
Large-scale synthesis of N-doped carbon spherical shells as high-performance cathode materials for Li–X (X = O2, S, Se) batteries†
Kailing
Sun
a,
Xiaocong
Deng
a,
Xian
Huang
a,
Shijun
Liao
 b,
Limei
Liu
a,
Mei
Yang
*c and
Tongye
Wei
*a
b,
Limei
Liu
a,
Mei
Yang
*c and
Tongye
Wei
*a
aDepartment of Physics, Hunan Institute of Advanced Sensing and Information Technology, Xiangtan University, Xiangtan 411105, P.R. China. E-mail: Weity@XTU.edu.cn
bThe Key Laboratory of Fuel Cell Technology of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510641, China
cKey Laboratory of Polymeric Materials & Application Technology of Hunan Province, Key Laboratory of Advanced Functional Polymeric Materials of College of Hunan Province, Key Lab of Environment-Friendly Chemistry and Application in Ministry of Education, Xiangtan University, Xiangtan 411105, Hunan, P. R. China. E-mail: yangmei@xtu.edu.cn
First published on 11th September 2024
Abstract
A porous carbon spherical shell (PCS) with an ordered pore structure is a promising electrode material for electrocatalysis and energy storage applications. However, the preparation of high-performance PCS on a large scale is complex and energy-consuming. We report a gram-scale synthesis of a hierarchical meso/macroporous carbon spherical shell (C–FN) through a facile spray-drying carbonization strategy. Systematic characterizations, including Raman and BET analysis, reveal that C–FN has a high degree of graphitization and a large specific surface area of 893.3 m2 g−1. In addition, a certain amount of doped N atoms in C–FN are beneficial in enhancing its electrocatalytic activity. When used as the cathode material in Li–O2 batteries, the optimized three-dimensional channels within C–FN not only can facilitate the transportation of oxygen, lithium-ion, and electrons but also accommodate the discharge product on both the inner and outer shells, which results in an ultrahigh discharge capacity of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 038 mA h g−1 or 7.85 mA h cm−2. Moreover, when assembling Li–S/Li–Se battery with S and Se infiltrated into C–FN, the nanocomposites obtained show favorable electrochemical performances in terms of specific capacity (Li–S: 1336.8 mA h g−1; Li–Se: 829.3 mA h g−1), cycling stability (after 270 cycle capacity retention of 661.5 mA h g−1 for Li–S; after 1000 cycle capacity retention of 150.1 mA h g−1 for Li–Se), and high-rate capability. Through rational and delicate design, the C–FN holds great promise for the development of Li–X (O2, S, Se) batteries with high power and energy densities.
038 mA h g−1 or 7.85 mA h cm−2. Moreover, when assembling Li–S/Li–Se battery with S and Se infiltrated into C–FN, the nanocomposites obtained show favorable electrochemical performances in terms of specific capacity (Li–S: 1336.8 mA h g−1; Li–Se: 829.3 mA h g−1), cycling stability (after 270 cycle capacity retention of 661.5 mA h g−1 for Li–S; after 1000 cycle capacity retention of 150.1 mA h g−1 for Li–Se), and high-rate capability. Through rational and delicate design, the C–FN holds great promise for the development of Li–X (O2, S, Se) batteries with high power and energy densities.
 Mei Yang | Yang Mei is an associate professor and master tutor in the School of Chemistry at Xiangtan University. Her research interests include the design and synthesis of functional nanomaterials and high-performance electrodes, surface-interface structure regulation, and its application in energy-related fields. Her representative publications are in Energy & Environmental Science, Advanced Functional Materials, Applied Catalysis B: Environmental, Energy Storage Materials, Journal of Materials Chemistry A, Carbon and other internationally renowned journals. |
Introduction
In the past decades, lithium-ion batteries (LiBs) have been widely used in portable electronic devices and emerging electric vehicles. To achieve higher energy density, Li–X (X = O2, S, Se) batteries are regarded as ideal candidates for the next generation of LIBs due to their high theoretical specific capacity of 3582, 2567, and 1627 W h kg−1.1 Although different in several ways, there are important similarities between Li–X (X = O2, S, Se) batteries. The anode material for Li–X (X = O2, S, Se) batteries is lithium, and the cathode reactants are O2, S, and Se belonging to group VI A in the periodic table. When reacting with lithium, the chalcogen elements can transfer a large amount of charge, but the conductivity of discharge products—such as Li2O2 for Li–O2 batteries, Li2S2 or Li2S for Li–S batteries, and Li2Se for Li–Se batteries—are poor. Moreover, the reaction barriers are very high during charging, which makes it difficult to achieve high reversibility. In particular, the gas–solid conversion involves Li–O2 batteries that need to break oxygen–oxygen covalent bonds during the discharge process. Therefore, the materials and structures of cathodes have a notable influence on the performance of these kinds of batteries.Recently, novel carbon-based nanostructures with advantageous chemical and physical properties, such as carbon nanotubes and graphene have been extensively researched as transistor materials, energy storage materials, catalysts, supports, and so on.2,3 In particular, porous carbon spheres and/or ordered porous carbon materials have huge potential applications as cathode materials for Li–X (X = O2, S, Se) because of their high conductivity, diverse morphologies, and compatibility with other materials. According to their pore sizes, porous carbon materials can be classified into three types: microporous (pore size < 2 nm), mesoporous (2 nm < pore size < 50 nm), and macroporous (pore size > 50 nm).4 Many research studies have suggested that the specific surface area and pore size distribution of porous materials are the key factors affecting their applications. For example, numerous micropores are beneficial for increasing specific surface area and pore volume; the mesoporous channels can effectively improve electrolyte immersion and facilitate Li+ diffusion and electron transfer. Macropores are beneficial for electrolyte infiltration and provide space for O2 diffusion and O2/Li2O2 conversion.5,6 In addition, a certain amount of impurity element doping (such as S, N, P, etc.) can change the electronic structure and surface energy of materials, thus enhancing electron transfer and improving hydrophilicity.7,8
Ideal new-generation porous carbon materials should have the following characteristics, low-cost material resources, simple, and environmentally friendly preparation, and superb performance.9 However, most carbon materials are synthesized by using petroleum-based chemical products that are unsustainable and may cause environmental pollution. Chitosan can be facilely fabricated from chitin, which is the second most abundant natural bio-polymer collected from the shells of shrimps, insects, fungi, algae, and so on. Furthermore, when combined with intrinsic nitrogen elements, chitosan can form N-self-doped carbon after carbonization without any complex doping modification steps. Based on the above advantages, chitosan-derived porous carbon has been extensively investigated as biosensors, water treatment, and battery electrode materials. Conventional pore-forming techniques, such as the active method (KOH, ZnCl, H3PO4), consume high amounts of energy and use the hard-templating method (nano-silicon spheres, polystyrene spheres). It is also difficult to ensure the uniform dispersion of the template in the carbon precursor. This makes it difficult to control the preparation of mesoporous carbon, especially on a large scale.10 It is of immense importance to achieve the mass production of advanced porous carbon materials with sustainable carbon sources.
Here, we realize a rapid, continuous, cost-effective, reproducible, and scalable production of meso- and macro-porous hollow carbon spheres (C–FN) by the spray-drying technique. By combining soft and hard templates, we have successfully prepared spherical sheath-structured nitrogen-doped carbon materials with interconnected meso-porous and macro-porous structures. The application of C–FN in Li–X batteries (Li–O2, Li–S, and Li–Se) is particularly attractive due to the following advantages: (i) hollow carbon sphere can host large amounts of discharge product; (ii) with an optimized loading amount, hollow carbon spheres can provide sufficient free space to accommodate the variation in volume during lithiation/delithiation; (iii) the pores on the shell of the hollow carbon sphere can ensure good accessibility of the lithium ion to the cathode; (v) combining sulfur or selenium with hollow porous carbon sphere enhances the electronic conductivity and traps the discharge product. When applied as cathode material in Li–O2 batteries, the material delivered a reversible specific capacity of 11038 mA h g−1 at 0.1 mA g−1. Meanwhile, it also displayed outstanding long-cycle stability (no capacity degradation after 138 cycles). In the case of Li–S/Li–Se batteries, a high reversible capacity of 1336.8 and 829.3 mA h g−1 at 0.1 mA g−1 was exhibited with improved cyclability. In our opinion, the meso- and macro-porous hollow carbon spheres fabricated here are promising cathode materials for Li–X (X = O2, S, Se) batteries.
