
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA super-tough plant oil based elastomer for UV-light assisted 3D printed soft robotics and shape-memory†
Agnija
Ritere
,
Maksims
Jurinovs
 *,
Oskars
Platnieks
,
Anda
Barkane
and
Sergejs
Gaidukovs
*
*,
Oskars
Platnieks
,
Anda
Barkane
and
Sergejs
Gaidukovs
*
Institute of Chemistry and Chemical Technology, Faculty of Natural Sciences and Technology, Riga Technical University, P. Valdena 3/7, Riga, LV-1048, Latvia. E-mail: Maksims.Jurinovs@rtu.lv; Sergejs.Gaidukovs@rtu.lv
First published on 7th June 2024
Abstract
Bio-based plant oil-derived elastomers are an attractive alternative to petroleum polymers due to a growing demand for flexible, high-strain materials in the 3D printable soft robotics field. So far, such solutions have been limited to slow and labor-demanding molding techniques, making them unable to achieve high design flexibility and exceptional resolution. Herein, we present a vat photopolymerization 3D printable and easily tailorable plant oil acrylate-based system with a bio-based carbon content ranging from 62% to 80%. By targeting the tuning of macromolecular design and post-processing conditions, a broad mechanical and functional contrast from soft and stretchable elastomers, with up to 180% elongation, to hard and ductile shape-memory polymers, is realized in 3D-printed parts. To further demonstrate the capability of the developed materials, we created a fully 3D-printed soft robotic actuator capable of fast and delicate movement. The proposed approach enables 3D printing of sustainable, high-resolution structures with targeted mechanical properties for application in various advanced fields.
Introduction
The ever-growing progress in numerous sophisticated disciplines, such as soft robotics, necessitates unique materials that can fulfill the demands of new applications. Materials suitable for such a specialized area must have flexible, highly elastic structures with high elongation to ensure proper functioning and delicate and dynamic movements.1 Elastomers, a subclass of polymers, are known for their weakly cross-linked, long polymer chains that enable significant reversible deformation under external pressure.2 This feature ensures their almost irreplaceable role in most soft robotic solutions.3 In addition, these materials are extensively used in various industries, including automotives, consumer goods, and medical devices, which further expands their undeniable value.4–6Additive manufacturing (AM) is a layer-by-layer fabrication process that allows for the rapid creation of highly customizable, complex structures. Vat photopolymerization (VP) of all AM technologies provides low-cost, rapid, efficient, and, most importantly, high-resolution 3D printing.7 In fact, VP has one of the highest 3D printing accuracies of any AM process, which may provide some advantages for soft robotics if intricate features are required. However, the printing of elastomers using a VP printer is a known challenge specific to the VP technique's operational conditions and the materials' limitations.8 During printing, precise control of the formulation and processing conditions, such as viscosity or shear forces, is mandatory. The aforementioned limitations influenced the market for commercially accessible VP resins, which are now confined to materials with moderate stretchability and toughness combined with a high modulus. Currently, there are a limited number of commercially available VP processable elastomers.9–12 Unfortunately, there are significant problems with most of them, including a closed system, limited usage outside of the manufacturer's system, high costs, and, worse – the exclusive use of petroleum-based feedstocks.
The search for environmentally friendly substitutes has given rise to a wide range of bio-based materials designed for multipurpose uses. Recently, gelatin-based materials have emerged as alternatives to commonly used silicones and polyurethanes, yet they suffer drawbacks such as a heavy reliance on relative humidity, impacting the actuator lifespan, and the labor-intensive molding processes used in their creation.13–15 Although direct ink writing enables 3D printing of gelatin systems through material extrusion, the resulting parts often lack dimensional stability and require specific 3D printer modifications for quick gelation.13 On the other hand, plant oil-based resins drew a lot of attention due to their low cost, ease of processing, and potential for chemical functionalization. Nevertheless, the majority of current vegetable oil acrylate-containing UV-curable resins reported in the literature often display glassy polymer specific – hard and brittle behavior,16–18 making them inappropriate for the requirements of soft robotics. While there are alternative plant oil-based elastomeric formulations, most of them are incompatible with VP printing methods or have elongation values limited to around 60%.19–22
To the best of our knowledge, there is a lack of literature reporting the use of plant oil-based high-strain acrylates for 3D printing and advanced applications, such as soft-robotics.23,24 This highlights the need for new formulations that can effectively tackle the particular difficulties presented by sustainable VP printing and advanced applications, which is what inspired us to perform this work.
Herein, we describe a viable and easily tailorable plant oil-based system with exceptional printability and a wide range of mechanical properties. We utilize three building blocks in the resin: acrylated rapeseed oil (ARO, derived from rapeseed oil, 89% biobased carbon content (BCC)) as a crosslinking agent, ensuring the creation of a highly elastic network; isobornyl acrylate (IBOA, derived from turpentine oil, 75% BCC) and hydroxyethyl acrylate (HEA) to form long, low-covalent crosslinked networks with high molecular mass and increased non-covalent bonding. Sustainable feedstocks, coupled with meticulous macromolecular chain design and UV and thermal post-processing optimization, can contain 62% to 80% BCC. 3D printed samples show elongation up to 180% and achieve a nearly 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times difference in toughness and 75 times difference in modulus between various compositions. To the best of our knowledge, no prior reports have documented the performance of the systems being provided with such a high bio-derived content.
000 times difference in toughness and 75 times difference in modulus between various compositions. To the best of our knowledge, no prior reports have documented the performance of the systems being provided with such a high bio-derived content.
Additionally, a fully 3D printed pneumatically operated soft robotic gripper capable of the previously mentioned delicate and dynamic movements was made possible by the created materials. Our systems' applicability is further enhanced by the expression of shape memory characteristics in formulations that underwent thermal processing. Thanks to the capacity to quickly create sophisticated parts with increased sustainability and a wide range of features, the presented material is a promising substitute for its counterparts based on petroleum chemicals in cutting-edge industries like soft robotics.
