
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuneable and efficient manufacturing of Li-ion battery separators using photopolymerization-induced phase separation†
Samuel
Emilsson
 a,
Göran
Lindbergh
b and
Mats
Johansson
*a
a,
Göran
Lindbergh
b and
Mats
Johansson
*a
aDivision of Coating Technology, Department of Fibre and Polymer Technology, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: matskg@kth.se
bDivision of Applied Electrochemistry, Department of Chemical Engineering, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden
First published on 12th October 2024
Abstract
In an effort to increase the thermomechanical stability of lithium-ion battery separators, thermoset membranes (TMs) are a viable alternative to commercial polyolefin separators. We present an efficient and scalable method to produce thin TMs via photopolymerization-induced phase separation (PIPS) in ambient conditions. The pore size is controllable and tuneable by varying the ratio between propylene carbonate (PC) and tetraethylene glycol (TEG) as porogens. The TMs maintain dimensional stability above 200 °C and display sufficient mechanical stiffness. By incorporating a small amount of a thiol monomer, the brittleness of the TMs was suppressed, and a high Young's modulus was achieved (880 MPa). The ionic conductivity of the optimized TMs was around 1 mS cm−2, with a low MacMullin number, NM (4.9). In symmetrical Li/Li cells, the TMs behaved similarly to the commercial PE reference, effectively suppressing short circuits for 1000+ hours although continuous overpotential build-up and electrolyte consumption eventually led to cell failure. In LiFePO4/Li half-cells, similar rate capabilities were achieved for the TMs compared to the reference showing its viability as a separator material.
1. Introduction
To meet the ambitious climate goals set up by the UN Paris Climate Accords, decarbonisation through electrification plays a key role. For this, the demand for batteries is expected to grow rapidly, with the automotive industry leading the way. This rapid expansion of the automotive industry puts heightened demands on batteries which should have high energy density, but also increased safety and longevity.1 Furthermore, the production of these batteries should be done in a sustainable and affordable way, at large scale. This puts rather heavy constraints on new battery materials development, where these factors should be considered.An often-overlooked aspect of materials development for batteries is the separator. The main purpose of the separator is to prevent electrical and physical contact between the electrodes while its porous structure allows an electrolyte (typically liquid) to transport ions. Conventionally, the separator is therefore a passive component. Despite this, it plays a vital role in the safety and performance of the battery. A separator should have low ionic resistance, high wettability, good mechanical and thermal stability.2,3 In this study we aim to develop a novel type of separator based on a thermoset polymer that meets these requirements, and can also be produced easily at scale.
The current state-of-the-art are microporous thermoplastic polyolefin separators (either polyethylene (PE) or polypropylene (PP) based). They are produced at large scale at a reasonable cost and display several advantages such as low thickness (≈20 μm) and high chemical and mechanical stability. However, they typically suffer from poor wettability and thermal stability due to their moderate melting point (<170 °C).4 To counteract this safety issue, tri-layer PP/PE/PP separators have been developed with a shutdown mechanism i.e. the middle PE layer melts first and closes the pores. However, this tri-layer setup leads to a decrease in power capabilities. In addition, the polyolefin separators that are power optimized (high ionic conductivity) have lower mechanical properties, meaning a compromise is typically made between these different properties.
A variety of alternative separator technologies have been reported in the literature.3,5,6 Firstly, multiple surface treatment strategies have been developed to increase electrolyte wettability and thermal stability of polyolefin separators, which has also been implemented industrially.5 Several ceramic coatings (such as aluminum oxide) have been employed, leading to lower thermal shrinkage at 150 °C and a higher affinity to the liquid electrolyte, boosting the ionic conductivity.5,7,8 High energy radiation-induced oxidation is also a technique that has been used to improve the surface properties of the separators.9 Several nonwoven membrane materials have also been explored. Cellulose-based separators can exhibit excellent ionic conductivities and thermal stability, but a challenge has been to control porosity for longer-term cycling stability in a scalable way.10 In addition, a multitude of separators developed via electrospinning (such as PET, PAN and PI) have been explored.5
Novel separators have also shown the possibility to enhance the performance of next generation batteries.11 For instance, by increasing cycle life of Li-metal batteries by suppressing lithium dendrite growth.12,13 A limitation with these studies is the use of traditional liquid electrolytes that ultimately degrade over time and where safety is still of concern. However, separators can also play a role as mechanical support in soft polymer or gel electrolytes, since thin separators can be achieved while still maintaining mechanical stability. Therefore, several studies have infused polymer electrolytes into separators, displaying better stability.14–17 For these applications, the dimensional stability at high temperatures increases in importance, allowing for safe high temperature operation.
A crucial aspect in the development of novel separator materials, is the scalability of their production. For polyolefin separators, two production pathways have been established, dry and wet processing. In wet processing, plasticizers are added to PE and films are biaxially stretched. Solvents are then used to remove the plasticizers, and the films are dried. Dry processing involves extruding polyolefin melts, annealing and uniaxially stretching. Celgard's PP and trilayer PP/PE/PP separators (often used in literature) are made this way.18 This method is attractive from a cost and environmental standpoint, since it doesn't require solvents. Despite this, wet processing makes up a majority of the market share thanks to the versatility of the process. In addition to this, both the wet and dry processes may require additional surface treatment steps to increase wettability and thermal stability.18,19 Therefore, developing novel methods for separator production that are versatile but still efficient and sustainable is highly attractive.
Bicontinuous polymer monoliths formed via polymerization-induced phase separation (PIPS) are a promising alternative. First developed for separation media, the principle behind PIPS involves the mixture of monomers with porogenic solvents that, when polymerized, phase separate to form a porous structure.20–22 By tuning the monomer(s) and porogenic solvent(s) structure and content, a wide variety of morphologies and porosities can be achieved.23,24 In addition, the resin mixture can be filled or infused into molds to achieve porous monoliths in a wide variety of geometries. By using crosslinking monomers, porous thermosets are obtained, which are known for their high chemical and thermal stabilities.25
In the context of batteries, several electrolyte systems have been developed using PIPS. Schulze et al. used PIPS to form polymer electrolytes with a structural phase and an ion-conducting phase.26 PIPS has also proven useful for creating electrolytes with good mechanical properties for structural power applications.27,28 Shirshova et al. has used it to form a structural supercapacitor electrolyte with a porous epoxy phase and an ionic liquid phase.29,30 In our research group, structural battery electrolytes have been developed by combining a dimethacrylate monomer with carbonate solvents, forming a high modulus electrolyte that has been used in structural batteries.31–36
PIPS has also been used in developing new separator materials. Sakakibara et al. developed an epoxy-based separator using low molecular poly(ethylene oxide) as a porogen.37 The pore size was also engineered by introducing a block copolymer surfactant. Li et al. used a similar system, but introduced ceramic particles (LLTO) into the structure to increase ionic transport properties.13 Recently, Manly et al. used UV-induced PIPS to form acrylate-based separators with ethylene carbonate (EC) as a porogen which could be used in a battery without washing away any solvent, but thereby requires production in inert conditions.38,39
In the present study, UV-induced PIPS is used to create methacrylate-based thermoset membranes (TM). A scalable process was developed in which UV-curing can be used in a continuous fashion at ambient conditions. In a first investigation, a combination of tetraethylene glycol (TEG) and propylene carbonate (PC) as cheap and recyclable porogens were used. By using different combinations of these porogens, a simple way of tuning the morphology of the TMs is shown. The TMs also display high thermomechanical stability, with a dimensional stability above 200 °C. In a second optimization, the addition of a small amount of a thiol monomer is presented to increase processability of the TMs. The toughness of the TMs increased significantly, making it possible to produce thin membranes (<30 μm). The TMs are compared to a commercial PE separator, displaying similar cycling performance in symmetrical Li/Li cells and towards commercial electrodes.