Experimental section
Materials preparation
In a typical synthesis, 10 g of chitosan was diluted with 500 mL of deionized water and stirred for 30 min. Next, 5 mL of CH3COOH was gradually added into the solution, and stirring was continued to yield a homogeneous sol–gel solution. Afterward, 10 g of NaCl and 2.5 g of F127 were added to the sol–gel solution and stirred continuously for another 10 h. The mixture was then subjected to spray-drying at 190 °C. Finally, the dried sample was transferred into a ceramic crucible and heat treated under an Ar atmosphere at 850 °C for 2 h. The heat-treated sample was cooled to room temperature and washed with deionized water for 5 h. The obtained sample was denoted as C–FN (carbon spheres prepared with F127 and NaCl). The C–F (carbon spheres prepared with F127) and C–N (carbon spheres prepared with NaCl) were also synthesized using the same procedure as described above but without the addition of NaCl or F127 precursors, respectively.To obtain the C–FN@S and C–FN@Se composite, C–FN was placed in a porcelain boat along with 0.5 g of sulfur/selenium powders upstream of the furnace. A quartz boat with 100 mg of C–FN was placed 10 cm downstream of the heating center. After being flushed with Ar, the center of the furnace was elevated to 500 °C at a ramp rate of 2 °C min−1 and kept at this temperature for 1 h. The mass loading of S was ∼65% after comparing the precursor and S-loading samples.
Characterization
The X-ray diffraction (XRD) measurements were collected using a TD-3500 powder diffractometer (Tongda, Cu–Kα radiation) at a scanning rate of 2° min−1 from 10 to 90°. Scanning electron microscopy (SEM) images were obtained on a Nova Nano 430 system (FEI, Netherlands) and transmission electron microscopy (TEM) images were obtained on a JEM-2100HR system (JEOL, Japan). Nitrogen adsorption–desorption measurements were performed on a Tristar II 3020 adsorption analyzer (Micromeritics, USA). X-ray photoelectron spectroscopy (XPS) analysis was conducted on an ESCALAB 250 X-ray photoelectron spectrometer (Thermo-VG Scientific, USA). Raman spectra were acquired using a Lab RAM Aramis Raman spectrometer (HJY, France).Electrochemical measurements
The Li–O2 battery was assembled using the following procedure. The synthesized composites (80 wt%) were mixed with poly (tetrafluoroethylene) (PTFE, 20 wt%) as a binder in ethanol to form a slurry. The slurry was then sprayed onto carbon paper (the load ratio of the catalyst was about 0.7 mg cm−2, the average diameter of the cathodes was 1.2 cm, and the area of the carbon paper was 1.131 cm2), which was dried at 80 °C for 24 h. The cell was then assembled in an Ar-filled glove box with lithium foil as the anode, polypropylene membrane as the separator, and 1.0 M LiN(CF3SO2)2 in tetraethylene glycoldimethyl ether (TEGDME) as the electrolyte. Finally, the assembled cells (CR 2032) were rested for 8 h under high-purity oxygen. The Li–S or Li–Se electrode was fabricated by mixing the prepared C–FN@S (or C–FN@Se), a conductive (super-P, Sigma-Aldrich), and PVDF (Sigma-Aldrich) in a weight ratio of 8:1:1. Next, the mixture was coated on an aluminum foil with a thickness of ca. 100 μm. The total mass of the electrode materials was approximately 2 mg according to measurements with an ultramicro analytical balance (Mettler Toledo XP2U, 0.1 mg resolution). After drying in air at 80 °C for 12 h, the electrodes were assembled into coin-like cells (CR2032) in an Ar-filled glove box with lithium foil as the anode, and glass fiber (Whatman GF/A) as the separator. The electrolytes used for Li–S and Li–Se batteries are 1.0 M LiTFSI + 2%LiNO3 in 1,3-dioxypolyalkane/1,2-dimethoxyethane (1:1 by volume) and 1.0 M LiPF6 in diethyl carbonate/ethylene carbonate (1:1 by volume), respectively. The galvanostatic charge–discharge measurements of the cells were tested on a Neware (Shenzhen, China) testing system in a 1 atm O2 atmosphere. CV analysis of the cells was conducted at a potential of 2.0–4.5 V at a rate of 0.3 mV s−1. All specific capacity data were normalized to the weight of the active material (synthesized catalyst) loaded on the oxygen cathode.
Results and discussion
Scheme 1 illustrates the experimental process. When chitosan was dispersed in an acetic acid solution, hydrogen bonds were formed between the ketonic oxygen of the acetate molecule and chitosan, producing a sol–gel solution. At the same time, some specific templates, such as NaCl or F127, were used to adjust the viscosity of the gel. The morphology and size of nanomaterials are important factors determining their properties. In spray drying, the empirical equation for controlling droplet diameter (Dd) is often represented as: Dd = Kf·Qn[ρa·σb·μc].11 The nature of the precursor solution (such as surface tension (r), viscosity (l), and density (q)) are key factors of the microstructure formed. As shown in Table 1, we adjusted the ratio of pore-forming agents and the concentration of precursor solutions to investigate the effect of dissolution viscosity on the formation of nanostructures. During the spray drying process, the sol–gel solution was transformed into a dry powder consisting of sphere particles. Because of the evaporation of moisture, NaCl nanocubes were uniformly distributed through the sphere, which created macropores after the calcination process and removal of the template. In addition, the triblock copolymer Pluronic F127 acted as a soft template to create meso-pores. The addition of the pore-forming agents, F127 and NaCl, could reduce the viscosity of the precursor which was beneficial to forming a hollow structure. Fig. 1 presents the SEM and TEM images of the