Experimental section
Materials
Unrefined rapeseed oil (RO) was purchased from a local producer, “Iecavnieks” (Latvia), and used as is. n-Hexane, boron trifluoride diethyl etherate (BF3·OEt2), NaHCO3, acrylic acid, and 2-hydroxyethyl acrylate (HEA) were supplied by Sigma-Aldrich and used as received. The photoinitiator (PI) ethyl(2,4,6-trimethylbenzoyl)phenylphosphinate (TPO-L) was purchased from FluoroChem. Isobornyl acrylate (IBOA, SARBIO 5102) was kindly supplied by ARKEMA Sartomer.Preparation and 3D printing of bio-based UV-curable resins
Rapeseed oil (RO) was combined with acrylic acid in a 1 to 0.5 (w/w) ratio; then BF3·O·Et2 was added to the original mixture in a 1 to 15 (w/v) ratio, followed by stirring at 80 °C for 5 h. After cooling the mixture, it was dissolved in n-hexane and washed with saturated NaHCO3 and NaCl solutions. The water layer was separated, the organic layer was dried over anhydrous Na2SO4, then filtered, and the solvent was removed using a rotary evaporator. The resulting acrylated rapeseed oil (ARO) was a low-viscosity, yellow liquid.16 ARO, IBOA, HEA, and PI were mixed in the ratios as shown in Table 1 for 2 h to prepare the UV-curable bio-based resins. To keep the bio-based carbon content in the formulations above 60% (Table 1), the HEA was only examined up to 30 wt%. During extended storage times of two months, no changes in the viscosities of the resins were noticed. Fig. S1(a)† displays the RIH3 viscosity dependency throughout the storage time. The samples were 3D printed using an Elegoo Mars 2PRO (M2P) digital light processing (DLP) printer. The printer utilizes a UV-LED wavelength of 405 nm and a 6-inch monochrome LCD screen with a 2560 × 1440 pixel resolution. Printing parameters were set with a layer height of 50 μm and a layer time of 20 seconds. The dimensions of the printed samples varied depending on the specific testing requirements and are detailed in the descriptions of respective experimental methods. Following printing, the samples underwent a rinsing process in propanol and were subsequently post-cured using a Prusa post-curing and washing machine (CW1, PRUSA Research, Czech Republic). This machine employs four UV-LED strips with a wavelength of 405 nm and a maximum power of 52.8 W. Thermally post-cured samples were cured for 30 min at 150 °C, and no changes in sample color were observed. The post-processing conditions and duration for each 3D printed formulation are provided in Table 1.| Sample name | ARO (wt%) | IBOA (wt%) | HEA (wt%) | PI (wt%) | Bio-based carbon (%) | Layer time (s) | UV post-curing (min) | Thermal post-curing (min, 150 °C) |
|---|---|---|---|---|---|---|---|---|
| ARO/IBOA ratio optimization | ||||||||
| 6_RIH2 | 60 | 20 | 20 | 2 | 74 | 20 | 1 | — |
| 4_RIH2 | 40 | 40 | 71 | |||||
| 3_RIH2 | 30 | 50 | 70 | |||||
| 2_RIH2 | 20 | 60 | 68 | |||||
|
||||||||
| HEA concentration optimization | ||||||||
| RIH0 | 25 | 75 | 0 | 2 | 80 | 20 | 1 | — |
| RIH1 | 22.5 | 67.5 | 10 | 74 | ||||
| RIH2 | 20 | 60 | 20 | 68 | ||||
| RIH3 | 17.5 | 52.5 | 30 | 62 | ||||
|
||||||||
| Post-curing process optimization | ||||||||
| RIH3_T | 17.5 | 52.5 | 30 | 2 | 62 | 20 | 1 | 30 |
| RIH3_10P | 10 | — | ||||||
| RIH3_20P | 20 | — | ||||||
| RIH3_20 PT | 20 | 30 | ||||||
Fourier transform infrared (FTIR) spectroscopy
A Fourier transform infrared spectrometer Nicolet 6700 (ThermoScientific, Germany) was used to examine the interactions and bonding between the printed and raw resins. A piece of the 3D-printed bio-based material, or a droplet of the raw material, was placed directly on a diamond crystal and pressed with constant pressure using a torque wrench already attached to the spectrometer. The spectrum was obtained as an average of sixteen separate spectra recorded in the 400–4000 cm−1 range with a resolution of 4 cm−1. Eqn (1) was used to calculate the double bond conversion rate (DBC%): | (1) |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond absorption intensity (at 810 cm−1) before and after polymerization, and Arb and Ara are the ester CO bond absorption intensity (at 1722 cm−1) before and after polymerization.
C bond absorption intensity (at 810 cm−1) before and after polymerization, and Arb and Ara are the ester CO bond absorption intensity (at 1722 cm−1) before and after polymerization.
Theoretical spectra in ESI Fig. S5 and S6† are calculated using in the Lambert–Beer law eqn (2):
| AXf = X × Af + (1 − X)Ap | (2) |
Rheology
An MCR302 rheometer from Anton Paar (Graz, Austria) was used for photorheology measurements. The rheometer was equipped with a plate/plate measuring system, a Peltier-controlled temperature chamber with a glass plate of 38 mm, and a metal top plate of 8 mm diameter. The resin samples were exposed to radiation using a UV/vis spot curing system, OmniCure S1500 (Excelitas Technologies Corp., USA), with the measurement gap set to 0.2 mm. Radiation was applied at room temperature (22 °C) at wavelengths between 250 and 450 nm with an intensity of 2 mW cm−2. The measurements were performed in dynamic oscillatory shear time mode with an angular frequency of 5 rad s−1 and 1% oscillatory strain. The UV/vis onset was set at 60 seconds. Storage modulus (G′), loss modulus (G′′), and complex viscosity (η*) were continuously recorded during the real-time photorheology measurement.The viscosity of the as prepared resin stored for 2 months was measured using a 25 mm diameter spindle with plate–plate geometry. The measurement was conducted in the shear rate range of 1 to 100 s−1 at 22 °C, and the measurement gap was set to 0.1 mm. The chosen optimal formulation (RIH3) was used.
The thermal curing behavior of the RIH3 formulation was studied in temperature ramp mode at a constant shear rate of 50 s−1 with a heating rate of 1.5 °C min−1 in the temperature range from 20 to 200 °C.
Differential scanning calorimetry (DSC)
The 3D-printed RIH3 sample was subjected to differential scanning calorimetry utilizing a DSC-1 (Mettler Toledo, USA) analyzer. Under nitrogen purge, the samples in aluminum pans (9–10 mg) were heated from −10 to 210 °C, cooled to −10 °C, then heated again to 210 °C. The heating and cooling rate was 3 °C min−1. Each sample was measured once.Density measurements