2. Experimental
2.1. Materials
Bisphenol A ethoxylate dimethacrylate (Mn = 540 g mol−1) was provided by Sartomer Company, Europe. Tris(2-(3-mercaptopropionyloxy)ethyl)isocyanurate (TEMPIC) was provided by Bruno Bock GmbH. Irgacure 651 (2,2-dimethoxy-1,2-diphenylethan-1-one) was purchased from BASF. Tetra(ethylene glycol) (99%) was purchased from Aldrich. Poly(vinyl alcohol) (PVA) (99+% hydrolyzed), propylene carbonate (PC) (99.7%) was purchased from Sigma-Aldrich. Battery grade 1 M lithium hexafluorophosphate (LiPF6) in ethylene carbonate (EC)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :diethyl carbonate (DEC) (1:1 v/v) and lithium ribbons (99.9%, 0.75 mm thick) were purchased by Sigma-Aldrich. A commercial PE separator was used as reference material.
:diethyl carbonate (DEC) (1:1 v/v) and lithium ribbons (99.9%, 0.75 mm thick) were purchased by Sigma-Aldrich. A commercial PE separator was used as reference material.
2.2. Membrane fabrication procedure
Prior to the fabrication, glass plates were coated with poly(vinyl alcohol) (PVA) solution. A PVA solution was prepared by dissolving 1 wt% PVA in deionized water for 2 hours at 90 °C. The glass was oxygen plasma treated for 2 minutes to increase wettability and an applicator was used to apply a thin layer (50 μm) of the PVA solution. The glass plates were then dried at 120 °C for 1 h.A precursor resin mixture was made by combining a fixed ratio of tetraethylene glycol (TEG) and propylene carbonate (PC) with the monomer bisphenol A ethoxylate dimethacrylate (BMA, Mn = 540 g mol−1) and the photo initiator Irgacure 651 (0.1 wt% of monomer). In some cases, the thiol monomer tris(2-(3-mercaptopropionyloxy)ethyl)isocyanurate (TEMPIC) was also added. See Table 1 for the different compositions that were investigated. The mixture was sonicated to remove bubbles.
| Sample | TEG | PC | BMA | TEMPIC |
|---|---|---|---|---|
| a Compositions of all formulations used in the initial screening test can be found in Table S1. | ||||
| TEGPC-1-40% | 0.2 | 0.2 | 0.6 | 0 |
| TEGPC-1-50% | 0.25 | 0.25 | 0.5 | 0 |
| TEGPC-2-40% | 0.27 | 0.13 | 0.6 | 0 |
| TEGPC-2-50% | 0.33 | 0.17 | 0.5 | 0 |
| TEGPC-4-40% | 0.32 | 0.08 | 0.6 | 0 |
| TEGPC-4-50% | 0.4 | 0.1 | 0.5 | 0 |
| TEGPC-4-40%-10T | 0.4 | 0.1 | 0.54 | 0.06 |
| TEGPC-2-50%-10T | 0.33 | 0.17 | 0.45 | 0.05 |
Using a syringe, a few droplets of the mixture was applied to the PVA-coated glass. Spacers were used to achieve the desired thickness (Kapton foil for the 20 μm membranes, clear tape for 50 μm membranes, and PTFE films for 100, 250 and 500 μm). A second PVA coated glass plate was added on top and the mixture, and two clamps were added to each side. The samples were cured using a FireJet FJ800 LED-UV lamp (60 W, 365 nm) for 120 seconds. Thicker films were flipped and cured for an additional 120 seconds.
The TMs were subsequently submerged in deionized water and gently detached from the glass plates. The TMs were kept in deionized water at 50 °C for 2 h followed by at least 12 h in room temperature to remove any residual solvent. Finally, the TMs were dried under vacuum for 16 h at 120 °C.
:PC ratio in the porogen mixture. This is followed by the porogen content (wt%) in relation to the monomer(s). Finally, the last number represents the content (wt%) of thiol monomer (see Table 1).
2.3. Materials characterization
The thermal stability of the TMs was evaluated using thermogravimetric analysis (TGA) on a TGA 1 from Mettler Toledo. The TMs (10 mg) were heated from 30 to 600 °C at a heating rate of 10 °C min−1 under nitrogen flow (50 mL min−1).
Uniaxial tensile tests were carried on a DMA Q800 from TA Instruments set at 25 °C. Specimens that were 20 × 4 mm were cut out from 100 μm thick TMs. Five specimens were tested for each formulation. An initial gap of 7 mm was used and a 10% min−1 strain rate was used. The Young's modulus was calculated from the linear region up to 0.5%.
 | (1) |
2.4. Electrochemical characterization
For capacity retention tests, LFP electrodes (1 × 2.1 cm) were cut and a slightly oversized Li metal (1.2 × 2.3) was used. For the long-term cycling tests, a 16 mm disk of LFP was used, and an 18 mm disk of Li. First, the LFP electrode was placed inside the pouch, and 100 μL of liquid electrolyte (LP40) was added to wet the electrode. A separator/TM was then placed on top of the electrode (2 × 3) and an additional 100 μL of liquid electrolyte was added. Subsequently, the lithium metal was placed on top and the pouch was vacuum sealed. The LFP electrodes were purchased from Custom Cells Itzehoe GmbH and had an areal capacity of 2 mA h cm−2, corresponding to an active mass loading of 13.3 mg cm−2 (90% active mass).
For capacity retention tests, the cells were cycled inside a climate-controlled room (22 °C) using a Neware battery testing system in a potential range of 2.6 and 3.9 V vs. Li+/Li. Two formation cycles at C/10 were followed by 5 cycles of C/5, C/2, and 1C followed by an additional 5 cycles at C/5. 1C corresponded to 150 mA g−1 based on supplier specifications.
For long-term cycling, the cells were cycled using a VMP3 potentiostat from Biologic was used. Cells were cycled in the potential range of 2.6 and 3.9 V vs. Li+/Li. Two formation cycles at C/10 were followed by cycling at C/5 for the subsequent cycles.
