
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating interfacial parameters of platinum–palladium bulk alloy single crystals†
Gabriel
Melle
 a,
Fabian
Scholten
b,
Juan M.
Feliu
a,
Enrique
Herrero
a,
Beatriz Roldan
Cuenya
b and
Rosa M.
Arán-Ais
*a
a,
Fabian
Scholten
b,
Juan M.
Feliu
a,
Enrique
Herrero
a,
Beatriz Roldan
Cuenya
b and
Rosa M.
Arán-Ais
*a
aInstituto de Electroquímica, Universidad de Alicante, Apdo. 99, E- 03080, Alicante, Spain. E-mail: rosa.aran@ua.es
bDepartment of Interface Science, Fritz-Haber Institute of the Max Planck Society, 14195 Berlin, Germany
First published on 14th May 2024
Abstract
The application of mixed catalysts in chemical to electrical energy transformation reactions can be a strategy for enhancing the catalysis of these reactions. Platinum and palladium exhibit significant catalytic activity in reactions of different natures, influenced not only by composition but also by structure. The present work presents a fundamental study of the characterization and electrochemical behavior of platinum-palladium bulk alloy single-crystal electrodes (Pt100−xPdx), prepared via a modified Clavilier protocol. Electrodes with varying mass ratios of Pt and Pd, compositions ranging from 0 to 15% Pd, and crystallographic orientations of (111) and (100) were manufactured. X-ray photoelectron spectroscopy (XPS) and low-energy electron diffraction (LEED) reveal a well-correlated bulk and surface composition with ordered crystalline structures. Cyclic voltammetry in HClO4 and H2SO4 electrolytes indicates stronger anion interactions compared to pure platinum. Charge displacement experiments by CO adsorption estimate the effect of surface orientation and Pd content on the potential zero total charge (pztc). For Pt100−xPdx(111) surfaces, the pztc is very close to that of Pt(111), while surfaces with (100) orientation show decreasing pztc values with higher Pd content, favoring OH adsorption at lower potentials. Finally, the CO oxidation reaction on these Pt–Pd bulk alloy electrodes is studied by cyclic voltammetry, providing a profoundly comparative discussion of this reaction with Pt(111), Pt(100), and Pt(111) modified with Pd sub-monolayers.
1. Introduction
The search for new electrode materials with better electrocatalytic activity and selectivity has led to the study of alloys and multicomponent systems, since in most cases, the activity of pure metals is not sufficient for practical applications. The electrochemical properties of a material are not only influenced by its composition but also by its surface structure. In this context, the use of catalysts with a well-defined surface, such as single-crystal electrodes, allows for the exploration of electrocatalytic reactions at the atomic level. Since the surface structure is well-defined, they facilitate the untangling of complex reaction mechanisms and set the basis for the development of advanced electrocatalysts.Metallic catalysts of mixed composition may present better catalytic activity. The bifunctional,1 third body,2 and electronic effects3 are the main phenomena that result in the synergistic interaction of different metals that make up an improved catalyst. These effects are widely explored in catalysts applied in transforming chemical energy into electrical energy in direct liquid fuel cells (DLFCs), powered by the oxidation of organic molecules at the anode and the reduction of oxygen at the cathode. Thermodynamically, organic molecules present attractive conditions for their use in DLFCs.4 However, the oxidation of these molecules occurs through more than one reaction pathway,5,6 leading to the formation of poisoning intermediates, and partially oxidized products. All these problems reduce the energetic efficiency of the DLFCs. CO and organic acids are one of the main undesirable by-products when it comes to the oxidation of organic molecules into platinum. Due to the strong interaction of these molecules with the surface, high overpotential values are necessary to oxidize them to CO2 and thus restore the activity of the catalyst and maintain a continuous reaction.
Platinum and palladium possess similar physicochemical properties and display good catalytic activity for the reactions used in a DLFC. Although platinum is considered the electrocatalyst per excellence, in some cases, palladium performs better than platinum in the oxidation of organic molecules. A typical example of this is the formic acid oxidation reaction.7,8 Contrary to what is observed for platinum, this reaction is not limited by surface poisoning by CO on palladium. Voltammetry studies combined with density functional theory (DFT) show that the oxidation of formic acid in a Pd monolayer deposited on Pt(111) occurs due to the formation of a large formate coating, hindering the formation of CO on the surface.9 However, one of the main problems with pure palladium is the absorption of hydrogen at low potentials, which may alter the properties of the electrode.10,11
The use of palladium monolayers on platinum basal planes as templates has been a way of studying the electrochemical behavior of Pd.12–18 Under these conditions, hydrogen absorption is not observed, allowing studies to be carried out in a lower potential region than those used in pure palladium electrodes. However, in these studies, the growth of 3D islands easily occurs without the complete formation of monolayers, compromising the good structural definition of the surface. An alternative approach to studying the Pt–Pd bimetallic system consists of preparing alloys with well-ordered surface structures. In this sense, Pt100−xPdx alloy single crystals can be used for fundamental electrochemical studies without the problem of hydrogen absorption and preserving their well-defined surface. In the case of Pt–Pd alloy single crystal surfaces, the amount of published work is small and many open questions about their interfacial properties have not been elucidated. The first reports of the use of a Pt100−xPdx alloy single-crystal surface in electrochemical studies were presented by Attard et al.19–21 Through the combination of different spectroscopy and voltammetry techniques, it was shown that regardless of the mixed composition of the single-crystal electrodes, the alloyed surfaces are well-defined, and electrochemical studies can be conducted at the atomic level on these surfaces.
The specific adsorption of species plays a crucial role in surface electrochemistry, exerting a significant influence on reaction kinetics by either promoting or inhibiting the process. Charge displacement experiments allow the determination of the potential of zero total charge (pztc) of platinum group metals and enable the discrimination of the different adsorption processes taking place at the surface of the electrode22 according to the sign of the associated total charge.
In the present work, a fundamental study of the characterization and electrochemical behavior of platinum–palladium bulk alloy single crystals is presented. Previous works in electrocatalysis have shown that the electrocatalytic behavior of the material depends not only on the surface structure and composition but also on the interfacial structure. Thus, surface charge,23 adsorbed species,24,25 pH, and water structure modify26 the activity of the electrode, and in some cases catalyze or deactivate reactions. A full characterization of the interfacial properties of these electrodes is required when their electrocatalytic behavior is to be understood. For this reason, Pt100−xPdx alloy single crystal electrodes with (111) and (100) orientation of bulk compositions ranging from 0 to 15% Pd were prepared and characterized by a combination of low-energy electron diffraction (LEED), X-ray photoelectron spectroscopy (XPS), and electrochemical techniques. The interaction of ion perchlorate and sulfate with Pt100−xPdx surface, as well as the adsorption of hydrogen and (hydro)oxygenated species, were studied under a voltammetric regime using reaction media based on perchloric or sulfuric acid. Finally, the potential of zero total charge and the electrocatalytic oxidation of CO were investigated by considering the effect of the surface orientation and composition.
2. Experimental section
2.1. Electrode preparation
Platinum single-crystal electrodes were manufactured following Clavilier's protocol27 and a brief modification of this procedure was introduced to prepare the Pt–Pd alloy single-crystal electrodes.19,21,28 Briefly, the end of an ultrapure (Mateck, 99.99%) Pt wire (0.5 mm diameter) is melted in a propane-oxygen flame to produce a single crystal bead of approximately 2–3 mm in diameter. By controlling the length of the melted Pt wire it is possible to control the amount of Pt in the single crystal bead. Then, a known amount of Pd wire (Good Fellow, 99.99%) is added to the melted Pt bead, and both elements are allowed to blend for several minutes in the molten solution. After careful and slow cooling of the bulk alloy, a single crystal bead with (100) and (111) microfacets is formed. The single crystal is then placed on a goniometer head at one end of an optical bench, where it is oriented, cut, and polished following the procedure described elsewhere.19 As the final step, the crystal is annealed in a Bunsen flame for 1 h to eliminate the surface damage from the polishing procedure. Pt–Pd alloy single crystal electrodes with (111) and (100) orientations of nominal bulk compositions ranging from 0 to 15% Pd were prepared following this methodology. The Pt composition and the geometrical area of the electrodes measured using XPS spectra and an optical microscope, respectively, are given in Table 1.| Pt(111) and Pt100−xPdx(111) surfaces | ||||
|---|---|---|---|---|
| Geometric area/cm2 | 0.0371 | 0.0468 | 0.0374 | 0.0505 |
| % Pt (XPS) | 100 | 97 | 96 | 88 |
| Pt(100) and Pt100−xPdx(100) surfaces | ||||
|---|---|---|---|---|
| Geometric area/cm2 | 0.0458 | 0.0469 | 0.0412 | 0.0472 |
| % Pt (XPS) | 100 | 99 | 93 | 85 |
Before each electrochemical experiment, the single-crystal electrodes were flame annealed in a Bunsen flame (propane–air) and cooled in a reductive atmosphere of an Ar and H2 mixture (N-50, Air Liquide) with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. The electrode surface is then protected by a drop of H2/Ar-saturated high-purity water (Millipore, Milli-Q system, 18.2 MΩ cm) and transferred to the electrochemical cell.
:1 ratio. The electrode surface is then protected by a drop of H2/Ar-saturated high-purity water (Millipore, Milli-Q system, 18.2 MΩ cm) and transferred to the electrochemical cell.