The densities of the 3D-printed bio-based materials were obtained by hydrostatic weighing. A Sartorius MC-1 analytical balance with a density measurement kit (Sartorius, Germany) was used to weigh the samples. Ten parallel sample measurements were performed for each material, and the sample size was 5 × 5 mm, with a thickness of 2 mm. First, the sample was weighed in air and afterward in ethanol. Ethanol density and temperature were also measured, and the density values for materials were calculated using eqn (3): | (3) |
Sol fraction measurements
The sol fraction was determined using the Soxhlet extraction method. Ten parallel samples were weighed (w0) and kept in acetone at boiling temperature for 72 h. The sol fraction is calculated according to eqn (4) by weighing the part of the sample not dissolved in acetone (w1). | (4) |
Thermogravimetric analysis (TGA)
The test was performed on a Mettler TG50 (Mettler Toledo, USA) thermogravimetric analyzer. The test results provided the degradation temperature and thermal stability of the 3D-printed bio-based polymer materials. A ceramic crucible containing approximately 10 mg of the bio-based polymer material was heated in a nitrogen environment at a flow rate of 60 mL min−1 at 10 °C min−1 between 25 and 750 °C.Dynamic mechanical analysis (DMA)
A dynamic mechanical analyzer, a Mettler DMA/SDTA861e (Mettler Toledo, USA), was used to obtain the storage and loss modulus as a function of temperature. The experiment was carried out in dual cantilever deformation mode at a heating rate of 3 °C min in the temperature range of −70 to 100 °C, an applied force of 10 N, a displacement of 20 m, and a frequency of 1 Hz. The sample size was 8.5 × 4 mm with 200 μm thickness (4 layers). All samples were stored in sealed plastic bags in a dark place before testing, and tests were conducted the next day after the samples were printed. The molecular weight between crosslinks Mc and cross-linking density N were calculated using the theory of entropic rubber elasticity using eqn (5) and (6): | (5) |
 | (6) |
Quasi-static tensile testing
A 25ST universal testing machine (Tinius Olsen, UK) was used to perform tensile tests on 75 mm long, 5 mm wide, and 2 mm thick, 3D-printed bio-based dog-bone (ISO 527-1BA) shaped samples. The modulus values were obtained at 1 mm min−1, while the maximal stress and strain values were measured at 5 mm min−1. All tests were performed at 22 °C. For each material, the values were determined using five parallel measurements. All tests were conducted the next day after printing. The samples were kept in a sealed bag in a dark place. The toughness values were calculated from areas under the stress–strain curves.Scanning electron microscopy (SEM)
A scanning electron microscope, Hitachi Tabletop Microscope TM3000 (Japan), was used for the resolution evaluation of 3D printed structures. No coating was applied to the 3D structures and a 5 kV accelerating voltage was used.Pneumatic actuator
The pneumatic actuator was printed using RIH3 resin, utilizing optimized parameters, and post-cured for 20 min. The tri-Y splitter was thermally cured to achieve a complex structure supporting the 3 actuators and avoiding deformation.Shape-memory
The shape memory effect of the 3D printed sample was tested using the RIH3_PT formulation. 3D printed samples were heated to Tg + 30 °C in water, and then the desired shape was fixed by deforming the sample and cooling it down to room temperature. The dog-bone-shaped sample was 75 mm long, 5 mm wide, and 2 mm thick. The butterfly model was 1.5 mm thick, 50 mm long, and 35 mm wide.Results and discussion
3D printing processing and accuracy
In VP, resin viscosity is essential in determining the flow characteristics of the material within the vat. Optimal viscosity promotes proper layer development and adhesion, influencing the printed products' dimensional accuracy and surface quality.25Fig. 1(a) and ESI Fig. S1(a) and (b)† show that all resins have low viscosities before curing, including those with a large proportion of ARO. This characteristic stems from the production of ARO directly in a single step, eliminating the need for an intermediate epoxidation process.16,18 The monomers only marginally reduce the viscosity of the resins, with no additional benefits conferred. | ||
| Fig. 1 Photo-rheology (a) complex viscosity and (b) dynamic time sweep curves (storage (solid) and loss (dashed) modulus vs. time), with varying ARO/IBOA ratios at 22 °C. (c) DSC first and second heating scan of 3D printed samples and the rheology temperature ramp curve for uncured resin. Images of a 3D printed (d) castle model with (e and f) close-up photos and SEM images of an in-cut of (g) 0.2 mm and (h) 0.5 mm and (i) a dome demonstrating separate layer lines. | ||
We investigated the influence of the ratio of ARO to IBOA on the resin's photopolymerization kinetics. Upon activation of the UV lamp, a rapid increase in the values depicted in Fig. 1(a) and (b) signifies cross-linking onset. Concurrently, the intersection of the storage modulus (G′) and loss modulus (G′′) in Fig. 1(b) marks the resin's gel point, indicating the reactive resin's transition into a solid state. This gel point is a critical juncture in forming the printed structure. Fig. 1(b) demonstrates that an increase in the ARO concentration in the resin extends both the induction time (tind) and the gel time (tgel), although the variations are moderate, with tind ranging from 4 to 7 seconds and tgel from 5 to 8 seconds. Similarly, varying the concentration of HEA up to 30 wt% resulted in no significant alteration in either tind or tgel, remaining within 4–5 seconds (Fig. S1(c)†). In addition, it is seen that a plateau is achieved after 20 seconds of irradiation, implying that the active crosslinking phase has ended. This time was chosen as an optimal layer curing time in VP printing. Complex viscosity graphs are provided for ARO/IBOA ratios and HEA concentration optimizations in Fig. 1(a) and S1(b)† to confirm these observations further. These observations suggest that the raw material ratios do not markedly influence the photopolymerization reaction's course within the resins examined in this study.
