dey, J. Lee, S. Britto, J. M. Stratford, E. N. Keyzer, M. T. Dunstan, G. Cibin, S. J. Cassidy, M. Elgaml and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 19588–19601 CrossRef CAS PubMed.
- K. Taniguchi, Y. Gu, Y. Katsura, T. Yoshino and H. Takagi, Appl. Phys. Express, 2016, 9, 11801 CrossRef.
- S. Hou, X. Ji, K. Gaskell, P.-F. Wang, L. Wang, J. Xu, R. Sun, O. Borodin and C. Wang, Science, 2021, 374, 172–178 CrossRef CAS PubMed.
- E. Lancry, E. Levi, Y. Gofer, M. D. Levi and D. Aurbach, J. Solid State Electrochem., 2005, 9, 259–266 CrossRef CAS.
- N. Phattharasupakun, M. M. E. Cormier, E. Lyle, E. Zsoldos, A. Liu, C. Geng, Y. Liu, H. Li, M. Sawangphruk and J. R. Dahn, J. Electrochem. Soc., 2021, 168, 90535 CrossRef CAS.
- S. G. Lim, M. S. Kwon, T. Kim, H. Kim, S. Lee, J. Lim, H. Kim and K. T. Lee, ACS Appl. Mater. Interfaces, 2022, 14, 33129 Search PubMed.
- P. Dotzel, H. Schäfer and G. Schön, Z. Anorg. Allg. Chem., 1976, 426, 260–268 CrossRef CAS.
- K. Wu, X. Su, Z. Yang and S. Pan, Dalton Trans., 2015, 44, 19856–19864 RSC.
- W. Klingen, G. Eulenberger and H. Hahn, Sci. Nat., 1968, 55, 229–230 CrossRef CAS.
- J. Glinnemann, H. E. King Jr, H. Schulz, Th. Hahn, S. J. La Placa and F. Dacol, Z. Kristallogr., 1992, 198, 177–212 CrossRef CAS.
- L. L. Wong, K. C. Phuah, R. Dai, H. Chen, W. S. Chew and S. Adams, Chem. Mater., 2021, 33, 625–641 CrossRef CAS.
- Y. Xu, X. Deng, Q. Li, G. Zhang, F. Xiong, S. Tan, Q. Wei, J. Lu, J. Li, Q. An and L. Mai, Chem, 2019, 5, 1194–1209 CAS.
- G. F. Koster, J. O. Dimmock and R. G. Wheeler, The Properties of the Thirty-Two Point Groups, The MIT Press, Cambridge, 1963 Search PubMed.
- S. L. Altmann and P. Herzig, Point-group Theory Tables, Clarendon Press, Oxford, 1994, 704 Search PubMed.
- J. Lee, B. J. Kim, Y. K. Chung, W. G. Lee, I. J. Choi, S. Chae, S. Oh, J. M. Kim, J. Y. Choi and J. Huh, J. Raman Spectrosc., 2020, 51, 1100–1107 CrossRef CAS.
- Y. Matsuda, N. Kuwata, T. Okawa, A. Dorai, O. Kamishima and J. Kawamura, Solid State Ionics, 2019, 335, 7–14 CrossRef CAS.
- J. F. Watts and J. Wolstenholme, An Introduction to Surface Analysis by XPS and AES, John Wiley & Sons, 2019 Search PubMed.
- C. Du, W. Younas, Z. Wang, X. Yang, E. Meng, L. Wang, J. Huang, X. Ma, Y. Zhu and C. Cao, J. Mater. Chem. A, 2021, 9, 3648–3656 RSC.
- T. G. Ulusoy Ghobadi, B. Patil, F. Karadas, A. K. Okyay and E. Yilmaz, ACS Omega, 2017, 2, 8319–8329 CrossRef CAS PubMed.
- S. Chae, C. Woo, G. H. Gu, T. Y. Kim, J. Jeon, H. J. Kwon, S. Oh, K. H. Choi, X. Dong, J. Ahn, G. Asghar, H.-S. Oh, H. K. Yu, H. W. Kim and J.-Y. Choi, ACS Appl. Nano Mater., 2023, 6, 16242–16252 CrossRef CAS.
- K.-Y. Hwa, A. Ganguly, A. Santhan and T. S. Kanna Sharma, Chemosphere, 2021, 282, 130874 CrossRef CAS PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy, Perkin-Elmer, 1981, vol. 3 Search PubMed.
- X. Wang, S. Zhang, X. Wang, Y. Su, Y. Qiu and Z. Zhang, Nat. Commun., 2014, 5, 1–7 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04469j |
| This journal is © The Royal Society of Chemistry 2024 |