prepared materials. The samples of C–N (carbon spheres prepared with NaCl) show a broken spherical shape with macropores (Fig. 1a–c and S1a–c†). The C–F (carbon spheres prepared with F127) are solid balls of varying size with lots of mesopores (Fig. 1d–f and S2a–c†). The C–FN is a hollow sheath structure with many meso- and macro-pores that are beneficial for ion transport and diffusion. As shown in Fig. 1g–k, the diameter of the carbon sheath is in the range of 1–4 μm with a thickness of ∼100 nm. Furthermore, the EDS mapping of C–FN (carbon spheres prepared with F127 and NaCl) has been tested, which showed that C, O, N, and Na are uniformly distributed in the material. The fuzzy distribution of Na indicated a minimal sodium content (below 0.5%) which is caused by the unclean removal of the NaCl template. In addition, with a diluted precursor solution, the viscosity of precursor solution is 13.4 mPa S for C–FN, which lowers to 3.6 mPa S for C–FN1/2, 2.7 mPa S for C–FN1/4, and 2 mPa S for C–FN1/10. As shown in Fig. S2d–l,† the diameter of the sheath became increasingly uneven, and the thickness of the sheath was thinned with the diluted precursor solution.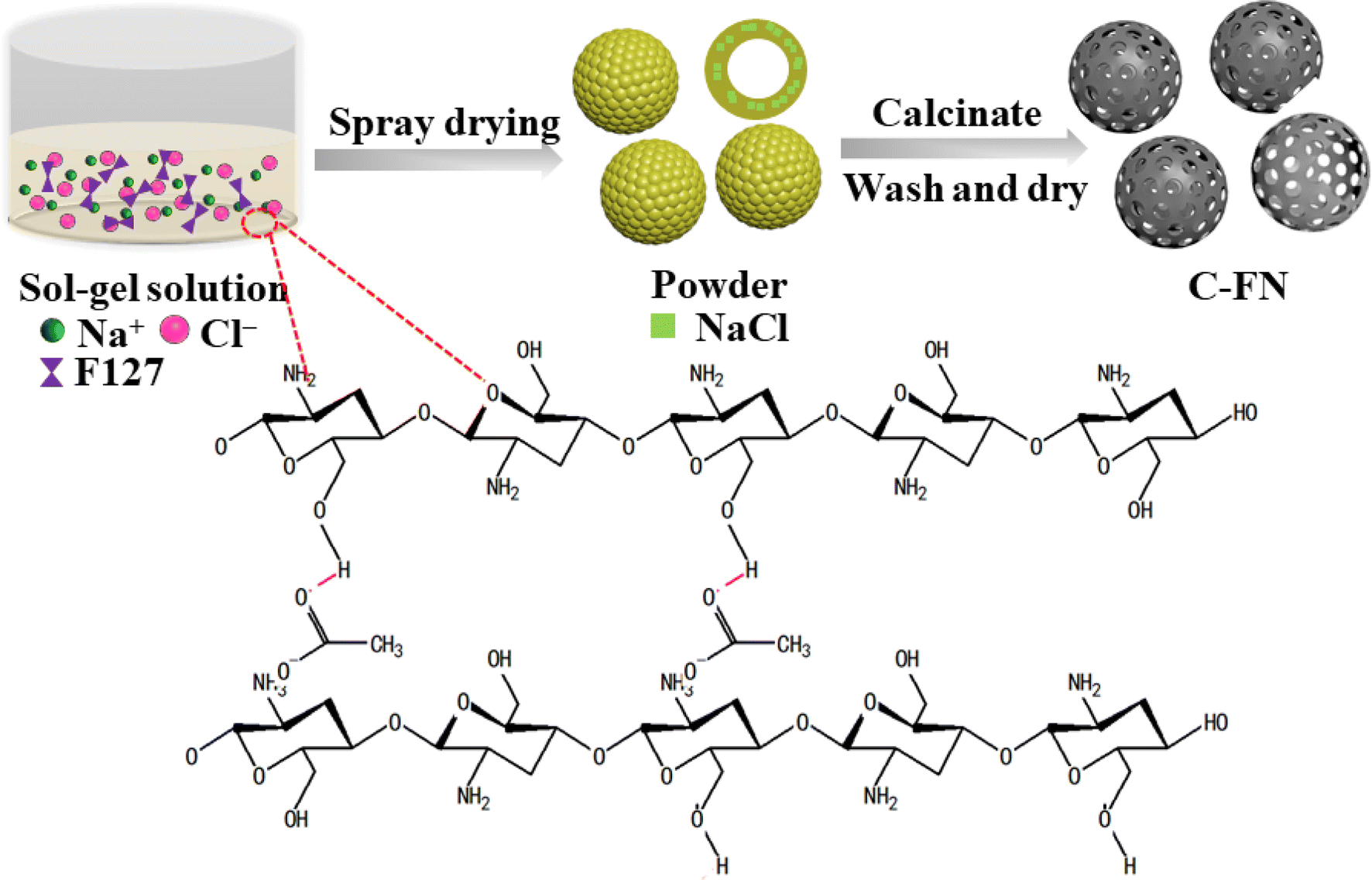 | ||
| Scheme 1 Schematic illustration of the synthesis of hollow porous carbon spheres. | ||
| C–F | C–N | C–FN | C–FN1/2 | C–FN1/4 | C–FN1/10 | ||
|---|---|---|---|---|---|---|---|
| Viscosity (mPa S) | 19.2 | 13.4 | 8.1 | 6.6 | 3.6 | 2.7 | 2 |
| F127 (%) | 0 | 1 | 0 | 1 | 0.5 | 0.25 | 0.1 |
| NaCl (%) | 0 | 0 | 1 | 1 | 0.5 | 0.25 | 0.1 |
| Chitosan (%) | 10 | 10 | 10 | 10 | 5 | 2.5 | 1 |
 | ||
| Fig. 1 Illustration of the fabrication of C–N, C–F, and C–FN, (a) SEM; (b and c) TEM images of C–N; (d) SEM and (e and f) TEM images of C–F; (g and j) SEM and (h, i and k) TEM images and; (l) the element mapping of C–FN. | ||
The XRD pattern of C–F, C–N, and C–FN is shown in Fig. 2a. There are two broad diffraction peaks at 25.2° and 43.4°, which are attributed to the (002) and (100) planes in the carbon structure.12 The peak intensity reflects some degree of graphitization in the samples. Fig. 2b displays the Raman spectrum of the three samples. The two peaks at 1343.2 and 1589.1 cm−1 belong to the D and G bands of the carbon. The IG/ID of the C–FN, C–N, and C–F are 1.036, 1.075, and 1.066, respectively, which reflect a relatively high degree of graphitization in the three samples.13 Based on the XRD and Raman analysis, different pore-forming agents have little effect on the degree of graphitization of the material.14 The BET-specific surfaces and porous structure of the C–F, C–N, and C–FN are obtained from the nitrogen adsorption–desorption isotherms (Fig. 2c and f). The result of C–N indicates type II isotherms that are typical of macroporous structures; the pore size distribution of C–N is around 100 nm. The C–F presents type IV isotherms that are typical of mesoporous materials with a diameter of ∼25 nm. The isothermal adsorption–desorption curve of C–FN reveals the composition of type II and IV.15,16 It is worth mentioning that the surface of C–FN, C–F, and C–N are 893.3, 405.6, and 403.2 m2 g−1. The C–FN not only possesses the highest BET surface but also has a large porous volume of 0.53 cm3 g−1 which is higher than that of C–N (0.27 cm3 g−1) and C–F (0.22 cm3 g−1). The high surface area and porous volume are beneficial to enhancing ion diffusion, as well as providing more reaction sites and space to store the discharge product. The surface bonding configurations of C and N of C–FN are revealed by XPS as shown in Fig. 2d and e. The C 1s peaks of C–FN contain three peaks that are centered at 284.8, 285.5, and 286.6 eV, due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, the C–O or C–N bonds, and the CO bond, respectively.17,18 The content of CC bonds is over 90%, indicating a high degree of graphitization that may be beneficial for electronic transmission. In addition, the N 1s spectrum of the C–FN exhibits four peaks at 398.5, 400.1, 401.2, and 403.1 eV, corresponding to pyridinic-N, pyrrolic-N, graphitic-N, and oxidized-N, respectively.19 The high content of pyridinic-N (31.0%) is beneficial to enhancing battery performance; the Lewis basicity of carbon atoms adjacent to pyridinic N is confirmed to offer stronger ORR active sites in N-doped carbon materials.20 Moreover, the high content of graphitic-N (42.2%) can accelerate charge transfer which is beneficial to reducing polarization voltage and improving battery stability. The XPS spectra and high-resolution XPS are shown in Fig. S3,† where the position and intensity of the peaks of C 1s, O 1s, and N 1s of the three samples are very close to each other. In addition, the infrared absorption spectra of the three samples are also very close (Fig. S4†), indicating that the influence of pore-forming agents on the composition of materials and the binding energy of elements is relatively small. The FTIR spectra (Fig. S4†) reveal the surface functionalization of the three materials. The three spectra present widened peaks of around 3400 cm−1 belonging to combined O–H and N–H stretching vibration bands. The peaks appear around 1720 cm−1 indicating stretching vibration of the carboxylic acid. In addition, some peaks at 1178 and 1090 cm−1 indicate the present C–OH and C–O–C functional groups, which are consistent with XPS testing.21 The contact angle at the base of the cathode on three materials is 125.4, 173.9, and 141.1° indicating that all three electrodes are hydrophobic (Fig. S5†).22 During the cycling process of organic Li–O2 batteries, the hydrophobic properties of electrodes can effectively reduce side reactions and enhance cycling stability.