Acrylates and acrylate/epoxy systems commonly undergo thermal post-curing to enhance mechanical properties.26,27 Typically, this requires an additional thermal initiator; however, the literature points to observed self-initiated thermal post-curing, characteristic of acrylate monomers and oligomers, that proceed without a secondary initiator and start polymerization reactions at elevated temperatures, typically above 110–130 °C.28 While the exact mechanism is still unclear,28,29 studies have shown this approach to improve mechanical performance.30
Rheology temperature ramp measurements on the RIH3 resin and DSC scans on RIH3's 3D printed counterparts were performed to assess the capability for self-initiated thermal post-curing. Fig. 1(c) reveals that both RIH3 and RIH3_20P samples display exothermic peaks at around 150 °C during the first heating scan, signifying the crosslinking reaction, which persists for approximately 25–28 minutes (calculated from the heating rate), commencing at 120 °C. Notably, RIH3 exhibits a smaller exothermic peak than RIH3_20P, attributed to the latter's extended UV post-curing time, resulting in fewer remaining unreacted acrylate groups. Subsequent (second) heating revealed no reaction, indicating that the curing process was complete. Rheology measurements corroborated these findings. At 149 °C, a pronounced increase in system viscosity signaled the onset of curing, which plateaued at around 190 °C after about 28 minutes (calculated from the heating rate), marking the end of the curing stage.
Fig. 1(d)–(f) present photos of a 3D-printed model (RIH3 resin, 4 × 4 × 6 cm) showcasing a highly detailed castle with an array of spires, towers, and intricate architectural nuances. The central portion of the structure is particularly notable for its refined windows, doors, and textured walls, highlighting the consistent and smooth layering throughout. Attention is drawn to the model's cylindrical towers and their adjacent features. These sections exhibit exceptional circular precision and layer uniformity – a characteristic hallmark for resin suitable VP. Additionally, the print is devoid of any bubbles or imperfections, which could stem from the resin. Our prior investigations have delved into various vegetable oil-based compositions, offering a comprehensive analysis of print resolution and the printability of resins employing the VP technique.17,31 Incorporating photo-absorbers, coloring agents, or diverse fillers can enhance printing precision. These additives improve the structural integrity and endow the prints with functional advantages, such as electroconductivity and luminescence.32,33 The well-resolved details are presented in the SEM analysis in Fig. 1(g)–(i). The original calibration model is depicted in ESI Fig. S2.† The cut-in cross patterns (Fig. 1(g) and (h)) with a resolution down to 0.2 mm can be achieved, showing the inherent high-resolution characteristics of VP printing. In addition, the dome (Fig. 1(i)) shows distinct layer lines specially designed to seek the correct calibration of printing times.
Macromolecular chain network design
Fig. 2(a) depicts a schematic representation of the sophisticated transformation of an elastomeric material's macrostructure through deliberate modulation of its molecular composition and targeted post-processing. This study delineates a methodical approach that harnesses both covalent and non-covalent interactions. This intricate balance depends on modulating the material's phase characteristics, transitioning adeptly between glassy and elastomeric states while finessing the crosslinking density to refine its mechanical qualities. We illustrated three main constituents: ARO, IBOA, and HEA, thereby elucidating their integral roles in the network's architecture (Fig. 2(b) and (c)). Furthermore, the incorporation of bio-based carbon content is meticulously documented, underscoring our commitment to advancing the frontiers of sustainable material design.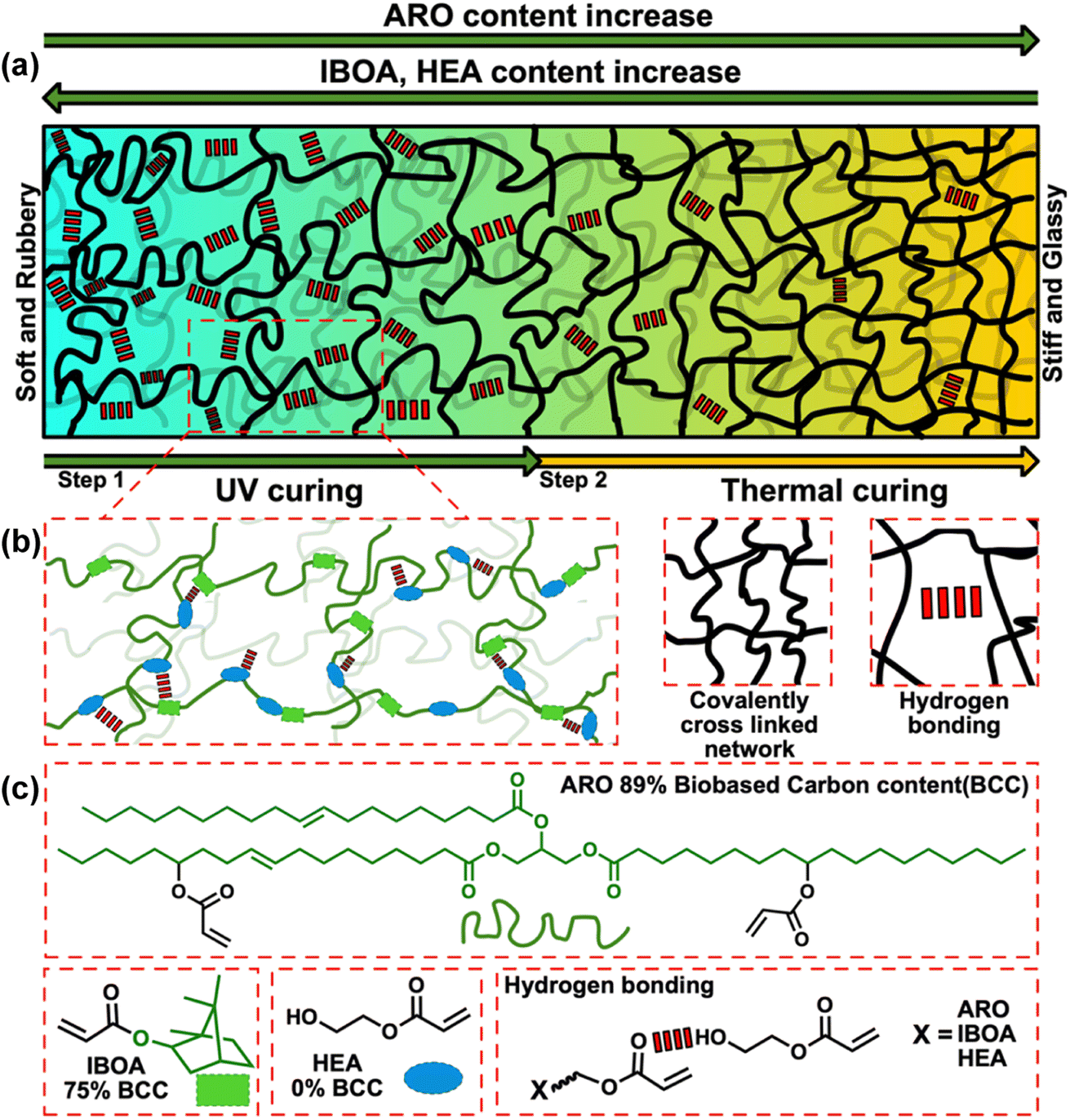 | ||
| Fig. 2 Schematic representation of (a) three distinct methods used to control crosslinked network development by adjusting formulation and post-processing factors and (b) possible crosslinked network and interactions consisting of (c) building blocks. | ||
The FTIR spectroscopy data presented in Fig. 3(a) and (b), along with Fig. S3,† elucidate the resin formulations' spectral characteristics before and after 3D printing. The spectra distinctly exhibit the characteristic peaks of the CC groups at 810, 1406, and 1632 cm−1, and that of the CO group at 1725 cm−1.34–36 Notably, the intensities of the vinyl group peaks are diminished after photopolymerization, indicating the occurrence of the crosslinking process during 3D printing. This trend is consistent across formulations with varying ratios of ARO to IBOA, as shown in Fig. S3.† An incremental increase in HEA content, ranging from 0 to 30 wt%, is traceable in Fig. 3(a) through the increase in the –OH group peak at 3462 cm−1.37 This peak intensity of the –OH group serves as the sole objective indicator for tracking the increase in HEA content. This specificity is due to overlapping of most other spectral peaks with those of IBOA, as shown in Fig. S4.†Fig. 3(b) reveals the further reduction of the vinyl peaks after post-processing. There are no discernible vinyl group peaks in the RIH3_20PT sample, which underwent the most extensive post-processing.
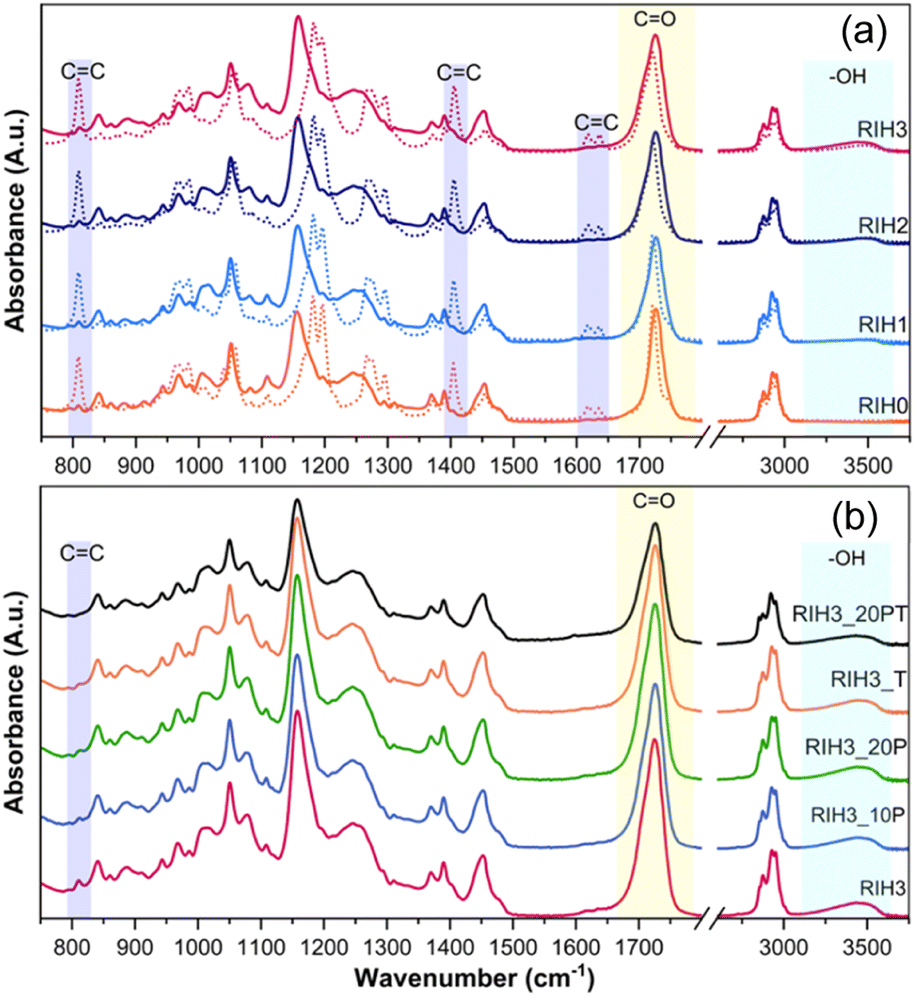 | ||
| Fig. 3 FTIR spectra for formulations with varying HEA ratios (a) (uncured resin (dots) and 3D printed sample (line)) and FTIR spectra of RIH3 with varying post-processing conditions (b). | ||
The evaluation of photopolymerization efficiency is predicated on the percentage of double bond conversion (DBC%), which is quantified using eqn (1). According to the data presented in Table 2, a trend is observed where an increase in ARO and HEA correlates with a slight decrease in total covalent bonding (DBC%). As anticipated, additional post-curing enhances DBC%, while numerical changes only show an increase from 86% to 89% when comparing RIH3 and RIH3_20PT. It is crucial to note that the covalent bonds resulting from the photopolymerization of vinyl groups do not solely account for all the crosslinks within the system.
