
Construction of nickel and sulfur co-doped carbon nanotubes derived from hydrogen-bonded organic frameworks for efficient biomass electrooxidation†
Yuting
Zhang
,
Junhua
Kuang
,
Jia
Yu
*,
Yangyang
Dong
,
Jiaran
Li
,
Tianwei
Xue
,
Jing
Wu
,
Junchi
Ma
,
Jinlong
Wan
,
Shiping
Zeng
,
Yong
Sun
 ,
Yue-Jiao
Zhang
,
Jin-Chao
Dong
,
Li
Peng
*,
Shuliang
Yang
* and
Jian-Feng
Li
*
,
Yue-Jiao
Zhang
,
Jin-Chao
Dong
,
Li
Peng
*,
Shuliang
Yang
* and
Jian-Feng
Li
*
College of Energy, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361102, Fujian, PR China. E-mail: yujia@xmu.edu.cn; li.peng@xmu.edu.cn; ysl@xmu.edu.cn; li@xmu.edu.cn
First published on 12th September 2024
Abstract
The electrochemical conversion of 5-hydroxymethylfurfural (HMF) into 2,5-furandicarboxylic acid (FDCA), powered by renewable electricity, provides an efficient strategy for upgrading biomass to produce high value-added products. In this work, sulfur-doped hollow metal–organic framework (MOF) nanotubes were synthesized in a scalable and controllable manner by using hydrogen-bonded organic frameworks (HOFs) as the template and precursor. Interestingly, sulfur doping of the catalysts could be simply achieved by replacing nickel nitrate with nickel sulfate at the synthesis stage, and the designed sulfur-doped catalysts demonstrated excellent electrocatalytic activity in the HMFOR and remarkable durability, achieving nearly 100% conversion of HMF and high FDCA faradaic efficiency. Moreover, the HMFOR activity could be further enhanced by additional sulfur doping of the prepared catalyst, while maintaining its unique tubular morphology. The combination of sulfur doping with hollow nanotubes greatly increases the electrochemically active surface area and improves electron transport. In situ Raman and electrochemical impedance spectroscopy disclose that the continuously generated Ni3+ active species act as intermediates to promote HMF oxidation. In addition, this facile fabrication method is more environmentally friendly and significantly expands the scope of preparing non-precious metal-based nanotubes for electrocatalytic applications.
 Shuliang Yang | Shuliang Yang is a full professor at the College of Energy, Xiamen University. He obtained his PhD from the Institute of Chemistry, Chinese Academy of Sciences, in 2016 under the supervision of Professor Weiguo Song. From 2017 to 2023, he conducted postdoctoral research at the Massachusetts Institute of Technology (supervisor: Professor Karthish Manthiram), Ecole Polytechnique Fédérale de Lausanne in Switzerland (supervisor: Professor Wendy L. Queen), and Northwestern University in the United States (supervisor: Professor J. Fraser Stoddart). Now, by leveraging innovative porous supports and graphdiyne-based materials, his research is dedicated to developing high-performance nanocatalysts for the efficient conversion and utilization of carbon dioxide, water, and plastics. |
Introduction
With mounting concerns regarding global climate change and the energy crisis, biomass, sourced from organisms such as animals, plants, and microorganisms, presents significant promise as a renewable and sustainable resource for producing valuable chemicals and fuels.1,2 Among these, 5-hydroxymethylfurfural (HMF) emerges as a pivotal biomass-derived platform chemical, garnering widespread industrial interest for its utility in polymer, pharmaceutical, and fine chemical production.3,4 2,5-furandicarboxylic acid (FDCA), obtained through the oxidation of HMF, exhibits excellent potential as a precursor for the production of bio-based polymers such as polyethylene furan dicarboxylate (PEF), which has the capability to replace traditional petroleum-based plastics like polyethylene terephthalate (PET) in various applications.5–7In contrast to the traditional thermochemical catalytic conversion method, which typically requires harsh reaction conditions (temperatures ranging from 50–160 °C and pressures of 1–40 bar oxygen) and precious metal catalysts like Pd and Ru, the electrochemical HMF oxidation reaction (HMFOR) to FDCA presents notable advantages.8,9 These include mild reaction conditions, controllable product distribution, and an environmentally friendly reaction process, rendering it highly appealing for practical applications. The HMFOR exhibits a lower theoretical potential compared to the oxygen evolution reaction (OER).10,11 When coupled with the hydrogen evolution reaction (HER), it could theoretically achieve high overall energy efficiency.12,13 However, catalysts for the HMFOR still suffer from low catalytic activity and unsatisfactory FDCA selectivity, which impedes the practical application of the HMFOR.14,15 Therefore, there is an urgent and highly demanding need to develop highly efficient electrochemical catalysts for HMF conversion in an efficient manner.16,17
Nickel-based electrocatalysts have emerged as promising candidates for the HMFOR due to the abundance of earth's reserves and their relatively high catalytic activity.18,19 Recent studies have demonstrated that the oxidation of HMF relies on the highly active Ni3+ species.20–22 Therefore, constructing catalysts with a higher concentration of high-valent nickel species is key to improving catalytic activity. Additionally, heteroatom doping (such as N, P, B, and S) has been identified as an effective strategy for enhancing the electrocatalytic activity for the HMFOR by adjusting the electronic structure of active sites.23–27 Recent studies have explored the use of sulfur-doped materials,28 including sulfonated carbon-based materials,29 sulfur-functionalized metal oxides, and sulfur-contained metal–organic frameworks (MOFs),30 for the selective oxidation of HMF. For instance, Yan et al. designed an S-modified catalyst by impregnating it in Na2S solution, achieving an FDCA yield of 97.1%.31 Qi et al. reported MOF-supported metal sulfide nanoclusters, showing a 99% yield of FDCA.32 Mu et al. prepared a Co9S8–Ni3S2@N, S, O triple-doped carbon (NSOC) catalyst by adding thiourea as a sulfur source via pyrolysis, achieving a 99.8% yield of FDCA.33 Despite progress in sulfur-doped catalysts for HMF oxidation, significant challenges remain. Firstly, these methods for fabricating S-doped Ni-based catalysts generally require the addition of extra sulfur sources, which complicates the process and raises environmental concerns. Additionally, the ability of a catalyst to adsorb organic molecules is critical for electrocatalytic conversion;34 however, the adsorption and activation capabilities of pure nickel species toward HMF molecules remain suboptimal, severely limiting the catalytic activity for the HMFOR. Furthermore, inadequate electrode stability during continuous operation hinders their practical applications.35 Therefore, the clear objective is to search for an innovative, environmentally friendly method for synthesizing a high-performance and high-stability Ni-based catalyst that could efficiently catalyze the HMFOR while minimizing environmental impacts.
Herein, we develop a straightforward and green method for synthesizing a nickel, sulfur-codoped carbon nanotubes catalyst (NiSO4@CNTs). This method utilizes HOFs with tubular morphology as the templates and NiSO4 salt as the source of both nickel and sulfur. Hydrogen-bonded organic frameworks (HOFs), composed of organic or metal–organic building blocks linked via intermolecular hydrogen bonding, feature ultrahigh surface areas, tubular morphology and easy coordination with metal ions, making them ideal templates for the preparation of metal catalysts.36–40 For comparison, a catalyst without sulfur, named Ni(NO3)2@CNTs, was also synthesized using only Ni(NO3)2 as the metal salt. Meanwhile, a catalyst with a higher sulfur content, denoted as Ni2.4S0.1@CNTs, was fabricated via introducing additional thiourea as the sulfur source. During the electrocatalytic HMFOR experiments, both NiSO4@CNTs and Ni2.4S0.1@CNTs exhibited higher FDCA yield (>96%) and Faraday efficiency (>99%) at 1.51 V vs. RHE compared to Ni(NO3)2@CNTs (with an FDCA yield ∼71% and a Faraday efficiency of ∼75.5%). Electrochemical tests revealed that sulfur doping promoted the formation of high-valent nickel species, which are highly active species for the HMFOR, enhanced the electron transfer rate, and increased the electrochemically active surface area, thereby accelerating the HMFOR kinetics. Furthermore, in situ electrochemical impedance spectroscopy (EIS) and in situ Raman analysis revealed that the high activity for the HMFOR, as opposed to the competitive OER, was attributed to the combined effects of the rapid formation of NiOOH active sites and the sluggish reaction kinetics of the OER. This work aims to develop a simple and environmentally friendly synthesis method for S-doped Ni-based catalysts by incorporating metal coordinate bonds into a hydrogen-bonded network, while also providing a comprehensive understanding of the role of sulfur doping in enhancing HMFOR performance. These findings could aid in the development of efficient electrocatalysts for the sustainable production of bio-based chemicals and materials.
