
Altering Na-ion solvation to regulate dendrite growth for a reversible and stable room-temperature sodium–sulfur battery†
Chhail Bihari
Soni
 a,
Saheb
Bera
b,
Sungjemmenla
a,
Mahesh
Chandra
a,
Vineeth
S. K.
ac,
Sanjay
Kumar
a,
Hemant
Kumar
*b and
Vipin
Kumar
*ac
a,
Saheb
Bera
b,
Sungjemmenla
a,
Mahesh
Chandra
a,
Vineeth
S. K.
ac,
Sanjay
Kumar
a,
Hemant
Kumar
*b and
Vipin
Kumar
*ac
aDepartment of Energy Science and Engineering, Indian Institute of Technology Delhi, Hauz Khas, 110016, New Delhi, India
bSchool of Basic Sciences, Indian Institute of Technology Bhubaneswar, 752050, Odisha, India. E-mail: hemant@iitbbs.ac.in
cUniversity of Queensland-IIT Delhi Academy of Research (UQIDAR), Indian Institute of Technology Delhi, Hauz Khas, New Delhi 110016, India. E-mail: vkumar@dese.iitd.ac.in
First published on 16th July 2024
Abstract
Unwarranted reactivity of sodium with electrolytes leads to their constant consumption and dendrite growth, causing sodium-metal batteries to fail prematurely. The interface and electrolytes are often engineered to boost stability and reversibility; however, designing and understanding the correlation between the interphase and electrolyte dynamics remained challenging. Here, we report an alloying-type electrolyte additive, i.e., bismuth triiodide (BiI3), to alter Na+-ion solvation and its redox dynamics on the sodium metal surface. Theoretical calculation assisted experimental characterization reveals that the additive changes the local solvation shell dynamics and improves the Na ion kinetics by reducing binding energy. In addition, the formation of an alloy interphase over a metal anode realizes a dendrite-free sodium-metal anode, which even retains its interfacial integrity after long-term cycling in both Na-symmetric cells (for over 1600 hours at 1 mA cm−2) and Na-metal batteries with sulfurized polyacrylonitrile (SPAN) as a cathode (for over 250 cycles). Electrolyte engineering through alloying additives alters both solvation dynamics and interfacial properties, pointing us toward a new direction to harness sodium metal as the most promising anode.
1. Introduction
Metal anode batteries are, undoubtedly, the best choice for high-energy batteries.1–3 The limited availability and high price of Li metal allowed researchers to look for other alternatives to develop metal anode batteries.4,5 Besides its high natural abundance (about 2.36% in the earth's crust), Na metal's high specific capacity (∼1166 mA h g−1) and sufficiently low reduction potential (−2.71 V vs. SHE) make it the most promising anode after Li metal.6–8 However, attempts to harness the Na metal anode have been prevented by its extreme reactivity with electrolytes (ether and carbonate-based), leading to irreversibility and low coulombic efficiency (CE).8–10 The irreversibility, caused by constant sodium metal and electrolyte consumption, triggers surface non-uniformity and leads to the development of notorious morphologies such as dendritic and dead Na metal.11 The highly reactive nature of sodium metal leads to continuous reactions with organic liquid electrolytes,12 forming a thick and uneven solid electrolyte interphase (SEI).13–16 This hinders the smooth transport of ions across the interface. Furthermore, the repetitive plating and stripping of sodium metal during the charging and discharging process cause significant volume changes.17,18 These volume changes, combined with the inherently fragile nature of the SEI, result in the fracturing of the SEI.19 This leads to uncontrolled side reactions and continuous reformation of the SEI, ultimately leading to low coulombic efficiency, the growth of dendritic sodium structures, and depletion of the electrolyte. In extreme cases, these dendrites can cause internal short circuits, posing a risk of battery explosions.20–23 Efforts to improve Na metal reversibility directly led us to the invention of the high-temperature sodium–sulfur battery (HT-Na/S). Over five decades after the commercial success of HT-Na/S, the pursuit for a safer, higher energy density and cost-effective battery has eventually forced us back to develop liquid electrolyte-based Na/S batteries.24,25 However, elemental sulfur, which is inherently electronically insulating, poses a two-fold challenge. First, it requires a conductive matrix to facilitate electron transfer, and second, it fails to hold the high-voltage discharge products, i.e., long-chain polysulfides.11,26 In liquid electrolytes, the polysulfides can readily shuttle and poison the anode, leading to capacity fade. Unlike the elemental sulfur cathode, SPAN is inherently electronically conductive and offers improved cycle stability by mitigating polysulfide dissolution. The SPAN matrix is a covalently bonded framework of S–C, which facilitates the solid–solid conversion reactions instead of liquid–liquid or liquid–solid conversion reactions.27,28In addition, constant efforts are in progress to prevent sodium metal reactivity, dendritic growth, and the formation of dead sodium. These efforts aim to design and develop extrinsic (formed before cell operation) and intrinsic (generated during cell operation) interphases to mitigate the uneven deposition of Na+-ions.29,30 Provided the electrochemical attributes of the additive/s, extrinsic or intrinsic interphases are formed chemically or electrochemically so that a better/smooth Na deposition morphology can be achieved. Since intrinsic interphases are being developed during electrochemical operations, they facilitate uniform and compact Na deposition. However, it promotes side reactions and irreversibility, and therefore, the cell survives only for hundreds of hours due to the consumption of sodium metal and electrolytes.
Most studies focused on designing intrinsic interphases could not count the effect of additives on the solvation dynamics of Na+-ions.31 Theoretical calculations/simulations have unveiled that, the deposition overpotential of Na+-ions is closely associated with the solvation dynamics of Na+-ions in the electrolyte.32 Independent experimental findings correlate deposition overpotential with the sodium deposition morphologies. Generally, deposition overpotential is considered a measure of the deposition morphologies; however, the role of solvation dynamics remained untouched in deciding their fates. With increased strength of the solvation shell, i.e., a higher de-solvation energy barrier for Na+-ions in the electrolyte, ionic transport slows down, which may favor uniform deposition. The de-solvation dynamics have a close connection with the structure of the Na+-ion solvation shell. Since the solvation structure depends on the nature of the salt and solvent, it can easily be affected by the foreign ions, i.e., additive salts. Thus, establishing a relationship between the solvation or de-solvation dynamics of Na+-ions and the nature of sodium deposition would be of great significance in broadening the spectrum of electrolyte design for high-energy sodium metal batteries.
In this work, we employed the BiI3 additive to alter the solvation shell of the Na+-ion in the electrolyte and examined its effect on the sodium deposition morphology. Besides that, an optimized amount of the additive facilitates the formation of a thin and compact layer of the Na–Bi alloy on the sodium metal surface which protects the sodium metal anode against parasitic reactions. Theoretical calculation-supported experimental findings reveal that the in situ formed Na–Bi layer facilitates rapid and uniform sodium plating underneath, leading to a dendrite-free morphology. In sharp contrast, dendritic and mossy structures are observed when sodium metal is cycled in the reference electrolyte. The sodium symmetric cell with an optimized additive could be cycled stably for over 1600 hours. Furthermore, we demonstrated superior performance in terms of both cycling stability and rate capability in full cells consisting of sodium, the BiI3-added electrolyte, and a sulfurized polyacrylonitrile (SPAN) cathode.