| Sample name | Density (g cm−3) | DBC (%) | Sol fraction (%) | M C (g mol−1) | N × 103 (mol cm−3) |
|---|---|---|---|---|---|
| 6_RIH2 | 1.087 ± 0.004 | 81 | 7.42 ± 0.42 | 513 | 2.120 |
| 4_RIH2 | 1.066 ± 0.002 | 86 | 6.39 ± 0.57 | 1337 | 0.797 |
| 3_RIH2 | 1.091 ± 0.002 | 87 | 6.89 ± 0.42 | 2554 | 0.427 |
| 2_RIH2 | 1.100 ± 0.003 | 87 | 7.14 ± 0.52 | 3504 | 0.314 |
| RIH0 | 1.058 ± 0.002 | 90 | 7.01 ± 0.50 | 1408 | 0.752 |
| RIH1 | 1.079 ± 0.002 | 87 | 7.34 ± 0.53 | 3102 | 0.348 |
| RIH2 | 1.100 ± 0.003 | 87 | 7.14 ± 0.52 | 3504 | 0.314 |
| RIH3 | 1.122 ± 0.003 | 86 | 6.85 ± 0.50 | 4231 | 0.265 |
| RIH3_T | 1.122 ± 0.003 | 89 | 4.25 ± 0.45 | 2394 | 0.469 |
| RIH3_10P | 1.121 ± 0.003 | 86 | 5.33 ± 0.40 | 2566 | 0.438 |
| RIH3_20P | 1.122 ± 0.002 | 87 | 4.74 ± 0.49 | 2481 | 0.453 |
| RIH3_20 PT | 1.123 ± 0.003 | 89 | 3.22 ± 0.51 | 2282 | 0.492 |
Beyond chemical crosslinks, the system also possesses physical crosslinks. Evidence of this can be derived from the molecular weight between crosslinks (Mc) and the crosslinking density (N), calculated using eqn (5) and (6), alongside sol fraction data presented in Table 2. The highest crosslinking density (2.120 mmol cm−3) is found in the 6_RIH2 sample, which contains the most considerable quantity of ARO, although it does not exhibit the lowest sol fraction. The lowest sol fraction (3.22%) is observed for the RIH3_20PT sample and is associated with the highest content of HEA and thermal post-curing. Given HEA's reported propensity to establish strong hydrogen bonding within the system,38 as well as the highest observed density of 1.123 g cm−3 in this sample, it is posited that the additional physical crosslinks and hydrogen bonds contribute significantly to this result. Moreover, physical interactions such as van der Waals forces may emanate from the aliphatic chains of ARO, coupled with a lower crosslinking density, culminating in a lengthy, entangled network depicted in Fig. 2.
Fig. S5 and S6† provide detailed visual insights into the peaks corresponding to the CO and OH bonds, commonly scrutinized for evidence of hydrogen bonding in acrylates.38 Deviations in peak intensity and shifts observed in the FTIR spectra, when contrasted with theoretical spectra (calculated with eqn (2) from the cured component spectra in Fig. S4†), further substantiate the existence of hydrogen bonding within the system.
Tensile properties
Fig. 4(a) shows that the sample 6_RIH2 composed of a high concentration of ARO exhibits linear stress–strain relationships, characterized by limited elasticity and low tensile strength, with the elongation at break reaching only 8% and the ultimate strength measuring a mere 0.5 MPa. The modulus values of vegetable oil-based acrylate resins are inherently low,18 which is a beneficial feature that will allow for soft and flexible 3D-printed structures. The tensile curves retain their linear elastic behavior with the addition of IBOA; while tensile strength and elongation values show a notable increase, and the modulus values decrease. As the ARO/IBOA ratio reaches 1/3 (sample RIH2), there is a sharp increase in tensile characteristics, demonstrating an astounding 1857% increase in elongation at break and 460% increase in ultimate strength compared with 6_RIH2. | ||
| Fig. 4 Stress–strain curves, modulus and toughness graph of 3D printed samples with variable (a and b) ARO/IBOA ratios, (c and d) variable HEA ratios, and (e and f) variable post-processing conditions. | ||
When coupled with high elongation and strength, low modulus values unlock significant potential for applications in soft robotics, notably in the development of pneumatic actuators.38,39 This can be attributed to changes in crosslinking density in the samples, which is depicted in a schematic representation of the crosslinked network in Fig. 2. The ARO contains, on average, 2.14 acrylate groups per molecule, as reported in our previous paper,16 thus leading to a more crosslinked network with brittle failure, low elongation, and higher crosslinking density (Table 2).
Incorporating a higher concentration of IBOA significantly shifts the equilibrium between non-covalent and covalent bonds within the polymer matrix, as shown in Fig. 2(a). In this case, ARO is acting as an oligomeric crosslinker, facilitating more efficient crosslinking due to long aliphatic chains and around 2 acrylate groups in the molecule (Fig. 2(c)); a similar effect was observed with petroleum-based urethane diacrylates reported elsewhere.38,40 At the same time, hard segments introduced from IBOA contribute to higher strength of 3D printed samples when an effective crosslinker/IBOA ratio is obtained. This underscores the critical role of meticulous formulation design, highlighting how the balance between hard and soft segments, along with the structure of the crosslinking network, significantly influences the material's properties and its applicability in advanced fields (Fig. 2(b)).
HEA, being a monofunctional monomer with a single acrylate group and a hydroxyl group, is known to enhance the elongation and strength of urethane acrylate polymer matrices due to its low glass transition temperature (Tg) and flexible molecular structure.38 Our study varied the HEA content in the formulation from 0 to 30%, aiming to maintain the bio-based characteristics of the formulation. As indicated in Fig. 4(c) and (d), including HEA significantly influences the tensile properties of the 3D printed specimens. By adjusting the HEA concentration, we could modify the mechanical behavior of the resins, resulting in samples ranging from hard and rigid to soft and pliable. The variations in strain, strength, and modulus between the samples RIH0 and RIH3 are notable, with changes of 167%, 45%, and 1540%, respectively. The addition of HEA results in a marked reduction in crosslink density, allowing for a greater degree of molecular mobility. The impact of HEA on the polymeric network is akin to that of isobornyl acrylate (IBOA), with the key distinction that HEA introduces additional hydroxyl groups into the formulation, contributing to the formation of a more flexible network.41
Altering post-processing conditions is a common strategy for tailoring the mechanical properties of acrylates produced using high-strain VP. A technique often used in VP printing involves under-curing characterized by low degrees of curing to create less densely crosslinked networks.38,42 A significant obstacle to this technique is the challenge of managing the UV-crosslinking process. Networks created in this manner are prone to further crosslinking upon exposure to any UV light source, unintentionally increasing the degree of cure. This propensity restricts their durability and range of applications, as it can lead to unpredictable performance over time. Therefore, formulating compositions with optimized post-curing is crucial. This foresight ensures that the materials are more resilient to unintended post-curing effects from accidental exposure to natural sunlight or other sources of low-level UV radiation, thereby enhancing their service life and reliability.
The data in Fig. 4(e) reveal that for the RIH3 system, the initially set 1 minute post-curing time is suboptimal. An extension of the curing period to 20 minutes resulted in a remarkable 263% enhancement in tensile strength and a 29% growth in elongation at break while preserving low modulus levels. In contrast, the differences observed between RIH3_10P and RIH3_20P were considerably less pronounced, leading to the conclusion that extending the curing time beyond 20 minutes would yield negligible benefits. Consequently, a 20 minute curing duration has been identified as the optimal period for achieving the desired properties of high elongation coupled with low stiffness.
Further investigations were carried out on the 3D-printed specimens using thermal post-curing. Fig. 4(e) illustrates a pronounced transformation in the material's response, transitioning from elastomeric to ductile behavior, evidenced by a 748% increase in modulus, as shown in Fig. 4(f). Comparable outcomes were observed for both RIH3_T and RIH3_20PT concerning the ultimate strength, elongation at break and modulus values, due to formation of similar macromolecular networks, as evidenced from Nc and DBC% values (Table 2). Ultimate strength values aligned with those of RIH3_10P and RIH3_20P, albeit with a significant reduction in strain by approximately 82%. At minor elongations (around 10%), the thermally post-cured samples began to demonstrate yield points and strain softening, after which the material could not return to its original configuration.43 This shift results in the formation of a highly crosslinked network, as shown in Fig. 2(a).
We opted to assess each formulation's toughness, which can determine application constraints and indicate the maximum energy a material can absorb before failure.44Fig. 4(b), (d) and (f) show that enhanced toughness can be achieved through tailored post-processing and formulation adjustments. Despite RIH0 exhibiting the highest toughness among formulations with varied HEA contents, the RIH3 sample presents a significantly broader performance spectrum, reaffirming its superior adaptability and suitability. We maintained the system's toughness by using modifying post-processing techniques while significantly altering material characteristics. With their high modulus and hardness, the thermally cured samples are deemed optimal for stiff structural components in 3D printing. Conversely, RIH3_20P, characterized by its low modulus and high durability, is well-suited for components undergoing dynamic, repetitive motions. Leveraging these insights, we developed a fully 3D-printed soft robotic gripper (discussed in the next section) as a proof of concept, demonstrating the practical application of our findings.