2.2. Ultra-high vacuum surface characterization
X-ray photoelectron spectroscopy (XPS) measurements were carried out using a sample-to-analyzer angle of 90°. The system employed was a commercial hemispherical analyzer (SPECS GmbH, Phoibos 150 equipped with a MCD-9 Detector) using a pass Energy of 15 eV and a monochromatic XRM X-ray source (SPECS GmbH) utilizing an Al anode (E = 1486.7 eV). A source power (P) of 300 W was used.Low energy electron diffraction (LEED) measurements were conducted using a commercial system (SPECS GmbH, ErLEED 150). After the optimization of the measurement conditions, electron energies of 132.5 and 80–95 eV were used for Pt(111) and Pt(100), respectively. Sputtering was carried out using Ar-ions (E = 2 keV) for 7 minutes at a chamber pressure of 3 × 10−5 mbar. Consecutive sample annealing was carried out using an electron source emission current (I) of 18 mA which equates to a sample temperature of roughly 850 °C in the employed system. Each sample was sputter-annealed three times.
Fig. S1† shows XPS C1s spectra of the Pt(100) electrode with different pretreatments. A significant amount of carbon species is present at the surface when various distinct pre-treatments are applied to the electrode, which highlights the high affinity of Pt for C species under ambient conditions. Upon flame-annealing in air and quick transfer to the UHV chamber, this amount is significantly reduced, but adventitious carbon is still found, which is expected even for small exposures to the ambient atmosphere. Only when the single crystals are cleaned in UHV using cycles of Ar ion sputtering and consecutive annealing, a fully carbon-free surface is obtained. LEED patterns were only attainable on the UHV-cleaned surfaces, which accounted for the carbon contaminations with the other preparation methods in UHV.
2.3. Electrochemical characterization
The electrochemical experiments were conducted at room temperature in a custom-made glass cell with a three-electrode configuration. The counter electrode (CE) consists of a platinum wire whose area is much larger than the working electrode (WE). The electrode potential was measured against a reversible hydrogen electrode (RHE) connected to the cell through a Lugging capillary. All potentials shown in this study are given on the RHE scale. The single crystal electrodes were characterized by cyclic voltammetry in meniscus configuration in argon-purged 0.1 M HClO4 (Sigma-Aldrich ACS reagent – 70–77%) and 0.1 M H2SO4 (Merck KGeA Suprapur – 96%) electrolytes prepared with ultrapure water (ElgaPurelab Ultra Analytic system, 18.2 MΩ cm). The voltammetric profiles of Pt(111) and Pt(100) were recorded at the beginning of each working session to ensure the cleanliness of the electrochemical system.21,29,30CO oxidation experiments were carried out to determine the properties of the electrodes for this process. CO adsorption on the electrode surface was performed under a potentiostatic regime, where the electrode was polarized at 0.1 V, and a CO flow was bubbled through the electrolyte until complete blockage of the surface (monitored by cycling the electrode between 0.05 and 0.3 V). Throughout this procedure, the electrode current was monitored to ensure that the electrode surface was fully saturated with adsorbed CO, according to the procedure described in ref. 31. Before the oxidation of the adsorbed CO, the CO present in the supporting electrolyte and the cell atmosphere were removed by argon flow for 15 minutes, maintaining the electrode potential in the potentiostatic regime. Then, two cyclic voltammograms at 0.02 V s−1 were recorded to register the electrochemical oxidation of adsorbed CO in the first one and the recovery of the initial profile in the second one.
Surface charge displacement experiments were performed by adsorbing CO on the surface in a potentiostatic regime, following the protocol previously used by our group.32 Before CO adsorption on the surface, a voltammetric profile of the electrode surface was recorded to ensure the cleanliness of the supporting electrolyte and the condition of the electrode surface. The electrode was polarized in potential where the oxidation of adsorbed CO does not occur (this information was obtained using the results of oxidation of adsorbed CO, previously described), being: 0.1, 0.2, or 0.3 V, and started measuring the current over time. A stream of CO was injected into the cell atmosphere until the system current remained at zero for a few seconds. With the integration of the current/time curve, the nature of the species adsorbed on the electrode was estimated at each studied potential, as well as its charge. All CO adsorption, as well as adsorbed CO oxidation experiments, were performed at least three times, and the results are presented in terms of their mean values with their associated error.
3. Results and discussion
3.1. UHV characterization of the Pt100−xPdx single-crystal electrodes