2. Experimental section
2.1 Materials
Sodium cubes, sodium triflate (NaCF3SO3), anhydrous diethylene glycol dimethyl ether (diglyme), bismuth(III) iodide, polyvinylidene fluoride, and N-methyl pyrrolidone were used as received from Sigma-Aldrich.2.2 Electrolyte and electrode preparation
The reference electrolyte (1 M NaOTf + diglyme) was prepared by dissolving sodium salt in diethylene glycol dimethyl ether. For the electrolyte with the additive, the same process was followed, but varying amounts of BiI3 additive were included. Here, ether-based electrolytes were chosen over esters due to their electrochemical compatibility with sodium metal anodes.33 A thin slice (∼1.0 mm) of sodium served as the auxiliary/reference anode in both symmetric- and full-cell configurations. For a full cell, SPAN was synthesized following a previously reported procedure.34 For cathode preparation, the active material (SPAN), conductive fillers (super P), and binding agents (PVDF) were mixed in 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:10 wt% in anhydrous N-methylpyrrolidone (NMP) and stirred for 10 hours to form the slurry. This slurry was coated onto carbon-coated aluminum foil (∼12 μm) using a bar coater and then dried overnight in a vacuum oven at 60 °C. The electrodes with a diameter of 15 mm were cut and transferred to an argon-filled glove box. The current density and capacity calculations were made based on an active material mass loading of approximately 2.4 mg cm−2. X-ray diffraction (XRD) analysis and surface morphologies with energy-dispersive X-ray spectroscopy (EDX) were examined, as shown in Fig. S9.†
:20:10 wt% in anhydrous N-methylpyrrolidone (NMP) and stirred for 10 hours to form the slurry. This slurry was coated onto carbon-coated aluminum foil (∼12 μm) using a bar coater and then dried overnight in a vacuum oven at 60 °C. The electrodes with a diameter of 15 mm were cut and transferred to an argon-filled glove box. The current density and capacity calculations were made based on an active material mass loading of approximately 2.4 mg cm−2. X-ray diffraction (XRD) analysis and surface morphologies with energy-dispersive X-ray spectroscopy (EDX) were examined, as shown in Fig. S9.†
2.3 Electrochemical measurements
All the electrochemical tests were conducted on 2032-type coin cells. These cells were assembled inside an argon-filled glovebox. In symmetric cells, two sodium electrodes of similar size (∼1 cm−2), with and without the additive, were used along with two layers of a Celgard separator (∼25 μm). The electrolyte volume was kept constant, i.e., 60 μl for various cell configurations. Battery cycling tests were performed on a NEWARE BTSClient 8.0.X battery tester at different current densities to evaluate the stability of the stripping and plating processes. Cyclic voltammetry (CV) measurements were conducted using a Corrtest (CS studio) single-channel potentiostat/galvanostat, employing a two-electrode cell setup with a scan rate of 5 mV s−1. Electrochemical impedance spectroscopy (EIS) was performed on sodium symmetric cells across a frequency range of 1 Hz to 100 kHz.2.4 In situ optical testing
Optical cell experiments were conducted to visualize the deposition of sodium with and without additives. For this purpose, a custom-made airtight cell was designed, utilizing stainless steel electrodes as current collectors for the sodium electrodes. The electrode spacing was maintained at 5 mm, and the empty region between the electrodes was filled with electrolytes containing 1 M NaOTf in diglyme, with and without additives. The experiments were conducted at a current density of 5 mA cm−2, and micrographs were obtained at different intervals to observe the sodium deposition process.2.5 Material characterization
Surface and cross-sectional microscope analysis of sodium metal was conducted using Field Emission Scanning Electron Microscopy (FESEM) with a JEOL JSM-7800F instrument. To ensure safe transportation of the samples for FESEM analysis, they were securely enclosed in a polyamide bag. The chemical composition of the interphase was investigated using X-ray Photoelectron Spectroscopy (XPS) with a Thermo Scientific Theta Probe Angle-Resolved Spectrometer equipped with a monochromatic Al Ka source (1486.7 eV). A specialized XPS holder was used to transport the samples from the laboratory to the test site, ensuring proper isolation from the open ambient environment. X-ray Diffraction (XRD) patterns were recorded using a Rigaku Ultima IV instrument. The chemical environment of the electrolyte solvation shell was investigated with a Bruker Ascend 500 MHz liquid probe. Deuterated DMSO is used as the reference solvent. The NMR sample was prepared in an argon filled glove box. An 8-inch NMR tube is filled with excess reference solvent, mixed with a minimal amount of 1 M electrolyte. 1H NMR is performed to examine the chemical shifts in CH2 molecules. A Renishaw Micro-Raman spectrometer is used with a 514 nm laser in the wavenumber range of 500 to 1500. All the samples were used in the liquid state.2.6 Theoretical calculations
The structural optimization of solvation shell configurations was carried out using density functional theory (DFT) with the hybrid exchange functional Becke3 and correlation functional PW91, as implemented in GAMESS.35–37 In all optimization and single-point energy calculations, the Gaussian basis set 3-21++G(d,p) was used to expand the Kohn–Sham orbitals of the molecules. Ground state energies for all solvation shells were determined under normal conditions, with thermal corrections at a temperature of T = 298.15 K and pressure of 1 atm.The Na plating mechanism on top of Na3Bi and Na metal surfaces was studied using density functional theory (DFT) simulations implemented in the Vienna ab initio simulation program (VASP) with a plane-wave basis set.38 To make our calculations efficient, we used projector-augmented wave (PAW) potentials with frozen core all-electron precision.39 The generalized gradient approximation (GGA) was used to approximate the electron–electron correlation.40 To accurately represent the ground state of the system, we set the cutoff energy for the plane waves at 400 eV. We sampled k-points on a 12 × 12 × 1 Monkhorst–Pack k-grid in the first Brillouin zone (BZ). To simulate the surface structure, a vacuum layer of 20 Å was introduced on top of the bulk-Na and Na3Bi slabs. The adsorption energy on bulk Na and Na3Bi surfaces and van der Waals (vdW) interaction are computed using the DFT-D3 method. Electronic relaxation was performed until the energy reached a self-consistency cutoff of 10−6 eV, and atom positions were adjusted until the force was less than 0.01 eV Å−1.
To investigate the impact of surface properties on sodium (Na) plating and the reduction decomposition of the solvation shell, we have identified the most stable surfaces of bulk Na metal and the Na3Bi alloy by calculating surface energy. The surface energies (σ) have been determined using the formula:
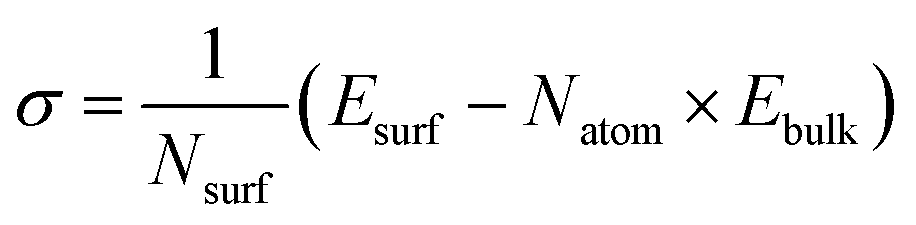 | (1) |
The formula utilized to compute the adsorption energy of the Na atom on top of a partially occupied Na3Bi and Na surface is expressed as follows:
| Eads = (EComplex − (EPristin + nENa))/n |
3. Results and discussion
In order to understand the solvation dynamics, the spectroscopic analysis was performed using Raman spectroscopy. The triflate anions surrounded by the diglyme molecules give rise to the appearance of peaks ranging from 920 to 800 cm−1. The deconvoluted Raman spectra for the reference electrolyte confirm the formation of Aggregated (AGG), Contact-Ion Pair (CIP), and free ion pairs, as shown in Fig. 1a. Upon addition of the additive salt, i.e., BiI3 to the reference system, the concentration of AGG is observed to increase sharply, indicating a decrease in the local polarity, as shown in Fig. 1b and e. A decreased polarity indicates improved voltage stability; see Fig. S1.† Moreover, electrolytes with reduced polarity are linked with reduced corrosiveness. In addition to that, the elevated AGG concentration strongly implies a surplus of anions, mimicking a high-concentration electrolyte, which shows the possible formation of an inorganic SEI, and corroborates the XPS findings.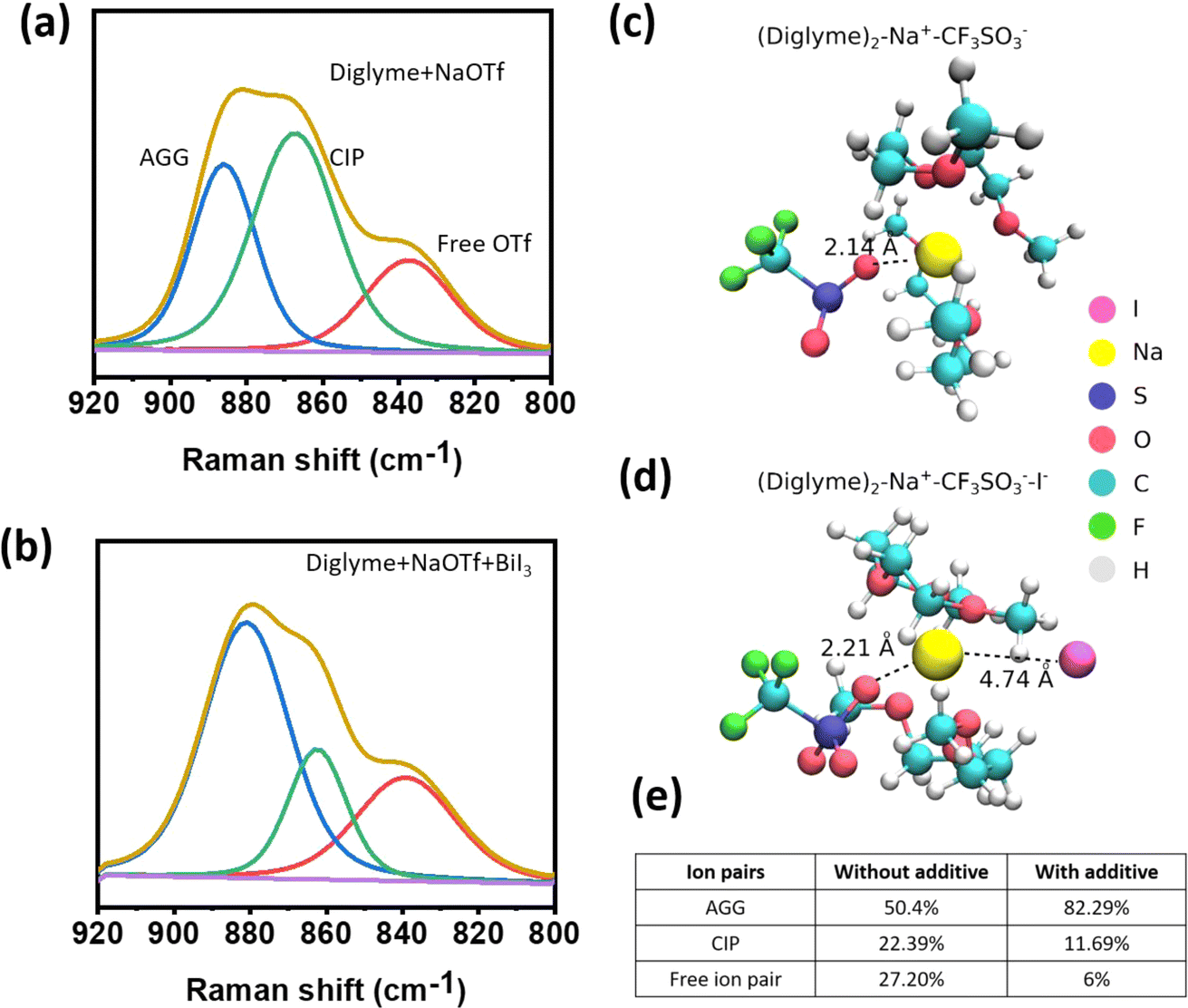 | ||
| Fig. 1 Raman spectra of solvation shell (a) reference electrolyte and (b) with additive ions, (c and d) model structure of the population of different ion pairs in the solvation shell of the electrolyte without an additive and with an additive, and (e) calculated relative distribution of different ion pairs in the electrolyte with and without an additive. The model structure of the solvation shell consists of one diglyme molecule and Na+ ion. | ||
The binding energy of the solvation shell is important because it determines the tendency for electrolyte decomposition and, hence, SEI formation on the anode surface. To calculate binding energy, we construct a model solvation shell containing two diglyme molecules, a CF3SO3− anion and Na+. The influence of the iodide ion (I−) on solvation shell stability was investigated by introducing an I− ion into this solvation shell. Our results show that the binding energy of the solvation shell in the presence of an I− anion is about 2.14 eV per diglyme, which is 0.29 eV per diglyme lower than that of the solvation shell without I− ions (2.43 eV per diglyme), as shown in Fig. 1c and d. A lower binding energy or reduced de-solvation energy barrier indicates fast sodium ion kinetics, which in turn improves the cycle life of the cell. In addition, the theoretical calculations show that the binding energy of the solvation shell decreases in the presence of I− in the solvation shell. This facilitates the removal of solvation molecules during the co-insertion of Na+ into the electrode. Furthermore, we observe an increase in bond length between O in CF3SO3− and Na+ from 2.14 Å to 2.21 Å in the presence of I− (Fig. 1c and d) in the Na+–CF3SO3− complex. This indicates that the presence of I− ions enhances the dissociation of NaCF3SO3 salt, consistent with a decrease in the CIP peak.