C bonds, the C–O or C–N bonds, and the CO bond, respectively.17,18 The content of CC bonds is over 90%, indicating a high degree of graphitization that may be beneficial for electronic transmission. In addition, the N 1s spectrum of the C–FN exhibits four peaks at 398.5, 400.1, 401.2, and 403.1 eV, corresponding to pyridinic-N, pyrrolic-N, graphitic-N, and oxidized-N, respectively.19 The high content of pyridinic-N (31.0%) is beneficial to enhancing battery performance; the Lewis basicity of carbon atoms adjacent to pyridinic N is confirmed to offer stronger ORR active sites in N-doped carbon materials.20 Moreover, the high content of graphitic-N (42.2%) can accelerate charge transfer which is beneficial to reducing polarization voltage and improving battery stability. The XPS spectra and high-resolution XPS are shown in Fig. S3,† where the position and intensity of the peaks of C 1s, O 1s, and N 1s of the three samples are very close to each other. In addition, the infrared absorption spectra of the three samples are also very close (Fig. S4†), indicating that the influence of pore-forming agents on the composition of materials and the binding energy of elements is relatively small. The FTIR spectra (Fig. S4†) reveal the surface functionalization of the three materials. The three spectra present widened peaks of around 3400 cm−1 belonging to combined O–H and N–H stretching vibration bands. The peaks appear around 1720 cm−1 indicating stretching vibration of the carboxylic acid. In addition, some peaks at 1178 and 1090 cm−1 indicate the present C–OH and C–O–C functional groups, which are consistent with XPS testing.21 The contact angle at the base of the cathode on three materials is 125.4, 173.9, and 141.1° indicating that all three electrodes are hydrophobic (Fig. S5†).22 During the cycling process of organic Li–O2 batteries, the hydrophobic properties of electrodes can effectively reduce side reactions and enhance cycling stability.
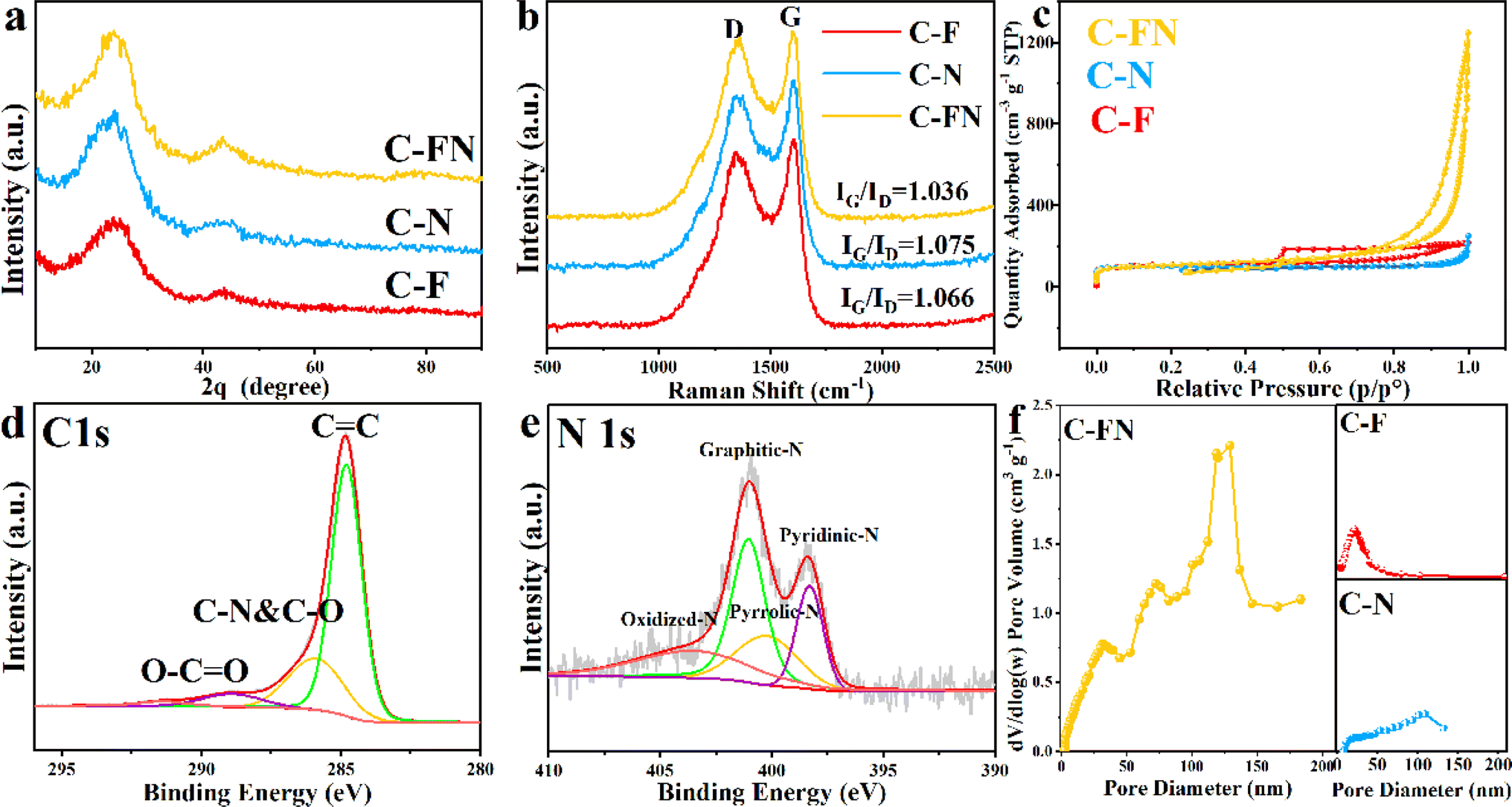 | ||
| Fig. 2 (a and b) The XRD and Raman spectra of C–FN, C–F, and C–N; (c) N2 adsorption–desorption isotherms curves of C–FN, C–F, and C–N; (d and e) the XPS spectra of C 1s and N 1s of C–FN; (f) the corresponding pore size distribution curves of C–FN, C–F, and C–N. | ||
The Li–O2 battery was assembled in a porous 2032 coin cell using the N-doped carbon spherical shell as a cathode. As reported in previous research, the reaction of lithium–oxygen battery with carbon-based catalysts involves the formation and decomposition of Li2O2 based on two-electron transfer reactions that can be described as O2 + 2Li+ + 2e− ↔ Li2O2 (E0 = 2.96 V versus Li/Li+).23 The cycle voltage (CV) curves were obtained at a scanning rate of 0.1 mV s−1 between 2–4.3 V.24 As shown in Fig. 3a, the reduction peak (ORR process) and the oxidation peak (OER process) of the C–FN cathode is higher than that of C–F and C–N.25 In addition, the area of the C–FN CV cures is the largest of the three samples, which indicates that C–FN possesses a higher specific capacity than the other two. Fig. 3b shows the first discharge capacity–voltage curves of Li–O2 batteries with C–FN, C–F, and C–N cathode at a current density of 100 mA g−1. The specific capacity of the batteries is in the order of C–FN > C–N > C–F. It is worth noting that the C–FN shows the highest initial discharge capacity of 11038 mA h g−1 (7.85 mA h cm−2 at a mass loading of 0.71 mg cm−2) which is 3 or 6 times higher than that of C–F and C–N cathode, respectively. In Fig. 3c, the C–FN cathode shows overpotentials of OER (0.62 V) and ORR (0.2 V) from an equilibrium potential (E0 = 2.96 V), which is much lower than that of C–N (0.73 and 0.38 V) and C–F (1.18 and 0.38 V).26 Owing to the high catalytic activity and smooth ion transmission channel, the C–FN cathode with connected macro-porous and meso-porous structures could effectively enhance battery capacity and coulombic efficiency. As shown in Fig. S7,† the lithium-ion coefficient of C–FN, C–F, and C–N for the Li–O2 is 3.75 × 10−13, 5.51 × 10−13, and 9.47 × 10−13 m2 s−1, respectively. The high lithium-ion coefficient is an advantage of the interconnecting macro- and mesopores.