Thermal and thermomechanical properties
Studies have found that IBOA degradation occurs at about 300 °C.45 HEA decomposes at around 449 °C,37 while our previous investigation showed that the most intensive degradation of ARO happens at around 350 °C and 420 °C.16 This information is consistent with the DTG curves shown in Fig. 5. Therefore, the first DTG peak, which is observable from 280 to 320 °C, depending on the composition, is attributed to IBOA. The first peak also tends to shift to higher temperatures at higher HEA concentrations, while the second DTG peak at around 350 °C is only visible in the composition without HEA (RIH0) and is attributed to ARO. The third and fourth DTG peaks merge; this overlap leads to the formation of a broad degradation peak, which encompasses the degradation signatures of both HEA and ARO. This overlap presents as single or two merged peaks depending on the composition. | ||
| Fig. 5 TGA curves of 3D printed samples with (a) varying ARO/IBOA ratios and (b) varying HEA ratios. | ||
The TGA clearly shows both the ARO/IBOA ratio (Fig. 5(a)) and HEA ratio (Fig. 5(b)) impact the degradation as the composition changes are reflected as clearly visible gaps in the curve profiles. Still, the only composition with a significantly altered maximum degradation temperature is 6_RIH2, which exhibits the most robust thermal resistance due to the highest crosslinking. When analyzing the effect of post-processing conditions on the material, there are no significant changes in the thermograms (Fig. S7†). The temperature of the initial peak for IBOA breakdown ranges from 295 to 297 °C. The RIH3_20PT sample is notable, which shows two distinct peaks at 295 °C and 305 °C. This could reflect the presence of a more highly crosslinked phase after the post-processing.
The thermomechanical properties of polymer networks are intrinsically tied to temperature variations. A comprehensive understanding of the transition zone, defining the glassy and rubbery states, is pivotal for tailoring the material properties within a specified temperature range. Notably, after a steep decline in storage modulus, as Fig. 6(a) illustrates, all initial compositions transition into soft elastomers in the 0 to 20 °C range. These compositions exhibit markedly low storage modulus values, characteristic of elastomeric materials. The glass transition temperature (Tg) is discernible from the peak in the loss modulus graphs (Fig. 6(b)), indicating that compositional variations significantly influence the Tg region, which spans from −26 °C to 7 °C.
 | ||
| Fig. 6 DMA storage modulus and loss modulus of 3D printed samples with variable (a and b) ARO/IBOA ratios, (c and d) variable HEA ratios, and (e and f) variable post-processing conditions and (g) Cole–Cole plot for RIH3 formulation. | ||
Consequently, the materials exhibit a broad spectrum of properties, necessitating further analytical scrutiny. Employing eqn (5) and (6), we quantified the molecular weight between crosslinks (Mc) and the crosslinking density (N), as shown in Table 2. Intriguingly, the 2_RIH2 composition exhibited the lowest crosslinking density coupled with the highest Mc while also possessing the highest Tg. These findings are consistent with data obtained from tensile testing, where we selected 2_RIH2 as our optimal choice due to high elongation values (Fig. 4(a)).
Optimization of the HEA component in our selected composition (RIH2) was undertaken. The storage modulus curves revealed an inverse correlation between values in the glassy state and the transition to the rubbery state. For instance, RIH3 displayed the highest modulus at −40 °C and the lowest modulus at 20 °C (Fig. 6(c)). While the glassy state properties are not direct predictors of elastomeric performance in the rubbery state, they offer insights into the compatibility and synergistic interactions of the components within the polymer network. In our study, this entails identifying the ratio that fosters optimal component mixing, thus ensuring a more stable network. The effect on Tg was notably subdued through HEA optimization, ranging from 3 to 13 °C (Fig. 6(d)). Mc and N calculations support our hypothesis that precise adjustments in crosslink weight are feasible, with RIH1 and RIH3 compositions exhibiting slightly higher and lower values, respectively, than our benchmark composition (RIH2). Despite all compositions exhibiting exceptional tensile performance (Fig. 4(c)), the marginally lower Tg favored the selection of RIH3.
Subsequent experiments focused on post-processing methods to induce a soft-to-rigid transition in our chosen composition (RIH3). This was achieved by manipulating the Tg to higher temperatures. Fig. 6(d) demonstrates that extended UV-post processing resulted in negligible increases in storage modulus at 20 °C among the tested approaches. Conversely, thermal curing led to a substantial enhancement, increasing the storage modulus from 23 MPa (RIH3) to 1354 MPa (RIH3_T) at 20 °C (Fig. 6(e)), alongside a more than 30 °C increase in Tg (from 3 °C to 41 °C) as can be seen in Fig. 6(f). The final comparison between RIH3_T and RIH3_20PT reveals that performance is relatively similar, with the former being slightly more densely crosslinked (higher N values), with a 1 °C higher Tg. Notably, RIH3_20PT clearly shows two overlaying glass transition (loss modulus) peaks, which are notable if shape memory properties are desirable.
Fig. 6(g) presents a Cole–Cole plot that illustrates alterations in the relaxation processes due to the modification of crosslinking conditions in the printed system. The pronounced flattening of the semi-circular trajectory, deviating from the ideal single relaxation time, signifies the presence of multiple (commonly two) relaxation phases within the system.46 The departure of any sample from the idealized semi-circular form reflects the intricate material composition and the crosslinking within the structure. The splitting of the loss peak for thermally cured samples and two glass transitions is reflected in the Cole–Cole curve.
Shape memory effect
Stimuli-responsive materials, specifically shape memory polymers (SMPs), hold significant promise for soft robotics across various advanced applications.47 These materials' shape memory effect (SME) is predominantly activated through thermal mechanisms.48 To evaluate the SME in our formulations, 3D printed samples were immersed in hot water (Tg + 30 °C), deformed, and then rapidly cooled down and reheated to (Tg + 30 °C) to initiate shape recovery. The SME timelines for RIH3_20PT, depicted in Fig. 7(a) and (b) and Supplementary Movies 1 and 2†, demonstrate recovery times on par with those of other rapid-response SMPs.49–51 It was observed that the samples' dimensional parameters and deformation significantly influence the recovery time. Furthermore, the samples exhibited excellent shape fixity, regaining their original form after multiple reheating cycles to Tg + 30 °C. The SME in these plant-based elastomers is consistent with mechanisms reported for formulations containing HEA and IBOA, attributed to their flexible chain structure rich in entanglements.40,52,53 During the programming stage at Tg + 30 °C, molecular mobility increases significantly, facilitating easy deformation within the rubbery state region, as evidenced by DMA analysis (Fig. 6). Upon cooling, the deformed molecular structure is ‘frozen’, storing the energy exerted during deformation, while molecular entanglements maintain the shape. When a trigger, such as heat, is applied, the molecular mobility accelerates, allowing polymer chains to revert to their initial state. The SME's stability is further enhanced by the reversible nature of hydrogen bonding.40,52 However, comprehensive studies are essential to fully understand the contribution of each component to the shape memory properties of these materials. | ||
| Fig. 7 Images of shape memory demonstration of the 3D printed (a) butterfly model, (b) dog-bone shaped sample, (c) tri-Y splitter, and assembled soft robotic gripper holding a (d) ball, (e) small 3D printed figure of irregular shape, and (f) large 3D printed object of irregular shape. | ||
Pneumatic soft robots
The rapidly evolving field of soft robotics requires quick and dependable processes for producing highly flexible components made of soft and high-strain materials. Nowadays, non-bio-based silicon rubbers, polyurethane acrylates, or thiol/acrylate systems are the primary materials of most reported soft robotics systems.38,39,54 While the application of additive manufacturing in creating fully 3D-printed soft robots is not unprecedented, the use of plant oil-based systems in this field remains limited.55 Recent research by Cornella et al. explored the use of Diels–Alder cycloaddition cross-linkable castor oil elastomers to create soft robotics.21 However, the actual pneumatically actuated gripper still demanded the creation of a mold and further complicated assembly procedures.In contrast, this study introduces a sustainable, fully 3D-printed soft robotic gripper that is simple to assemble. As the dimensional stability of the printed structures is essential for soft robotic applications, additional tests were conducted on printed 5 × 5 × 5 mm cubes (Fig. S8†). After performing 5 parallel measurements, the dimensional stability of the printed structures appeared to be 4.99 ± 0.03 mm, which demonstrates exceptional resolution of the developed material. Fig. 7(c) demonstrates the parts of the gripper and a fully assembled prototype. The gripper consists of 3 actuators (from RIH3_20P) and a thermally post-cured tri-Y splitter (from RIH3_20PT), ensuring uniformity and limited joint flexibility. All parts are easily joined together, while the flexibility and soft nature of the gripper material ensure a tight and enclosed fit. As demonstrated in Fig. 7(c) and (f) and Supplementary Movie 3,† the gripper effectively handles a variety of objects, including balls, irregularly shaped small models, and a large, heavy castle model previously discussed. The essential advantage of this system lies in its adaptability and ability to target and customize each aspect of the 3D printed parts, leveraging the versatility of additive manufacturing and the broad performance spectrum of the developed materials.
