
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring bismuth defects in Bi2WO6 nanosheets for photocatalytic C–H activation†
Xinye
Li‡
abc,
Luteng
Luo‡
 a,
Hele
Guo
c,
Bo
Weng
*cde,
Li
Sun
f,
Gangamallaiah
Velpula
f,
Imran
Aslam
c,
Maarten B. J.
Roeffaers
c,
Qinghua
Chen
ab,
Lingxing
Zeng
*ab,
Min-Quan
Yang
b and
Qingrong
Qian
*ab
a,
Hele
Guo
c,
Bo
Weng
*cde,
Li
Sun
f,
Gangamallaiah
Velpula
f,
Imran
Aslam
c,
Maarten B. J.
Roeffaers
c,
Qinghua
Chen
ab,
Lingxing
Zeng
*ab,
Min-Quan
Yang
b and
Qingrong
Qian
*ab
aEngineering Research Center of Polymer Green Recycling of Ministry of Education, College of Environmental Science and Engineering, Fujian Normal University, Fuzhou, Fujian 350007, China. E-mail: zenglingxing@fjnu.edu.cn; qrqian@fjnu.edu.cn
bFujian Key Laboratory of Pollution Control & Resource Reuse, Fuzhou, Fujian 350007, China
ccMACS, Department of Microbial and Molecular Systems, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium
dCAS Key Laboratory of Urban Pollutant Conversion, Institute of Urban Environment, Chinese Academy of Sciences, 1799 Jimei Road, Xiamen 361021, China. E-mail: bweng@iue.ac.cn
eUniversity of Chinese Academy of Sciences, 19A Yuquan Road, Beijing 100049, China
fDivision of Molecular Imaging and Photonics, Department of Chemistry, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium
First published on 29th April 2024
Abstract
Functionalizing sp3 C–H bonds in toluene through photocatalysis poses a significant challenge in organic synthesis, garnering substantial research attention. Nevertheless, the inert nature of the C–H bond restricts the photocatalytic conversion efficiency. Here, we construct bismuth defect modified ultrathin Bi2WO6 nanosheets (BT-48) for the conversion of toluene into benzaldehyde under light irradiation. Density functional theory (DFT) calculations validate the effectiveness of Bi defects in Bi2WO6 in activating the C–H bond in toluene. Moreover, a comprehensive physiochemical analysis shows that the introduction of bismuth defects in ultrathin Bi2WO6 nanosheets could promote the separation of photoexcited charge carriers. As a result, the optimal BT-48 sample exhibits an impressive benzaldehyde formation (6781 μmol g−1 h−1) with a high selectivity of 96%, which is 6 times higher than that of Bi2WO6 (1109 μmol g−1 h−1) and surpasses those of all the previously reported Bi2WO6-based photocatalysts. It is expected that this work could enhance our understanding of designing efficient photocatalysts for the activation of C–H bonds under light illumination.
The activation of the C–H bond, especially the sp3 C–H bond, stands as a crucial focus within the realm of organic synthesis, which holds immense significance in the synthesis of pharmaceutical intermediates and fine chemicals.1,2 Nevertheless, traditional C–H bond activation systems commonly face challenges, such as requiring harsh process conditions (high temperature and pressure) and producing hazardous byproducts.3,4 Consequently, developing efficient and environmentally friendly synthetic protocols for direct activation of the C–H bond has become a crucial facet of green chemistry.5 Semiconductor-based photocatalytic conversion of the C–H bond, powered by solar energy under mild reaction conditions, has garnered significant interest due to their gentler reaction conditions and potential for sustainability.6–8
Among various semiconductors, Bi2WO6 stands out as a promising photocatalyst for C–H activation due to its non-toxic nature, robust oxidation potential, and suitable band gap for solar activation.9 Numerous Bi2WO6-based photocatalysts have been developed for activating the sp3 C–H bond.10–13 However, the inherent activity of bulk Bi2WO6 is relatively limited due to a scarcity of reactive sites and the rapid recombination of electron–hole pairs.14,15 Hence, extensive efforts have been dedicated to enhancing the efficiency of C–H bond activation using Bi2WO6 through different methods such as morphology control, introducing co-catalysts, constructing heterostructures, and more.16–19 For example, Yin et al. fabricated flower-like Bi2WO6, which exhibited improved photocatalytic performance towards selective oxidation of toluene into benzaldehyde as compared to Bi2WO6 nanoparticles.20 However, the efficiency of Bi2WO6-based photocatalysts for the C–H bond activation reaction is still unsatisfactory due to the lack of sufficient surface active sites.