3. Results and discussion
3.1. Separator manufacturing
When designing the manufacturing process and formulations in this study, the scalability was of particular interest. Firstly, the choice of bisphenol A ethoxylate dimethacrylate (BPA-DMA) monomer stemmed from its combination of attractive properties, but also since it is produced at scale. The monomer has also been used for both UV and thermally-initiated PIPS previously, showing promising performance as hybrid electrolyte.31,32 BPA-DMA contains a rigid aromatic structure which provides high mechanical rigidity, an important property when designing separators. The ethoxylate chains, provide some flexibility, which leads to the resin having a lower viscosity, making it easier to work with. In addition, the ethoxylate chains have the potential to interact with the liquid electrolyte, potentially increasing the electrolyte uptake. Finally, the use of methacrylates instead of e.g. acrylates stems from the higher glass transition temperature (Tg) typically seen for methacrylates, giving the separator a higher thermomechanical stability.41 For polymerization-induced phase separation (PIPS) the choice of porogen(s) are key for the pore structures that can be obtained. Tetra(ethylene glycol) (TEG) has previously shown to form large pore morphologies when combined with epoxy systems.37 Propylene carbonate (PC) has also been used, but generally leading to smaller pore size.29 Thereby, a combination of these porogens were used to tune the morphology. Furthermore, both TEG and PC are considered safe and cheap solvents.42,43 The high boiling points (240 °C for PC and 314 °C for TEG) also makes recovery of the solvents by distillation of the water during the washing process feasible.42,43 UV-curing is also a scalable method which is suitable for manufacturing of films and coatings in a continuous process.44Fig. 2 shows a process schematic. An initial mixture of monomer, initiator and porogens is spread and sandwiched between two glass plates with a spacer to control the thickness. To ensure membranes with porous interfaces, a PVA layer was coated onto the glass. This strategy was previously used by Sakakibara et al. when making membranes from an epoxy resin combined with polyethylene glycol 200 (PEG200), where the polar hydroxyl groups on PVA was proposed to attract PEG200 to the surface.37 They noted that when membranes were made without coating the glass, a non-porous skin was obtained. Interestingly, despite both changing polymer system (from epoxy to methacrylate) and curing method (from thermal to UV-curing), a similar non-porous skin was obtained for our system as well (see Fig. S2†). Previous work on UV-driven PIPS has also suggested asymmetric porosity when comparing the top and bottom of the film.45 For this study, symmetrical porosity was observed when using PVA-coated glass as substrates.
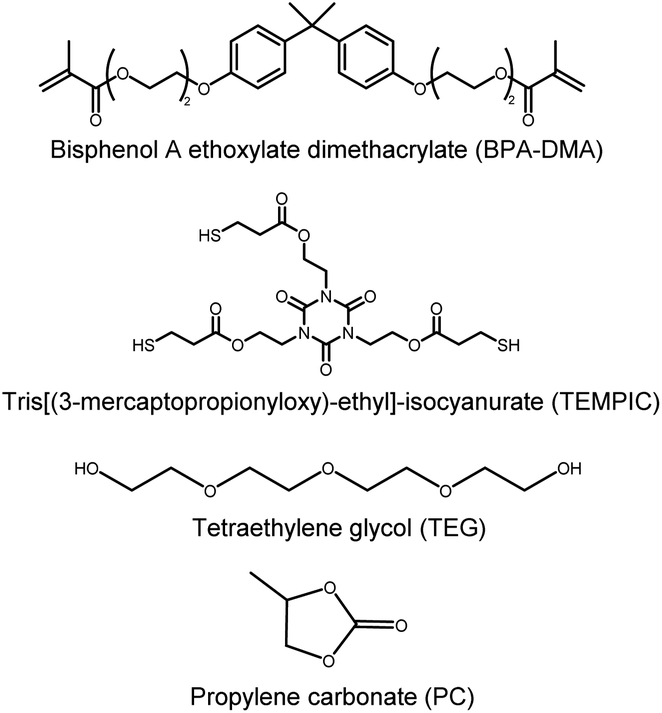 | ||
| Fig. 1 Molecular structures of monomers and porogens used in the present study. | ||
 | ||
| Fig. 2 Process schematic of thermoset membrane fabrication. | ||
The resin mixtures were rapidly cured (2 × 120 s) at ambient conditions. An opaque film was obtained, implying that a phase separation had occurred. Fig. 3 shows the FTIR spectra of one formulation, TEGPC-4-40%, before and after curing. A clear reduction of the vinyl stretching mode at 1637 cm−1 between the resin and cured film can be seen. A monomer conversion of ∼80% was calculated, similar to a previous study when using the same monomer.32 The limited conversion is likely due to vitrification occurring, due to the high Tg of the dimethacrylate, lying far above the curing temperature. Unreacted monomer could potentially affect the electrochemical stability of the separator, and therefore a high conversion is preferred. However, since each monomer contains two methacrylate groups, it is likely that a majority of the remaining unreacted groups (∼20%) are on partially reacted pendant monomers rather than free monomers.46,47 After curing, the membranes detached from the glass plates in water and the porogens could be washed away. After drying under vacuum, uniformly porous membranes of different thickness were obtained, depending on the spacer used during curing. The absence of the peak at 1800 cm−1 related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in PC can be seen in Fig. 3, and the disappearance of the peak at 3400 cm−1 related to the –OH groups in TEG can be seen in Fig. S3,† showing that the washing and drying process were successful i.e. no porogen was trapped in closed voids.
O group in PC can be seen in Fig. 3, and the disappearance of the peak at 3400 cm−1 related to the –OH groups in TEG can be seen in Fig. S3,† showing that the washing and drying process were successful i.e. no porogen was trapped in closed voids.
 | ||
| Fig. 3 FTIR spectra of TEGPC-4-40% before and after curing, and after washing and drying the membrane. The peak at 1637 cm−1 represents the vinyl stretching mode. The peak at 1800 cm−1 represents the carbonyl stretching on PC. | ||
3.2. Effect of porogen structure and content
A screening of various compositions was performed to see how the effect of porogen content and ratio of the respective porogens affected the morphology of the porous TMs, as seen in Fig. 4. From this, two clear trends can be observed. Firstly, when increasing the porogen content, the TMs become less transparent, indicating that they contain larger pores in a dimension that scatter light. This is an expected phenomenon, well in line with literature.48,49 Secondly, the TMs become less transparent when increasing the concentration of TEG in the composition, indicating that larger pores are formed when using TEG as a porogen compared to PC. This suggests that TEG is a rather poor solvent for BPA-DMA compared to PC. Indeed, when mixing BPA-DMA purely with TEG, an immiscible solution was obtained already before curing. However, when combining the two solvents in different composition, a variety of morphologies could be obtained, some of which can be seen in the micrographs in Fig. 4. | ||
| Fig. 4 (Left) Digital photographs of different porous membranes made using varying contents the porogens TEG and PC. (Right) Cross-sectional SEM micrographs of the freeze fractured films of selected formulations, showing a variation in pore size. | ||
At the lowest porogen content (30 wt%) and TEGPC-1-30%, a completely transparent film is obtained which suggests that no proper phase separation has occurred. When increasing the TEG content to TEGPC-4-30%, a porous film is obtained (see micrograph in Fig. S5†). However, the porosity of all these samples are too low to be of relevance as separator materials. On the other hand, the highest porogen content (60 wt%) results in brittle films which fell apart when removing them from the glass. These compositions could be useful for applications where high porosities are important, e.g. absorbants. Ultimately, the compositions containing 40 and 50 wt% were therefore used for further analysis as separator materials.