To further investigate the effect of BiI3 additives on the Na+-ion solvation dynamics, we conducted 1H NMR spectroscopy on the electrolytes. 1H NMR experiments were performed at a field strength of 11.7 T and a Larmor frequency of 500 MHz. Fig. 2a depicts the 1H NMR spectrum of neat diglyme solvent without a sodium salt or additive. The NMR spectrum of the solvent is in good agreement with the literature.41 For this study, three different proton environments are arbitrarily labeled A–C, from the lowest-to-highest chemical shift in a pure solvent. For instance, “A” corresponds to the end methyl (CH3) groups, and “B” is for the CH2 groups nearer to the terminating methyl groups, while “C” represents the middle methylene (CH2) groups. The “C” middle CH2 groups are assigned the highest chemical shift due to proximity to nearby electronegative oxygen atoms, unlike the other groups that lie closer to the end methyl groups. With the addition of the sodium salt, the resonance spectrum for A and C type protons shifted to a higher magnetic field than that of the pure solvent, as shown in Fig. 2b. This behaviour suggests that the solvation of sodium salt occurs through the terminal and the central oxygen, i.e., oxygen atoms in the solvent molecules interact with Na+ cations.
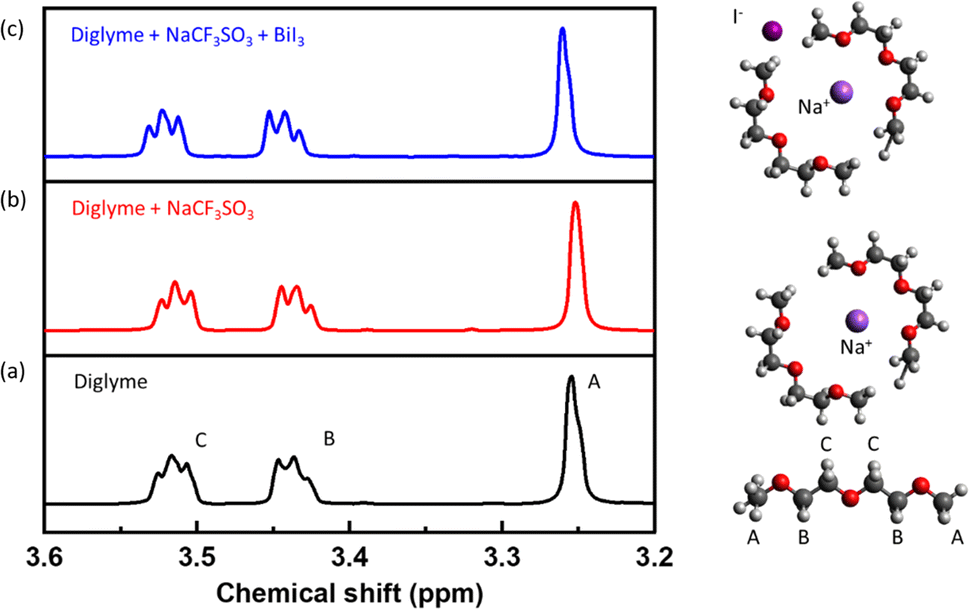 | ||
| Fig. 2 1H NMR spectra of (a) pure diglyme solvent, (b) diglyme with sodium salt (NaCF3SO3), and (c) diglyme with sodium salt (NaCF3SO3) and additives (BiI3). Deuterated DMSO is used as the reference solvent for all three testing studies. | ||
Upon addition of the additive, the NMR spectrum experiences a downfield chemical shift, as shown in Fig. 2c. This downshift indicates that the protons (1H) in the solvating diglyme interact with the I− anions. The presence of I−, an electron-rich species, contributes to a de-shielding effect on the 1H nuclei in the diglyme's CH2 groups. This characteristic of the I− anions reduces the electron deficiency of the CH2 groups, thereby weakening the Na+ solvation by the solvent molecules. This weakening of the Na+ solvation shell implies that the sodium ions can be more readily de-solvated, facilitating their movement. This finding aligns well with the theoretical predictions.
It is worth noting that the favorable redox potential of the additive cation, i.e., Bi3+, facilitates a spontaneous galvanic reaction (ΔG < 0, 3Na + Bi3+ → Na3Bi, ΔE = 3.09 V) with the sodium metal anode. The physical appearance of the sodium metal surface is observed to change instantly from silvery-white to dark brown, suggesting the formation of a sodium metal alloy. The formation of the alloy phase is confirmed by X-ray diffraction, as shown in Fig. 3a. To understand the role of I ions in the interphase, the NaI additive is used. It is observed that the presence of iodine alone did not cause any color change in the sodium metal surface upon contact with the electrolyte, as shown in Fig. S2(b), and S2(a)† corresponds to bare Na metal for reference. Though iodine does not facilitate the color change, it partakes in the solvation, as supported by theoretical calculations (Fig. 1).