Experimental
Materials and chemicals
Nickel nitrate hexahydrate (Ni(NO3)2·6H2O, 98%), nickel sulfate hexahydrate (NiSO4·6H2O, 98%), trimesic acid (BTC, 98%), furfural, 5-hydroxymethyl-2-furaldehyde (HMF, >98%), 2,5-furandicarboxylic acid (FDCA, >98%), 2,5-diformylfuran (DFF, >98%), 5-hydroxymethyl-2-furancarboxylic acid (HMFCA, >98%) and 5-formyl-2-furancarboxylic acid (FFCA, >98%) were purchased from Aladdin Reagents Ltd. Melamine (MA, 99%) was bought from Sigma-Aldrich (Shanghai) Trading Co., Ltd. Thiocarbamide (thiourea, 98%) was provided by Sahn Chemical Technology (Shanghai) Co., Ltd. Potassium hydroxide (KOH, >98%) was purchased from Sinopharm Chemical Reagent Co., Ltd. Nafion solution (5 wt% in water and isopropanol) was obtained from Meryer (Shanghai) Chemical Technology Co., Ltd. Conductive hydrophobic carbon paper was provided by Suzhou Sinero Technology Co., Ltd. Unless otherwise noted, deionized water (18.2 MΩ cm) was used throughout this work.Synthesis of NiSO4@CNTs
Typically, 1.2 mmol of BTC and 1.2 mmol of MA were dispersed in 35 mL of methanol and ultrasonicated for about 2 h until a uniform milky suspension was formed. To synthesize HOF templates with larger diameters, the milky suspension was transferred into a 100 mL Teflon autoclave and heated to 150 °C for 12 h. After that, 2.4 mmol of NiSO4·6H2O was added to the previously formed suspension and stirred for 15 min to obtain a uniform colloidal suspension. The green mixture was then transferred into a 100 mL Teflon autoclave and heated to 150 °C for 24 h. The green powder was collected by centrifugation, washed several times with ethanol and dried under vacuum at 60 °C. Finally, NiSO4@CNTs was obtained by pyrolyzing the green powder in a tubular furnace at 500 °C for 4 h with a heating rate of 5 °C min−1 under an argon atmosphere.Synthesis of Ni(NO3)2@CNTs
Ni(NO3)2@CNTs was synthesized following a procedure similar to that for NiSO4@CNTs, except that NiSO4·6H2O (2.4 mmol) was replaced with Ni(NO3)2·6H2O (2.4 mmol) as the metal precursor.Synthesis of Ni2.4Sx@CNTs
Ni2.4Sx@CNTs was synthesized using the same method as NiSO4@CNTs, with the modification of replacing NiSO4·6H2O (2.4 mmol) with a mixture of NiSO4·6H2O (2.4 mmol) and thiourea (x mmol). Ni2.4Sx@CNTs samples with different contents of S were obtained by varying the amount of thiourea. Through this approach, Ni2.4S0.1@CNTs, Ni2.4S0.3@CNTs, Ni2.4S0.6@CNTs, Ni2.4S1.2@CNTs, and Ni2.4S1.8@CNTs with different sulfur contents were prepared successfully.Characterization of materials
Powder X-ray diffractometer patterns were recorded on a Rigaku Smart Lab-SE powder X-ray diffractometer with CuKα radiation (λ = 1.5418 Å) at 30 mA and 40 kV. SEM images were acquired using a field emission scanning electron microscope (Zeiss GeminiSEM 500) working at 5 kV. EDS mappings of the materials were conducted using an Oxford super EDS ultim Extreme detector. TEM images and HRTEM images were obtained using a transmission electron microscope (FEI Tecnai F30 TWIN). The nickel contents of different samples were measured using a SPECTROBLUE FMX36 inductively coupled plasma optical emission spectrometer (ICP-OES, SPECTRO, Germany). X-ray photoelectron spectroscopy (XPS) measurements were performed on a Thermo ESCALAB 250XI electron spectrometer with monochromatic AlKα radiation (hv = 1486.8 eV) and the spot size of all spectra was 650 μm. Brunauer–Emmett–Teller (BET) surface area and pore size measurements were carried out on a Micromeritics ASAP 2020M instrument at 77 K. The Raman spectra were collected on an XploRA PLUS Raman spectrometer using a 638 nm laser.Electrochemical measurements
All the electrochemical measurements were performed in a standard three-electrode system on a CHI-660E electrochemical workstation. The working electrode was carbon paper (1 × 1 cm2) coated with the catalysts, while a Pt plate (1 × 1 cm2) and Hg/HgO electrode served as the counter electrode and the reference electrode, respectively. 5 mg of catalyst was dispersed in 1 mL of ethanol/ultrapure water (V![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V = 7:3) and 15 μL of Nafion, followed by ultrasonication for 1 h at room temperature. Then, 200 μL of catalyst ink was drop-coated onto the hydrophobic carbon paper (geometric area: 1 × 1 cm2). The catalyst loading was about 1 mg cm−2. Linear sweep voltammetry (LSV) measurements were performed at room temperature with a scanning rate of 5 mV s−1 in 0.1 M KOH electrolyte solution. The potential values were converted to potential versus the reversible hydrogen electrode (RHE) using the following equation:
:V = 7:3) and 15 μL of Nafion, followed by ultrasonication for 1 h at room temperature. Then, 200 μL of catalyst ink was drop-coated onto the hydrophobic carbon paper (geometric area: 1 × 1 cm2). The catalyst loading was about 1 mg cm−2. Linear sweep voltammetry (LSV) measurements were performed at room temperature with a scanning rate of 5 mV s−1 in 0.1 M KOH electrolyte solution. The potential values were converted to potential versus the reversible hydrogen electrode (RHE) using the following equation:| ERHE = EHg/HgO + 0.059 × pH + 0.098 V |
All LSV data were recorded without iR compensation. Electrochemical impedance spectroscopy (EIS) tests were conducted in the frequency range of 0.1 Hz to 100 kHz with 10 mV amplitude at 1.46 V vs. RHE. Double-layer capacitance values (Cdl) were calculated using cyclic voltammetry (CV) curves with scan rates of 20, 40, 60, 80, 100, and 120 mV s−1 in the non-faradaic region. In situ EIS was performed at a potential of 0.15 V ∼0.95 V vs.Hg/HgO with a frequency ranging from 100 kHz to 0.1 Hz at an amplitude of 10 mV.
Quantitative analysis of products
The products were analyzed using a high-performance liquid chromatography (HPLC) equipped with an HPX-87H column (column chamber at 60 °C) and a UV-vis detector. The mobile phase for HPLC was 5 mM H2SO4 aqueous solution (0.6 mL min−1). All products were analyzed at the maximum absorption wavelength (λ = 285 nm for HMF, DFF and FFCA; λ = 260 nm for HMFCA and FDCA).The conversion of HMF, selectivity and yield of FDCA were calculated using the following equations:
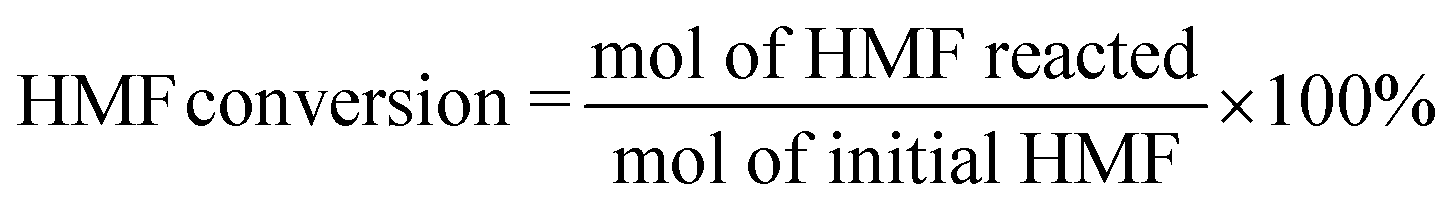

The faradaic efficiency of FDCA formation was calculated using the following equation:
 485 C mol−1).
485 C mol−1).
In situ electrochemical Raman measurements
The electrochemical Raman measurements were carried out on a Raman microscope, including EC-Raman (Xiamen SHINs Technology Co., Ltd) and XploRA (HORIBA). The excitation wavelength of the He–Ne laser was 638 nm, and a 50× microscope objective was used. Before each Raman experiment, the Raman frequencies were calibrated using a Si wafer (638 nm). In situ electrochemical Raman experiments were carried out in a commercial Raman cell (Xiamen SHINs technology Co., Ltd) equipped with a three-electrode system: a carbon paper electrode coated with the catalyst served as the working electrode, Pt wire functioned as the counter electrode, and an Ag/AgCl electrode was employed as the reference electrode. The applied potential was controlled using a potentiostat from Xiamen Qunji Instrument Co. Ltd. Raman spectra were collected in the potential range of 1.30–1.70 V (vs. RHE), and no less than 30 seconds should be spent before acquiring each Raman spectrum.Results and discussion
Nickel and sulfur co-doped carbon nanotubes (NiSO4@CNTs) were prepared via a simple solvothermal-annealing approach, as illustrated in Fig. 1a. Firstly, NiSO4, BTC, and MA were mixed in a methanol solution and reacted in a hydrothermal reactor at 150 °C for 24 h to obtain Ni-based MOF nanotubes. In this process, the initially formed HOF nanowires, resulting from the self-assembly of BTC and MA, were transformed into MOF nanotubes while simultaneously coordinating with Ni species (Fig. S1†). The formation of tubular catalysts through a hydrothermal process utilized HOF nanowires as templates. During the hydrothermal process, Ni ions diffused into the HOF template, replacing the weak hydrogen bonds with strong N–Ni–O coordination bonds. This process gradually led to the formation of a Ni-based MOF shell on the surface of the HOF, creating a core–shell hybrid nanostructure.40 As the MOF shell thickened, the diffusion of Ni ions became increasingly hindered, shifting the transformation process from being chemical reaction-driven to diffusion-controlled. The dense MOF shell, combined with the release of melamine (MA) molecules, created gaps at the interface between the MOF shell and the HOF core, leading to the formation of a yolk–shell structure. Finally, the HOF core dissolved, leaving behind the tubular MOF shell, which maintained its tubular morphology after the subsequent annealing step. Subsequently, the Ni-based MOF nanotubes were annealed at 500 °C for 5 h under an argon atmosphere, resulting in the formation of NiSO4@CNTs. By substituting nickel sulfate with nickel nitrate or introducing thiourea during the solvothermal process, catalysts with different sulfur contents could be prepared, designated as Ni(NO3)2@CNTs and Ni2.4Sx@CNTs (where x represents the molar amount of thiourea added), respectively. Scanning electron microscopy (SEM) was employed to characterize the morphological structure of the three samples. As displayed in Fig. 1b–d, NiSO4@CNTs, Ni(NO3)2@CNTs, and Ni2.4S0.1@CNTs exhibited similar morphologies, all appearing as hollow nanotubes with a diameter of approximately 60 nm. TEM images show that the tube's appearance became more regular after sulfur doping, which could contribute to enhanced morphological stability (Fig. S2†). HRTEM analysis revealed the well-resolved lattice fringes with interplanar spacings of 0.237 nm and 0.183 nm, corresponding to the (003) and (113) planes of Ni3S2, respectively (Fig. S2c†). The influence of the added amount of thiourea and nickel salt on catalyst morphology was further investigated. As depicted in Fig. S3,† the hollow nanotubes transformed into spheres as the content of thiourea increased from 0.1 mmol to 1.8 mmol. Additionally, with an increase in the nickel salt content, the tubular morphology collapsed and led to the formation of irregular flakes (Fig. S4†). Therefore, to eliminate the impact of morphology on catalytic performance, 2.4 mmol of nickel salt and 0.1 mmol of thiourea were added during the solvothermal process to prepare the Ni2.4S0.1@CNTs catalyst. SEM-EDS elemental mappings (Fig. 1e and S5†) confirmed that the characteristic Ni, S, C, N, and O elements were homogeneously distributed within the nanotubes. The BET specific surface area, determined from nitrogen adsorption–desorption isotherms at 77 K, increased slightly after sulfur doping with Ni2SO4, and significantly increased after the addition of thiourea (Fig. S6†). Raman spectra revealed a decrease in the degree of graphitization and an increase in defects in the catalysts after sulfur doping (Fig. S7†). The high defect content could enhance the exposure of active sites and improve catalytic performance.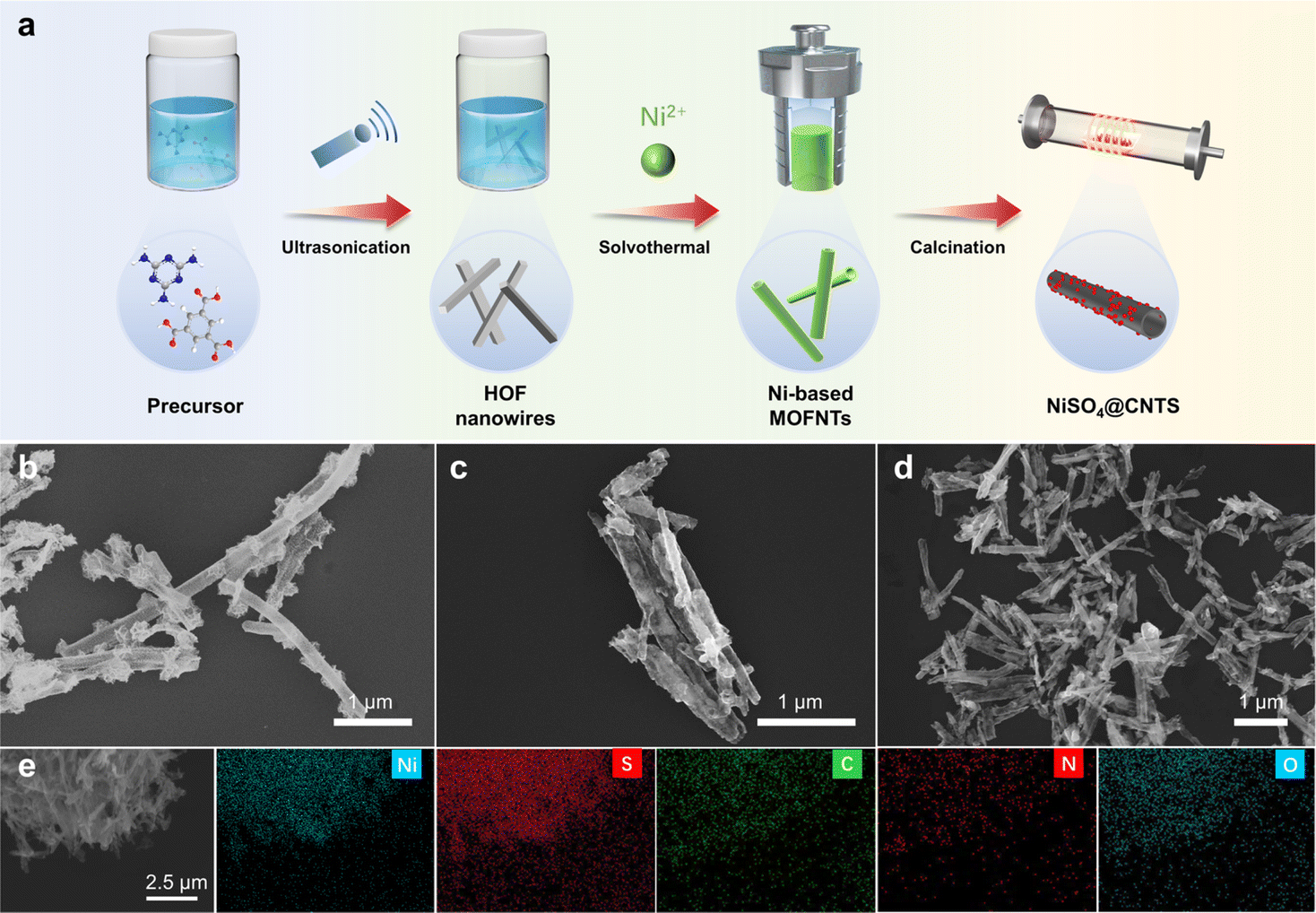 | ||