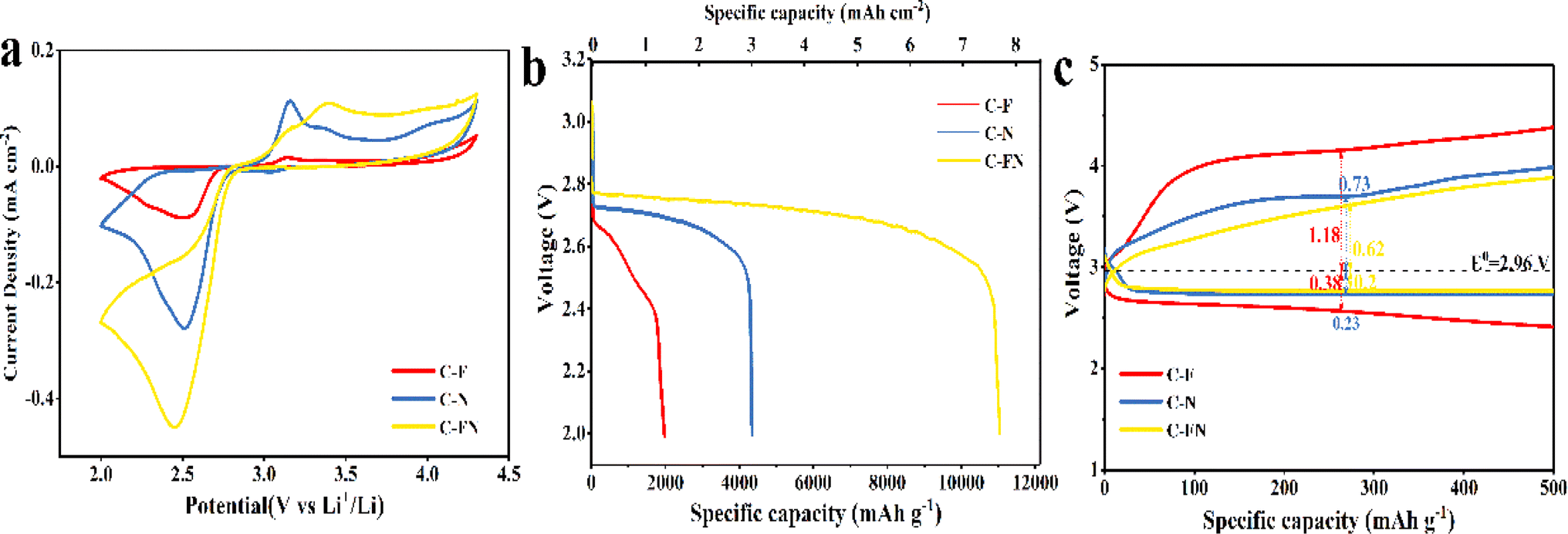 | ||
| Fig. 3 (a) CV curves; (b) the initial discharge profiles; (c) discharge–charge profiles at a cut-off capacity of 500 mA h g−1 of the battery with C–FN, C–F, and C–N cathode. | ||
The cycling stability profiles of the C–FN, C–F, and C–N batteries were studied at a current density of 100 mA g−1 and a cut-off capacity of 500 mA h g−1. The selected charge–discharge (CD) profiles are shown in Fig. 4a–c. Before the first 20 cycles, the charge plateau of the C–FN cathode is stabilized at ∼3.5 V (Fig. 4c), and the discharge plateau is higher than 2.8 V. Up to the 100th cycle, the discharge voltage plateau is stabilized around 2.7 V, indicating good cycle stability. However, when cycling to the 138th cycle, the discharge platform quickly reduces to ∼2.5 V and the charging platform rises to 4.3 V. On the other hand, the CD profiles of C–N and C–F show a quick increase of the over-potential in the initial cycles. The charging platform of C–F is upper 4 V in the second cycle (Fig. 4a), and the charging platform of C–N is ∼4 V after the 20th cycle (Fig. 4b). As shown in Fig. 4d, the three cathodes are investigated at 100 mA g−1 at 2.0–4.3 V. The C–FN can maintain a stable circulation of 500 mA h g−1 for 138 cycles compared with the C–N and C–F for a few cycles.
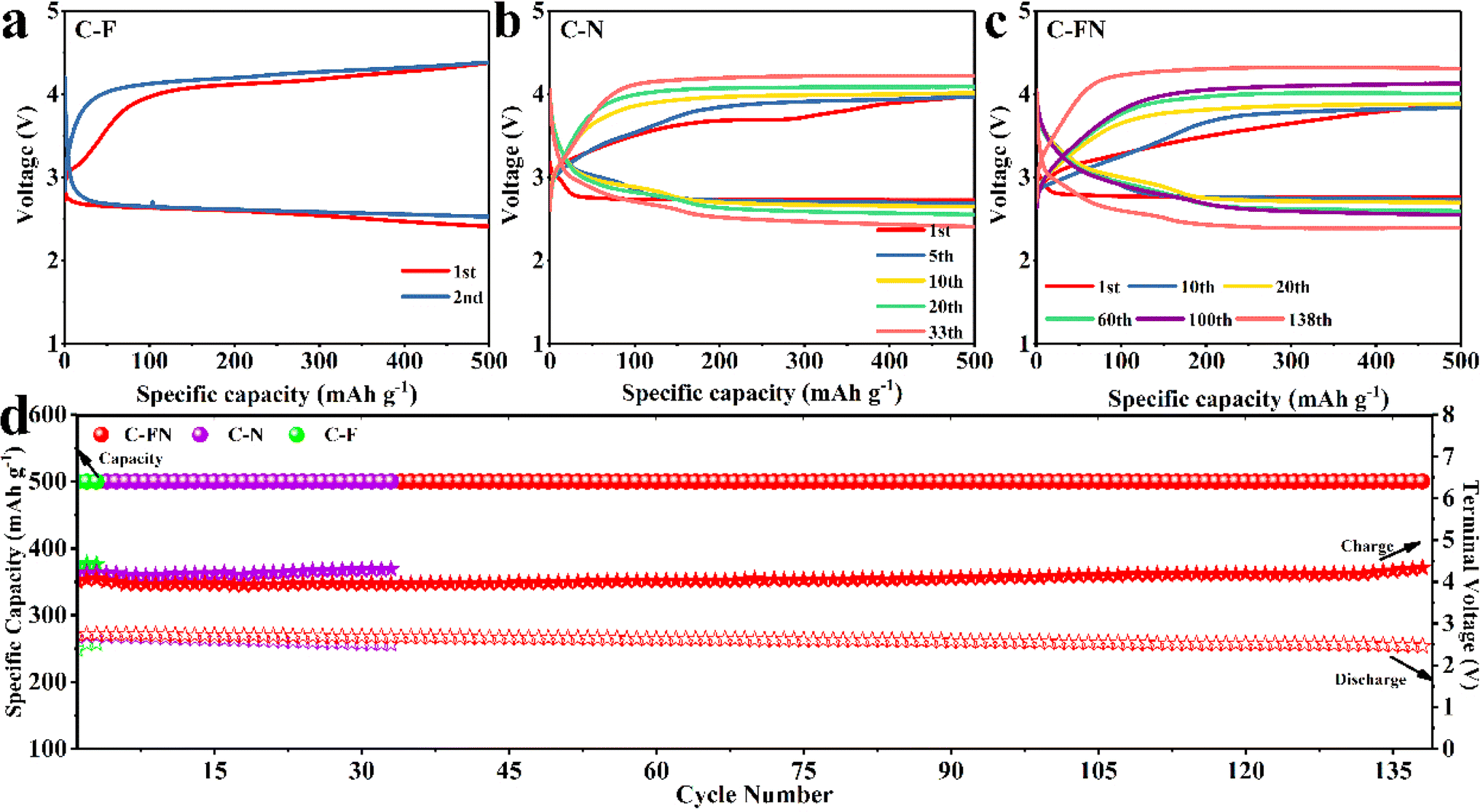 | ||
| Fig. 4 (a) The different cycle discharge–charge profiles of the Li–O2 battery with C–F cathode in the potential range from 2.0–4.3 V; (b) the different cycle discharge–charge profiles of Li–O2 battery with C–N cathode in the potential range of 2.0–4.3 V; (c) the different cycle discharge–charge profiles of the Li–O2 battery with C–FN cathode in the potential range from 2.0–4.3 V; (d) the cycling performance of the Li–O2 battery with the C–F, C–N, and C–FN cathode. | ||
To better study the mechanism of cycle stability, the morphology of discharge products of the three cathodes at different discharge and charge stages are carefully characterized. As shown in Fig. 5a and b, when the C–FN is discharged to 2.0 V, the fine particle discharge product is uniformly deposited on the surface of C–FN. As the XRD pattern shows (insets in Fig. 5b and S10†), three peaks appeared at 32.9°, 35.0°, and 58.7° that index to the 100, 101, and 110 crystal planes of Li2O2 (PDF#09-0355), indicating that the discharge produced by Li–O2 batteries is Li2O2. However, the broadened and weak characteristic diffraction peaks indicate the low crystallinity of discharge products, which is beneficial to the reversible cycle of the battery. As shown in Fig. 5c and S10a,† when recharged to 4.3 V, these peaks weaken or even disappear and no obvious discharge product is observed on the surface of C–FN—indicating the complete decomposition of the product. At the same time, the XRD peaks at 25.2°, 43.4°, and 54° remain unchanged whether in charged or discharged state, which indicates the structural stability of carbon materials. The Raman diagram is presented in Fig. S10b.† Additionally, the radio of IG/ID also remains almost the same, showing that the carbon material remains stable during the charging and discharging process; these observations match the results from XRD. As a comparison, the discharge products at 2.0 V deposited film-like structures on the surface of the C–N cathode (Fig. 5d and e) and large nubby structures on the C–F (Fig. 5g and h). When recharging to 4.3 V, the large particles in Fig. 5i could not be completely decomposed, causing an increase in the charging voltage platform and terrible cyclic stability. In 0.1 M KOH solution, the improved OER performance of C–FN compared with the C–F and C–N samples is shown in Fig. S6.† As in a high specific surface area and excellent catalytic activity, the C–FN-based Li–O2 battery shows excellent rechargeability.