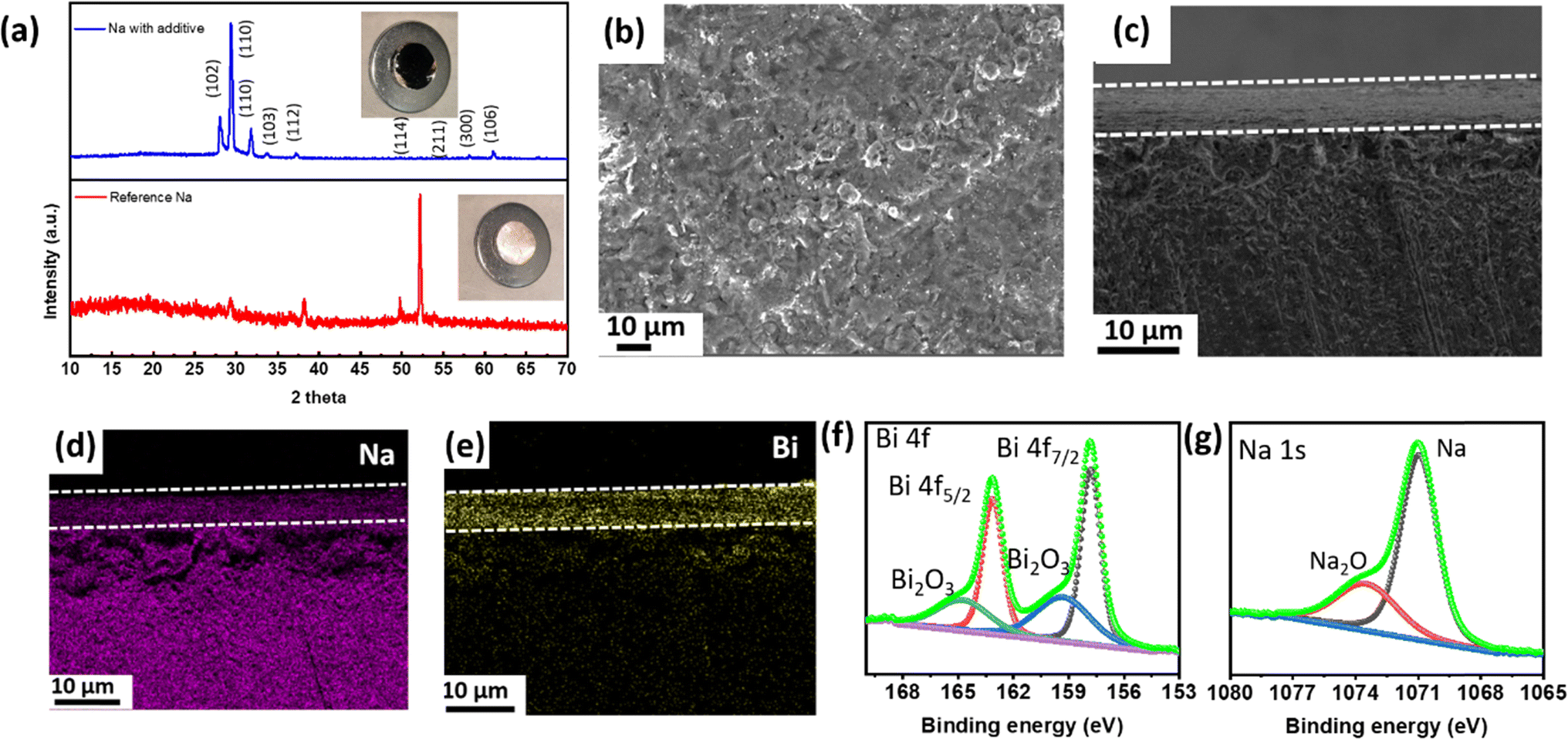 | ||
| Fig. 3 (a) X-ray diffraction of a sodium metal anode with and without an electrolyte additive. The inset shows digital micrographs of sodium metal with and without an electrolyte additive: (b) top view and (c) cross-sectional view after reaction with BiI3-added ether electrolyte. (d and e) Cross-sectional EDX mapping images of BiI3-treated Na metal with (d) Na and (e) Bi elemental signals. High-resolution XPS (f) Bi 4f and (g) Na 1s spectra of BiI3-treated Na metal. | ||
To examine the formation of the alloy phase, XRD analysis of the Na metal anode treated with a BiI3 additive was conducted. A set of distinct diffraction peaks appeared, which corresponds to the Na3Bi phase of the sodium–bismuth alloy,42 as shown in Fig. 3a. XRD of pure sodium metal was also carried out for comparison. While the second peak (2θ = 29.4°) in Fig. 3a corresponds to the (110) plane of elemental sodium, which arises from unreacted sodium, the third peak (2θ = 32.8°) corresponds to the (110) plane of Na3Bi.42,43 In the reference Na sample, the second peak (2θ = 29.4°) is less prominent, likely due to the formation of a sodium oxide layer on the bare Na surface. The formation of the Na–Bi phase is thermodynamically favorable due to spontaneous reactions between Na and Bi3+ ions. The reaction between Na and 3I− is not thermodynamically favorable and requires additional energy to form NaI. Since XRD was conducted before electrochemical tests, the formation of NaI could not be realized. The observed peaks confirm the successful formation of the Na3Bi phase, indicating the interaction between sodium and bismuth from the BiI3 treatment.
In order to gain a deeper understanding of the structural modifications induced by the BiI3-added electrolyte, FESEM micrographs of Na metal after the reaction were obtained. The surface micrograph of sodium after the reaction is shown in Fig. 3b. When analyzing the cross-sectional micrographs of the metal anode, the presence of an additional layer, densely packed with particles, becomes evident, as denoted by the dotted line, as shown in Fig. 3c. This layer demonstrates a uniform coverage over the entire Na metal surface. The corresponding X-ray mapping is presented in Fig. 3d and e, respectively. These mapping results unequivocally establish a robust and conformal distribution of the elements within the as-formed layer. This spatial arrangement of Na and Bi species provides compelling evidence supporting the successful incorporation of these constituents in the newly formed layer. Fig. S3(a and b)† exhibits the EDX mappings of Na and Bi elements (top view). Here, Na comprises 68% and Bi 23% of the elemental composition on the metal anode.
To analyze the chemical composition of the newly formed layer on the Na metal surface, X-ray photoelectron spectroscopy (XPS) was conducted. Fig. 3f displays the XPS spectrum of Bi, revealing distinct peaks at 163.64 eV and 158.4 eV. These peaks can be attributed to Bi 4f5/2 and Bi 4f7/2 respectively. Additionally, peaks at 166.72 eV and 161 are observed, corresponding to Bi2O3 of Bi 4f5/2 and Bi 4f7/2 respectively, further confirming the presence of Bi species. These findings contribute to a comprehensive understanding of the chemical transformations taking place at the interface between BiI3 and Na metal. In addition, Na XPS spectra show typical peaks from Na metal as shown in Fig. 3g.
The comprehensive findings of this study reveal that introducing BiI3 into the electrolyte solution leads to the uniform formation of a Na–Bi alloy on the surface of sodium metal and alters the solvation shell in the electrolyte. This formation has significant implications for enhancing the stability of sodium metal as an anode material in batteries.
Coulombic efficiency, a pivotal metric gauging the reversibility of the electrochemical processes, is assessed by quantifying the ratio of stripped Na capacity to plated Na capacity. The reference cell manifests persistent fluctuations in coulombic efficiency, as shown in Fig. 4a, indicative of an irregular and non-uniform deposition morphology (Fig. S4(c, and d)†). In contrast, the cell incorporating the additive demonstrates a sustained average coulombic efficiency of approximately 99% even after 350 cycles, as shown in Fig. 4a, underscoring the significant improvement in the uniformity and reversibility of the Na stripping/plating processes (Fig. S4(a, and b)†). Voltage vs. times plots indicate the effectiveness of stripping plating, which is essential for smooth deposition, as shown in Fig S5.†
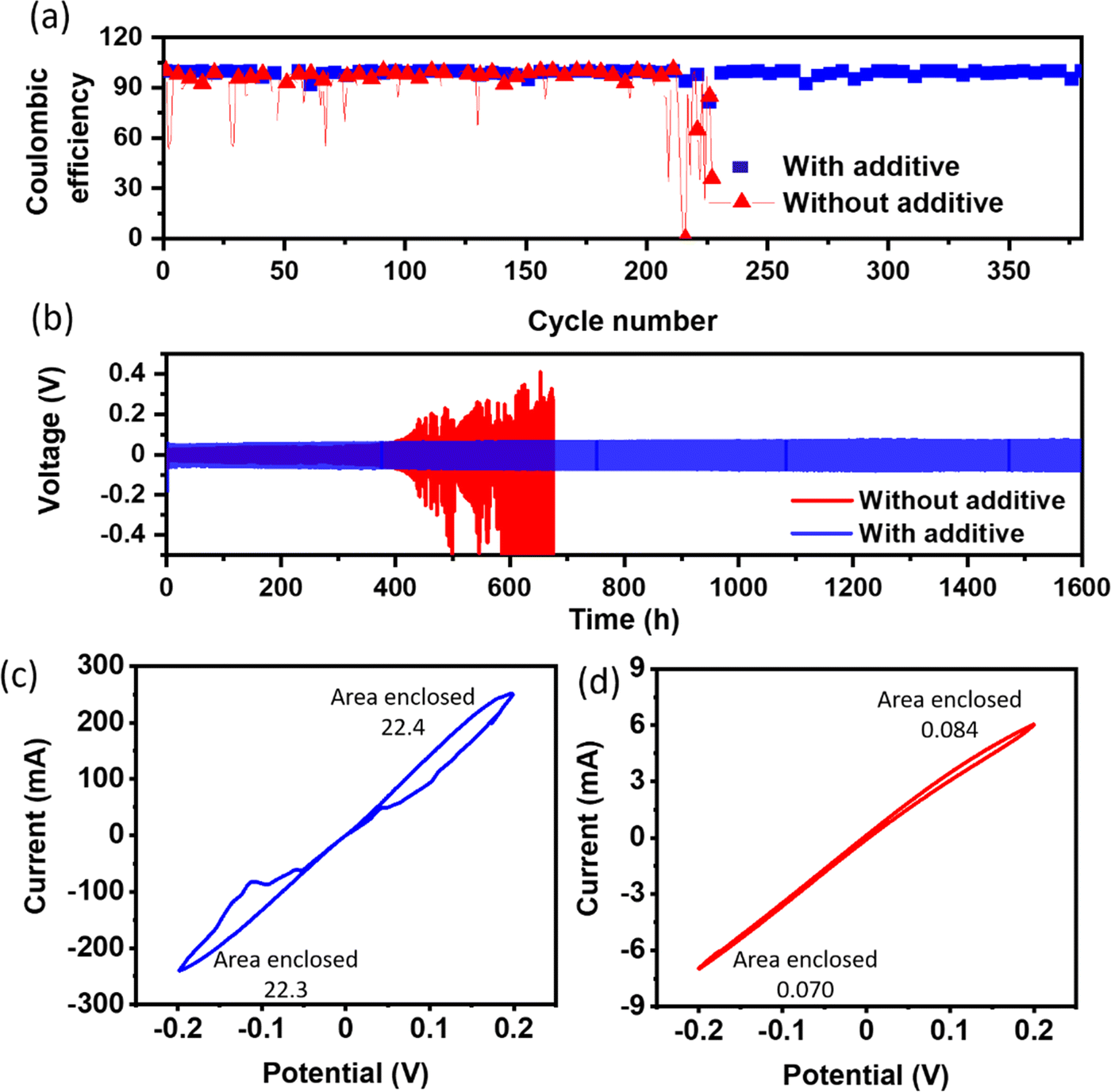 | ||
| Fig. 4 (a) Coulombic efficiency of Na//Cu half cells with and without an additive at 1 mA cm−2 current density and 1 mA h cm−2 capacity. (b) Stripping plating cycling performance of a Na//Na symmetric cell at 1 mA cm−2 current density and 1 mA h cm−2 capacity, and the cyclic voltammogram of a Na//Na symmetric cell (c) with and (d) without an additive. The CV scans were conducted at a sweep rate of 5 mV s−1. | ||
Fig. 4b presents the voltage profiles obtained from Na/Na symmetric cells during repeated plating and stripping cycles, wherein a current density of 1 mA cm−2 and a capacity of 1 mA h cm−2 were applied. Remarkably, the addition of only 100 mM BiI3 to the electrolyte resulted in a longer cycle life (∼1600 h) and with a slightly higher overpotential (∼90 ± 5 mV) as compared to the reference cell (60 ± 5 mV initially). The introduction of additives leads to a noticeable increase in overpotential. Besides de-solvation kinetics, the stripping/plating overpotential is closely related to the surface uniformity. Since Na3Bi forms spontaneously in the presence of Bi3+ ions, it leads to a non-uniform surface (Fig. 3b). A non-uniform and irregular surface causes an increased nucleation overpotential, which eventually contributes to the stripping/plating overpotential. Though the observed overpotential on the Na3Bi surface is slightly higher than that of the reference system, it effectively suppresses dendrite formation, leading to an unprecedented cycle-life of the cell. The enhanced cycle life reported in the additive-based cell is primarily due to reduced electrolyte breakdown and less dendritic growth. In contrast, the reference cell initially shows a lower overpotential due to a decreased desolvation energy barrier. Nevertheless, a further rise in overpotential, presumably caused by the formation of dendrites and increased electrolyte breakdown during repeated cycles, finally results in a short circuit. These findings highlight the complex relationship between the solvation dynamics and the growth of sodium dendrites. During the study of plating and stripping behavior at high current densities, Na//Na symmetric cells were exposed to current densities of 5 mA cm−2 and 10 mA cm−2 while retaining an areal capacity of 1 mA h cm−2, as shown in Fig. S6a and S6b.† Consistent with prior findings at 1 mA cm−2, introducing the additive led to a little rise in overpotential while also demonstrating a prolonged cycle life.