Although the cross-sectional micrographs seen in Fig. 4 give a good indication of the morphologies of the different compositions relative to each other, it is important to note that they are not fully representative of the true porosity. A previous study on similar systems showed that the morphology of freeze fractured samples, like the samples in this study, differed extensively from cross sectional images of samples that have been broad ion beam milled and then analysed with SEM.35 Furthermore, as previously mentioned, the surfaces of the TMs are of vital importance to ensure good ion transport at the interfaces. Therefore, the surfaces of selected TMs were also examined (Fig. 5). The surface of TEGPC-2-40% exhibits smaller pores than TEGPC-4-40%, and both display uniformly distributed pores that are similar in scale to the commercial PE reference.
 | ||
| Fig. 5 Top view SEM micrographs of the surface of (a) PE reference material (b) TEGPC-2-40% and (c) TEGPC-4-40% at 10000× magnification. | ||
3.3. Thermal and mechanical properties
Apart from the morphology, the thermal and mechanical properties of the TMs are of significant importance when developing separator materials. The dimensional integrity of the separator when the temperature increases is important since it ensures that a short circuit does not occur. DSC measurements were performed to examine the thermal behaviour of one the TMs, TEGPC-4-40%, compared to a commercial PE reference. Fig. 6a shows a clear melting point at 141 °C during the heating cycle, typical for separators based on PE with a high molecular weight.5 During the cooling cycle, a crystallization peak is seen at 116 °C. In contrast, no thermal transitions can be observed within the measured range for TEGPC-4-40%, due to its amorphous structure and the high crosslinking density. This indicates that the integrity of TEGPC-4-40% is maintained, even at temperatures up to 200 °C. This is also higher than common commercial PP and PP/PE/PP separators which have been analysed elsewhere.50 Previous studies have also shown that, particularly when looking at larger battery packs, the shutdown mechanism in PP/PE/PP separators is insufficient for halting a thermal runaway. Therefore, it is attractive to instead “close the gap” between the separator shrinkage/melting temperature and the battery runaway temperature (typically above 200 °C).50,51 The close relationship between the separator breakdown temperature and thermal runaway is further described by Feng et al.52Fig. 6b shows the thermal degradation of the separators using TGA analysis. TEGPC-4-40% has a lower degradation onset point (337 °C) than PE (459 °C), possibly due to the presence of heteroatoms in the polymer structure of TEGPC-4-40%. However, the degradation starts at a temperature that is higher than the battery runaway temperature, and therefore considered sufficiently high. | ||
| Fig. 6 (a) DSC thermograph with a heating rate of 10 °C min−1 and cooling rate of 5 °C min−1 and (b) TGA thermograph with a heating rate of 10 °C min−1 of a representative thermoset membrane. | ||
To gain a better understanding of the mechanical integrity of the separators, dynamic mechanical analysis (DMA) was also performed on the reference PE separator and several formulations of TMs. This is of particular relevance when characterizing highly crosslinked materials, since typically a broad glass transition temperature (Tg) is obtained which are difficult to determine using DSC. The storage modulus also gives an indication of the elastic modulus of the material. Fig. 7 shows the PE reference exhibits a relatively high storage modulus at ambient temperatures (above 1000 MPa). This is higher than several other commercial separators that have been tested previously.53 The separator also exhibits anisotropic properties, with almost double the modulus in one direction. Accurate determination of the modulus of the PE separator proved difficult due to the low thickness (20 μm), with a large spread between samples (see Fig. S6†). In all cases, however, the storage modulus decreases rapidly and a drop-off occurs already at 115 °C, below the melting point of PE. In contrast, all TM formulations exhibit a storage modulus above 100 MPa at 150 °C, which is a good indication of maintained mechanical integrity even at elevated temperatures.
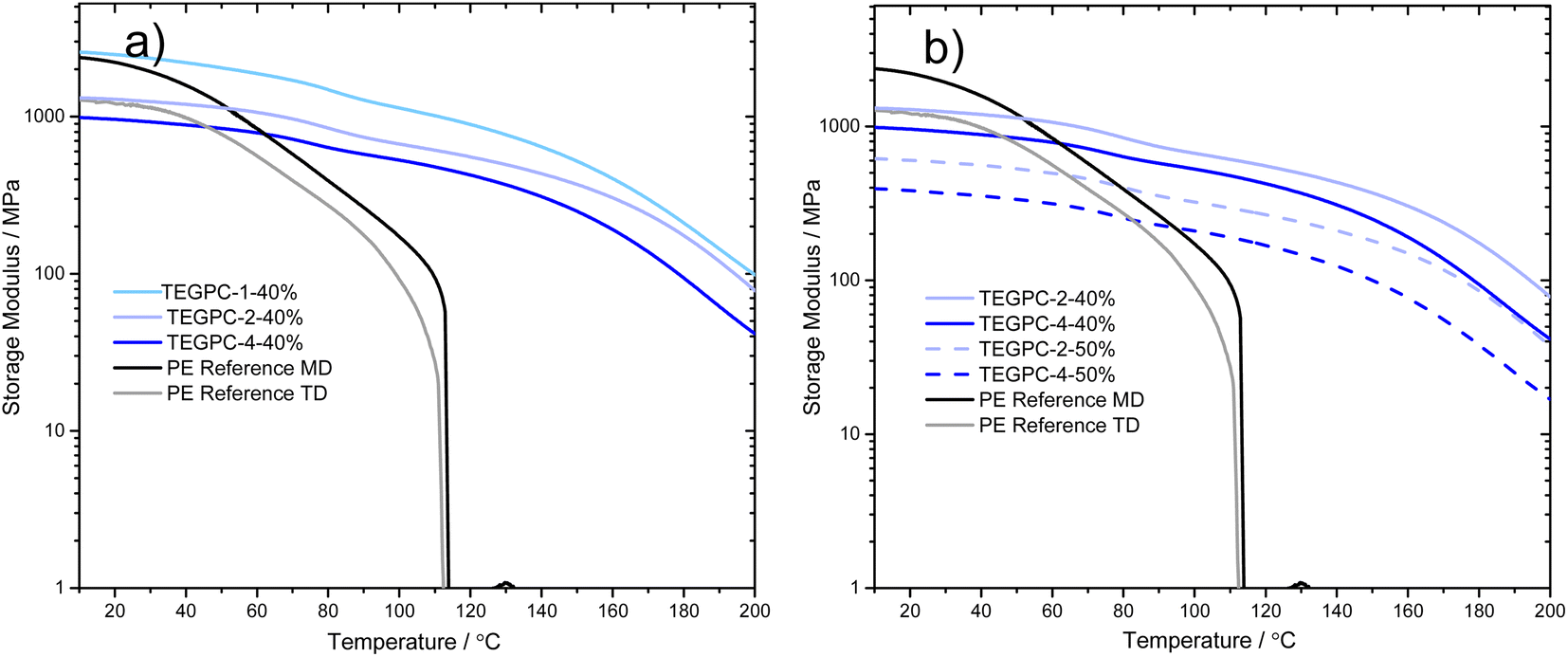 | ||
| Fig. 7 DMA thermographs for thermoset membranes made with (a) different TEG content and (b) different TEG and total porogen content. The PE reference is added for both the transversal (TD) and machine direction (MD). | ||
Fig. 7a shows various compositions where the TEG content has been varied, keeping a constant porogen content. In Fig. 7b, the effect of increasing porogen content is instead displayed. In both cases, the curves of the different compositions lie relatively parallel, mainly shifting in storage modulus. This indicates that while the different compositions affect the morphology, the polymer network remains relatively unchanged. A clear correlation between larger pore sizes (in Fig. 4) and a lower storage modulus (or elastic modulus) can also be noted, well in line with previous studies using PIPS.13,29,34,38 In Table 2, the storage modulus at 25 °C of selected compositions are listed.