To validate the preparation of the Pt100−xPdx alloy single crystals, the surface composition and structure of the electrodes were assessed using XPS and LEED, respectively. Although complete miscibility of Pt and Pd has been reported elsewhere33 and no surface segregation was calculated for this alloy,34 earlier works showed that Pd can segregate and that this separation is structure-dependent.30 However, our XPS data reveal a good correlation between bulk and surface compositions. To evaluate the surface composition, Pt 4f and Pd 3d XPS spectra were acquired on the UHV-prepared single-crystal surfaces, Fig. 1(A). Both metals were found to be in their metallic state (EB,Pt = 70.76 eV and EB,Pd = 335.04 eV),35 irrespective of the amount of Pd in the alloy and the surface orientation. The measured surface composition, determined by the relative integrated peak area ratio of the Pt 4d to Pd 3d core level region, fits the expected relative nominal values and is summarized in Table 1. It should be noted that although XPS is a surface-sensitive technique, the signal acquired typically probes up to 5–10 nm below the surface. Nonetheless, the contribution of the different layers to the spectra diminishes with the distance to the surface. Although enrichment of Pd has been proposed for the topmost layer, the values obtained here are clearly an averaged value of all the sampled layers and therefore, do not only reflect the very last surface layer.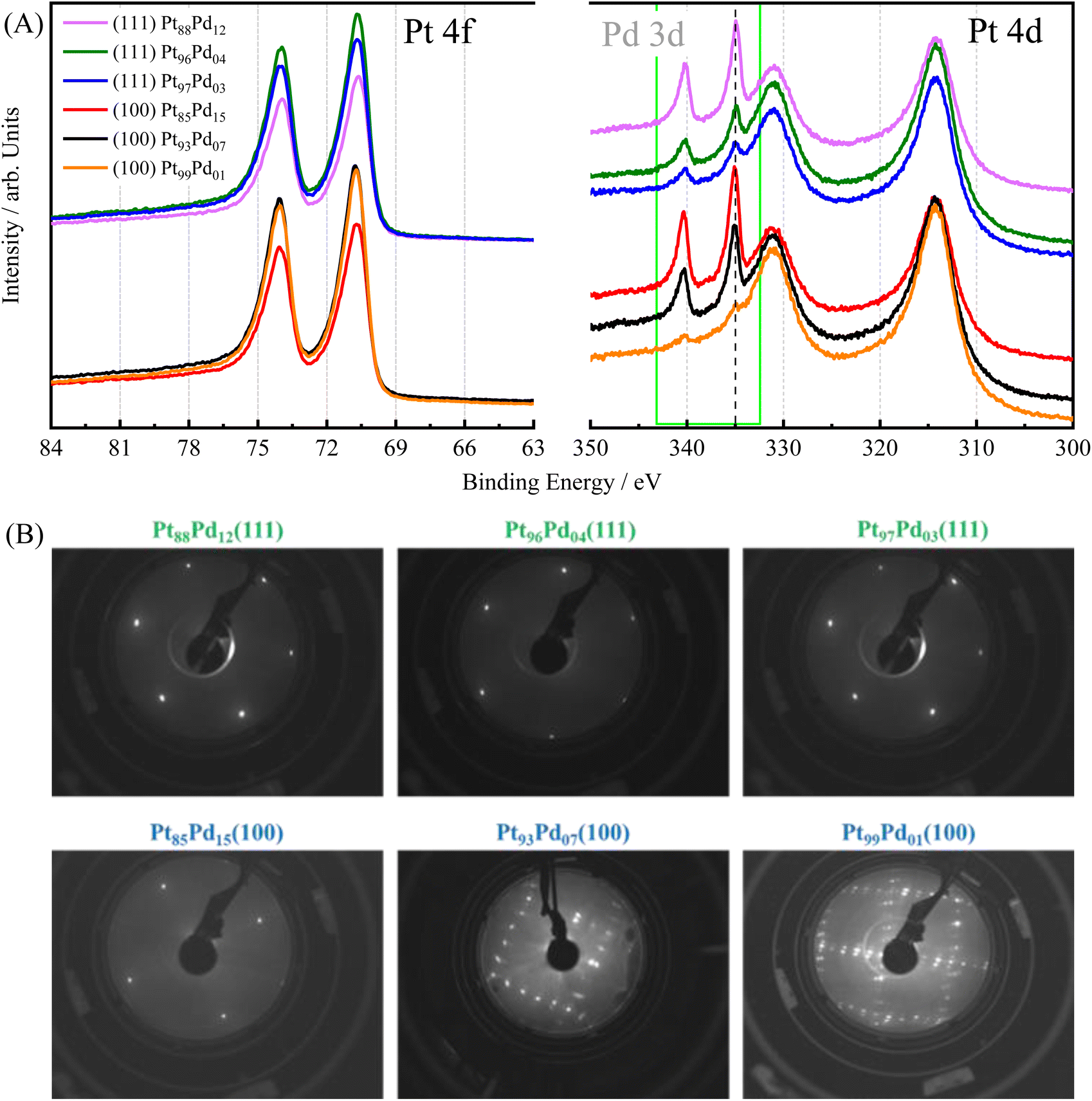 | ||
| Fig. 1 (A) Pt 4f and Pd 3d XPS spectra for Pt100−xPdx(111), and Pt100−xPdx(100) single crystal surfaces. (B) LEED patterns acquired on Pt100−xPdx(111), and Pt100−xPdx(100) single crystalline surfaces after UHV sputter-annealing treatment. The electron energies were 132.5 and 80–95 eV (111) and (100) structures, respectively. | ||
Fig. 1(B) shows the corresponding LEED patterns of the Pt100−xPdx(111), and Pt100−xPdx(100) surfaces. The low background and sharpness of the diffraction spots point out the well-ordered surface structure of all samples. All of the (111) crystals yield a (1 × 1) structure with no indication of any other ordered overlayer phase present, independently of the amount of Pd in the electrode.36 For the (100) surfaces containing a low amount of Pd, a Pt-hex-R0.7° structure is observed, which is consistent with previous studies on pure Pt(100) and Pt(100) with small amounts of foreign adatoms.30,37,38 When increasing the Pd content up to 15%, as obtained from the integrated XPS areas (Table 1), the LEED shows a (1 × 1) structure pattern. It is known that Pd(100) does not reconstruct,39 which leads to the suppression of the hexagonal (hex) reconstruction once the Pd content is sufficiently high.30
3.2. Voltammetric characterization of the Pt100−xPdx single crystal surfaces
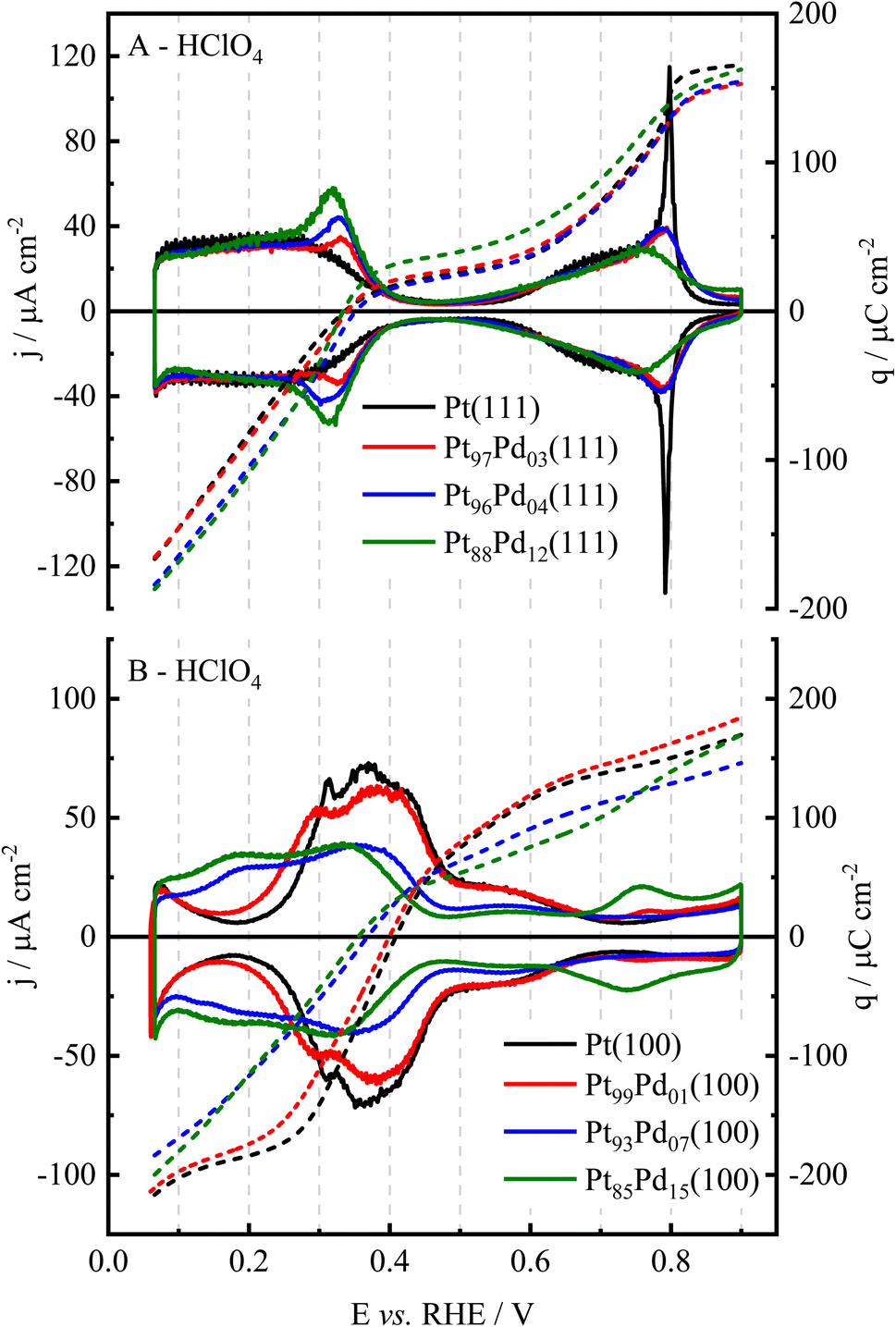
Cyclic voltammograms of a Pt-based surface on a given electrolyte provide important correlations between surface structure and composition since the voltammetric profiles serve as surface fingerprints. Also, information on the adsorption processes that take place on the electrode surface can be achieved. This information is very relevant when electrocatalytic processes are studied since adsorbed molecules can activate or hinder the electrochemical reactions.24,40,41Fig. 2(A) shows the voltammetric profiles of Pt(111), and Pt100−xPdx(111) surfaces, in both 0.1 mol L−1 H2SO4 and 0.1 mol L−1 HClO4. The potential range was set in both electrolytes between 0.065 and 0.900 V for all surfaces except for those (100)-oriented and measured in sulfuric acid medium. For these electrodes the profile was recorded up to 0.80 V, avoiding the formation of the soluble palladium sulfate salt.42–45 Unlike pure Pd electrodes, Pt100−xPdx surfaces do not absorb hydrogen, which allows reaching potentials lower than 0.2 V.30 | ||
| Fig. 2 Voltammetric profiles of (A1 and A2) Pt(111) and Pt100−xPdx(111) surfaces in (A1) 0.1 mol L−1 HClO4 and (A2) 0.1 mol L−1 H2SO4, registered at 0.05 V s−1. Voltametric profiles of (B1 and B2) Pt(100) and Pt100−xPdx(100) surfaces in (B1) 0.1 mol L−1 HClO4 and (B2) 0.1 mol L−1 H2SO4, registered at 0.05 V s−1. | ||
Pt100−xPdx(111) surfaces in perchloric acid share common features with Pt(111) electrode (Fig. 2(A1)). Thus, there is a clear distinction between hydrogen and anion adsorption regions. However, the addition of Pd brings about some changes to the voltammetric profile. Currents in the hydrogen region nicely overlap below 0.2 V for all the electrodes, although a slightly smaller current is observed for the surfaces with a higher content of Pd. This behavior is also reported for studies carried out in different acid, neutral, and alkaline electrolytes46–48 because this potential region is a consequence of the predominant reversible reaction of hydrogen adsorption. When Pd is added, a pair of reversible peaks appears in the potential region between 0.2 and 0.4 V, and their peak potential moves from 0.335 V (red curve) to 0.320 V (green curve) with increasing Pd loading. Also, the charge under the peak increases. On the other hand, some modifications also appear in the OH adsorption region. First, the sharp spike observed at 0.80 V for the Pt(111) disappears as the Pd concentration increases, and the onset for OH adsorption seems to shift to lower potential values. However, the general shape of the process is mainly preserved.
These voltammograms can also be compared to those obtained for Pt(111) electrodes modified with Pd sub-monolayers (Pdsub–Pt(111)).22 Although the qualitative behavior is similar, that is, additional peaks appear in the hydrogen adsorption region, and the OH adsorption region is displaced to lower potential values, quantitative differences can be observed. First, for the Pd submonolayers, the new peak appears at 0.28 V for very low Pd coverages and splits into two as the coverage increases. On the other hand, the shape in the OH region is also significantly different. For very low coverages, the sharp peak for Pt(111) disappears and a new peak appears at 0.71 V. All these facts clearly indicate that the energetics and probably the distribution of the Pd atoms on the surface are significantly different on both surfaces: Pt100−xPdx(111) and Pdsub–Pt(111).