| Fig. 1 Preparation and characterization of the prepared catalysts. (a) Schematic illustration of the NiSO4@CNTs electrode synthesis. (b–d) SEM images of Ni(NO3)2@CNTs, NiSO4@CNTs, and Ni2.4S0.1@CNTs. (e) Elemental mapping images of Ni2.4S0.1@CNTs with its characteristic Ni, S, C, N, and O elements. | ||
The X-ray diffraction patterns of NiSO4@CNTs, Ni(NO3)2@CNTs, and Ni2.4S0.1@CNTs are shown in Fig. 2a. The diffraction peaks of Ni(NO3)2@CNTs, located at 44.4°, 51.8°, and 76.3°, corresponded to the (111), (200), and (220) planes of metallic Ni (PDF#70-1849), respectively. The diffraction peaks detected in NiSO4@CNTs and Ni2.4S0.1@CNTs at 21.7°, 31.1°, 37.7°, 49.7°, and 55.1° corresponded to the (100), (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10), (111), (210), and (21) planes of Ni3S2 (PDF#85-1802), respectively. X-ray photoelectron spectroscopy (XPS) was employed to investigate the elemental compositions and electronic structure of the synthesized catalysts (Fig. 2b–d). The survey spectrum of NiSO4@CNTs revealed the coexistence of Ni, C, N, O, and S elements (Fig. S8†). The high-resolution XPS spectrum of Ni 2p could be deconvoluted into three spin–orbit doublet peaks, with binding energies at 852.6 eV (Ni 2p3/2) and 869.7 eV (Ni 2p1/2) for Ni, 855.4 eV (Ni 2p3/2) and 872.8 eV (Ni 2p1/2) for Ni2+, and 857.4 eV(Ni 2p3/2) and 875.5 eV (Ni 2p1/2) for Ni3+ along with two satellite peaks (denoted as “Sat.”).41,42 Noticeably, a higher content of Ni3+ species was observed in NiSO4@CNTs (22.1%) than that of Ni(NO3)2@CNTs (10.1%). And the Ni3+ content was further improved for Ni2.4S0.1@CNTs (22.3%) with the introduction of higher sulfur content. Moreover, as compared to those in Ni(NO3)2@CNTs, the peaks related to Ni2+ and Ni0 in NiSO4@CNTs and Ni2.4S0.1@CNTs shifted to higher energy. The interaction between Ni and S was further confirmed from the S 2p XPS spectra of NiSO4@CNTs and Ni2.4S0.1@CNTs. The S 2p XPS spectra could be divided into three peaks, such that the peaks at 162.3 eV and 163.7 eV were assigned to the Ni–S bonds.42,43 These results indicated that the presence of S could modify the electronic structure of Ni species, and improve the content of Ni3+ due to the stronger electronic interaction between Ni and S. In the O 1s spectra, three main peaks located at 529.6 eV, 531.5 eV and 533.8 eV are associated with the lattice O2− bonds (Ni–O, OL), hydroxyl groups, and oxygen-species absorbed on the surface (Oads), respectively.44–46 Overall, both XRD and XPS results clearly demonstrated that sulfur doping induced the transformation of Ni species to Ni3S2 with electron transfer between Ni species and S. Moreover, the strong electronic interaction between S and Ni increased the concentration of Ni3+ in Ni2.4S0.1@CNTs, leading to a great enhancement in the subsequent catalytic performance.
10), (111), (210), and (21) planes of Ni3S2 (PDF#85-1802), respectively. X-ray photoelectron spectroscopy (XPS) was employed to investigate the elemental compositions and electronic structure of the synthesized catalysts (Fig. 2b–d). The survey spectrum of NiSO4@CNTs revealed the coexistence of Ni, C, N, O, and S elements (Fig. S8†). The high-resolution XPS spectrum of Ni 2p could be deconvoluted into three spin–orbit doublet peaks, with binding energies at 852.6 eV (Ni 2p3/2) and 869.7 eV (Ni 2p1/2) for Ni, 855.4 eV (Ni 2p3/2) and 872.8 eV (Ni 2p1/2) for Ni2+, and 857.4 eV(Ni 2p3/2) and 875.5 eV (Ni 2p1/2) for Ni3+ along with two satellite peaks (denoted as “Sat.”).41,42 Noticeably, a higher content of Ni3+ species was observed in NiSO4@CNTs (22.1%) than that of Ni(NO3)2@CNTs (10.1%). And the Ni3+ content was further improved for Ni2.4S0.1@CNTs (22.3%) with the introduction of higher sulfur content. Moreover, as compared to those in Ni(NO3)2@CNTs, the peaks related to Ni2+ and Ni0 in NiSO4@CNTs and Ni2.4S0.1@CNTs shifted to higher energy. The interaction between Ni and S was further confirmed from the S 2p XPS spectra of NiSO4@CNTs and Ni2.4S0.1@CNTs. The S 2p XPS spectra could be divided into three peaks, such that the peaks at 162.3 eV and 163.7 eV were assigned to the Ni–S bonds.42,43 These results indicated that the presence of S could modify the electronic structure of Ni species, and improve the content of Ni3+ due to the stronger electronic interaction between Ni and S. In the O 1s spectra, three main peaks located at 529.6 eV, 531.5 eV and 533.8 eV are associated with the lattice O2− bonds (Ni–O, OL), hydroxyl groups, and oxygen-species absorbed on the surface (Oads), respectively.44–46 Overall, both XRD and XPS results clearly demonstrated that sulfur doping induced the transformation of Ni species to Ni3S2 with electron transfer between Ni species and S. Moreover, the strong electronic interaction between S and Ni increased the concentration of Ni3+ in Ni2.4S0.1@CNTs, leading to a great enhancement in the subsequent catalytic performance.