| Sample | Storage modulus/MPa (25 °C) | Electrolyte uptake/% | Ionic conductivity/mS cm−1 (25 °C) | N M |
|---|---|---|---|---|
| a Calculated based on measured ionic conductivity of liquid electrolyte (EC/DEC 1 M LiPF6) – 4.9 mS cm−1. | ||||
| PE reference | 1095 (TD) | 103 | 0.44 ± 2% | 10.9 |
| TEGPC-2-40% | 1320 | 85 | 0.48 ± 2% | 10.2 |
| TEGPC-4-40% | 943 | 92 | 0.99 ± 4% | 4.9 |
| TEGPC-2-50% | 594 | 128 | 1.4 ± 9% | 3.3 |
| TEGPC-4-40%-10T | 799 | 88 | 0.99 ± 11% | 4.9 |
| TEGPC-2-50%-10T | n.m. | 125 | 1.4 ± 6% | 3.4 |
3.4. Ion transport properties
To study the ion transport properties through the membranes, they were first submerged in liquid electrolyte (LP40) to evaluate their electrolyte uptake. Proper electrolyte uptake is essential for ensuring high and uniform ion transport. The electrolyte uptake is partly due to the affinity of the polymer to the polar solvents in the electrolyte. The presence of polar ethoxylate chains in BPA-DMA are expected to improve the affinity to the electrolyte. However, a high crosslinking density generally restricts solvent absorption, due to the restricted mobility of the polymer chain. More importantly, the porosity plays a crucial role in the electrolyte uptake, as it allows electrolyte to fill the voids in the membranes. Table 2 indeed shows that the formulations made with 40% porogen have a slightly lower electrolyte uptake than the reference. However, when increasing to 50% porogen, an electrolyte uptake above 120% by weight is obtained.The effective ionic conductivity (σeff) of the electrolyte-soaked separators is presented in Table 2. TEGPC-2-40% has a similar σeff to the PE reference, while TEGPC-4-40% displays a doubling in σeff. This large increase in σeff is observed despite having a lower electrolyte uptake, suggesting that the pore size and shape lead to lower ionic resistance. TEGPC-2-50% displays the highest σeff at 1.4 mS cm−1, which is sufficient for high-power applications. As σeff is highly dependent on the liquid electrolyte conductivity, a more relevant comparison is the MacMullin number, NM, defined as:
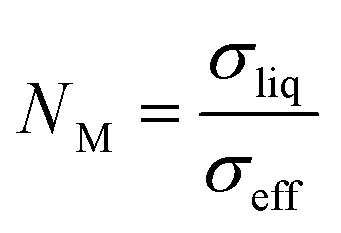 | (2) |
3.5. Effect of incorporating thiol monomer
The formulations using purely BPA-DMA as a monomer show promising characteristics, such as high thermomechanical stability and high storage modulus. The compositions with larger pores also display excellent ionic conductivities. However, the formulations with the largest pores (TEGPC-4-40% and TEGPC-2-50%) proved difficult to make thinner than 50 μm, as the films became too brittle. Ideally, the separators should be 25 μm to become competitive with commercial separators.5 To counteract this issue, the thiol co-monomer tris[(3-mercaptopropionyloxy)-ethyl]-isocyanurate (TEMPIC) was introduced (see Fig. 1). Thiol–ene photopolymerization has become an established alternative to pure (meth)acrylate photopolymerization, exhibiting some key advantages. Firstly, the addition of thiols often leads to faster reaction kinetics. The thiol–ene reaction also occurs via step-growth propagation, as opposed to chain-growth, which delays the gel point and generally leads to higher monomer conversion and a more homogeneous polymer network.54,55 Incorporating a small amount of thiol to methacrylate systems has also been shown to be beneficial in lowering shrinkage stresses during polymerization as well as the thiol-ether linkage increasing network flexibility.56 Using TEMPIC combines the advantages of thiol–ene reactions while the rigid core ensures that the modulus remains high. Thiol–ene based polymers have also been demonstrated to be chemically stable under the conditions present in lithium-ion batteries.57–59Initial tests were performed by replacing 5, 10 and 20 wt% of BPA-DMA with TEMPIC. A clear increase in monomer conversion was observed with TEGPC-4-40%-10T (containing 10 wt% TEMPIC) exhibiting >90% conversion, compared to 80% for TEGPC-4-40% (see Fig. S4†). Interestingly, despite a partial change in the monomer and polymerization mechanism, no significant changes in morphology could be observed when varying the TEMPIC content (see Fig. S8†). As a consequence, the electrolyte uptake and the ionic conductivity of formulations containing TEMPIC are very similar to the same formulations without TEMPIC (see Table 2). DMA data in Fig. S7† shows a clear decrease in Tg upon increasing TEMPIC content, due to the added flexibility of the thiol-ether linkage. Nevertheless, the sample containing 10 wt% TEMPIC still displays a Tg = 148 °C with a storage modulus above 10 MPa at 200 °C, which is expected to be sufficient to maintain dimensional stability. It should also be noted that the tanδ peak is narrower with the addition of TEMPIC (Fig. S7†), indicating that a more homogenous polymer network is obtained.54,60 This is expected to have a beneficial effect on the mechanical properties.61
For TEGPC-4-40%, the addition of 10 wt% TEMPIC made it less brittle and significantly easier to handle. This is underlined by preliminary uniaxial tensile tests were performed for TEGPC-4-40% and TEGPC-4-40%-10T, shown in Fig. 8. A clear enhancement in the tensile strength and elongation at break can be observed. A rather large spread in elongation at break is observed, which could be expected for thin porous materials where crack initiation points are built into the material. The enhancement of the tensile strength with the addition of TEMPIC is likely due to multiple factors. The flexible thiol-ether linkage and more homogenous polymer network should decrease brittleness. Rather surprisingly, the Young's modulus also increases from 631 ± 45 MPa to 882 ± 21 MPa with the addition of TEMPIC. For this, the pore homogeneity may also play a role, although a deeper analysis of the morphology is needed. Nevertheless, TEGPC-4-40%-10T exhibits excellent mechanical properties, well above commercial requirements.3 A consequence of the increased tensile strength and toughness for TEGPC-4-40%-10T is also that films could be made which were 20–30 μm thin, making them more commercially viable.