To explore the effect of increasing the concentration of BiI3 in the electrolyte to 150 or 200 mM, further investigations were conducted, as shown in Fig S7.† Surprisingly, no additional improvement in performance was observed beyond the 100 mM concentration. This finding suggests that concentrations higher than 100 mM of BiI3 do not provide additional benefits in terms of promoting sodium ion diffusion or suppressing the formation of dendritic structures within the cell. Upon increasing the additive content beyond 100 mM, the electrolyte turned into a turbid solution with apparent undissolved particles. Besides that, electrolytes with higher additive concentrations were not used because firstly, the electrolyte with higher additive content is difficult to disperse on the separator, and secondly, the undissolved particles are likely to settle down unevenly on the electrode surface. Due to the aforementioned issues, the additive concentration was not increased beyond 100 mM. Consequently, the subsequent characterization and experiments were conducted using a fixed BiI3 concentration of 100 mM. The stripping/plating reversibility of sodium in a sodium symmetric cell is also investigated in EC/DMC electrolyte. A highly unstable and non-uniform voltage profile, suggesting its incompatibility with Na metal, is shown in Fig S8.† In addition, ethers are comparatively less prone to dendrite growth.44 Esters, although offering high dielectric constants, are less stable and prone to decomposition with reactive sodium metal, leading to enhanced dendrite growth, and adversely impacting battery performance and safety. The cycling performance of Na//Na symmetric cells is also compared with the additive based literature for sodium metal anodes, and this work stands well comparatively, as shown in Table S1.†
CV scans were performed to investigate the electrochemical behavior of the Na metal electrode in the presence of the BiI3-added electrolyte. The CV results, presented in Fig. 4c and d, provide valuable insights into the electrochemical processes occurring at the electrode–electrolyte interface. In the CV scans, two distinct peaks were observed near 0.15 ± 0.02 V and −0.15 ± 0.02 V, which can be attributed to the alloying-dealloying process of Na metal. These peaks represent the reversible electrochemical reactions involving the insertion and extraction of sodium ions into and from the formed Na–Bi alloy on the Na metal electrode surface. Notably, the CV analysis revealed a significantly higher current density in the presence of the BiI3-added electrolyte than that of the control sample. The higher current density signifies more efficient alloying-dealloying processes, which are essential factors in achieving enhanced electrochemical performance and stability in sodium metal-based batteries. However, no peak is observed in the reference system.
Theoretical studies were conducted to investigate the influence of the Na3Bi/Na metal interface on improving the efficiency of Na metal batteries compared to bulk Na metal when used as an anode. The optimized structures of Na and Na3Bi have lattice parameter values of 4.16 Å and 3.83 Å, respectively. Comparison of different surfaces reveals that the (110) surface exhibits minimal surface energy for both Na metal and Na3Bi, consistent with previously reported studies.45,46
To understand Na plating on anode surfaces, we compare Na adsorption energy in two plating modes: (1) horizontal plating, where Na atoms are adsorbed in the same plane, and (2) vertical plating, where Na atoms are adsorbed on top of a partially filled plane, as shown in Fig. 5a and b. In both plating modes, the adsorption energy of Na atoms is found to be negative for Na3Bi and Na metal surfaces. This negative value suggests that both plating configurations are possible on Na3Bi and Na metal surfaces. Furthermore, horizontal plating is energetically preferable to vertical plating for Na and Na3Bi surfaces across various Na deposition concentrations, as shown by the relatively lower Na adsorption energy. However, the difference in the adsorption energy of Na atoms between these plating types determines the relative preference over two plating modes, i.e., a lower energy difference indicates a higher probability of vertical plating and hence, dendrite growth. As shown in Fig. 5c and d, the Na adsorption energy difference between horizontal and vertical plating on the Na metal surface is lower for high Na deposition concentrations than the Na3Bi surface. This suggests that dendrite formation is less likely for the Na3Bi surface, indicating potential for an extended cycle life.
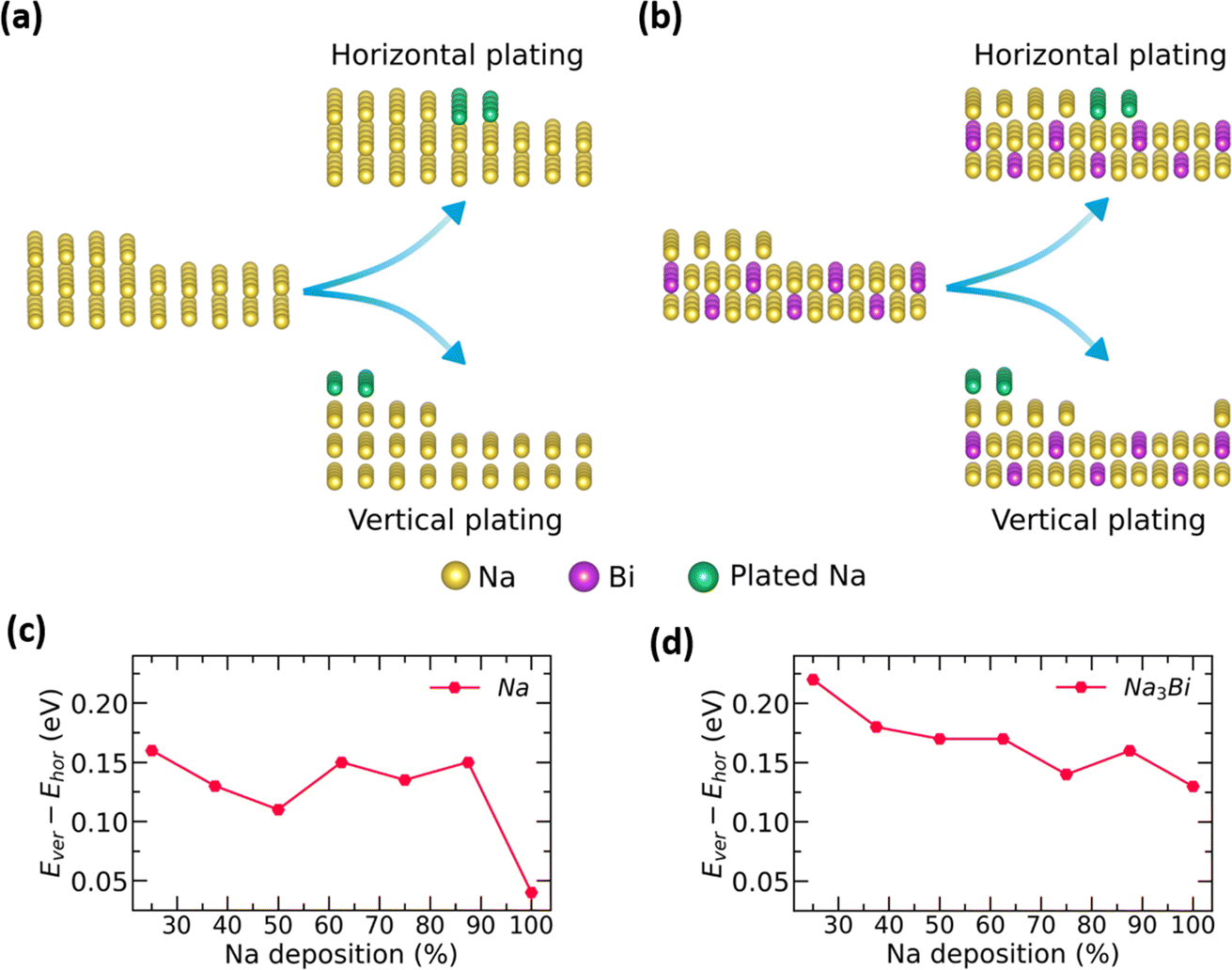 | ||
| Fig. 5 Schematic representation illustrating the horizontal and vertical plating mechanisms of Na on (a) bulk Na-metal and (b) Na3Bi surfaces. The graphs below show the variation in the energy difference between vertical and horizontal plating in response to varying Na deposition concentrations for (c) bulk Na-metal and (d) Na3Bi surfaces. | ||
Optical cell studies were performed to visually evaluate the kinetics of sodium deposition with and without additives. We utilized an air-tight optical cell with stainless steel electrodes serving as current collectors. The electrode gap was set at a fixed spacing of 5 mm, ensuring an accurate alignment of the electrodes. The space between the electrodes was filled with electrolytes, specifically a solution of 1 M NaOTf in diglyme, both with and without additives. An applied current density of 5 mA cm−2 was maintained, and micrographs were consistently obtained at different time intervals. The digital micrographs demonstrate the quick formation of sodium metal dendrites in the reference cell, as shown in Fig. S9a.† These dendrites initially appear as mossy structures and thereafter cause significant breakdown of the electrolyte and the release of gas within the cell. This process plays a key role in the reduction of battery capacity over time and the decrease in the number of charge–discharge cycles owing to the ongoing breakdown of the electrolyte. In contrast, the cell that contains additives shows no or negligible dendritic growth and essentially no electrolyte breakdown, as shown in Fig S9b.† This indicates that adding the additive serves a dual purpose by reducing the breakdown of electrolytes and simultaneously preventing dendrite growth. The results highlight the crucial function of the additive in improving the stability and longevity of the cell and are in agreement with theoretical findings.