Conclusions
In the present investigation, we tackled the challenge of formulating bio-based materials suitable for additive manufacturing with targeted elastomeric properties for application in soft robotics. The developed series of 3D printable materials showcased remarkable versatility, demonstrating a diverse array of mechanical characteristics, including an elongation of 8–180%, ultimate tensile strength of 0.4–14 MPa, and modulus of 5–190 MPa. Furthermore, other adjusted properties include thermal transitions, viscosity, and curing times, among others. The distinction between UV-cured and thermally cured materials was particularly pronounced, facilitating the fabrication of both flexible, soft models ideal for pneumatically actuated soft robotics and harder, flexible models endowed with a shape memory effect.The elastomers were synthesized from bio-based precursors, notably rapeseed oil acrylate and isobornyl acrylate. The addition of up to 30 wt% of 2-hydroxyethyl acrylate tailored the mechanical properties while maintaining a bio-based carbon content of over 60%. Our methodology underscores the synergy between the developed resins and vat polymerization techniques, broadening the scope of potential applications and promoting sustainable practices within additive manufacturing. Future investigations will focus on refining these formulations for specific uses, including the potential integration of additives or modifiers to fine-tune the material properties for targeted applications in soft robotics and other domains.
This investigation substantially contributes to developing and applying sustainable bio-based materials in cutting-edge sectors. We envisage that our findings will encourage the adoption of more environmentally friendly material development strategies and heighten the focus on bio-based raw materials, thereby advancing a more sustainable trajectory for additive manufacturing, soft robotics, and related fields.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. A. R.: investigation; formal analysis; visualization; writing – original draft. M. J.: conceptualization; investigation; formal analysis; visualization; validation; writing – original draft, review & editing. O. P.: investigation; formal analysis; writing – original draft, review & editing. A. B.: investigation; formal analysis; writing – original draft. S. G.: conceptualization; methodology; resources; supervision; writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Latvian Council of Science in the framework of the FLPP “Printed biopolymer 4D TENG device for mechanical energy harvesting” (lzp-2022/1-0485).References
- E. Sachyani Keneth, A. Kamyshny, M. Totaro, L. Beccai and S. Magdassi, Adv. Mater., 2021, 33, 2003387, DOI:10.1002/adma.202003387.
- R. A. Shanks and I. Kong, in Advances in Elastomers I. Adv. Struct. Mater., ed. P. M. Visakh, S. Thomas, A. K. Chandra and A. P. Mathew, Springer, Berlin, Heidelberg, 2013, vol. 2, pp. 11–45 Search PubMed.
- C. Majidi, Adv. Mater. Technol., 2019, 4, 1800477, DOI:10.1002/admt.201800477.
- S. Utrera-Barrios, R. Verdejo, M. Á. López-Manchado and M. Hernández Santana, Eur. Polym. J., 2023, 190, 112023, DOI:10.1016/j.eurpolymj.2023.112023.
- S. Li, X. Zhou, Y. Dong and J. Li, Macromol. Rapid Commun., 2020, 41, 2000444, DOI:10.1002/marc.202000444.
- S. Han, Q. Wan, K. Zhou, A. Yan, Z. Lin, B. Shu and C. Liu, ACS Appl. Nano Mater., 2021, 4, 8273–8281, DOI:10.1021/acsanm.1c0145.
- A. Jandyal, I. Chaturvedi, I. Wazir, A. Raina and M. I. Ul Haq, Sustainable Oper. Comput., 2022, 3, 33–42, DOI:10.1016/j.susoc.2021.09.004.
- J. Herzberger, J. M. Sirrine, C. B. Williams and T. E. Long, Prog. Polym. Sci., 2019, 97, 101144, DOI:10.1016/j.progpolymsci.2019.101144.
- Elastomer-X, https://www.liqcreate.com/product/elastomer-x/, accessed February 2024.
- Flexible80A, https://formlabs.com/eu/materials/flexible-elastic/, accessed February 2024.
- Tango, https://www.stratasys.com/en/materials/materials-catalog/polyjet-materials/tango/, accesed February 2024.
- EPU40, https://www.carbon3d.com/materials/epu-40, accessed February 2024.
- A. Heiden, D. Preninger, L. Lehner, M. Baumgartner, M. Drack, E. Woritzka, D. Schiller, R. Gerstmayr, F. Hartmann and M. Kaltenbrunner, Sci. Robot., 2022, 7, eabk2119, DOI:10.1126/scirobotics.abk2119.
- M. Baumgartner, F. Hartmann, M. Drack, D. Preninger, D. Wirthl, R. Gerstmayr, L. Lehner, G. Mao, R. Pruckner, S. Demchyshyn, L. Reiter, M. Strobel, T. Stockinger, D. Schiller, S. Kimeswenger, F. Greibich, G. Buchberger, E. Bradt, S. Hild, S. Bauer and M. Kaltenbrunner, Nat. Mater., 2020, 19, 1102–1109, DOI:10.1038/s41563-020-0699-3.
- R. Chen, X. Li, Q. Xiong, X. Zhu, H. Wang, W. Wang, G. Bao, Z. Chen, C. Cao and J. Luo, Mater. Des., 2023, 227, 111783, DOI:10.1016/j.matdes.2023.111783.
- S. Briede, M. Jurinovs, S. Nechausov, O. Platnieks and S. Gaidukovs, Mol. Syst. Des. Eng., 2022, 7, 1434–1448, 10.1039/D2ME00085G.
- M. Jurinovs, A. Barkane, O. Platnieks, S. Beluns, L. Grase, R. Dieden, M. Staropoli, D. F. Schmidt and S. Gaidukovs, ACS Appl. Polym. Mater., 2023, 5, 3104–3118, DOI:10.1021/acsapm.3c00245.
- C. Vazquez-Martel, L. Becker, W. V. Liebig, P. Elsner and E. Blasco, ACS Sustainable Chem. Eng., 2021, 9, 16840–16848, DOI:10.1021/acssuschemeng.1c06784.
- M. Fei, T. Liu, B. Zhao, A. Otero, Y.-C. Chang and J. Zhang, ACS Appl. Polym. Mater., 2021, 3, 2470–2479, DOI:10.1021/acsapm.1c00063.
- Z. Wang, P. Tang, S. Chen, Y. Xing, C. Yin, J. Feng and F. Jiang, Carbohydr. Polym., 2023, 305, 120577, DOI:10.1016/j.carbpol.2023.120577.