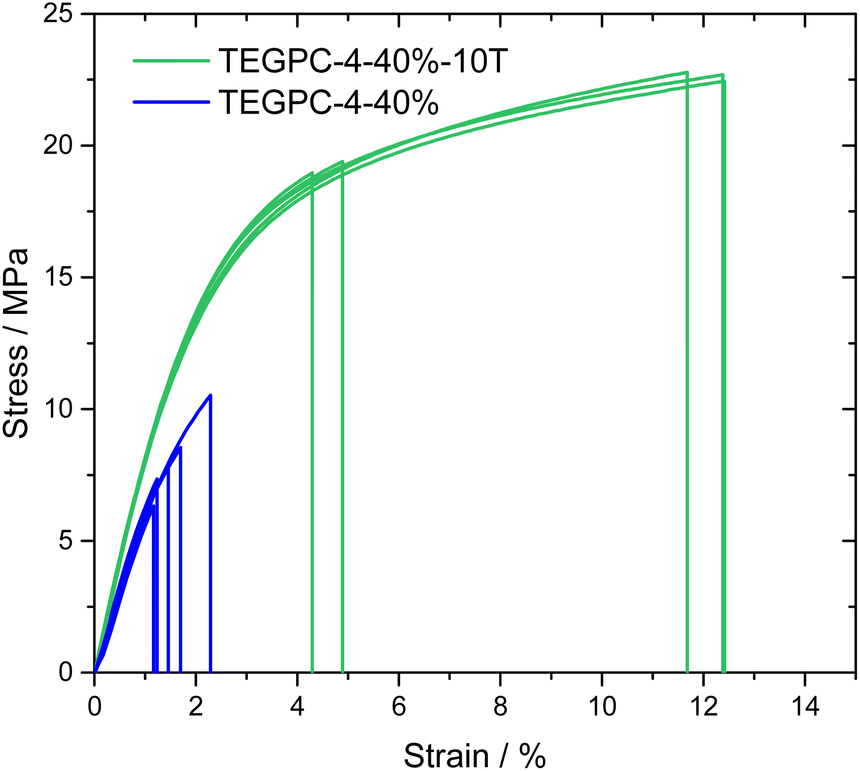 | ||
| Fig. 8 Stress–strain curves of uniaxial tensile tests for TEGPC-4-40% and TEGPC-4-40%-10T samples. | ||
3.6. Performance in Li/Li cells
For further electrochemical characterization, TEGPC-4-40% and TEGPC-4-40%-10T were compared to the PE reference. Inspired by previous work which have shown that design of separators can improve lithium plating, symmetrical Li/Li cells were constructed to analyse the plating efficiency of the separators.12,13,62 These tests are also useful to see if the TMs are sufficiently mechanically robust to suppress short circuiting via dendrite growth through the separator.Fig. 9 shows the voltage profiles for Li/Li cells containing either TEGPC-4-40% or TEGPC-4-40%-10T compared to the commercial PE reference. During the first few cycles (Fig. 9b), the initial overpotential is significantly higher for TMs, particularly TEGPC-4-40%, but decreases rapidly before stabilizing after about 10 cycles. This has previously been attributed to SEI formation,63,64 but could also indicate that there are some residual impurities (moisture, monomer, etc.) left in the TMs. The large difference between TEGPC-4-40% and TEGPC-4-40%-10T is rather unexpected, but may be due to a combination of a thicker TM and lower monomer conversion. Such a high initial overpotential could be problematic in full cells, and would require several formation cycles. After stabilizing, all three separators display stable overpotentials with a “peaking” voltage profile which has been suggested to imply that minimal dead lithium is formed.65 TEGPC-4-40% stills displays the highest overpotential, 63 mV at the middle of the 10th cycle, compared to 48 mV for TEGPC-4-40%-10T and 37 mV for PE reference. The thickness of the respective TMs is one source for the added overpotential, however, given their higher ionic conductivity compared to PE this cannot be the whole explanation. Another source of this overpotential may be the different SEI formations, leading to a higher resistance. Fig. 9c shows that the voltage profiles remain stable over 200 hours, but a slow transfer from a “peaking” voltage profile to a “arching” voltage profile seems to occur, indicating a built-up of dead lithium.65 This becomes even more clear in duplicate TEGPC-4-40%-10T and PE reference cells that were cycled for 1600 hours (Fig. S9†). These results indicate that the TM is sufficiently mechanically robust to prevent internal short-circuiting via dendrite growth when cycling at the set conditions 0.5 mA cm−1, 0.5 mA h cm−1 for 1600 hours. However, the plating of lithium ultimately still leads to depletion of the liquid electrolyte and likely growth of mossy lithium, as in the PE reference.64,65 Increasing the current density further is likely to speed up this process, as shown in previous literature.65 To summarize, the TMs seem to exhibit a slightly higher overpotential to lithium, however it does not show signs of being more susceptible to short circuiting compared to the PE reference.
 | ||
| Fig. 9 Voltage profiles of Li/Li symmetrical cell cycling at 0.5 mA cm−1 (0.5 mA h cm−1) with PE reference and two thermoset membrane formulations for (a) first 240 hours, (b) first 20 hours and (c) hours 220–240. The cells were stopped after 245 hours. | ||
3.7. Performance in LFP/Li cells
To further test the compatibility of the TMs as separators, half cells were constructed with lithium iron phosphate (LFP) electrodes. Rather often, a low cathode mass loading is used to test of novel electrolyte or separator materials in literature, which can overestimate rate capabilities and cycling stability.66 The areal capacity of the LFP electrodes used in this study was 2 mA h cm−2 (mass loading: 13 mg cm−2) which is more commercially relevant. Fig. 10a shows the capacity retention when increasing the current rate from C/10 to 1C. The obtained capacities at low C-rates are similar for both TMs and the PE reference. At 1C, the discharge capacity drops to about 125 mA h g−1 for TEGPC-4-40%-10T and the PE reference cells. TEGPC-4-40% shows a slightly lower capacity (115 mA h g−1) at 1C, which is likely due to the thickness of the TM. Nevertheless, the TMs manage to maintain a reasonable capacity at 1C and a 2 mA h cm−2, which corresponds to the rather high lithium plating current rate of 2 mA cm−2. | ||
| Fig. 10 Cycling performance of LFP/Li half cells cycled from 2.6 to 3.9 V at 22 °C (cathode mass loading = 13.3 mA h g−1). (a) Capacity retention test at different C-rates with 1C = 150 mA g−1. (b) Voltage vs. specific capacity at different C-rates, the final cycle of each step was taken (cycle 7, 12 and 17). | ||
Throughout the capacity retention test, the difference between the PE reference and TEGPC-4-40%-10T are marginal as demonstrated by the voltage profiles at different C-rates seen in Fig. 10b. Fig. S10† also shows duplicate cells that were constructed at another time point. The batch-to-batch difference is larger than the difference between the different membranes, indicating that particularly TEGPC-4-40%-10T is on par with the reference material. Finally, when returning to a lower C-rate (C/5), the capacity returns to 150 mA h g−1 which indicates that no significant irreversible capacity loss occurred during faster charging. The low initial capacity of some of the TMs in Fig. S10† indicates that the TMs are more sensitive to small variations in the preparation conditions.