Changes in the cyclic voltammograms of Pt100−xPdx(111) are more obvious in sulfuric acid solutions (Fig. 2(A2)). The voltammetric profiles in perchloric and sulfuric acid overlap for the same Pd concentration below 0.3 V, which necessarily implies that hydrogen adsorption/desorption processes are responsible for the voltammetric signals in this region (Fig. S2†). However, the signals appearing between 0.25 and 0.4 V significantly increase their intensity as compared to perchloric acid solutions. This fact means that the sulfate adsorption/desorption process is responsible for the current increase in this region. In parallel, the signals at E > 0.4 V diminish. Thus, sulfate adsorption, which on the Pt(111) takes place between 0.35 and 0.60 V, is displaced at significantly lower potentials as the Pd content in the electrode increases. One important feature that disappears is the sharp peak associated with the order/disorder transition for the sulfate adlayer on Pt(111) at 0.5 V. Scanning tunneling microscopy (STM) studies have revealed that sulfate forms an ordered adlayer with a (√7 × √3)R19.1° on several fcc (111) facets.49–54 The phase transition from the disordered to the ordered structure is marked by the presence of sharp spikes, as those observed at the completion of the adsorption process on Pt(111),49 Au(111),50,51 and Pd(111).52 The initial adsorption process leads to the formation of very small, ordered domains, which grow progressively as the adsorption proceeds. This results in the formation of a large number of rotational and translational domain boundaries with a sulfate coverage lower than that expected for a perfectly ordered adlayer. At a certain potential, the different ordered domains coalesce to form large, ordered domains. In this process, the coverage increases abruptly, giving rise to the observed spikes in the voltammogram. For this process to take place, the adsorbed sulfate molecules should be able to move freely across the surface, so that adsorbed species at a domain boundary move from one domain to the neighboring one, so that an ordered domain grows at the expense of the second one. Furthermore, the mobility of sulfate requires that the adsorption energy of all the different surface sites should be the same. Because of this, the spike is very sensitive to the presence of defects or contaminants on the surface since they alter adsorption energies and restrict the movements of species on the surface. In fact, for Pt(111) and Au(111) their sharpness is indicative of the surface order (quality of the electrode and surface cleanliness). The disappearance of the spike is clearly a consequence of the different adsorption energies of sulfate on the different sites. The comparison between Pt(111), Pt100−xPdx(111), and Pd(111) electrodes indicates that sulfate is more strongly adsorbed on Pd sites. The adsorption of sulfate will involve several atoms, and owing to the low concentration of Pd in the prepared alloys, sulfate will interact mainly with two types of ensembles: pure Pt ensembles and ensembles containing one Pd atom. In this latter type of ensembles, sulfate adsorption will be stronger. Then, the movement of sulfate across the surface is hindered because of this energy difference between the adsorption sites, which prevents the disorder/order transition.
When these voltammograms are compared to those of Pdsub–Pt(111), significant differences are also observed. On Pdsub–Pt(111), there is a sharp spike at ca. 0.24 V, whose full width at half maximum (fwhm) is smaller than 3 mV and the spike corresponding to the phase transition of sulfate on Pt(111) is still visible (although significantly smaller) for coverages close to 1.55 These facts indicate that the Pd adlayers on the Pt(111) have an island growth mode. Thus, the sharp spike is due to the competitive adsorption of hydrogen and sulfate on the Pd island, whereas the size of the free-Pd domains is large enough to give rise to a phase transition. Clearly, the situation for Pt100−xPdx(111) is different. The disappearance of the peak related to the phase transition even for low Pd concentrations indicates that the distribution of Pd on the surface is homogeneous. Moreover, the wide peaks related to the competitive adsorption of hydrogen and sulfate at E > 0.3 V suggest the presence of different Pd–Pt environments.
Pt(100) is a more complex surface than Pt(111) in terms of electrochemical characterization since different voltammetric profiles can be obtained for Pt(100) depending on the cooling conditions after the flame-annealing treatment.56 In this work, reductive cooling conditions have been used for all surfaces to avoid oxygen adsorption and obtain two-dimensionally ordered terraces. The reconstruction of the Pt(100) surface, which is eliminated upon the immersion of the electrode in the electrolyte, gives rise to the formation of mesas on the electrode surface with the excess of atoms in the hex reconstruction with respect to the (1 × 1) structure. The absence of the reconstruction on the Pt100−xPdx(100) electrodes with moderate Pd content (Fig. 1(B)) suggests that the surface order of these electrodes should be higher than that of the pure Pt(100) electrode. It should be noted that, in the case of the reconstructed surfaces, the reconstruction is lifted upon immersion in water, and the resulting surface structure is stable between 0.0 and 0.9 V.
Fig. 2(B) displays the voltammetric profiles of Pt(100) and Pt100−xPdx(100) surfaces in perchloric and sulfuric acid solutions, illustrating a different and characteristic profile depending on the Pd content of the alloy and the electrolyte used. On Pt(100) electrodes, the signals below 0.4 V are assigned mainly to hydrogen adsorption, whereas the wave between 0.45 and 0.6 V corresponds to adsorbed OH. As the Pd content increases, there is a shift of the hydrogen adsorption to lower potentials, as can be seen in Fig. 2(B). On the other hand, the signals in the OH adsorption region also diminish. In this case, two different scenarios can be considered: either OH adsorption is disfavored on the Pt100−xPdx(100) alloy electrodes or OH adsorption takes place at lower potentials overlapping with the final stages of hydrogen adsorption and thus, the signals in the voltammogram between 0.3 and 0.4 V can also contain contributions from OH adsorption. This issue will be revisited when CO displacement experiments are analyzed. Again, this profile is different from that obtained for Pdsub–Pt(100) in this media.13
The presence of sulfuric acid in the electrolyte brings about significant changes in the voltammogram due to the specific adsorption of sulfate. Below 0.3 V, the voltammograms for Pt(100) in sulfuric and perchloric acid solutions overlap (Fig. S2†), clearly indicating that the signals up to 0.3 V are related to the adsorption/desorption of hydrogen. However, above this value, a sharp peak is observed in sulfuric acid. This peak is due to the competitive adsorption of hydrogen and sulfate on the surface. A similar peak is also observed for Pd(100) surfaces.45 It has been shown that the replacement of one species by the other gives rise to sharp peaks.57 As the Pd content increases, this sharp peak diminishes and finally disappears. From the comparison between the voltammograms in perchloric and sulfuric acid media (Fig. S2†), it can be seen that although the voltammograms overlap at the low potential limit, there is a significant increase in the currents between 0.2 and 0.4 V in the latter. This increase should be ascribed to the adsorption of sulfate. The absence of the sharp peak should be related to the heterogeneity of the surface sites and the different site requirements for hydrogen and sulfate adsorption. Whereas hydrogen adsorption occupies only one site, adsorbed sulfate is coordinated to several sites. Thus, changes in the surface energy induced by the presence of Pd affect more importantly hydrogen adsorption, because it depends only on the specific site composition. For sulfate adsorption, due to the small Pd content, this energy change is less important due to the involvement of several sites.
3.3. CO displacement experiments on the Pt100−xPdx single crystal surfaces
To obtain more information on the nature of adsorbed species on the single-crystal Pt100−xPdx surfaces, CO displacement experiments were carried out and the results were compared with those obtained for pure Pt electrodes with the same surface orientation. Due to the strong interaction of the sulfate ion with Pt and Pt100−xPdx surfaces, the experiments were performed in HClO4 solution by dosing gaseous CO in the cell atmosphere at a fixed potential. Fig. S3† shows the current transients for the CO displacement experiments on Pt100−xPdx(111) surfaces, at 0.1, 0.2, and 0.3 V. The experimentally measured displacement charges correspond to oxidation processes associated with the oxidative displacement of adsorbed hydrogen, according to:| Pt − H + CO → Pt − CO + H+ + e− |
Charge density–potential curves can be represented by combining the displaced charge and the charges calculated from the voltammetric profile in Fig. 2(A), using the equation:58,59
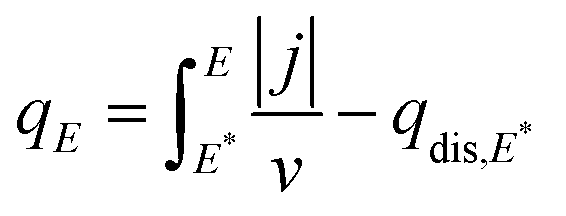
Fig. 3 shows the voltammetric profile of the Pt88Pd12(111) electrode together with the charge density–potential curves. The displaced charge density measured at 0.3 V has been used as the integration constant to obtain this curve. The experimental charges displaced at 0.1 and 0.2 V nicely overlap with the integrated curve (see Table S1†), validating the use of the CO displacement experiments to measure the total charge on the electrode.60 As expected, increasing the electrode potential results in a rise in the charge density.