 | ||
| Fig. 5 (a, b, d, e, g, and h) SEM images of C–F, C–N, and C–FN cathode that were discharged to 2.0 V; (c, f and then i) charged to 4.3 V. | ||
To further leverage its advantage and expand its usefulness, the C–FN is used as carbon substrates to load S and Se. The Li–S and Li–Se batteries are assembled with C–FN@S and C–FN@Se as cathodes, respectively. As observable in Fig. S8,† the lithium-ion coefficient of the C–FN-based cathode for Li–S and Li–Se battery is 2.35 × 10−11 and 1.58 × 10−11 m2 s−1 respectively. In addition, as shown in Fig. S9,† the lithium-ion coefficient of the C–FN cathode for Li–S ranges from 10−11 to 10−13 m2 s−1 which is in line with the result calculated with EIS, i.e., 2.35 × 10−11 m2 s−1. The DLi+ for Li–Se battery ranges from 10−10 to 10−12 m2 s−1 which is consistent with the finding from EIS, i.e., 1.5 × 10−11 m2 s−1. These results indicate the high ion diffusion rate in the C–FN-based cathode that proves to be a favorable factor for the high performance of the Li–S and Li–Se batteries. Fig. 6 shows the performance profile of Li–S batteries based on the C–FN@S cathodes. The CV curves (Fig. 6a) show that at a scan rate of 0.1 mV s−1, the actual position of the open circuit voltage is 2.3 V, and there are two obvious redox peaks on the first cycle curve. After the first cycle, the curves present four typical peaks: two anodic peaks (2.05 and 2.33 V) and two cathodic peaks (2.31 and 2.40 V), suggesting relatively low polarization and fast reaction kinetics. The two anodic peaks correspond to the conversion of high-valence lithium–sulfur compounds (such as Li2S8) to Li2Sn (8 > n ≥ 4) and then to Li2S2 or Li2S. The cathodic peaks correspond to Li2S, which gradually oxidized to Li2S8.27,28Fig. 6b illustrates the first 10 discharge–charge cycle curves, where the discharge capacity is as high as 1336.8 mA h g−1 for the first cycle. After a few cycles, the capacity gradually decreases until the tenth lap (962 mA h g−1). However, the polarization voltage decreases from 0.160 V for the first cycle to 0.101 V after cycling. This may be caused by the side reactions between sulfur and electrolyte to form a CEI (cathode–electrolyte interface).29,30 As shown in Fig. 6c, the specific capabilities of C–FN@S cathode are 920, 810, 738, 642, 504 mA h g−1 at different current densities of 0.1, 0.2, 0.5, 1.0, and 2.0 A g−1, indicating its high rate-specific capabilities. Fig. 6d revealed the long cycling stabilities of C–FN@S at 0.5 A g−1. After 270 cycles, it maintained a high capacity of 663.6 mA h g−1.
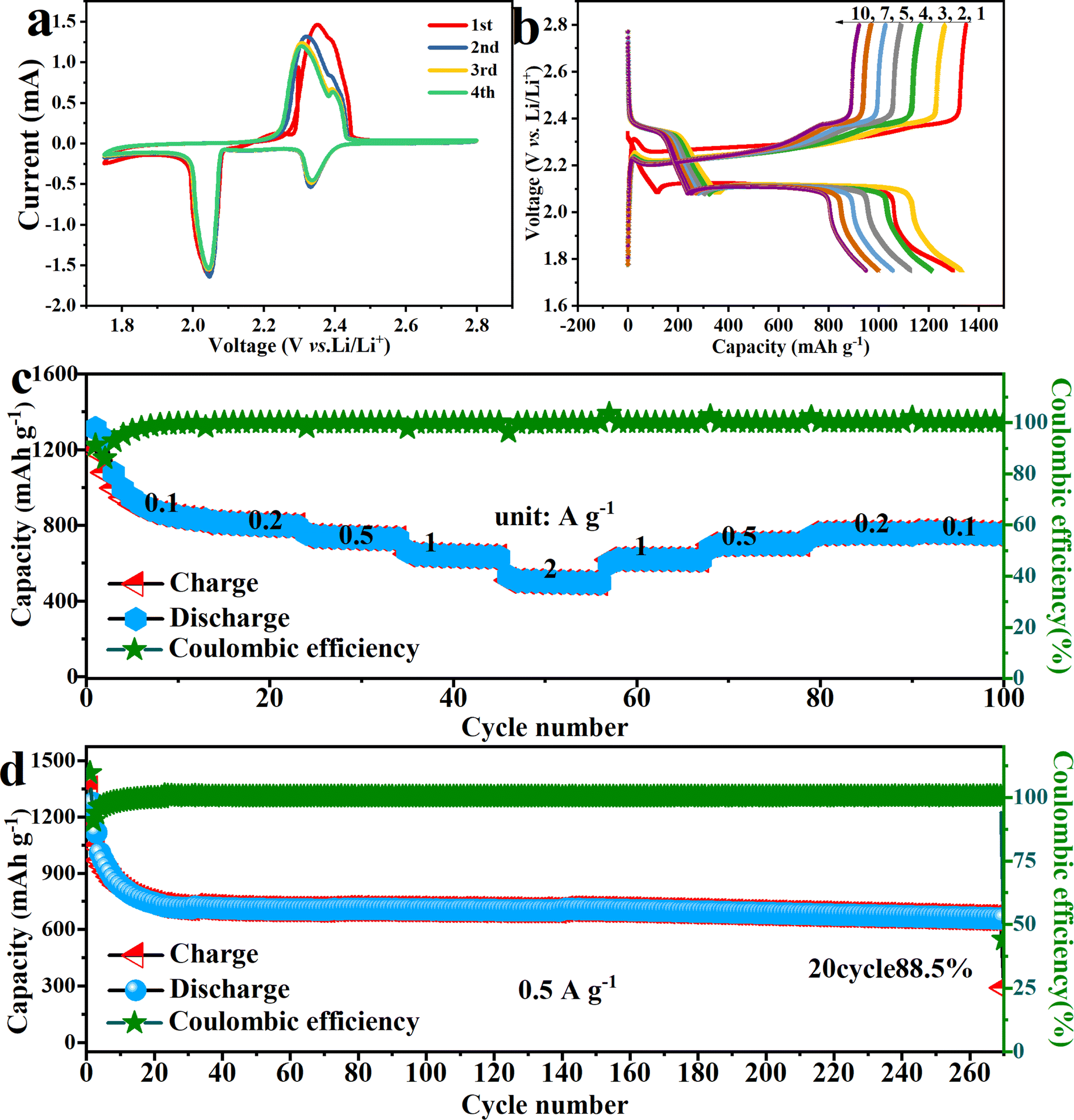 | ||
| Fig. 6 (a) The initial four cycles of CV curves; (b) the first 10 cycles of charge–discharge profiles; (c) rate performance at 0.1, 0.2, 0.5, 1.0, and 2.0 A g−1; (d) cycling performance at a current density of 0.5 A g−1 of Li–S batteries based on C–FN@S cathode. | ||
Selenium, as a member of the oxygen-sulfur group, is also loaded onto carbon spheres to prepare Li–Se batteries. The charge–discharge reaction mechanism is similar to that of the Li–S batteries. Selenium loading on C–FN is reduced to Li2Sen (n ≥ 4), Li2Se2, and finally to Li2Se during discharging. When charging, Li2Se is directly oxidized to Li2Sen (n ≥ 4) and then Se.31,32 As shown in Fig. 7a, the C–FN@Se cathodes were tested in the voltage range of 0.5–3.0 V at different scan rates of 0.1–0.5 mV s−1. A pair of oxidation/reduction peaks were observed at 1.75 and 2.01 V at a scan rate of 0.1 mV s−1. The shape was well maintained with an increasing scan rate, indicating good reversibility of charging and discharging. Fig. 7b shows charge/discharge cycling curves at 50 mA g−1 across a voltage window of 0.5–3.0 V. For the first cycle, the discharge capacitance is 840 mA h g−1. There are discharge and charge platforms at 1.7 and 1.9 V, respectively, which is in accordance with the