TEGPC-4-40%-10T and the reference were also used in an LFP/Li half-cell that was cycled at C/5 for 50 cycles. Fig. 11 shows the discharge capacity as a function of cycle number. The cells showed similar initial capacity (150–155 mA h g−1), decreasing steadily to 125–130 mA h g−1 over 50 cycles. The capacity retention was similar, around 83% for TEGPC-4-40%-10T and 84% for the reference. Fig. 11 also shows that the coulombic efficiency (CE) for most cycles is around 98%, which is rather low, but not uncommon when using lithium metal as a counter electrode.67 This is likely mainly due to degradation of the liquid electrolyte and non-optimized experimental conditions (no additives used, CC/CC charging, etc.). Although similar, the CE is slightly lower for TEGPC-4-40%-10T, which could indicate some instabilities in this membrane. Since the difference is so marginal, this is more likely due to residual contaminants from the fabrication process. Further investigation into the long-term stability is the subject of future work.
 | ||
| Fig. 11 Cycling performance of LFP/Li half cells using a TEGPC-4-40%-10T separator (green) or PE reference (red), cycled from 2.6 to 3.9 V at 22 °C. Two formation cycles at C/10 were followed by 50 cycles at C/5. | ||
4. Conclusions
In the present study, the use of photopolymerization-induced phase separation as a scalable process to produce methacrylate-based thermoset membranes is investigated. A combination of PC and TEG as safe and cheap porogens are used. When increasing the TEG content and the total porogen content, the pore size increases. By tuning the ratio between PC and TEG, different morphologies are achieved. Due to the high crosslinking density of the methacrylate thermoset, the membranes display high thermomechanical properties. The membranes maintain dimensional stability above 200 °C, well above the temperature at which the commercial PE reference starts to melt. The study also shows that the elastic modulus of the membranes clearly decreases with increasing pore size and the formulations with the highest pore size become brittle, making it difficult to make thin films (<50 μm). To counteract this problem, a small amount of a thiol monomer (TEMPIC) was added which significantly increases the toughness of the material and made it possible to produce thinner membranes. An improvement of the Young's modulus is also achieved (880 MPa), above the industry requirement. The ionic conductivities of the different membrane formulations were similar or better than the commercial reference, with TEGPC-4-40%-10T displaying a conductivity of 1 mS cm−2 and a NM = 4.9. Selected membranes were tested in Li/Li symmetrical cells, showing sufficient mechanical stability to avoid short circuits. A slow overpotential build up due to electrolyte depletion was seen, similar to the PE reference. LFP half cells with an areal capacity of 2 mA h cm−2 were cycled at different C-rates, with TEGPC-4-40%-10T displaying a similar performance to the PE reference. In summary, this study contributes to further development of thermoset membranes as battery separators by presenting a scalable and efficient way to manufacture membranes using photopolymerization-induced phase separation in ambient conditions.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization, S. E., M. J., investigation, S. E., funding acquisition, M. J., G. L. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge financial support from BASE Battery Sweden (Vinnova grant: 2019-00064). The research group Kombatt and colleagues at the Division of Electrochemistry at KTH are acknowledged for helpful discussions.References
- A. A. Tidblad, K. Edström, G. Hernández, I. de Meatza, I. Landa-Medrano, J. Jacas Biendicho, L. Trilla, M. Buysse, M. Ierides, B. P. Horno, Y. Kotak, H.-G. Schweiger, D. Koch and B. S. Kotak, Energies, 2021, 14, 4223 CrossRef CAS.
- M. F. Lagadec, R. Zahn and V. Wood, Nat. Energy, 2018, 4, 16–25 CrossRef.
- P. Arora and Z. Zhang, Chem. Rev., 2004, 104, 4419–4462 CrossRef CAS.
- F. Wang, X. Ke, K. Shen, L. Zhu and C. Yuan, Adv. Mater. Technol., 2022, 7, 2100772 CrossRef.
- N. Lingappan, W. Lee, S. Passerini and M. Pecht, Renewable Sustainable Energy Rev., 2023, 187, 113726 CrossRef CAS.
- X. Huang, J. Solid State Electrochem., 2011, 15, 649–662 CrossRef CAS.
- C. Shi, P. Zhang, L. Chen, P. Yang and J. Zhao, J. Power Sources, 2014, 270, 547–553 CrossRef CAS.
- J. Moon, J. Y. Jeong, J. I. Kim, S. Kim and J. H. Park, J. Power Sources, 2019, 416, 89–94 CrossRef CAS.
- K. J. Kim, Y. H. Kim, J. H. Song, Y. N. Jo, J.-S. Kim and Y.-J. Kim, J. Power Sources, 2010, 195, 6075–6080 CrossRef CAS.
- H. Chen, Z. Wang, Y. Feng, S. Cai, H. Gao, Z. Wei and Y. Zhao, Chem. Eng. J., 2023, 471, 144593 CrossRef CAS.
- Y. Pan, S. Chou, H. K. Liu and S. X. Dou, Natl. Sci. Rev., 2017, 4, 917–933 CrossRef CAS.
- R. Pan, R. Sun, Z. Wang, J. Lindh, K. Edström, M. Strømme and L. Nyholm, Nano Energy, 2019, 55, 316–326 CrossRef CAS.
- M. Li, H. Li, J.-L. Lan, Y. Yu, Z. Du and X. Yang, J. Mater. Chem. A, 2018, 6, 19094–19106 RSC.
- Y. H. Chen, P. Lennartz, K. L. Liu, Y. C. Hsieh, F. Scharf, R. Guerdelli, A. Buchheit, M. Grünebaum, F. Kempe, M. Winter and G. Brunklaus, Adv. Funct. Mater., 2023, 33, 2300501 CrossRef CAS.
- C. Fu, G. Homann, R. Grissa, D. Rentsch, W. Zhao, T. Gouveia, A. Falgayrat, R. Lin, S. Fantini and C. Battaglia, Adv. Energy Mater., 2022, 12, 2200412 CrossRef CAS.
- Q. Liang, L. Chen, J. Tang, X. Liu, J. Liu, M. Tang and Z. Wang, Energy Storage Mater., 2023, 55, 847–856 CrossRef.
- J. Wan, J. Xie, X. Kong, Z. Liu, K. Liu, F. Shi, A. Pei, H. Chen, W. Chen, J. Chen, X. Zhang, L. Zong, J. Wang, L.-Q. Chen, J. Qin and Y. Cui, Nat. Nanotechnol., 2019, 14, 705–711 CrossRef CAS.
- V. Deimede and C. Elmasides, Energy Technol., 2015, 3, 453–468 CrossRef.
- H. Heimes, A. Kampker, C. Offermanns, N. Lackner, T. Elliger, V. Mussehl, S. Michaelis and J. Zienow, Production of Lithium-Ion Battery Cell Components, 2nd edn, 2023 Search PubMed.
- F. Svec, J. Chromatogr. A, 2010, 1217, 902–924 CrossRef CAS.
- C. Viklund, F. Svec, J. M. J. Fréchet and K. Irgum, Chem. Mater., 1996, 8, 744–750 CrossRef CAS.
- F. Svec and J. M. J. Frechet, Anal. Chem., 1992, 64, 820–822 CrossRef CAS.
- N. Tsujioka, N. Ishizuka, N. Tanaka, T. Kubo and K. Hosoya, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3272–3281 CrossRef CAS.
- K. Nakanishi and N. Tanaka, Acc. Chem. Res., 2007, 40, 863–873 CrossRef CAS.
- J. P. Pascault and R. J. J. Williams, Overview of thermosets: present and future, Elsevier, 2018, pp. 3–34, DOI:10.1016/b978-0-08-101021-1.00001-0.
- M. W. Schulze, L. D. McIntosh, M. A. Hillmyer and T. P. Lodge, Nano Lett., 2014, 14, 122–126 CrossRef CAS.
- L. E. Asp, M. Johansson, G. Lindbergh, J. Xu and D. Zenkert, Funct. Compos. Struct., 2019, 1, 042001 CrossRef CAS.