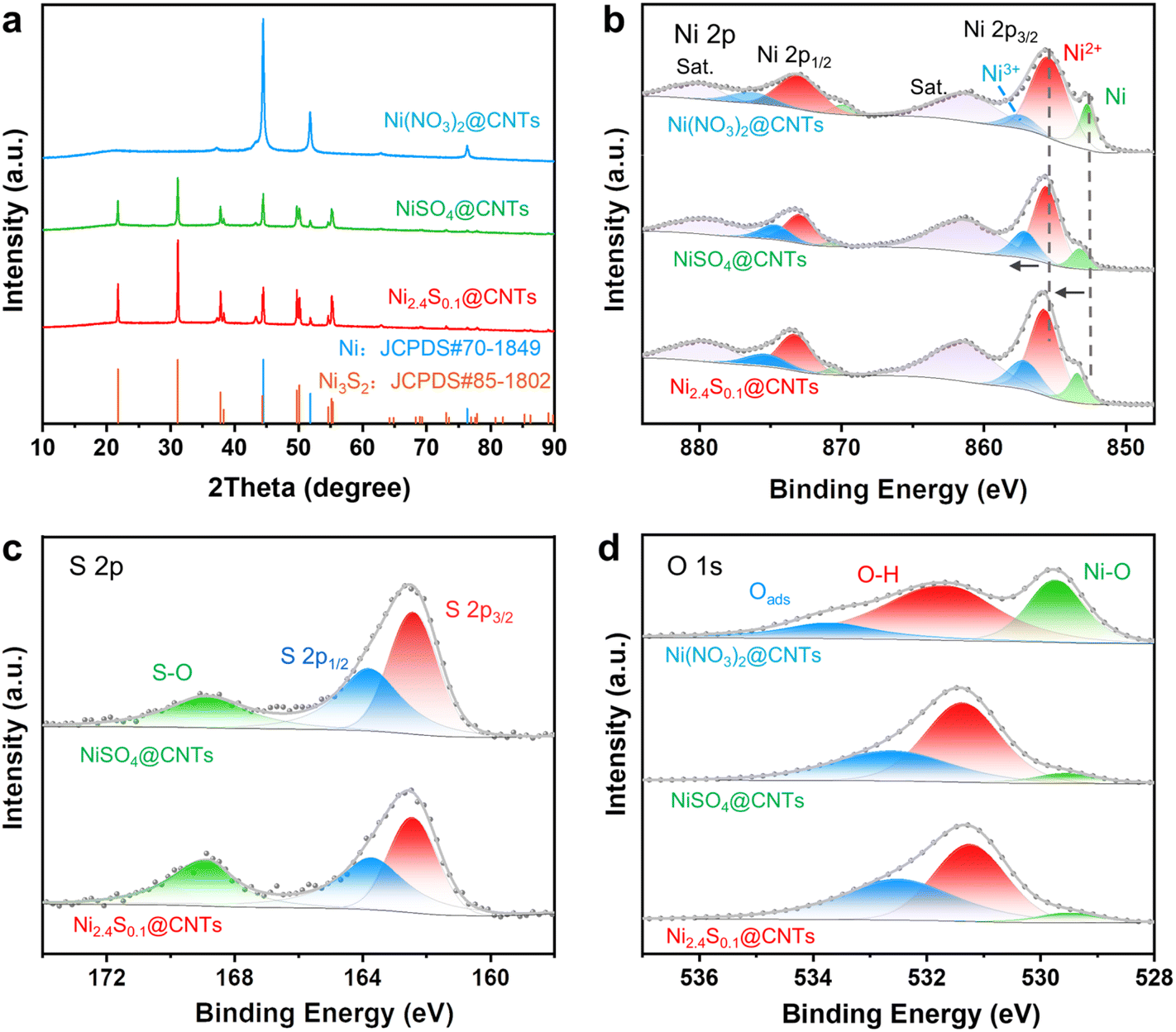 | ||
| Fig. 2 (a) XRD patterns of Ni(NO3)2@CNTs, NiSO4@CNTs, and Ni2.4S0.1@CNTs. (b–d) High-resolution XPS spectra of Ni 2p, S 2p, and O 1s for Ni(NO3)2@CNTs, NiSO4@CNTs, and Ni2.4S0.1@CNTs samples. | ||
The electrocatalytic performance of the prepared catalysts was tested in an H-cell with 0.1 M KOH as the electrolyte under ambient conditions. The catalysts were deposited on carbon paper with a loaded mass of 1 mg cm−2. Linear sweep voltammetry (LSV) was conducted on Ni(NO3)2@CNTs, NiSO4@CNTs, and Ni2.4S0.1@CNTs with or without 10 mM HMF to evaluate the relationship between HMF oxidation activities and the catalysts' structure (Fig. 3a). As observed, the current densities significantly increased upon the addition of 10 mM HMF into the electrolyte, indicating a preference for HMF oxidation over the OER on all three catalysts. Compared to Ni(NO3)2@CNTs, NiSO4@CNTs exhibited a higher current density at the same overpotential, indicating that sulfur doping could improve the charge transfer rate. The current density was further enhanced for Ni2.4S0.1@CNTs with higher sulfur content, proving the positive effect of sulfur introduction on catalyst activity (Fig. 3b). The LSV curves of catalysts synthesized with higher thiourea content were also tested (Fig. S9†). Remarkably, when normalized to nickel loading, both Ni2.4S0.1@CNTs and NiSO4@CNTs exhibited superior mass activity toward HMF oxidation compared to Ni(NO3)2@CNTs, while the Ni2.4S0.1@CNTs possessed the best performance among the tested catalysts. The content of Ni in all catalysts were detected via inductively coupled plasma optical emission spectroscopy (ICP-OES; Table S1†). For example, at 1.51 V vs. RHE, the mass activities of Ni2.4S0.1@CNTs (14.3 mA mg−1) and NiSO4@CNTs (10.4 mA mg−1) were 5.5 and 4.0 times higher than that of Ni(NO3)2@CNTs (2.6 mA mg−1), respectively (Fig. 3c).
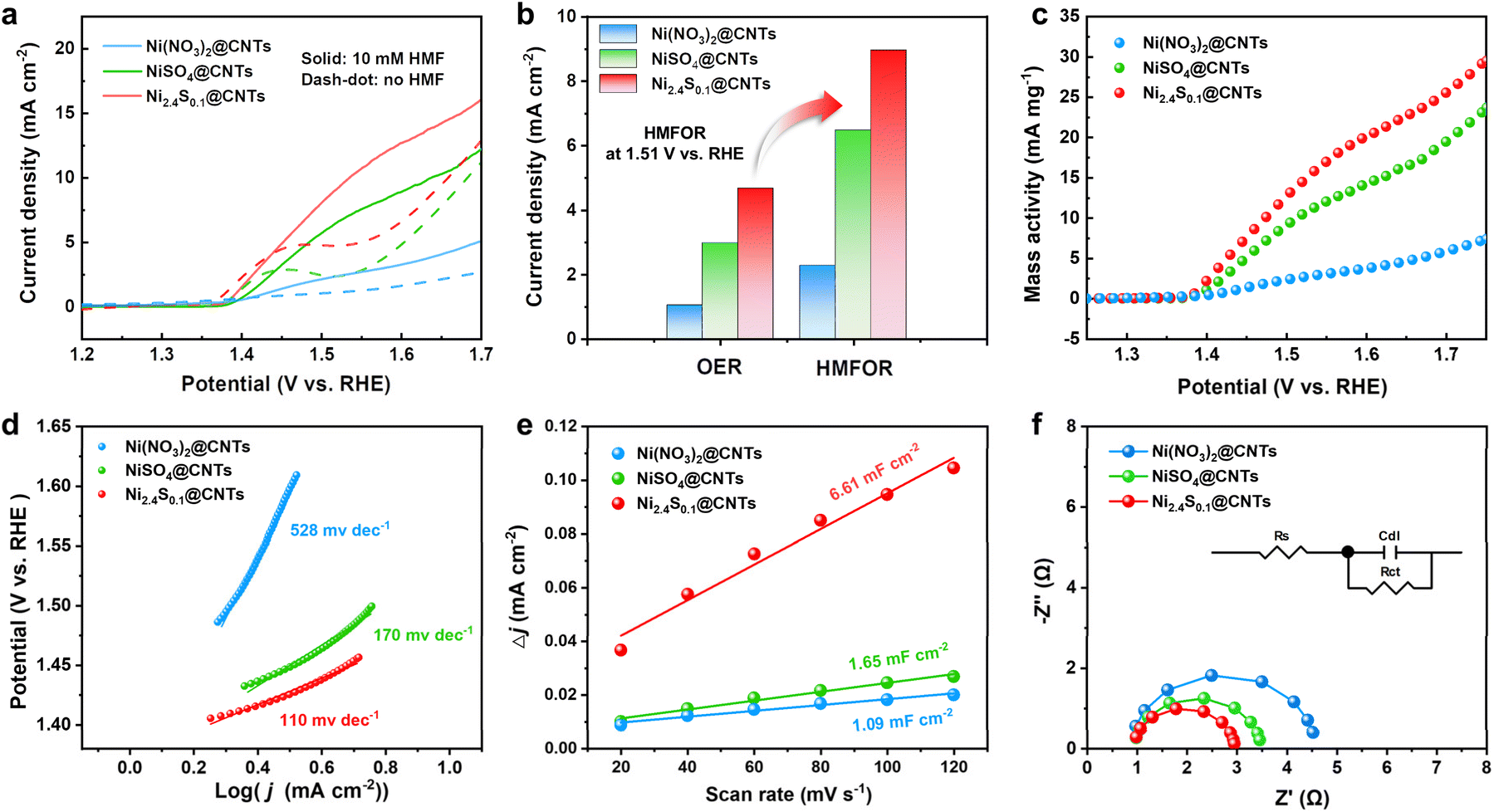 | ||
| Fig. 3 Catalytic performance of HMFOR and electrochemical characterization of Ni(NO3)2@CNTs, NiSO4@CNTs, and Ni2.4S0.1@CNTs electrodes. (a) Linear sweep voltammetry (LSV) curves. (b) The corresponding current densities for the OER and HMFOR. (c) Mass activity, (d) Tafel plots, (e) capacitive currents, and (f) EIS Nyquist plots with different catalysts. | ||
The intrinsic activity of different samples in the presence of HMF was further assessed using the Tafel slopes (Fig. 3d). Ni2.4S0.1@CNTs exhibited the lowest Tafel slope (110 mV dec−1) compared to Ni(NO3)2@CNTs (528 mV dec−1) and NiSO4@CNTs (170 mV dec−1), demonstrating its favorable HMF electrooxidation kinetics. The electrochemically active surface area (ECSA) of the three catalysts was estimated by determining the double-layer capacitance (Cdl), calculated by performing cyclic voltammetry (Fig. S10†) in the non-faradaic region. The Cdl of Ni2.4S0.1@CNTs was determined to be 6.61 mF cm−2, which is larger than that of NiSO4@CNTs (1.65 mF cm−2) and Ni(NO3)2@CNTs (1.09 mF cm−2) (Fig. 3e), revealing the largest ECSA of Ni2.4S0.1@CNTs. Clearly, these positive results also revealed that the introduction of sulfur could provide more exposed active sites for the catalytic conversion of HMF to FDCA. Furthermore, electrochemical impedance spectroscopy (EIS) was also conducted (Fig. 3f) to investigate the reaction kinetics of catalysts for HMF oxidation. Nyquist plots were fitted with an equivalent circuit model (Fig. 3f, inset). Compared with Ni(NO3)2@CNTs, NiSO4@CNTs and Ni2.4S0.1@CNTs showed smaller charge-transfer resistance (Rct) indicating the improved charge-transfer behavior at the catalyst interface (Table S2†). All the aforementioned results revealed that the interaction between S and Ni species improved the ECSA, reduced mass transfer resistance, and facilitated charge-transfer kinetics. As a result, these improvements contribute to the enhanced electrocatalytic performance for HMF oxidation.