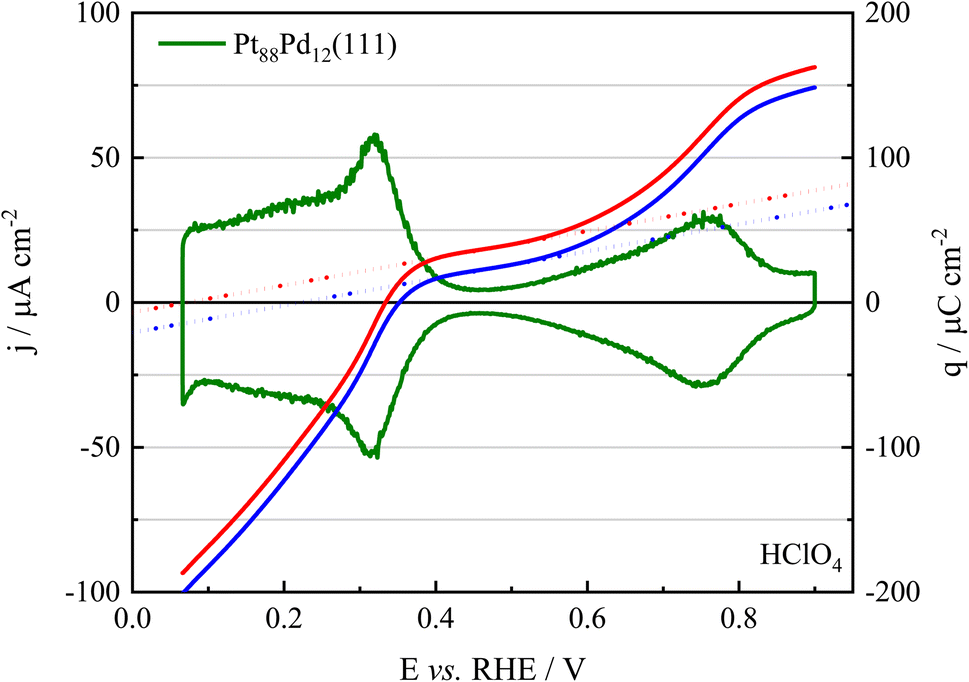 | ||
| Fig. 3 Cyclic voltammetric profile (green line) and total charge curve (red line) for the Pt88Pd12(111) surface in 0.1 mol L−1 HClO4 as a function of the potential recorded at a scan rate of 50 mV s−1. The blue line is a corrected charge density taking into account the remaining charge on the CO-covered surface (see ref. 47). | ||
The charge curves for Pt(111), and all the Pt100−xPdx(111) surfaces are shown in Fig. 4(A). Several conclusions can be drawn from these measurements. First, the charge density at 0.1 V increases in absolute value with the Pd content. The same trend is observed at 0.2 V, showing that, for a given potential, the hydrogen coverage increases with the Pd content. For the Pt(111) electrode, the charge measurements indicate that the hydrogen coverage at 0.06 V, the onset of hydrogen evolution, is ca. 2/3. In this case, the increase of the charge at 0.06 V for Pt88Pd12(111) allows the estimation of a coverage value of ca. 0.75. It should be noted that, due to the small difference in the lattice parameters of Pt and Pd (ca. 1%), the expected difference in the charge of a process exchanging 1 e per surface atom on Pt(111) and Pd(111) is ca. 2%, which is significantly smaller than the value measured for the charge increase at the onset of hydrogen evolution. For this reason, the increase in the charge value should be assigned to a change in the hydrogen coverage related to the energetics of the hydrogen adsorption process. On the other hand, the comparison at 0.06 V with the Pd(111) electrode is not possible due to the hydrogen absorption on Pd which takes place at E < 0.2 V. However, the measured charge density for the Pd(111) electrode in perchloric acid solution at 0.225 V is ca. – 125 μC cm−2,45 which is higher (in absolute value) than that measured for the Pt100−xPdx(111) surface. Thus, as the Pd content increases, the hydrogen coverages at a given potential increase with the Pd content for (111) surfaces.
 | ||
| Fig. 4 Cyclic voltammetric profile (full lines) and the total charge curve (dotted lines) for (A) Pt100−xPdx(111), and (B) Pt100−xPdx(100) surfaces in 0.1 mol L−1 HClO4 as a function of the potential recorded at a scan rate of 50 mV s−1. | ||
The charge curves allow for obtaining the pztc of the different electrodes, which is the potential at which the total charge displayed in Fig. 4(A) is zero. A more accurate value is obtained when the charge curves are corrected by the residual charge of the CO-covered electrode, as shown in ref. 47 and 61. For that, it will be assumed that the correction is the same one as that used for the Pt(111) electrode. The charge curve after correction is shown in Fig. 3. The values of the pztc for the different electrodes are summarized in Table 2 and the comparison of the corrected charge curves for all the electrodes is shown in Fig. S3.† The values for the Pt(111) electrode are the same as those reported in the literature for this electrolyte solution.47 Regarding the evolution of the potential of zero total charge for the different (111) surfaces (Table 2), for very low Pd content the values are constant within the experimental error of the measurements. On the other hand, for the Pt88Pd12(111) surface, a significant diminution is observed. In fact, for the Pd(111) electrode in the same solution, the uncorrected pztc is ca. 0.28 V.45 Moreover, a significant positive charge density, ca. 50 μC cm−2, is measured in the apparent double layer region of the Pd(111) electrode (E > 0.35 V). This fact indicates that OH is already adsorbed on the Pd(111) electrode at 0.35 V and that the signals corresponding to the final stages of the hydrogen desorption overlap with those of the OH adsorption. A similar situation is observed for the Pt88Pd12(111), for which the total charge at 0.4 V is significantly higher than that observed for the other electrodes. Owing to the capacity of Pd(111) for adsorbing OH at low potentials, it can be proposed that the peak centered at ca. 0.32–0.36 V for the alloy electrodes is due to the competitive adsorption/desorption of H and OH on the Pd atoms.
| Surface | Pztc/V (uncorrected) | Pztc/V (corrected) | Pzfc/V (uncorrected) | Pzfc/V (corrected) |
|---|---|---|---|---|
| Pt(111) | 0.3365 ± 0.0005 | 0.385 ± 0.0005 | 0.160 ± 0.010 | 0.341 ± 0.010 |
| Pt97Pd03(111) | 0.346 ± 0.001 | 0.385 ± 0.001 | 0.164 ± 0.010 | 0.350 ± 0.010 |
| Pt96Pd04(111) | 0.352 ± 0.001 | 0.395 ± 0.001 | 0.204 ± 0.010 | 0.375 ± 0.010 |
| Pt88Pd12(111) | 0.334 ± 0.003 | 0.352 ± 0.003 | 0.073 ± 0.010 | 0.222 ± 0.010 |
Other important data that can be obtained from the total charge curves is the OH coverage. As can be seen, for the two alloy electrodes with low Pd content, the charge curves almost overlap with that of the Pt(111) electrode up to the potential at which the sharp peak for the Pt(111) surface is observed (ca. 0.8 V). From that point, the charge for the Pt(111) electrode is higher than that of the two other electrodes, since the sharp peak is absent for the Pd alloy electrodes. The behavior is similar to that observed for the phase transition in the sulfate layer. Thus, it can be proposed that the sharp peak at ca. 0.8 V for the Pt(111) electrode is due to a phase transition in the OH layer. It should be noted that, in those processes, other solution species, such as cations62,63 or other anions,64 are involved, indicating that the interfacial structure is much more complicated than a simple layer of adsorbed anions. For the Pt88Pd12(111) electrode, the final OH coverage is similar to that of the Pt(111) electrode, owing to the significant adsorption of OH on the Pd sites, which takes place on the peak at 0.32 V.
For the Pt(111) electrode, as the process of OH and H adsorption are separated by ca. 0.25 V in a region where there are no specifically adsorbed species on the surface, it was possible to estimate the potential of zero free charge of the electrode (pzfc).47 The pzfc is the potential at which the surface charge of the electrode is zero and can be related to the electronic properties of the material.65 On the other hand, the pztc contains the contribution of the charge exchanged due to the adsorbed species on the surface and that corresponding to the charge at the metal surface. To estimate the pzfc, the behavior of the charge curve in the region where no species are adsorbed on the electrode surface (the so-called double layer region) has to be extrapolated, to the point where a charge zero is obtained. The corrected curve, the extrapolated line, and the pzfc for the Pt88Pd12(111) electrode are shown in Fig. 3, and the values of the pzfc for the different electrodes are given in Table 2. As before, the values obtained for the Pt(111) electrode agree with those of the literature.47 On the other hand, for the electrodes with a low Pd content, the pzfc increases. This increase has been observed for Pt(111) surfaces modified with adatoms, due to the disruption of the water layer on the Pt(111) surface induced by the presence of a foreign species.66,67 On the other hand, the estimated value of the pzfc for the Pt88Pd12(111) electrode is significantly lower. Two possible reasons can be foreseen. First, the work function of Pd(111) is lower than that of Pt(111). Due to the linear relationship between the work function and the pzfc,68 it is expected that an increase in the Pd content results in a diminution of the pzfc. For the very low Pd coverages, the effect of the disruption of the water adlayer dominates over the change in the electronic properties, as has been shown for adatoms on Pt(111) with similar electronic effects to Pd.67 On the other hand, it is also possible that the determination of the pzfc on the Pt88Pd12(111) surface is biased by the presence of some adsorbed OH in the region used for the extrapolation, resulting in lower values.
CO displacement experiments were also carried out for the (100) electrodes. Transients for the different electrodes can be found in Fig. S4† and the charge curves for the different (100) electrodes are shown in Fig. 4(B). Unlike what is observed for the (111) electrodes, the hydrogen coverage at constant potential diminishes as the Pd content increases. The diminution is significant at 0.2 V because, at this potential value the hydrogen coverage has almost reached the expected value of 1 monolayer for the Pt(100) electrode. On the other hand, the differences at 0.06 V, the onset of hydrogen evolution, are less important, and the values of the hydrogen charge are close to 1 monolayer for the studied electrodes.