Surface composition analysis of the cycled Na metal electrode was conducted using XPS to gain insights into the chemical species present on the surface. Fig. 6a–f depict the XPS spectra of C 1s, O 1s, Na 1s, I 3d, Bi 4f–S 2p, and F 1s, respectively, on the surface of the Na metal anode in the presence of BiI3-added electrolyte after 20 plating/stripping cycles in a Na//Na symmetric cell.
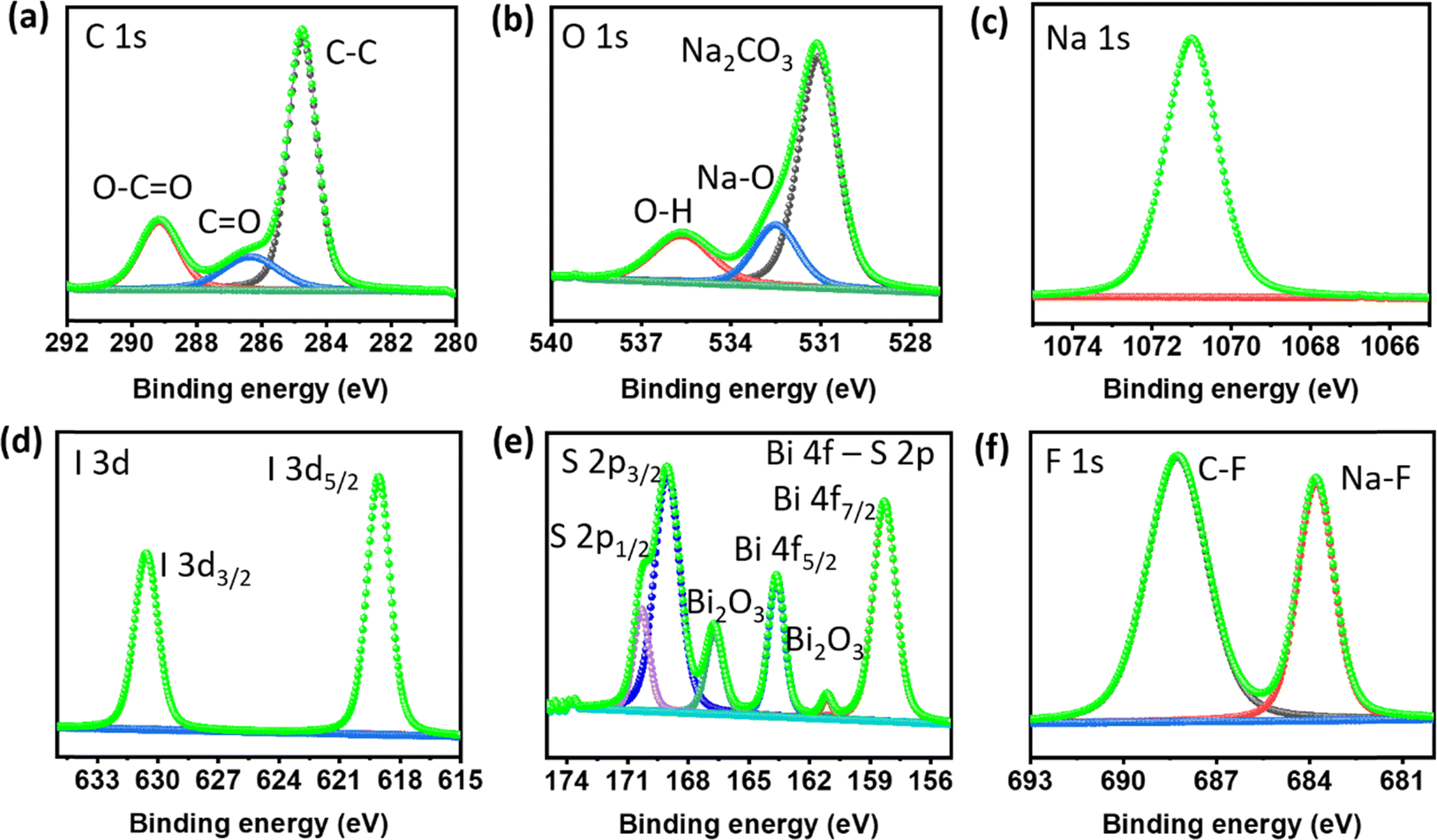 | ||
| Fig. 6 High-resolution XPS spectra of Na metal cycled in 100 mM BiI3-added electrolyte for 20 cycles. (a) C 1s, (b) O 1s, (c) Na 1s, (d) I 3d, (e) Bi 4f–S 2p and (f) F 1s spectra. | ||
The C 1s spectrum was deconvoluted into three distinct peaks with corresponding binding energies. These peaks were observed at 289.1 eV (ROCO2Na), 286.4 eV (–C–O–), and 284.8 eV (–C–C–), indicating the presence of typical decomposition products of the electrolyte. These products arise from the breakdown of electrolyte components during the cycling process. The O 1s spectrum displayed binding energies at 535.6 eV, 532.5 eV, and 531.1 eV, representing Na auger, Na–O, and NaOH/Na2CO3, respectively. This spectrum further confirms the presence of species resulting from electrolyte decomposition within the surface layer.
Regarding studying the I 3d spectra, the low-intensity peak at 619.09 eV in the I 3d5/2 spectrum suggests trace amounts of sodium iodide (NaI). This finding indicates that traces of NaI are being incorporated into the surface layer, specifically concentrated in the solid electrolyte interphase (SEI) layer. Simultaneously, the Bi peaks, notably Bi 4f5/2 and Bi 4f7/2, correspond to the presence of bismuth (Bi) compounds, providing evidence for the presence of a Bi–Na alloy phase. In addition, the binding energy for S 2p and Bi 4f overlaps, with both forming doublet peaks within the same range. The energy difference (spin–orbit coupling) between the doublet peaks for S 2p is typically around 1–1.16 eV,47 whereas for Bi 4f, it ranges between 5 and 5.5 eV,48 depending upon the chemical environment. Here, the energy difference of the Bi doublet peaks is around 5.1 eV (Bi 4f5/2 and Bi 4f7/2), indicating that these peaks correspond to Bi 4f.49,50 In addition, other deconvoluted doublet peaks correspond to S 2p with a binding energy difference of around 1 eV and an intensity ratio of around 0.5.
Significantly, the Na 1s spectrum displays expected peaks that are indicative of sodium. Notably, the F 1s spectrum displays distinct peaks correlating with C–F and Na–F interactions. The detection of Na–F peaks signifies the development of a durable SEI layer that is rich in inorganic compounds. This SEI layer is crucial for maintaining the stability of battery performance. In contrast, the XPS findings of the reference cell without additives reveal the poor formation of a NaF-rich SEI, as shown in Fig. S10 and S11.† These findings provide valuable insights into the surface composition of the Na metal electrode after cycling with the BiI3-added electrolyte, as shown in Table S2.† The presence of NaI and decomposition products of the electrolyte indicates the formation of a complex and protective surface layer, contributing to the improved stability and performance of the sodium metal anode.