- M. Y. Tan, D. Safanama, S. S. Goh, J. Y. C. Lim, C. H. Lee, J. C. C. Yeo, W. Thitsartarn, M. Srinivasan and D. W. H. Fam, Chem.–Asian J., 2022, 17, e202200784 CrossRef CAS.
- N. Shirshova, A. Bismarck, E. S. Greenhalgh, P. Johansson, G. Kalinka, M. J. Marczewski, M. S. P. Shaffer and M. Wienrich, J. Phys. Chem. C, 2014, 118, 28377–28387 CrossRef CAS.
- N. Shirshova, A. Bismarck, S. Carreyette, Q. P. V. Fontana, E. S. Greenhalgh, P. Jacobsson, P. Johansson, M. J. Marczewski, G. Kalinka, A. R. J. Kucernak, J. Scheers, M. S. P. Shaffer, J. H. G. Steinke and M. Wienrich, J. Mater. Chem. A, 2013, 1, 15300 RSC.
- N. Ihrner, W. Johannisson, F. Sieland, D. Zenkert and M. Johansson, J. Mater. Chem. A, 2017, 5, 25652–25659 RSC.
- L. M. Schneider, N. Ihrner, D. Zenkert and M. Johansson, ACS Appl. Energy Mater., 2019, 2, 4362–4369 CrossRef CAS.
- W. Johannisson, N. Ihrner, D. Zenkert, M. Johansson, D. Carlstedt, L. E. Asp and F. Sieland, Compos. Sci. Technol., 2018, 168, 81–87 CrossRef CAS.
- S. Emilsson, V. Vijayakumar, J. Mindemark and M. Johansson, Electrochim. Acta, 2023, 449, 142176 CrossRef CAS.
- M. Cattaruzza, Y. Fang, I. Furó, G. Lindbergh, F. Liu and M. Johansson, J. Mater. Chem. A, 2023, 11, 7006–7015 RSC.
- S. Duan, M. Cattaruzza, V. Tu, R. M. Auenhammer, R. Jänicke, M. K. G. Johansson, F. Liu and L. E. Asp, Commun. Mater., 2023, 4, 49 CrossRef CAS.
- K. Sakakibara, H. Kagata, N. Ishizuka, T. Sato and Y. Tsujii, J. Mater. Chem. A, 2017, 5, 6866–6873 RSC.
- A. J. Manly and W. E. Tenhaeff, J. Mater. Chem. A, 2022, 10, 10557–10568 RSC.
- A. J. Manly and W. E. Tenhaeff, Electrochim. Acta, 2022, 425, 140705 CrossRef CAS.
- J. Landesfeind, J. Hattendorff, A. Ehrl, W. A. Wall and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, A1373–A1387 CrossRef CAS.
- D. W. Van Krevelen and K. Te Nijenhuis, in Properties of Polymers, ed. D. W. Van Krevelen and K. Te Nijenhuis, Elsevier, Amsterdam, 4th edn, 2009, pp. 129–188, DOI:10.1016/B978-0-08-054819-7.00006-6.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef.
- J. Fowles, M. Banton, J. Klapacz and H. Shen, Toxicol. Lett., 2017, 278, 66–83 CrossRef CAS.
- M. D. Soucek and X. Ren, in Photocured Materials, ed. A. Tiwari and A. Polykarpov, The Royal Society of Chemistry, 2014, 10.1039/9781782620075-00015.
- H. Yoshihara and M. Yamamura, J. Appl. Polym. Sci., 2019, 136, 47867 CrossRef.
- W. D. Cook, Polymer, 1992, 33, 2152–2161 CrossRef CAS.
- J. W. Stansbury, Dent. Mater., 2012, 28, 13–22 CrossRef CAS.
- M. H. Mohamed and L. D. Wilson, Nanomaterials, 2012, 2, 163–186 CrossRef CAS.
- H. M. J. Boots, J. G. Kloosterboer, C. Serbutoviez and F. J. Touwslager, Macromolecules, 1996, 29, 7683–7689 CrossRef CAS.
- C. J. Orendorff, T. N. Lambert, C. A. Chavez, M. Bencomo and K. R. Fenton, Adv. Energy Mater., 2013, 3, 314–320 CrossRef CAS.
- E. P. Roth, D. H. Doughty and D. L. Pile, J. Power Sources, 2007, 174, 579–583 CrossRef CAS.
- X. Feng, X. He, M. Ouyang, L. Wang, L. Lu, D. Ren and S. Santhanagopalan, J. Electrochem. Soc., 2018, 165, A3748 CrossRef CAS.
- C. T. Love, J. Power Sources, 2011, 196, 2905–2912 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS.
- T. Y. Lee, Z. Smith, S. K. Reddy, N. B. Cramer and C. N. Bowman, Macromolecules, 2007, 40, 1466–1472 CrossRef CAS.
- H. Lu, J. A. Carioscia, J. W. Stansbury and C. N. Bowman, Dent. Mater., 2005, 21, 1129–1136 CrossRef CAS.
- C. Zuo, B. Zhou, Y. H. Jo, S. Li, G. Chen, S. Li, W. Luo, D. He, X. Zhou and Z. Xue, Polym. Chem., 2020, 11, 2732–2739 RSC.
- S. Park, B. Jeong, D.-A. Lim, C. H. Lee, K. H. Ahn, J. H. Lee and D.-W. Kim, ACS Appl. Mater. Interfaces, 2020, 12, 19553–19562 CrossRef CAS.
- J. Hu, W. Wang, B. Zhou, J. Sun, W. S. Chin and L. Lu, Small, 2024, 20, 2306622 CrossRef CAS.
- B. H. Jones, T. M. Alam, S. Lee, M. C. Celina, J. P. Allers, S. Park, L. Chen, E. J. Martinez and J. L. Unangst, Polymer, 2020, 205, 122783 CrossRef CAS.
- A. Shundo, M. Aoki, P. Wang, T. Hoshino, S. Yamamoto, S. Yamada and K. Tanaka, Macromolecules, 2023, 56, 3884–3890 CrossRef CAS.
- R. Pan, R. Sun, Z. Wang, J. Lindh, K. Edström, M. Strømme and L. Nyholm, Energy Storage Mater., 2019, 21, 464–473 CrossRef.
- G. Bieker, M. Winter and P. Bieker, Phys. Chem. Chem. Phys., 2015, 17, 8670–8679 RSC.
- B. Wu, J. Lochala, T. Taverne and J. Xiao, Nano Energy, 2017, 40, 34–41 CrossRef CAS.
- K.-H. Chen, K. N. Wood, E. Kazyak, W. S. Lepage, A. L. Davis, A. J. Sanchez and N. P. Dasgupta, J. Mater. Chem. A, 2017, 5, 11671–11681 RSC.
- P. Lennartz, B. A. Paren, A. Herzog-Arbeitman, X. C. Chen, J. A. Johnson, M. Winter, Y. Shao-Horn and G. Brunklaus, Joule, 2023, 7, 1471–1495 CrossRef CAS.
- G. M. Hobold, J. Lopez, R. Guo, N. Minafra, A. Banerjee, Y. Shirley Meng, Y. Shao-Horn and B. M. Gallant, Nat. Energy, 2021, 6, 951–960 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03701d |
| This journal is © The Royal Society of Chemistry 2024 |