- A. Costa Cornellà, S. K. Tabrizian, P. Ferrentino, E. Roels, S. Terryn, B. Vanderborght, G. Van Assche and J. Brancart, ACS Sustainable Chem. Eng., 2023, 11, 3437–3450, DOI:10.1021/acssuschemeng.2c06874.
- Z. Wang, L. Yuan, N. M. Trenor, L. Vlaminck, S. Billiet, A. Sarkar, F. E. Du Prez, M. Stefik and C. Tang, Green Chem., 2015, 17, 3806–3818, 10.1039/C5GC00822K.
- H. Soleimanzadeh, B. Rolfe, M. Bodaghi, M. Jamalabadi, X. Zhang and A. Zolfagharian, Adv. Sustainable Syst., 2023, 7, 2300289, DOI:10.1002/adsu.202300289.
- F. Hartmann, M. Baumgartner and M. Kaltenbrunner, Adv. Mater., 2021, 33, 2004413, DOI:10.1002/adma.202004413.
- A. Vyas, V. Garg, S. B. Ghosh and S. Bandyopadhyay-Ghosh, Mater. Today Proc., 2022, 62, 1435–1439, DOI:10.1016/j.matpr.2022.01.172.
- S. Nechausov, A. Ivanchenko, O. Morozov, A. Miriyev, I. Must, M. Jurinovs, O. Platnieks, S. Gaidukovs, A. Aabloo, M. Kovač and B. Bulgakov, Addit. Manuf., 2022, 56, 102895, DOI:10.1016/j.addma.2022.102895.
- A. C. Uzcategui, A. Muralidharan, V. L. Ferguson, S. J. Bryant and R. R. McLeod, Adv. Eng. Mater., 2018, 20, 1800876, DOI:10.1002/adem.201800876.
- S. Srinivasan, A. M. Rappe and M. Soroush, in Computational Quantum Chemistry, ed. M. Soroush, Elsevier, Amsterdam, 2019, 4, pp. 99–134 Search PubMed.
- S. Laki, A. A. Shamsabadi, H. Riazi, M. C. Grady, A. M. Rappe and M. Soroush, Ind. Eng. Chem. Res., 2020, 59, 2621–2630, DOI:10.1021/acs.iecr.9b05050.
- J. Bauer, A. G. Izard, Y. Zhang, T. Baldacchini and L. Valdevit, Opt. Express, 2020, 28, 20362–20371, DOI:10.1364/OE.395986.
- A. Barkane, O. Platnieks, M. Jurinovs, S. Kasetaite, J. Ostrauskaite, S. Gaidukovs and Y. Habibi, Polymers, 2021, 13, 1195, DOI:10.3390/polym13081195.
- H. Y. Jeong, E. Lee, S.-C. An, Y. Lim and Y. C. Jun, Nanophotonics, 2020, 9, 1139–1160, DOI:10.1515/nanoph-2019-0483.
- M. Jurinovs, A. Barkane, O. Platnieks, L. Grase and S. Gaidukovs, ACS Appl. Polym. Mater., 2023, 5, 7120–7131, DOI:10.1021/acsapm.3c01136.
- A. R. Mahendran, G. Wuzella, N. Aust, A. Kandelbauer and U. Müller, Prog. Org. Coat., 2012, 74, 697–704, DOI:10.1016/j.porgcoat.2011.09.027.
- Y. Lu, C. Du, Y. Shao and J. Zhou, J. Food Sci. Eng., 2014, 4, 244–249, DOI:10.17265/2159-5828/2014.05.004.
- S. Yu, Y. Li, S. Wu, Z. Tang, L. Zhang and B. Guo, SusMat, 2023, 3, 320–333, DOI:10.1002/sus2.122.
- E. Vargün and A. Usanmaz, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3957–3965, DOI:10.1002/pola.20867.
- L. Yue, S. Macrae Montgomery, X. Sun, L. Yu, Y. Song, T. Nomura, M. Tanaka and H. Jerry Qi, Nat. Commun., 2023, 14, 1251, DOI:10.1038/s41467-023-36909-y.
- E. F. Gomez, S. V. Wanasinghe, A. E. Flynn, O. J. Dodo, J. L. Sparks, L. A. Baldwin, C. E. Tabor, M. F. Durstock, D. Konkolewicz and C. J. Thrasher, ACS Appl. Mater. Interfaces, 2021, 13, 28870–28877, DOI:10.1021/acsami.1c06419.
- B. Zhang, H. Li, J. Cheng, H. Ye, A. H. Sakhaei, C. Yuan, P. Rao, Y.-F. Zhang, Z. Chen, R. Wang, X. He, J. Liu, R. Xiao, S. Qu and Q. Ge, Adv. Mater., 2021, 33, 2101298, DOI:10.1002/adma.202101298.
- Z. Wan, L. Wai Hin, A. Shegiwal and D. Haddleton, Eur. Polym. J., 2023, 196, 112324, DOI:10.1016/j.eurpolymj.2023.112324.
- X. Kuang, J. Wu, K. Chen, Z. Zhao, Z. Ding, F. Hu, D. Fang and H. J. Qi, Sci. Adv., 2019, 5, eaav5790, DOI:10.1126/sciadv.aav5790.
- E. F. Abdewi, Mechanical Properties of Reinforcing Steel Rods Produced by Zliten Steel Factory, Elsevier, Amsterdam, 2017 Search PubMed.
- M. D. Kiran, H. K. Govindaraju, B. R. Lokesh Yadhav and N. Kumar, Mater. Today Proc., 2023, 92, 1–5, DOI:10.1016/j.matpr.2023.03.129.
- S. Ozlem, E. Aslan-Gürel, R. M. Rossi and J. Hacaloglu, J. Anal. Appl. Pyrolysis, 2013, 100, 17–25, DOI:10.1016/j.jaap.2012.10.024.
- J. Jyoti, B. P. Singh, A. K. Arya and S. R. Dhakate, RSC Adv., 2016, 6, 3997–4006, 10.1039/C5RA25561A.
- Y. Zhao, M. Hua, Y. Yan, S. Wu, Y. Alsaid and X. He, Annu. Rev. Control Rob., Auton. Syst., 2022, 5, 515–545, DOI:10.1146/annurev-control-042920-014327.
- M. Lei, Z. Chen, H. Lu and K. Yu, Nanotechnol. Rev., 2019, 8, 327–351, DOI:10.1515/ntrev-2019-0031.
- X. Wang, J. Sparkman and J. Gou, Compos. Sci. Technol., 2017, 141, 8–15, DOI:10.1016/j.compscitech.2017.01.002.
- S. S. Mahapatra, S. K. Yadav, H. J. Yoo, M. S. Ramasamy and J. W. Cho, Sens. Actuators, B, 2014, 193, 384–390, DOI:10.1016/j.snb.2013.12.006.
- Y. Feng, Y. Hu, L. Man, T. Yuan, C. Zhang and Z. Yang, Eur. Polym. J., 2019, 112, 619–628, DOI:10.1016/j.eurpolymj.2018.10.025.
- A. Cosola, M. Sangermano, D. Terenziani, R. Conti, M. Messori, H. Grützmacher, C. F. Pirri and A. Chiappone, Appl. Mater. Today, 2021, 23, 101060, DOI:10.1016/j.apmt.2021.101060.
- N. Zeggai, Z. Bouberka, F. Dubois, L. Bedjaoui, T. Bouchaour, C. E. Gherdaoui, J. Potier, P. Supiot and U. Maschke, Compos. Sci. Technol., 2022, 219, 109213, DOI:10.1016/j.compscitech.2021.109213.
- Y.-F. Zhang, C. J.-X. Ng, Z. Chen, W. Zhang, S. Panjwani, K. Kowsari, H. Y. Yang and Q. Ge, Adv. Mater. Technol., 2019, 4, 1900427, DOI:10.1002/admt.201900427.
- Y. Xin, X. Zhou, H. Bark and P. S. Lee, Adv. Mater., 2023, 2307963, DOI:10.1002/adma.202307963.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta02218a |
| This journal is © The Royal Society of Chemistry 2024 |