redox peaks in the CV curves. The rate capability is presented in Fig. 7c, where the specific capacities are 288.4, 264.4, 257.7, 238.8, 212.4, and 163.4 mA h g−1 at 0.05, 0.1, 0.2, 0.5, 1.0, and 2.0 A g−1, respectively. The high rate performance could be ascribed to the stabilization and continuous structure of the C–FN cathodes throughout. As seen in Fig. 7d, the cycling performance of C–FN@Se cathodes is recorded up to 1000 cycles at a current density of 1.0 A g−1. In the initial cycles, the discharge capacity of the battery rapidly drops from 262.5 to 222.0 mA h g−1, which is due to a severe mismatch in the initial charging and discharging of the battery. We believe that this may be caused by the formation of the SEI (solid–electrolyte interface) film.33 Subsequently, the battery stabilizes gradually, and after 1000 cycles, the capacity retention rate remains around 80%. These observations confirm that the materials tested here possess excellent electrochemical stabilities.
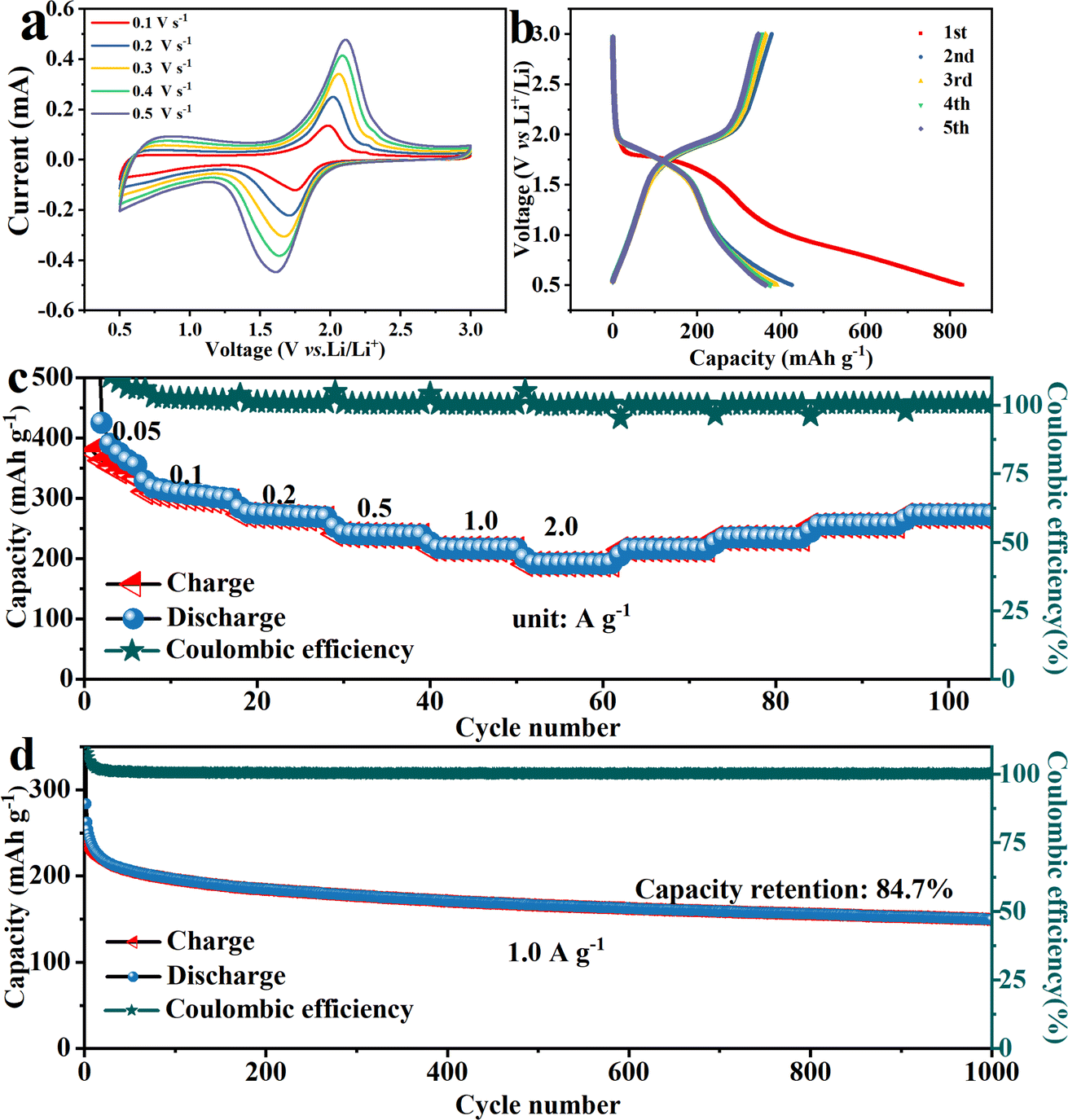 | ||
| Fig. 7 (a) The CV curves at a scan rate of 0.1, 0.2, 0.3, 0.4, and 0.5 mV s−1, (b) the charge–discharge profile of the first five cycles, (c) rate performance at 0.05, 0.1, 0.2, 0.5, 1.0, and 2.0 A g−1, (d) cycling performance at a current density of 1.0 A g−1 of Li–Se batteries based on the C–FN@Se cathode. | ||
Conclusion
We have developed a facile and efficient method for large-scale production of porous spherical carbon material and demonstrated its potential applications as cathode material for Li–X (O2, S, and Se) batteries with high reversible capacity and good cycling stability. The excellent electrochemical performance of the C–FN-based cathode can be attributed to the following reasons: firstly, the high specific surface and large pore volume can provide more active sites and storage spaces for discharge products; secondly, the excellent electronic conductivity of 3D spheroidal sheath structures framework will significantly shorten the time required for electron transport during the cycling, while the interpenetrating macro and mesoporous structures can shorten the ion transport path; finally, a certain amount of nitrogen doping can improve the catalytic activity and decrease the decomposition energy barrier of the discharge product during the charging process. All these are essential for the effective performance of the C–FN-based cathode.Data availability
Data for this article, including [description of data types] are available at [URL – format https://doi.org/DOIA-ART-04-2024-002466.R2]. The data supporting this article have been included as part of the ESI.†Author contributions
Kailing Sun: methodology, investigation, data curation, writing, original draft. Xiaocong Deng, Limei Liu: investigation, data curation, original draft. Xian Huang: investigation, data curation, original draft. Shijun Liao: investigation, supervision, writing, review & editing. Tongye Wei: investigation, supervision, validation, resources, writing, review & editing. Mei Yang: investigation, supervision, validation, resources, writing, review & editing.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52402313), the Science and Technology Innovation Program of Hunan Province (2022JJ40447, 2023JJ20036), the Hunan Science and Technology Department, China (2021RC5007), the Hunan Provincial Education Office Foundation of China (21B0146, 23C0042), the Xiangtan City Science and Technology Plan Project (GX-YB20231008), and the Guangdong Basic and Applied Basic Research Foundation (2022A1515111170, 2021A1515110347).References
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. J. N. m. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- Y. Gong, W. Ding, Z. Li, R. Su, X. Zhang, J. Wang, J. Zhou, Z. Wang, Y. Gao, S. Li, P. Guan, Z. Wei and C. Sun, ACS Catal., 2018, 8, 4082–4090 CrossRef CAS.