Post-mortem SEM characterization was conducted to assess the stability of the formed layers. Fig. S12a† illustrates the surface of the Na metal electrode cycled in a blank electrolyte for 50 cycles without an additive, revealing a rough morphology with visible fractures and dendrites. This porous and rough surface structure increases the surface area and exacerbates the reaction with the electrolyte, leading to adverse effects on the electrochemical performance. In contrast, the Na metal electrode cycled in the 100 mM BiI3-added electrolyte maintained a dense and uniform surface morphology without detectable dendritic growth or fractures (Fig. S12b†). This observation can be attributed to the homogeneous transport of ions at the interface facilitated by the formed layers. The effective surface protection provided by Na–Bi contributes to maintaining a smooth and intact surface, preventing the formation of detrimental morphological features. The dense and uniform morphology observed without dendritic growth or fractures indicates the effective mitigation of side reactions and the preservation of a stable interface. These findings highlight the importance of the formed layers in enhancing the stability and overall performance of the sodium metal anode.
To evaluate the practicality of the BiI3-added electrolyte in combination with a Na metal anode and SPAN cathode, full cells were fabricated and subjected to testing. The SPAN cathode material was synthesized using a solid–state reaction method, as described in the Experimental section, and shown in Fig. S13.†
The full cells were cycled at a rate of 160 mA g−1 within a voltage range of 0.6 to 2.6 V. Initially, both electrolytes demonstrated almost similar discharge capacities, as shown in Fig. 7a and a little decay in capacity after the 1st cycle (Fig. 7b). However, as cycling progressed, the cell using the blank electrolyte exhibited poor capacity retention, with only 320 mA h g−1 of capacity remaining after 100 cycles, as shown in Fig. 7c. In contrast, the cell employing the 100 mM BiI3-added electrolyte demonstrated significantly improved capacity retention of more than 500 mA h g−1. This comparison highlights the notable performance advantage conferred by the BiI3-added electrolyte.
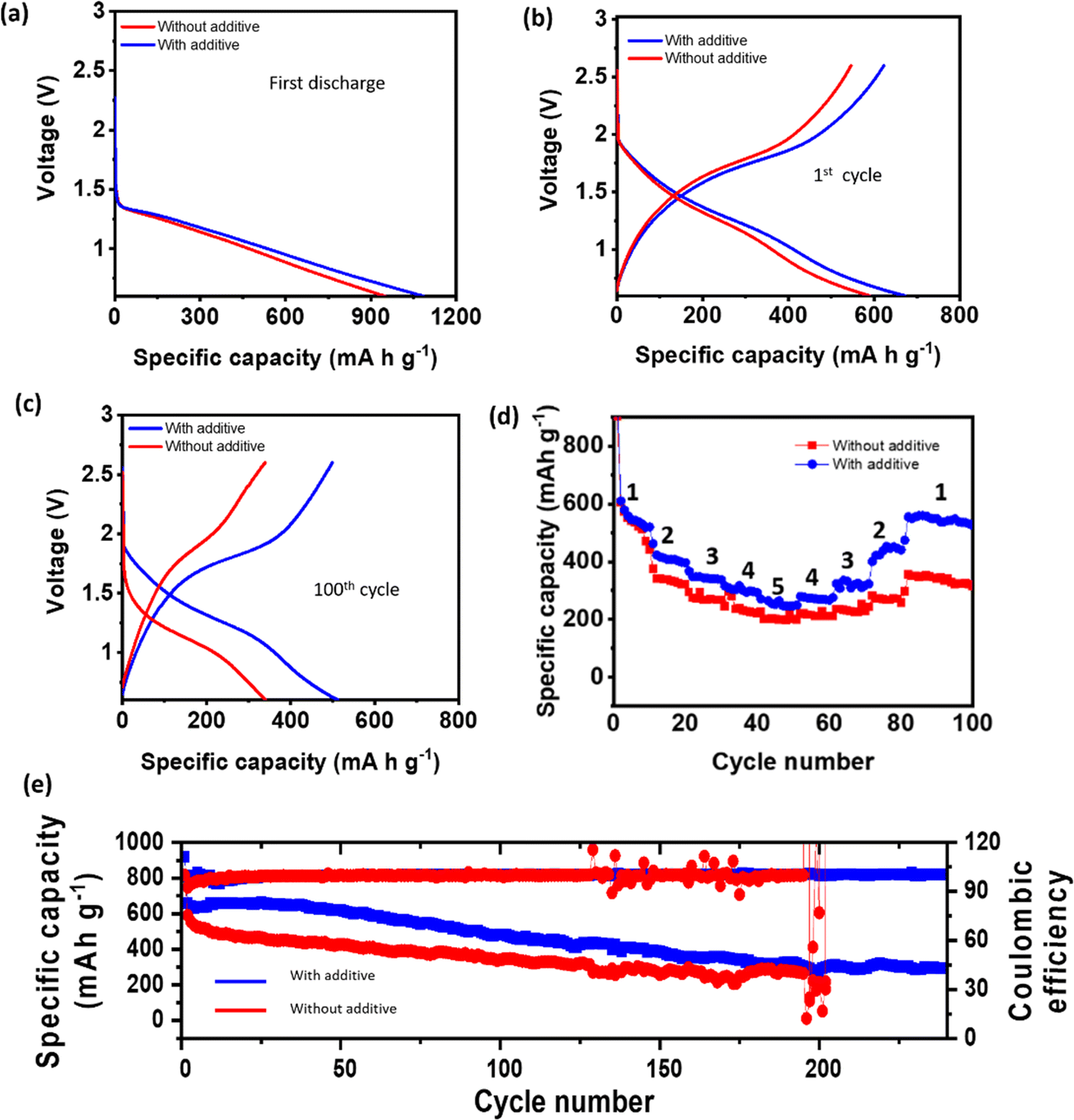 | ||
| Fig. 7 Galvanostatic charging/discharging of a Na//SPAN cell operated in a potential window of 0.6 to 2.6 V vs. Na/Na+ at 160 mA g−1, (a) first discharge with an additive and without an additive and (b, and c) voltage profile of the cells at 160 mA g−1 showing the 1st and 100th cycle with an additive and without an additive. (d) Rate performance analysis for Na//SPAN full cells, where 1–5 corresponds to 160 mA g−1, 320 mA g−1, 640 mA g−1, 800 mA g−1, and 1000 mA g−1 current density, respectively. (e) The specific capacity and coulombic efficiency of cells with respect to the cycle number. | ||
The voltage profiles of the cells further substantiated the advantages of incorporating BiI3 into the electrolyte. The cell utilizing the BiI3-added electrolyte demonstrated large polarization without an additive and smaller polarization with an additive, indicating smooth deposition (Fig. 7c). Furthermore, the higher discharge capacities achieved at various rates (Fig. 7d) demonstrate the improved rate capability of the cell incorporating BiI3. Moreover, the full cell employing the BiI3-added electrolyte exhibited consistently high coulombic efficiency (CE) over 230 cycles, in contrast to the control cell that experienced rapid fading after only 190 cycles and ultimately shorted. This improvement in CE signifies a higher utilization of charge and discharge capacity, as shown in Fig. 7e. These observations highlight the crucial role of anode protection provided by the BiI3 additive in improving the overall performance and stability of the full cells. The BiI3-added electrolyte not only enhances capacity retention and reduces polarization but also ensures a high coulombic efficiency, improved discharge performance at different rates and faster kinetics (Fig. S14†). In addition, the role of iodine ions in improving the rate capability of RT-Na–S batteries is also investigated. The equimolar NaI additive was used in the electrolyte. The findings suggest that iodine ions do indeed improve the rate performance of the cell. However, the rate performance with BiI3 shows significant improvement due to its ability to facilitate the formation of an alloying interphase leading to improved charge transfer kinetics, as shown in Fig. S15.† These results underscore the significance of anode protection in achieving high-performance sodium-based energy storage systems.
4. Conclusion
In conclusion, the addition of a small amount of BiI3 in the reference electrolyte resulted in a decrease in binding energy through the modulation of the solvation shell. Theoretical calculation-assisted experimental findings unveiled that the additive facilitates the formation of an alloy phase directly over the sodium metal anode, leading to suppressed dendrite growth. The Na//Na symmetric cells with a BiI3 additive demonstrated remarkable cycling stability over an unprecedented duration of 1600 hours. The improved performance was not limited to symmetric cells but was also observed in full cells, where high-capacity retention and limited cell polarization could be achieved even at high charging and discharging rates. These findings strongly suggest that Na3Bi inhibits not only dendritic growth but also module solvation shells, improving kinetic behavior near the anode surface. Therefore, using a BiI3 additive appears to be a promising option for future development of the Na-metal batteries, demonstrating potential for better performance and durability in practical applications.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
V. K and M. C acknowledge ReNeW Power for the financial support under the Research Grant (MI02550NC/Sub-project under RP03421). S. K. V., C. B. S., S. K., and Sungjemmenla acknowledge the scholarship granted by the University of Queensland-IIT Delhi Academy of Research (UQIDAR), the Indian Institute of Technology Delhi (IIT Delhi), and the Prime Minister Research Fellowship (PMRF), respectively. H. K. would like to thank SERB, India, for financial support under SRG and SAMKHYA: HPC Facility at IoP, Bhubaneswar, for the computational facility. SB thanks CSIR for the financial support. C. B. S. would further like to acknowledge the central research facility (CRF), Nanoscale research facility (NRF) IIT Delhi for providing the characterization facilities.References
- F. Shi, X. Guo, C. Chen, L. Zhuang, J. Yu, Q. Qi, Y. Zhu, Z. L. Xu and S. P. Lau, Nano Lett., 2023, 23, 7906–7913 CrossRef CAS PubMed.