The presence of both hydroxymethyl and aldehyde groups in HMF allows for oxidation to aldehydes and carboxylic acids. Thus, there are two possible pathways for the HMF oxidation reaction (Fig. 4a). If the aldehyde group in HMF is oxidized initially, it yields 5-hydroxymethyl-2-furanformic acid (HMFCA), whereas if the hydroxymethyl group is oxidized first, 2,5-diformylfuran (DFF) will be produced. Both HMFCA and DFF can undergo further oxidation to generate the FFCA intermediate. Subsequently, the FFCA is further oxidized to produce the desired FDCA. To identify the real reaction path in our catalytic system, the concentration of substrates, intermediates and products changing with time was quantified in 0.1 M KOH with 10 mM HMF at a constant potential of 1.51 V vs. RHE by high-performance liquid chromatography (HPLC). As displayed in Fig. 4b, only two kinds of intermediates, HMFCA and FFCA, were detected by HPLC during the HMF oxidation process, demonstrating that HMF oxidation followed the aldehyde oxidation pathway (path I). This result is consistent with the mechanism reported in the literature. To compare the electrocatalytic HMF oxidation performance of the three catalysts, the faradaic efficiency (FE) and yield of FDCA of Ni(NO3)2@CNTs, NiSO4@CNTs and Ni2.4S0.1@CNTs were tested at the same potential (1.51 V vs. RHE). As shown in Fig. 4c, both NiSO4@CNTs and Ni2.4S0.1@CNTs showed superior FDCA yield (>96%) and higher FE (>99%) at 1.51 V vs. RHE compared to Ni(NO3)2@CNTs (FDCA yield ∼71%, FE ∼75.5%). Moreover, for the optimized Ni2.4S0.1@CNTs, both FE and FDCA yields were over 90% from 1.45 to 1.51 V vs. RHE, indicating a wide range of applied potentials (Fig. S11†). As the reaction progressed (Fig. S12†), the current density gradually decreased to zero due to HMF consumption. It is noteworthy that both NiSO4@CNTs and Ni2.4S0.1@CNTs exhibited excellent electrocatalytic activities for the HMFOR, highlighting the unique contribution of sulfur doping in the HMFOR. The additional sulfur doping using thiourea in Ni2.4S0.1@CNTs further improved the reaction activity and shortened the reaction time. Therefore, Ni2.4S0.1@CNTs was selected as the best catalyst for subsequent stability tests and in situ characterization studies. To evaluate the stability of Ni2.4S0.1@CNTs for the HMFOR, continuous electrolysis experiments were performed (Fig. 4d). The results of the stability tests were presented as time-dependent current density plots (Fig. S13†). The performance remained almost unchanged over five successive cycles, further confirming the excellent stability of Ni2.4S0.1@CNTs for the HMFOR.
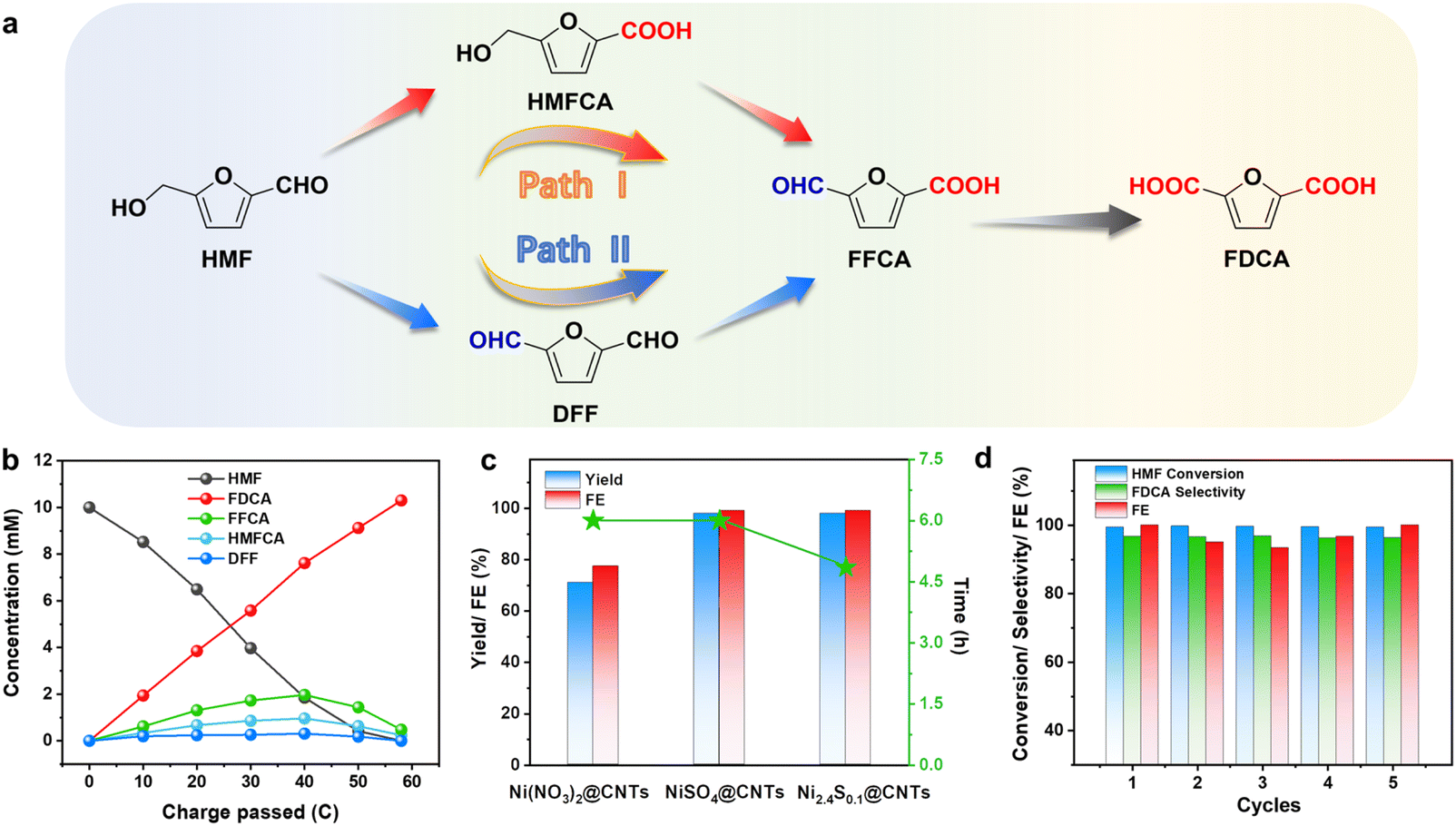 | ||
| Fig. 4 Electrocatalytic oxidation of HMF to FDCA with the Ni2.4S0.1@CNTs catalyst: (a) two possible pathways of HMF oxidation to FDCA. (b) Concentration evolution of products during the HMFOR process. (c) FDCA yield (%) and Faraday efficiency (FE, %) over different catalysts. (d) The HMFOR performance of Ni2.4S0.1@CNTs over five continuous cycles. | ||
To study the relationship between potential, structure, and activity, in situ electrochemical impedance spectroscopy (EIS), an electronic technique, was adopted to explore the interfacial evolution of the Ni2.4S0.1@CNTs catalyst. The low-frequency region (10−1–101 Hz) is related to the oxidation of species at the electrode interface, such as the OER and HMFOR, while the high-frequency region (101–105 Hz) corresponds to the oxidation of the electrode catalysts.47–49 As shown in Fig. 5a, the peak in the low-frequency region from 1.60 V vs. RHE suggested that the OER occurred with sluggish kinetics. In contrast, after adding HMF, the peak belonging to the HMFOR could be observed at the potentials of 1.35 and 1.40 V vs. RHE, where no signals were observed for the OER, suggesting that the HMFOR occurred instead of the OER (Fig. 5b). These results unraveled the sluggish OER kinetics and high HMFOR oxidation activity on Ni2.4S0.1@CNTs. To further quantify the relationship between the structural evolution of the catalyst and electrode interface reaction, EIS data (Table S3†) were fitted through the model as shown in Fig. S14.† The Rp which represented the resistance of the electrode catalyst to oxidation and interface oxidation, decreased when the potential reached 1.25 V vs. RHE, indicating changes in the catalyst structure.49,50 The resistance of interface oxidation (Rct) decreased when potential reached 1.35 V vs. RHE (Fig. 5c). These results indicated that the catalyst was first oxidized to real active sites and then participated in the HMFOR.