- G.-H. Lee, M.-C. Sung, J.-C. Kim, H. J. Song and D.-W. Kim, Adv. Energy Mater., 2018, 8, 1801930 CrossRef.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed. Engl., 2008, 47, 3696–3717 CrossRef CAS.
- G. Jiang, R. A. Senthil, Y. Sun, T. R. Kumar and J. Pan, J. Power Sources, 2022, 520, 230886 CrossRef CAS.
- W. Xiong, M. Liu, L. Gan, Y. Lv, Y. Li, L. Yang, Z. Xu, Z. Hao, H. Liu and L. Chen, J. Power Sources, 2011, 196, 10461–10464 CrossRef CAS.
- T. P. Mofokeng, Z. N. Tetana and K. I. Ozoemena, Carbon, 2020, 169, 312–326 CrossRef CAS.
- W. Wei, Z. Chen, Y. Zhang, J. Chen, L. Wan, C. Du, M. Xie and X. Guo, J. Energy Chem., 2020, 48, 277–284 CrossRef.
- X. Chen, M. Zhou, Y. Zhao, W. Gu, Y. Wu, S. Tang and G. Ji, Green Chem., 2022, 24, 5280–5290 RSC.
- S. Sundriyal, V. Shrivastav, H. D. Pham, S. Mishra, A. Deep and D. P. Dubal, Resour., Conserv. Recycl., 2020, 169, 105548 CrossRef.
- A. B. D. Nandiyanto and K. Okuyama, Adv. Powder Technol., 2011, 22, 1–19 CrossRef CAS.
- J. Hong, S. Hyun, M. Tsipoaka, J. S. Samdani and S. Shanmugam, ACS Catal., 2022, 12, 1718–1731 CrossRef CAS.
- Z. Tong, C. Lv, Y. Zhou, P. F. Zhang, C. C. Xiang, Z. G. Li, Z. Wang, Z. K. Liu, J. T. Li and S. G. Sun, Small, 2022, 18, e2204836 CrossRef PubMed.
- X. Tian, Z. Chen, J. Hou and Z. Li, J. Cleaner Prod., 2022, 363, 132524 CrossRef CAS.
- D. Tang, T. Wang, W. Zhang, Z. Zhao, L. Zhang and Z. A. Qiao, Angew. Chem., Int. Ed. Engl., 2022, 61, e202203967 CrossRef CAS PubMed.
- M. Shi, X. Hong, C. Liu, H. Qiang, F. Wang and M. Xia, Chem. Eng. J., 2023, 453, 139764 CrossRef CAS.
- F. Wang, Y. Liu, H. Zhao, L. Cui, L. Gai, X. Han and Y. Du, Chem. Eng. J., 2022, 450, 138160 CrossRef CAS.
- C. Ke, R. Shao, Y. Zhang, Z. Sun, S. Qi, H. Zhang, M. Li, Z. Chen, Y. Wang, B. Sa, H. Lin, H. Liu, M. S. Wang, S. Chen and Q. Zhang, Adv. Funct. Mater., 2022, 32, 2205635 CrossRef CAS.
- X. Zhang, X. Xu, S. Yao, C. Hao, C. Pan, X. Xiang, Z. Q. Tian, P. K. Shen, Z. Shao and S. P. Jiang, Small, 2022, 18, e2105329 CrossRef.
- H. Chen, Y. Ye, X. Chen, L. Zhang, G. Liu, S. Wang and L.-X. Ding, Chin. J. Catal., 2022, 43, 1511–1519 CrossRef CAS.
- R. Atchudan, T. N. J. I. Edison, S. Perumal, N. Muthuchamy and Y. R. Lee, Fuel, 2020, 275, 117821 CrossRef CAS.
- J. W. Song and L. W. Fan, Adv. Colloid Interface Sci., 2021, 288, 102339 CrossRef CAS.
- E. J. Askins, M. R. Zoric, M. Li, R. Amine, K. Amine, L. A. Curtiss and K. D. J. N. C. Glusac, Nat. Chem., 2023, 15, 1247–1254 CrossRef CAS PubMed.
- S. Guo, J. Wang, Y. Sun, L. Peng and C. Li, Chem. Eng. J., 2023, 452, 139317 CrossRef CAS.
- L. Ren, F. Kong, X. Wang, Y. Song, X. Li, F. Zhang, N. Sun, H. An, Z. Jiang and J. Wang, Nano Energy, 2022, 98, 107248 CrossRef CAS.
- X. X. Wang, D. H. Guan, F. Li, M. L. Li, L. J. Zheng and J. J. Xu, Adv. Mater., 2022, 34, e2104792 CrossRef PubMed.
- Q. Liang, S. Wang, Y. Yao, P. Dong and H. J. A. F. M. Song, Adv. Mater., 2023, 33, 2300825 CAS.
- C. Zhao, K. Amine and G.-L. Xu, Acc. Chem. Res., 2023, 56, 2700–2712 CrossRef CAS PubMed.
- M. Sun, X. Wang, J. Wang, H. Yang, L. Wang and T. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 35175–35183 CrossRef CAS PubMed.
- L. Zhang, M. Ling, J. Feng, L. Mai, G. Liu and J. Guo, Energy Storage Mater., 2018, 11, 24–29 CrossRef.
- Y. Cui, A. Abouimrane, J. Lu, T. Bolin, Y. Ren, W. Weng, C. Sun, V. A. Maroni, S. M. Heald and K. Amine, J. Am. Chem. Soc., 2013, 135, 8047–8056 CrossRef CAS PubMed.
- C. Luo, Y. Xu, Y. Zhu, Y. Liu, S. Zheng, Y. Liu, A. Langrock and C. J. Wang, ACS Nano, 2013, 7, 8003–8010 CrossRef CAS PubMed.
- J. T. Lee, H. Kim, N. Nitta, K.-s. Eom, D.-C. Lee, F. Wu, H.-T. Lin, B. Zdyrko, W. I. Cho and G. Yushin, J. Mater. Chem. A, 2014, 2, 18898–18905 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02466d |
| This journal is © The Royal Society of Chemistry 2024 |