- F. Shi, L. Zhai, Q. Liu, J. Yu, S. P. Lau, B. Y. Xia and Z. L. Xu, J. Energy Chem., 2023, 76, 127–145 CrossRef CAS.
- F. Shi, J. Yu, C. Chen, S. P. Lau, W. Lv and Z. L. Xu, J. Mater. Chem. A, 2022, 10, 19412–19443 RSC.
- F. Duffner, N. Kronemeyer, J. Tübke, J. Leker, M. Winter and R. Schmuch, Nat. Energy, 2021, 6, 123–134 CrossRef CAS.
- A. Y. S. Eng, C. B. Soni, Y. Lum, E. Khoo, Z. Yao, S. K. Vineeth, V. Kumar, J. Lu, C. S. Johnson, C. Wolverton and Z. W. Seh, Sci. Adv., 2022, 8, eabm2422 CrossRef CAS PubMed.
- T. Liu, X. Yang, J. Nai, Y. Wang, Y. Liu, C. Liu and X. Tao, Chem. Eng. J., 2021, 409, 127943 CrossRef CAS.
- L. Zhu, Y. Li, J. Zhao, J. Liu, L. Wang and J. Lei, Green Energy Environ., 2022, 8, 1279–1307 CrossRef.
- L. Lin, C. Zhang, Y. Huang, Y. Zhuang, M. Fan, J. Lin, L. Wang, Q. Xie and D. Peng, Small, 2022, 18, 2107368 CrossRef CAS PubMed.
- V. Kumar, A. Y. S. Eng, Y. Wang, D. T. Nguyen, M. F. Ng and Z. W. Seh, Energy Storage Mater., 2020, 29, 1–8 CrossRef.
- V. Kumar, Y. Wang, A. Y. S. Eng, M.-F. Ng and Z. W. Seh, Cell Rep. Phys. Sci., 2020, 1, 100044 CrossRef.
- C. B. Soni, Sungjemmenla, S. K. Vineeth and V. Kumar, Energy Storage, 2022, 4, e264 CrossRef CAS.
- S. K. Vineeth, C. B. Soni, Sungjemmenla, C. Sanjaykumar, Y. Yamauchi, M. Han and V. Kumar, J. Energy Storage, 2023, 73, 108780 CrossRef.
- S. K. Vineeth, M. Tebyetekerwa, H. Liu, C. B. Soni, N. Sungjemmenla, X. S. Zhao and V. Kumar, Mater. Adv., 2022, 2, 6415–6440 RSC.
- C. Bao, B. Wang, P. Liu, H. Wu, Y. Zhou, D. Wang, H. Liu and S. Dou, Adv. Funct. Mater., 2020, 52, 2004891 CrossRef.
- Y. Gu, W.-W. Wang, Y.-J. Li, Q.-H. Wu, S. Tang, J.-W. Yan, M.-S. Zheng, D.-Y. Wu, C.-H. Fan, W.-Q. Hu, Z.-B. Chen, Y. Fang, Q.-H. Zhang, Q.-F. Dong and B.-W. Mao, Nat. Commun., 2018, 9, 1339 CrossRef PubMed.
- Y. Zhang, D. Krishnamurthy and V. Viswanathan, J. Electrochem. Soc., 2020, 167, 070554 CrossRef.
- Y. Zhao, X. Yang, L. Y. Kuo, P. Kaghazchi, Q. Sun, J. Liang, B. Wang, A. Lushington, R. Li, H. Zhang and X. Sun, Small, 2018, 14, 1–8 CAS.
- N. Yabuuchi, Y. Matsuura, T. Ishikawa, S. Kuze, J. Y. Son, Y. T. Cui, H. Oji and S. Komaba, ChemElectroChem, 2014, 1, 580–589 CrossRef.
- C. B. Soni, V. Kumar and Z. W. Seh, Batteries Supercaps, 2022, 5, e202100207 CrossRef CAS.
- H. Sun, G. Zhu, X. Xu, M. Liao, Y. Y. Li, M. Angell, M. Gu, Y. Zhu, W. H. Hung, J. Li, Y. Kuang, Y. Meng, M. C. Lin, H. Peng and H. Dai, Nat. Commun., 2019, 10, 1–11 CrossRef PubMed.
- E. Matios, H. Wang, C. Wang and W. Li, Ind. Eng. Chem. Res., 2019, 58, 9758–9780 CrossRef CAS.
- L. Zhou, Z. Cao, J. Zhang, Q. Sun, Y. Wu, W. Wahyudi, J. Y. Hwang, L. Wang, L. Cavallo, Y. K. Sun, H. N. Alshareef and J. Ming, Nano Lett., 2020, 20, 3247–3254 CrossRef CAS PubMed.
- W. Li, H. Wang, E. Matios and J. Luo, Chem. Soc. Rev., 2020, 49, 3783–3805 RSC.
- Sungjemmenla, C. B. Soni, S. K. Vineeth and V. Kumar, Mater. Adv., 2021, 2, 4165–4189 RSC.
- Sungjemmenla, C. B. Soni, S. K. Vineeth and V. Kumar, Adv. Energy Sustainability Res., 2022, 3, 2100157 CrossRef CAS.
- Sungjemmenla, C. B. Soni and V. Kumar, Nanoscale Adv., 2021, 3, 1569–1581 RSC.
- S. Tan, M. M. Rahman, Z. Wu, H. Liu, S. Wang, S. Ghose, H. Zhong, I. Waluyo, A. Hunt, P. Liu, X. Q. Yang and E. Hu, ACS Energy Lett., 2023, 8, 2496–2504 CrossRef CAS.
- S. Wang, B. Lu, D. Cheng, Z. Wu, S. Feng, M. Zhang, W. Li, Q. Miao, M. Patel, J. Feng, E. Hopkins, J. Zhou, S. Parab, B. Bhamwala, B. Liaw, Y. S. Meng and P. Liu, J. Am. Chem. Soc., 2023, 145, 9624–9633 CrossRef CAS PubMed.
- C. B. Soni, Sungjemmenla, S. K. Vineeth, C. S. Kumar and V. Kumar, Sustainable Energy Fuels, 2023, 7, 1908–1915 RSC.
- C. B. Soni, N. Arya, Sungjemmenla, S. K. Vineeth, V. Balakrishnan and V. Kumar, Energy Technol., 2022, 10, 2200742 CrossRef CAS.
- S. K. Vineeth, C. B. Soni, Y. Sun, V. Kumar and Z. W. Seh, Trends Chem., 2022, 4, 48–59 CrossRef CAS.
- C. Bihari, S. Bera, S. K. Vineeth, H. Kumar and V. Kumar, J. Energy Storage, 2023, 71, 108132 CrossRef.
- Z. W. Seh, J. Sun, Y. Sun and Y. Cui, ACS Cent. Sci., 2015, 1, 449–455 CrossRef CAS PubMed.
- J. Wang, J. Yang, J. Xie and N. Xu, Adv. Mater., 2002, 14, 963–965 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- J. P. Perdew and K. Burke, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 16533–16539 CrossRef CAS PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- Z. Zhang, A. Kitada, S. Gao, K. Fukami, N. Tsuji, Z. Yao and K. Murase, ACS Appl. Mater. Interfaces, 2020, 12, 43289–43298 CrossRef CAS PubMed.
- C. H. Lim, B. Selvaraj, Y. F. Song, C. C. Wang, J. T. Jin, S. S. Huang, C. H. Chuang, H. S. Sheu, Y. F. Liao and N. L. Wu, J. Mater. Chem. A, 2017, 5, 21536–21541 RSC.
- D. Shi, X. Lv, Y. Yang, X. Zhang, Z. Tao, C. Xu and X. Rui, Small, 2024, 2402206 CrossRef PubMed.
- Y. Han, Y. Jie, F. Huang, Y. Chen, Z. Lei, G. Zhang, X. Ren, L. Qin, R. Cao and S. Jiao, Adv. Funct. Mater., 2019, 29, 1–7 Search PubMed.
- H. Wan, Z. Wang, W. Zhang, X. He and C. Wang, Nature, 2023, 623, 739–744 CrossRef CAS PubMed.
- E. Aghemenloh, J. O. Umukoro, S. O. Azi, S. Yusuf and J. O. A. Idiodi, Comput. Mater. Sci., 2011, 50, 3290–3296 CrossRef CAS.
- O. Baseggio, D. Toffoli, M. Stener, G. Fronzoni, M. de Simone, C. Grazioli, M. Coreno, A. Guarnaccio, A. Santagata and M. D'Auria, J. Chem. Phys., 2017, 147, 244301 CrossRef CAS PubMed.
- S. Singh, R. K. Sahoo, N. M. Shinde, J. M. Yun, R. S. Mane, W. Chung and K. H. Kim, RSC Adv., 2019, 9, 32154–32164 RSC.
- R. Zalecki, W. M. Woch, M. Kowalik, A. Kolodziejczyk and G. Gritzner, Acta Phys. Pol., A, 2010, 118, 393–395 CrossRef.
- D. A. Zatsepin, D. W. Boukhvalov, N. V. Gavrilov, E. Z. Kurmaev and I. S. Zhidkov, Appl. Surf. Sci., 2016, 387, 1093–1099 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03187c |
| This journal is © The Royal Society of Chemistry 2024 |