The curves also allow for calculating the pztc, which can be used to determine whether the signals observed at potentials higher than 0.3 V are related to the OH adsorption process. Again, the values for the Pt(100) electrode agree with those reported previously.31 For these electrodes, only the pztc can be determined since the estimation of the pzfc requires a region in which no adsorbed species are present on the surface. As will be shown, for the Pt100−xPdx(100) surfaces, the hydrogen, and OH adsorption/desorption processes overlap, preventing any reasonable estimation of the pzfc. The values of the pztc obtained from Fig. 4(B) are given in Table 3. As can be seen, the pztc is located between 0.4 and 0.3 V, in a region where important current signals are observed in the voltammograms. Thus, the final part of the signals observed in the voltammograms, are due to the OH adsorption process, and the final stages of hydrogen desorption coincide with the initial stages of OH adsorption. On the other hand, the pztc shifts to lower values as the Pd content increases. This means that OH adsorption is favored at lower potentials with the addition of Pd. However, the OH coverage at 0.5 or 0.6 V for the different electrodes diminishes with the Pd content, as the lower charge density for these electrodes indicates. Thus, in the presence of Pd, the initial adsorption of OH is favored, probably on the Pd sites, likely due to the Pd effect on the surrounding Pt atoms, the completion of the OH adsorption adlayer on Pt is not favored.
| Surface | Pztc/V (uncorrected) |
|---|---|
| Pt(100) | 0.407 ± 0.003 |
| Pt99Pd01(100) | 0.400 ± 0.006 |
| Pt93Pd07(100) | 0.369 ± 0.005 |
| Pt85Pd15(100) | 0.355 ± 0.006 |
3.4. CO oxidation on the electrodes
The electrooxidation of CO on platinum is a well-explored reaction due to its frequent occurrence as a poisoning species in various electrochemical reactions, particularly during the oxidation of organic compounds or fuels. This reaction is highly sensitive to surface structure variations, and in this study, it has been used to further characterize the properties of the single-crystal electrodes. Fig. 5 shows the voltammetric profiles for CO oxidation on Pt(111) and Pt100−xPdx(111) surfaces. Studies on Pd(111) showed that CO oxidation occurs at potentials higher than 0.9 V,45 unlike what is observed here for Pt100−xPdx(111) surfaces, where COad is fully oxidized at potentials lower than 0.83 V. From these curves, two different parameters can be extracted: the CO oxidation charge and the peak potential. For the CO oxidation charge, it should be noted that the oxidation peak contains charge contributions not only from the CO oxidation process but also those related to the changes in the double-layer charge from a CO-covered electrode to a clean surface. Thus, during the oxidation process, the total charge of the electrode changes from that obtained for a CO-covered electrode to that registered on the clean surface. This correction is equal to the uncorrected total charge measured at 0.9 V. Thus, the integrated charge for the CO oxidation peak between the initial potential and 0.9 V should be corrected by the uncorrected total charge at 0.9 V. Peak potential values and CO oxidation charges are given in Table S3.† As can be seen, the charge values for the (111) surfaces are around 330 μC cm−2, which results in a coverage of ca. 0.69. For the Pt(111) electrode, in the absence of CO in the solution, STM results show that the CO adlayer forms an ordered √19 × √19 − 13 CO with a CO coverage of 0.68.69 Thus, the results indicate that the CO coverage is nearly independent of the Pd content, and probably, the same CO adlayer structure is obtained. However, a significant diminution of the peak potential is observed when Pd is introduced in the adlayer. Model calculations for the CO oxidation process in voltammetry on the Pt(111) electrode show that the peak potential is very dependent on the number of initial defects on the CO adlayer.70 Since CO oxidation on Pt takes place according to a Langmuir–Hinshelwood mechanism between adsorbed CO and adsorbed OH, the initial stages require the presence of some defects in the adlayer where OH adsorption can occur. As mentioned before, the presence of Pd creates sites with different adsorption energy and thus, the mobility of the adsorbed species to form ordered structures is hindered. Thus, it is expected that the CO adlayer in the Pd-containing electrodes has more defects than that on a perfect Pt(111) surface. The consequence of the higher number of defects in the CO adlayer is the lower onset and peak potential for the CO oxidation process.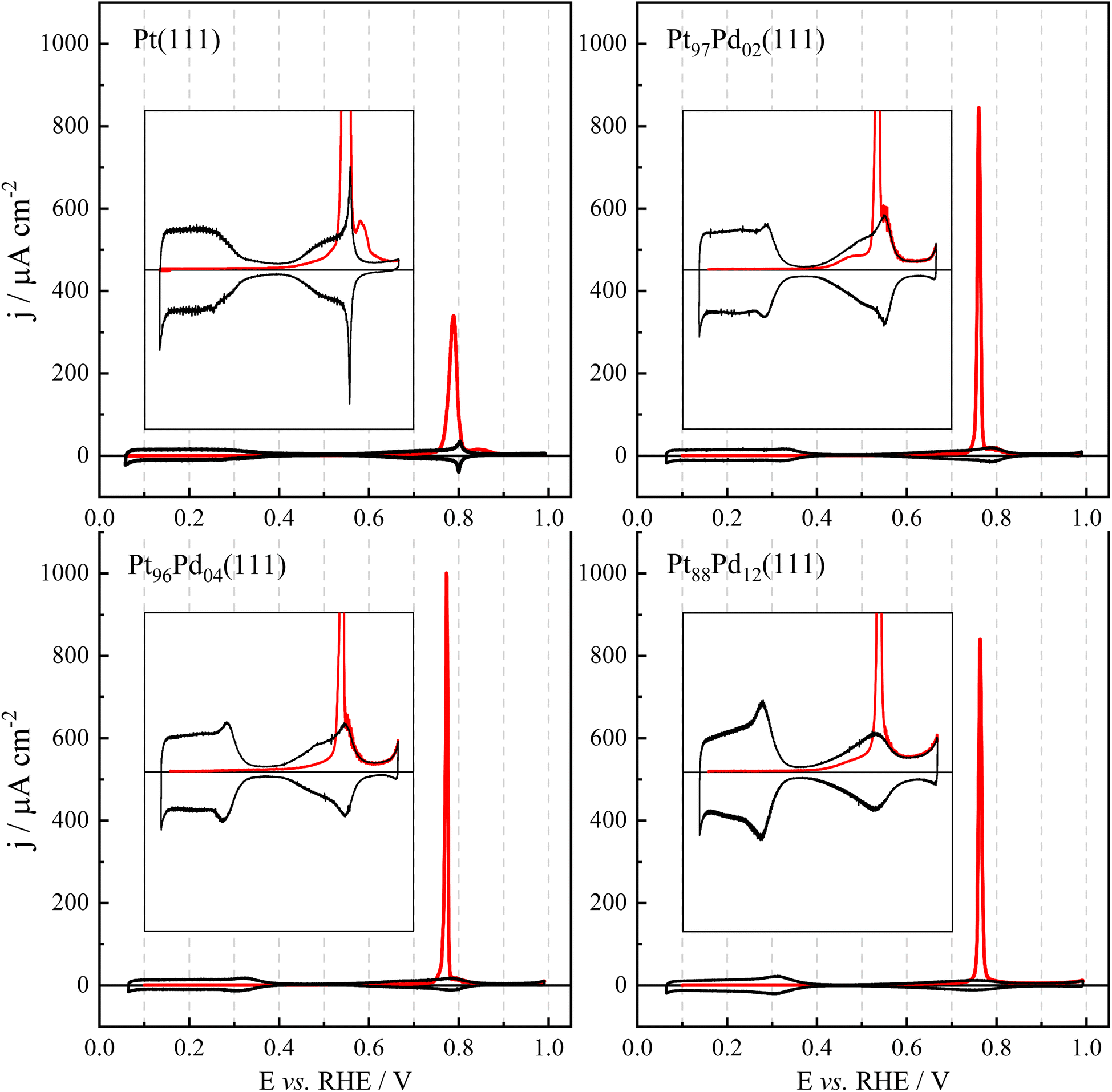 | ||
| Fig. 5 Cyclic voltammetric profile for CO oxidation, at 0.02 V s−1, on Pt100−xPdx(111) surfaces. The insets show a magnification of the voltammetric profiles. | ||
The CO oxidation study was also carried out on Pt(100) and Pt100−xPdx(100) surfaces under the same experimental conditions. The cyclic voltammetric profiles for CO oxidation on (a) Pt(100) or (b-d) Pt100−xPdx(100) surfaces are shown in Fig. 6. For this family of electrodes, CO has been completely oxidized at 0.80 V45 (maximum potential used in the studies of the previous topics for these surfaces), and the charges were integrated up to 0.85 V. Values of the integrated charges and peak potentials are given in Table S4.† The value for the Pt(100) electrode agrees with previous results.71,72 The increase of the Pd content leads to changes in the CO oxidation behavior. First, the CO oxidation charge increases from 330 to ca. 350 μC cm−2. Thus, the CO coverage increases from 0.75 to 0.83. Another observed change in the voltammetric profiles is the presence of a pre-wave, as shown in the insets of Fig. 6. As can be seen, the currents in the pre-wave region diminish with increasing Pd content. These pre-oxidation waves have been assigned to the presence of surface defects.73,74 According to this, the presence of defects on the CO adlayer diminishes with the content of Pd. As mentioned before, the lifting of the reconstruction when the (100) electrodes are immersed in the electrolyte solution leads to the formation of mesas, whose borders act as surface defects. The absence of reconstruction for the Pt85Pd15(100) electrode should give rise to a much-ordered surface structure, with a much lower number of defects, justifying the absence of the pre-wave for this electrode.