 | ||
| Fig. 5 (a and b) Bode phase plots of the in situ EIS for Ni2.4S0.1@CNTs at different potentials with and without HMF. (c) The resistance of the electrode interface reaction (Rct) and electrode inner oxidation (Rp) on Ni2.4S0.1@CNTs. (d) Schematic diagram of the in situ Raman device. (e and f) In situ Raman spectroscopy of Ni2.4S0.1@CNTs at different potentials with and without HMF. | ||
An in situ Raman device was employed to directly investigate the oxidized species during the HMFOR (Fig. 5d). In a 0.1 M KOH aqueous solution, two distinct vibration peaks at 472 cm−1 and 553 cm−1 assigned to the Ni–O stretching modes of Ni3+OOH were observed,51,52 confirming that the high valence Ni3+OOH species were formed in the OER (Fig. 5e). The intensity of both peaks increased as the potential increased from 1.35 to 1.70 V vs. RHE. After adding HMF, the characteristic peaks corresponding to Ni3+OOH vanished at potentials ranging from 1.30 to 1.55 V vs. RHE (Fig. 5f), suggesting that the in situ generated Ni3+OOH immediately reacted with HMF and was reduced to Ni2+. When the potential reached 1.60 V vs. RHE, the peaks related to Ni3+OOH reappeared, which was due to the faster kinetics of the OER than that of the HMFOR.19,41 Thus, based on the in situ EIS and in situ Raman results, it is reasonable to infer that during the HMFOR, Ni2+ was initially oxidized to Ni3+OOH, which then facilitated the conversion of HMF to FDCA accompanied by the reduction of Ni3+OOH to Ni2+. The continuously generated Ni3+ active species on the surface of the Ni2.4S0.1@CNTs catalyst are responsible for the highly efficient oxidation of HMF to FDCA.
To gain deeper insight into the catalytic process and mechanism, we conducted further analyses and summarized the findings based on the above discussions. For the HMFOR using Ni2.4S0.1@CNTs the catalyst, HMF oxidation to FDCA followed a stepwise pathway: HMF was first oxidized to HMFCA, then to FFCA, and finally to FDCA, as confirmed by HPLC analysis (Fig. 4b). In situ Raman and EIS techniques revealed that the catalyst was initially oxidized to NiOOH, the active form for HMF oxidation. Sulfur doping facilitated the formation of NiOOH and enhanced the adsorption of HMF onto the Ni3S2 surface. The aldehyde group in HMF was selectively oxidized to form HMFCA, which was further oxidized to FFCA by removing hydrogen from the hydroxyl group. Finally, FFCA was oxidized to FDCA at the NiOOH active sites. The desorption of FDCA from the catalyst surface completed the catalytic cycle, which was facilitated by the reduced surface energy imparted by sulfur doping.
Following the continuous HMFOR experiments at constant potentials, we conducted a series of characterization studies to investigate the possible changes in the morphology and structure of the catalyst during the stability testing process. The SEM image (Fig. S15†) revealed that the tubular morphology of the catalysts remained intact, indicating good structural stability. XRD patterns (Fig. S16†) confirmed that the crystallinity and Ni3S2 phase of the catalyst were preserved, suggesting minimal structural degradation. Furthermore, XPS analysis (Fig. S17†) revealed the changes in the chemical composition and oxidation states of the catalysts. The disappearance of Ni0 was observed, indicating a complete transformation of metallic nickel to higher-valent nickel states. The retention of catalytic activity despite the absence of Ni0 suggested that metallic nickel did not play a significant role in the catalytic mechanism for the oxidation of HMF to FDCA. Instead, the higher-valent nickel species were the active centers responsible for the catalytic conversion.
Conclusions
In summary, we have developed a simple method for producing a carbon nanotube-supported Ni3S2 catalyst (NiSO4@CNTs) by using tubular-shaped HOFs as templates, and NiSO4 salt as both the nickel and sulfur source. The catalysts without sulfur (Ni(NO3)2@CNTs) and with high sulfur content (Ni2.4S0.1@CNTs) were also fabricated for comparison. The use of NiSO4 as the sole sulfur source in NiSO4@CNTs showed excellent catalytic performance for the HMFOR. Further sulfur doping using thiourea emphasized the unique contribution of sulfur in enhancing the catalytic activity for the HMFOR. Both NiSO4@CNTs and Ni2.4S0.1@CNTs demonstrated superior FDCA yield (>96%) and higher Faraday efficiency (>99%) at 1.51 V vs. RHE than Ni(NO3)2@CNTs (FDCA yield ∼71% and Faraday efficiency ∼75.5%). Sulfur doping could facilitate the formation of high-valent nickel species which are active species for the HMFOR, enhance electron transfer rates and increase the electrochemically active surface area, thus improving catalytic activity. Both in situ EIS and in situ Raman analysis indicated that the enhanced HMFOR activity, rather than the competitive OER, was attributed to the rapid formation of Ni3+ active sites for the HMFOR and sluggish kinetics of the OER over the Ni2.4S0.1@CNTs catalyst. This work not only presents a facile and economical route for fabricating sulfur-doped Ni-based catalysts, but also provides a rational pathway for improving HMFOR catalytic activity through heteroatom doping.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Y. T. Z., J. Y., L. P., S. L. Y., and J. F. L. proposed the project, designed the experiments, and wrote the manuscript. Y. T. Z. performed the whole experiment. J. H. K., Y. Y. D., and J. R. L. performed the analysis of experimental data. T. W. X, J. C. M., and J. W. conducted a part of the characterization studies. J. L. W., S. P. Z., Y. S., Y. J. Z., and J. C. D. participated in the discussions. J. Y., L.P., S. L. Y., and J. F. L. supervised the whole project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors sincerely thank all co-workers involved in the development of this work. This work was financially supported by the Natural Science Foundation of Fujian Province, China (2022J05009), the National Natural Science Foundation of China (22373080 and 21925404) and the Fundamental Research Funds for the Central Universities (20720240054). L. P. and S. Y. sincerely acknowledge the financial support from the Nanqiang Youth Scholar Program of Xiamen University. We express sincere gratitude to Zhifeng He at Xiamen University for help in SEM and TEM analyses.References
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- A. H. Motagamwala, W. Won, C. Sener, D. M. Alonso, C. T. Maravelias and J. A. Dumesic, Sci. Adv., 2018, 4, eaap9722 CrossRef PubMed.
- R.-J. Van Putten, J. C. Van Der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- S. Rajendran, R. Raghunathan, I. Hevus, R. Krishnan, A. Ugrinov, M. P. Sibi, D. C. Webster and J. Sivaguru, Angew. Chem., Int. Ed., 2015, 54, 1159–1163 CrossRef CAS PubMed.
- Y. Lu, T. Liu, C. L. Dong, Y. C. Huang, Y. Li, J. Chen, Y. Zou and S. Wang, Adv. Mater., 2021, 33, 2007056 CrossRef CAS PubMed.
- A. J. J. E. Eerhart, A. P. C. Faaij and M. K. Patel, Energy Environ. Sci., 2012, 5, 6407 RSC.
- N. K. Gupta, S. Nishimura, A. Takagaki and K. Ebitani, Green Chem., 2011, 13, 824 RSC.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- Y. Yan, J.-Y. Zhang, X.-R. Shi, Y. Zhu, C. Xia, S. Zaman, X. Hu, X. Wang and B. Y. Xia, ACS Nano, 2021, 15, 10286–10295 CrossRef CAS PubMed.