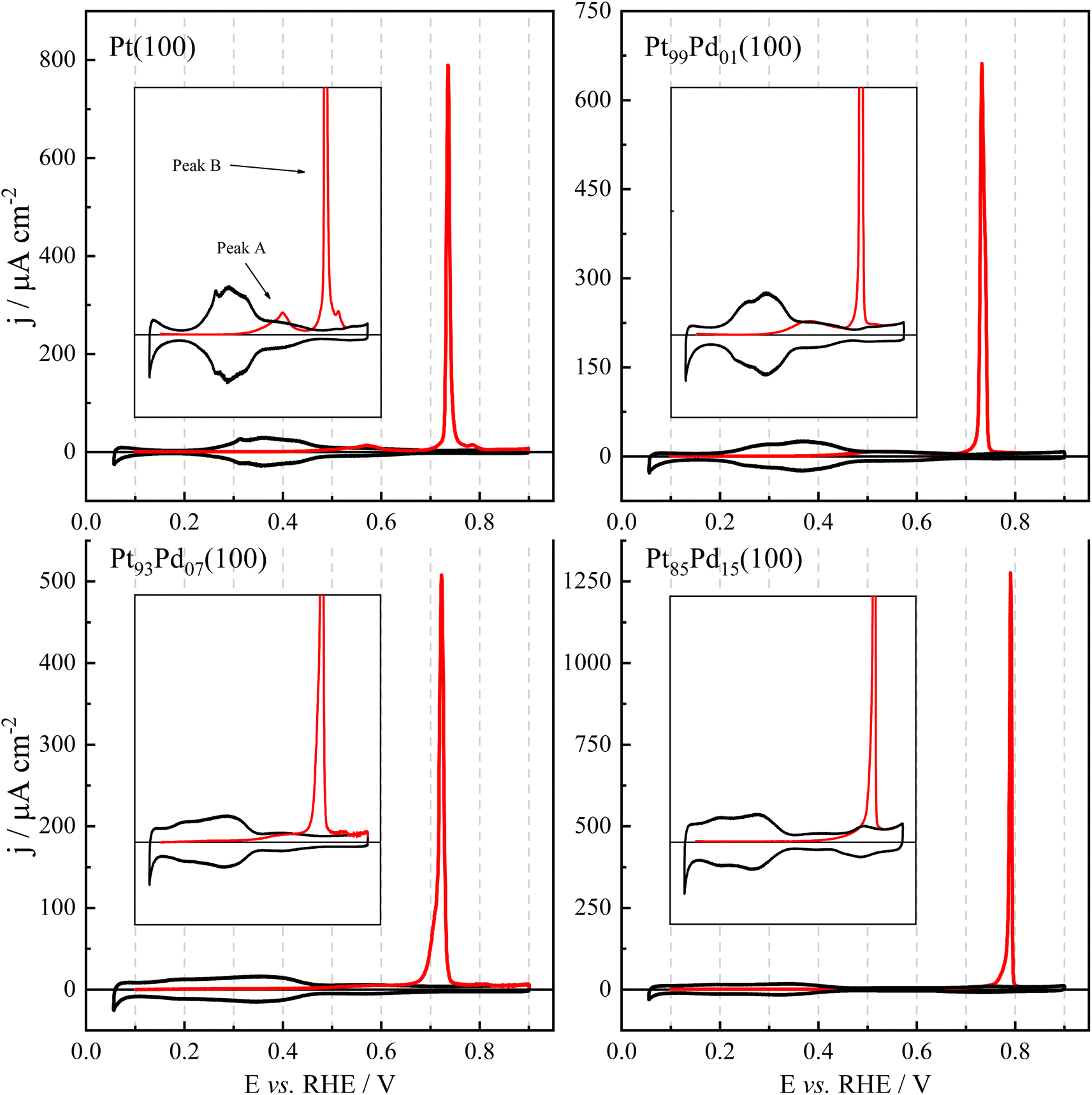 | ||
| Fig. 6 Cyclic voltammetric profile for CO oxidation, at 0.02 V s−1, on Pt100−xPdx(100) surfaces. The insets show a magnification of the voltammetric profiles. | ||
The Pd content also affects the peak potential. As shown in Table S4,† the peak potential for the CO oxidation peak first diminishes to increase for the Pt85Pd15(100). According to the Langmuir–Hinshelwood model for CO oxidation, the peak potential for CO oxidation is governed by three factors:70 CO coverage, number of defects on the adlayer, and energetics of OH adsorption. Normally, an increase in the coverage and a diminution of defects in the adlayer, as observed for the Pt99Pd01(100) and Pt93Pd07(100), are associated with a higher peak potential. However, for these electrodes, a diminution of the peak potential is observed. Then, the reason for the diminution should be a more favorable OH adsorption, as observed from the CO displacement experiments, which show that the onset for OH adsorption is displaced to lower potential values. On the other hand, a significant increase in the peak potential is observed from the Pt85Pd15(100). In this case, the potential increase should be assigned to the significant diminution of the number of defects in the adlayer, which overcompensates the more favorable OH adsorption.
4. Conclusions
The validation of Clavilier's method for manufacturing well-defined Pt–Pd alloy single-crystal surfaces has been achieved through a combination of electrochemical and UHV surface science characterization methods. Insights into the nature of the adsorbed species in different acidic electrolytes, namely, perchloric and sulfuric acids, have been attained via the analysis of the voltammetric profiles and the determination of the pztc. Our results reveal that anion interaction with the alloyed surfaces is stronger in comparison to pure platinum. Although the pztc of Pt100−xPdx(111) surfaces remains almost constant with increasing Pd content, it has been found that hydrogen desorption overlaps with the adsorption of OH, which takes place at already 0.35 V. On Pt100−xPdx(100) surfaces, the OH adsorption is favored at lower potentials, as pointed out by the shift of the pztc to lower values as the Pd content increases. CO oxidation experiments indicate that the CO coverage and adlayer structure on the electrodes with (111) orientation are similar to those on Pt(111). However, a lower onset for the CO oxidation process is observed due to the earlier adsorption of OH. Similar behavior for the onset of CO oxidation was found for Pt100−xPdx(100) surfaces, although higher CO coverages are achieved on these electrodes due to the well-ordered surface structure induced by the inclusion of Pd on the Pt(100) structure. Various reaction mechanisms are significantly influenced by the presence of OHads species, including the oxidation reactions of organic molecules and oxygen reduction reaction, with OHads being identified as the principal intermediate. Therefore, the fundamental insights here reported on platinum–palladium electrodes are of particular importance for the proper interpretation of the catalytic properties of these materials.Author contributions
Gabriel Melle: investigation, methodology, writing – review & editing. Fabian Scholten: investigation, methodology, writing – review & editing. Juan M. Feliu: writing – review & editing, Enrique Herrero: writing – review & editing, supervision. Beatriz Roldan Cuenya: writing – review & editing. Rosa M. Arán-Ais conceptualization, methodology, investigation, writing – review & editing, supervision.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank the funding by the Max Planck Society through the funding of the Max Planck Partner Group on “Mechanistic insights into electrocatalytic reactions on well-defined bimetallic surfaces”. Financial support from the Ministerio de Ciencia e Innovación (project PID2022-137350NB-I00) is also acknowledged.Notes and references
- C. Roth, A. J. Papworth, I. Hussain, R. J. Nichols and D. J. Schiffrin, J. Electroanal. Chem., 2005, 581, 79–85 CrossRef CAS.
- E. Herrero, A. Fernández-Vega, J. M. Feliu and A. Aldaz, J. Electroanal. Chem., 1993, 350, 73–88 CrossRef CAS.
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Phys. Rev. Lett., 1998, 81, 2819–2822 CrossRef.
- G. B. Melle, T. Altair, R. L. Romano and H. Varela, Energy Fuels, 2021, 35, 6202–6209 CrossRef CAS.
- A. Capon and R. Parson, J. Electroanal. Chem. Interfacial Electrochem., 1973, 44, 1–7 CrossRef CAS.
- R. Parsons and T. VanderNoot, J. Electroanal. Chem. Interfacial Electrochem., 1988, 257, 9–45 CrossRef CAS.
- C. A. Rice, A. Bauskar and P. G. Pickup, in Electrocatalysis in Fuel Cells, 2013, vol. 9, pp. 69–87 Search PubMed.
- C. A. Rice and A. Wieckowski, Electrocatalysis of Formic Acid Oxidation, in Electrocatalysis in Fuel Cells: A Non- and Low-Platinum Approach, ed. M. Shao, SpringerLondon, London, 2013, pp. 43–67 Search PubMed.
- X. Chen, L. P. Granda-Marulanda, I. T. McCrum and M. T. M. Koper, Nat. Commun., 2022, 13, 38 CrossRef CAS PubMed.
- N. Z. M. J. N. de Pourbaix and J. Van Muylder, Platinum Met. Rev., 1959, 3, 47 CrossRef.
- M. Grdeń, M. Łukaszewski, G. Jerkiewicz and A. Czerwiński, Electrochim. Acta, 2008, 53, 7583–7598 CrossRef.
- B. A. F. Previdello, E. Sibert, M. Maret and Y. Soldo-Olivier, Langmuir, 2017, 33, 2087–2095 CrossRef CAS PubMed.
- M. J. Llorca, J. M. Feliu, A. Aldaz and J. Clavilier, J. Electroanal. Chem., 1993, 351, 299–319 CrossRef CAS.
- R. Gómez, A. Rodes, J. M. Pérez, J. M. Feliu and A. Aldaz, Surf. Sci., 1995, 344, 85–97 CrossRef.
- B. Álvarez, A. Berná, A. Rodes and J. M. Feliu, Surf. Sci., 2004, 573, 32–46 CrossRef.
- X. Chen, K. Ojha and M. T. M. Koper, JACS Au, 2023, 3, 2780–2789 CrossRef CAS PubMed.
- C. Ye, F. Dattila, X. Chen, N. López and M. T. M. Koper, J. Am. Chem. Soc., 2023, 145, 19601–19610 CrossRef CAS PubMed.
- X. Chen, L. P. Granda-Marulanda, I. T. McCrum and M. T. M. Koper, Chem. Sci., 2020, 11, 1703–1713 RSC.
- D. J. Watson and G. A. Attard, Electrochim. Acta, 2001, 46, 3157–3161 CrossRef CAS.
- D. Watson and G. Attard, Surf. Sci., 2002, 515, 87–93 CrossRef CAS.
- T. J. Schmidt, N. M. Markovic, V. Stamenkovic, P. N. Ross, G. A. Attard and D. J. Watson, Langmuir, 2002, 18, 6969–6975 CrossRef CAS.
- B. Álvarez, V. Climent, A. Rodes and J. M. Feliu, Phys. Chem. Chem. Phys., 2001, 3, 3269–3276 RSC.
- R. Rizo, E. Herrero, V. Climent and J. M. Feliu, Curr. Opin. Electrochem., 2023, 38, 101240 CrossRef CAS.
- D. S. Mekazni, R. M. Arán-Ais, A. Ferre-Vilaplana and E. Herrero, ACS Catal., 2022, 12, 1965–1970 CrossRef CAS.
- Y. Yang, R. G. Agarwal, P. Hutchison, R. Rizo, A. V. Soudackov, X. Lu, E. Herrero, J. M. Feliu, S. Hammes-Schiffer, J. M. Mayer and H. D. Abruña, Nat. Chem., 2023, 15, 271–277 CrossRef CAS PubMed.
- V. Briega-Martos, E. Herrero and J. M. Feliu, Electrochim. Acta, 2017, 241, 497–509 CrossRef CAS.
- J. Clavilier, D. Armand, S. G. Sun and M. Petit, J. Electroanal. Chem. Interfacial Electrochem., 1986, 205, 267–277 CrossRef CAS.
- D. J. Watson and G. A. Attard, Surf. Sci., 2002, 515, 87–93 CrossRef CAS.
- F. J. Vidal-Iglesias, A. Al-Akl, D. Watson and G. A. Attard, J. Electroanal. Chem., 2007, 611, 117–125 CrossRef CAS.
- D. J. Watson and G. A. Attard, Surf. Sci., 2002, 515, 87–93 CrossRef CAS.
- R. M. Arán-Ais, M. C. Figueiredo, F. J. Vidal-Iglesias, V. Climent, E. Herrero and J. M. Feliu, Electrochim. Acta, 2011, 58, 184–192 CrossRef.
- J. M. Orts, R. Gómez, J. M. Feliu, A. Aldaz and J. Clavilier, Electrochim. Acta, 1994, 39, 1519–1524 CrossRef CAS.
- T. B. Massalski, H. Okamoto and A. S. M. International, Binary Alloy Phase Diagrams, ASM International, Materials Park, Ohio SE, 2nd edn, 1990 Search PubMed.
- A. V Ruban, H. L. Skriver and J. K. Nørskov, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15990–16000 CrossRef.
- Z. Liu, Y. Li, X. Zhang, S. Rao, J. Li, W. Wang, Z. Sun and J. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 28816–28825 CrossRef CAS PubMed.
- S. A. Krasnikov, S. Murphy, N. Berdunov, A. P. McCoy, K. Radican and I. V Shvets, Nanotechnology, 2010, 21, 335301 CrossRef CAS PubMed.
- M. Wakisaka, M. Sugimasa, J. Inukai and K. Itaya, J. Electrochem. Soc., 2003, 150, E81 CrossRef CAS.
- T. E. Felter and A. T. Hubbard, J. Electroanal. Chem. Interfacial Electrochem., 1979, 100, 473–491 CrossRef CAS.
- G. W. Simmons, Y. N. Wang, J. Marcos and K. Klier, J. Phys. Chem., 1991, 95, 4522–4528 CrossRef CAS.
- R. Rizo, S. Pérez-Rodríguez and G. García, ChemElectroChem, 2019, 6, 4725–4738 CrossRef CAS.
- A. Ferre-Vilaplana, J. V. Perales-Rondón, C. Buso-Rogero, J. M. Feliu and E. Herrero, J. Mater. Chem. A, 2017, 5, 21773–21784 RSC.
- T. Solomun, J. Electroanal. Chem. Interfacial Electrochem., 1987, 217, 435–441 CrossRef CAS.
- T. Solomun, J. Electroanal. Chem. Interfacial Electrochem., 1988, 255, 163–177 CrossRef CAS.
- T. Solomun, J. Electroanal. Chem. Interfacial Electrochem., 1991, 302, 31–46 CrossRef CAS.
- M. Hara, U. Linke and T. Wandlowski, Electrochim. Acta, 2007, 52, 5733–5748 CrossRef CAS.
- R. Gisbert, G. García and M. T. M. Koper, Electrochim. Acta, 2010, 55, 7961–7968 CrossRef CAS.
- R. Rizo, E. Sitta, E. Herrero, V. Climent and J. M. Feliu, Electrochim. Acta, 2015, 162, 138–145 CrossRef CAS.
- K. N. da Silva and E. Sitta, J. Solid State Electrochem., 2020, 24, 1921–1926 CrossRef CAS.
- U. Stimming and R. Vogel, DOI:10.1016/S0022-0728(96)05051-6.
- A. Cuesta, M. Kleinert and D. M. Kolb, Phys. Chem. Chem. Phys., 2000, 2, 5684–5690 RSC.
- K. Sato, S. Yoshimoto, J. Inukai and K. Itaya, Electrochem. Commun., 2006, 8, 725–730 CrossRef CAS.
- L. Wan, T. Suzuki, K. Sashikata, J. Okada, J. Inukai and K. Itaya, J. Electroanal. Chem., 2000, 484, 189–193 CrossRef CAS.
- Y. G. Kim, J. B. Soriaga, G. Vigh and M. P. Soriaga, J. Colloid Interface Sci., 2000, 227, 505–509 CrossRef CAS PubMed.
- L. J. Wan, S. L. Yau and K. Itaya, J. Phys. Chem., 1995, 99, 9507–9513 CrossRef CAS.
- B. Alvarez, J. M. Feliu and J. Clavilier, Electrochem. Commun., 2002, 4, 379–383 CrossRef CAS.
- A. Rodes, M. A. Zamakhchari, K. El Achi and J. Clavilier, J. Electroanal. Chem. Interfacial Electrochem., 1991, 305, 115–129 CrossRef CAS.
- N. Garcia-Araez, J. J. Lukkien, M. T. M. Koper and J. M. Feliu, J. Electroanal. Chem., 2006, 588, 1–14 CrossRef CAS.
- V. Climent, N. García-Araez, E. Herrero and J. Feliu, Russ. J. Electrochem., 2006, 42, 1145–1160 CrossRef CAS.
- R. M. Arán-Ais, M. C. Figueiredo, F. J. Vidal-Iglesias, V. Climent, E. Herrero and J. M. Feliu, Electrochim. Acta, 2011, 58, 184–192 CrossRef.
- R. Rizo, J. Fernández-Vidal, L. J. Hardwick, G. A. Attard, F. J. Vidal-Iglesias, V. Climent, E. Herrero and J. M. Feliu, Nat. Commun., 2022, 13, 2550 CrossRef CAS PubMed.
- M. J. Weaver, S.-C. Chang, L.-W. H. Leung, X. Jiang, M. Rubel, M. Szklarczyk, D. Zurawski and A. Wieckowski, J. Electroanal. Chem., 1992, 327, 247–260 CrossRef CAS.
- N. Garcia-Araez, V. Climent, P. Rodriguez and J. M. Feliu, Phys. Chem. Chem. Phys., 2010, 12, 12146 RSC.
- V. Climent, N. García-Araez and J. M. Feliu, Electrochem. commun., 2006, 8, 1577–1582 CrossRef CAS.
- A. Berna, V. Climent and J. Feliu, Electrochem. commun., 2007, 9, 2789–2794 CrossRef CAS.
- S. Trasatti and E. Lust, in Modern Aspects of Electrochemistry, 2002, pp. 1–215 Search PubMed.
- V. Climent, Electrochem. commun., 2001, 3, 590–594 CrossRef CAS.
- N. García-Aráez, V. Climent and J. M. Feliu, J. Am. Chem. Soc., 2008, 130, 3824–3833 CrossRef PubMed.
- W. M. Haynes, Handbook of Chemistry and Physics, CRC Press, 97th edn, 2017, p. 2554 Search PubMed.
- I. Villegas and M. J. Weaver, J. Chem. Phys., 1994, 101, 1648–1660 CrossRef CAS.
- C. A. Angelucci, E. Herrero and J. M. Feliu, J. Phys. Chem. C, 2010, 114, 14154–14163 CrossRef CAS.
- K. Domke, E. Herrero, A. Rodes and J. M. Feliu, J. Electroanal. Chem., 2003, 552, 115–128 CrossRef CAS.
- C. K. Rhee, J. M. Feliu, E. Herrero, P. Mrozek and A. Wieckowski, J. Phys. Chem., 1993, 97, 9730–9735 CrossRef CAS.
- A. López-Cudero, A. Cuesta and C. Gutiérrez, J. Electroanal. Chem., 2005, 579, 1–12 CrossRef.
- M. J. S. Farias, B. A. V. Lima, G. Tremiliosi-Filho and E. Herrero, J. Electroanal. Chem., 2021, 896, 115382 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01771d |
| This journal is © The Royal Society of Chemistry 2024 |