- W. Guan, Y. Zhang, Y. Wei, B. Li, Y. Feng, C. Yan, P. Huo and Y. Yan, Fuel, 2020, 278, 118362 CrossRef CAS.
- G.-R. Xu, M. Batmunkh, S. Donne, H. Jin, J.-X. Jiang, Y. Chen and T. Ma, J. Mater. Chem. A, 2019, 7, 25433–25440 RSC.
- M. Park, M. Gu and B.-S. Kim, ACS Nano, 2020, 14, 6812–6822 CrossRef CAS PubMed.
- M. Li, L. Chen, S. Ye, G. Fan, L. Yang, X. Zhang and F. Li, J. Mater. Chem. A, 2019, 7, 13695–13704 RSC.
- J. Ren, J. Wang, Z. Li, C. Ning, W. Cao, S. Li, G. I. N. Waterhouse, L. Zheng, D. O'Hare and Y. Zhao, J. Mater. Chem. A, 2024, 12, 4333–4342 RSC.
- Y. Lu, C. L. Dong, Y. C. Huang, Y. Zou, Z. Liu, Y. Liu, Y. Li, N. He, J. Shi and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 19215–19221 CrossRef CAS PubMed.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- M. J. Kang, H. Park, J. Jegal, S. Y. Hwang, Y. S. Kang and H. G. Cha, Appl. Catal., B, 2019, 242, 85–91 CrossRef CAS.
- Z. Cheng, D. Fu, W. Zhou, W. Deng, Y. Tan and M. Ma, J. Mater. Chem. A, 2023, 11, 25983–25991 RSC.
- D. Chen, Y. Ding, X. Cao, L. Wang, H. Lee, G. Lin, W. Li, G. Ding and L. Sun, Angew. Chem., Int. Ed., 2023, 62, e20230947 Search PubMed.
- X. Lu, K. H. Wu, B. Zhang, J. Chen, F. Li, B. J. Su, P. Yan, J. M. Chen and W. Qi, Angew. Chem., Int. Ed., 2021, 60, 14528–14535 CrossRef CAS PubMed.
- B. Zhang, H. Fu and T. Mu, Green Chem., 2022, 24, 877–884 RSC.
- W.-J. Liu, L. Dang, Z. Xu, H.-Q. Yu, S. Jin and G. W. Huber, ACS Catal., 2018, 8, 5533–5541 CrossRef CAS.
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS PubMed.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS PubMed.
- S. Barwe, J. Weidner, S. Cychy, D. M. Morales, S. Dieckhöfer, D. Hiltrop, J. Masa, M. Muhler and W. Schuhmann, Angew. Chem., Int. Ed., 2018, 57, 11460–11464 CrossRef CAS PubMed.
- M. Zhang, Y. Liu, B. Liu, Z. Chen, H. Xu and K. Yan, ACS Catal., 2020, 10, 5179–5189 CrossRef CAS.
- Y. Han, D. Yan, Z. Ma, Q. Wang, X. Wang, Y. Li and G. Sun, Int. J. Biol. Macromol., 2023, 244, 125363 CrossRef CAS PubMed.
- S. Chandrasekaran, L. Yao, L. Deng, C. Bowen, Y. Zhang, S. Chen, Z. Lin, F. Peng and P. Zhang, Chem. Soc. Rev., 2019, 48, 4178–4280 RSC.
- C. Chen, J. Chai, M. Sun, T. Guo, J. Lin, Y. Zhou, Z. Sun, F. Zhang, L. Zhang, W. Chen and Y. Li, Energy Environ. Sci., 2024, 17, 2298–2308 RSC.
- C. Liu, X. R. Shi, K. Yue, P. Wang, K. Zhan, X. Wang, B. Y. Xia and Y. Yan, Adv. Mater., 2023, 35, 2211177 CrossRef CAS PubMed.
- Y. X. Feng, K. Yang, R. L. Smith and X. H. Qi, J. Mater. Chem. A, 2023, 11, 6375–6383 RSC.
- Y. Zhang, Z. Xue, X. Zhao, B. Zhang and T. Mu, Green Chem., 2022, 24, 1721–1731 RSC.
- R.-Q. Huang, W.-P. Liao, M.-X. Yan, S. Liu, Y.-M. Li and X.-W. Kang, J. Appl. Electrochem., 2023, 29, 2203081 Search PubMed.
- M.-Y. Ding, W.-J. Jiang, T.-Q. Yu, X.-Y. Zhuo, X.-J. Qin and S.-B. Yin, J. Appl. Electrochem., 2023, 29, 2208121 Search PubMed.
- B. Zhou and D. Yan, Adv. Funct. Mater., 2019, 29, 1807599 CrossRef.
- P. Giusto, H. Arazoe, D. Cruz, P. Lova, T. Heil, T. Aida and M. Antonietti, J. Am. Chem. Soc., 2020, 142, 20883–20891 CrossRef CAS PubMed.
- M. Igarashi, T. Nozawa, T. Matsumoto, F. Yagihashi, T. Kikuchi and K. Sato, Nat. Commun., 2021, 12, 7025 CrossRef CAS PubMed.
- J. Zhang, Y. Feng, R. J. Staples, J. Zhang and J. N. M. Shreeve, Nat. Commun., 2021, 12, 2146 CrossRef CAS PubMed.
- Z.-X. Cai, Y. Xia, Y. Ito, M. Ohtani, H. Sakamoto, A. Ito, Y. Bai, Z.-L. Wang, Y. Yamauchi and T. Fujita, ACS Nano, 2022, 16, 20851–20864 CrossRef CAS PubMed.
- L. Zheng, Y. Zhao, P. Xu, Z. Lv, X. Shi and H. Zheng, J. Mater. Chem. A, 2022, 10, 10181–10191 RSC.
- S. Li, S. Wang, Y. Wang, J. He, K. Li, Y. Xu, M. Wang, S. Zhao, X. Li, X. Zhong and J. Wang, Adv. Funct. Mater., 2023, 33, 2214488 CrossRef CAS.
- Y. Feng, R. L. Smith, J. Fu and X. Qi, Green Chem., 2023, 25, 8698–8705 RSC.
- B. Zhang, Z. Yang, C. Yan, Z. Xue and T. Mu, Small, 2023, 19, 2207236 CrossRef CAS PubMed.
- M. Hou, S. Gong, L. Ji, J. Huang, M. Xu and Z. Chen, Chem. Eng. J., 2021, 419, 129575 CrossRef CAS.
- G. Yang, Y. Jiao, H. Yan, Y. Xie, A. Wu, X. Dong, D. Guo, C. Tian and H. Fu, Adv. Mater., 2020, 32, 2000455 CrossRef CAS PubMed.
- W. Chen, C. Xie, Y. Wang, Y. Zou, C.-L. Dong, Y.-C. Huang, Z. Xiao, Z. Wei, S. Du, C. Chen, B. Zhou, J. Ma and S. Wang, Chem, 2020, 6, 2974–2993 CAS.
- R. Lin, L. Kang, T. Zhao, J. Feng, V. Celorrio, G. Zhang, G. Cibin, A. Kucernak, D. J. L. Brett, F. Corà, I. P. Parkin and G. He, Energy Environ. Sci., 2022, 15, 2386–2396 RSC.
- Y. Lu, T. Liu, C. L. Dong, C. Yang, L. Zhou, Y. C. Huang, Y. Li, B. Zhou, Y. Zou and S. Wang, Adv. Mater., 2022, 34, 2107185 CrossRef CAS PubMed.
- W. Chen, L. Xu, X. Zhu, Y. C. Huang, W. Zhou, D. Wang, Y. Zhou, S. Du, Q. Li, C. Xie, L. Tao, C. L. Dong, J. Liu, Y. Wang, R. Chen, H. Su, C. Chen, Y. Zou, Y. Li, Q. Liu and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 7297–7307 CrossRef CAS PubMed.
- P. Zhang, X. Sheng, X. Chen, Z. Fang, J. Jiang, M. Wang, F. Li, L. Fan, Y. Ren, B. Zhang, B. J. J. Timmer, M. S. G. Ahlquist and L. Sun, Angew. Chem., Int. Ed., 2019, 58, 9155–9159 CrossRef CAS PubMed.
- C. Huang, Y. Huang, C. Liu, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 12014–12017 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02469a |
| This journal is © The Royal Society of Chemistry 2024 |