Since the C–H bond activation energy can be significantly affected by surface active sites of catalysts, the strategies of nano-architecture aiming at exposing more active sites of Bi2WO6 to promote C–H bond activation are urgently in need.21 Considering the anisotropic structure and weak interlayer forces of Bi2WO6, exfoliating bulk structures into ultrathin nanosheets with larger specific surface areas and abundant surface atoms can establish reliable active sites.22 Moreover, engineering defects, including an oxygen vacancy (VO) and/or a Bi vacancy (VBi) of Bi2WO6, within the ultrathin nanosheets can further significantly tailor its electronic state and local coordination environment by regulating the surface atomic structure for promoting C–H bond activation.23 In general, the presence of VO serves to broaden the light response range, whereas VBi enhances the adsorption and activation of reactants on the surface. For example, Liu et al. demonstrated that the incorporation of VBi in Bi2WO6 improves the adsorption and mineralization of gaseous toluene.24 However, it is noteworthy that most studies have primarily concentrated on VO, with the engineering of metal Bi vacancies into Bi2WO6, especially for C–H bond activation, being scarcely reported.
Herein, we propose a meticulously optimized defect-doping strategy to construct bismuth defect modified ultrathin Bi2WO6 nanosheets (BT-48) for the activation of the sp3 C–H bond in toluene under light irradiation. The BT-48 catalyst characterized by a blend of theoretical simulations and comprehensive physiochemical analyses could not only enhance the adsorption and activation of the benzylic C–H bond in toluene but also boost the effective desorption of the benzaldehyde product. Additionally, the existence of bismuth defects in Bi2WO6 customizes the material's surface electronic structure, thereby promoting the separation and transfer of photogenerated electron–hole pairs while impeding their recombination. Consequently, under simulated solar light irradiation, the benzaldehyde formation rate in toluene employing BT-48 achieves an impressive 6781 μmol g−1 h−1 with a high selectivity of 96%, which exhibits a 6-fold improvement compared to that of bulk Bi2WO6 (1109 μmol g−1 h−1) and surpasses those of all of the previously reported Bi2WO6-based photocatalysts.
Using density functional theory (DFT), we first explore the role of Bi defects in Bi2WO6 materials for activation of the sp3 C–H bond in toluene. We modelled Bi2WO6 surfaces with and without bismuth defects, as well as investigated their interaction with toluene. Fig. 1a and b illustrate that the absorption energies of toluene are −1.607 eV for bulk Bi2WO6 (denoted as Bulk-BT) and −2.174 eV for the BT-48 sample. The improved absorption energy is ascribed to the presence of Bi defects that strengthen the interaction between the O atom in BT-48 and the H atom from toluene,25 as supported by the shortened O–H bond length as shown in Fig. S1 (see the ESI† for details). Furthermore, the electron state of toluene experiences significant alteration owing to the intense interaction between BT-48 and toluene (Fig. 1c–f). The C–H bond length of toluene absorbed on BT-48 slightly increases compared to that on Bulk-BT (Fig. S1†). This implies that activating the C–H bond will be easier with the BT-48 sample than with the Bulk-BT material.26 Additionally, the adsorption energy between the catalyst and product, benzaldehyde, was also evaluated. The absorption energies of benzaldehyde are −1.343 eV and −0.646 eV for Bulk-BT and BT-48 (Fig. S2†), respectively, suggesting that the produced benzaldehyde detaches more easily on the BT-48 surface as compared with Bulk-BT, which is beneficial for enhancing the photocatalytic activity and selectivity of the product.27 The above calculation results theoretically indicate that, with the introduction of VBi defects in Bi2WO6 materials, the photocatalytic efficiency of toluene C–H activation can be ultimately improved.
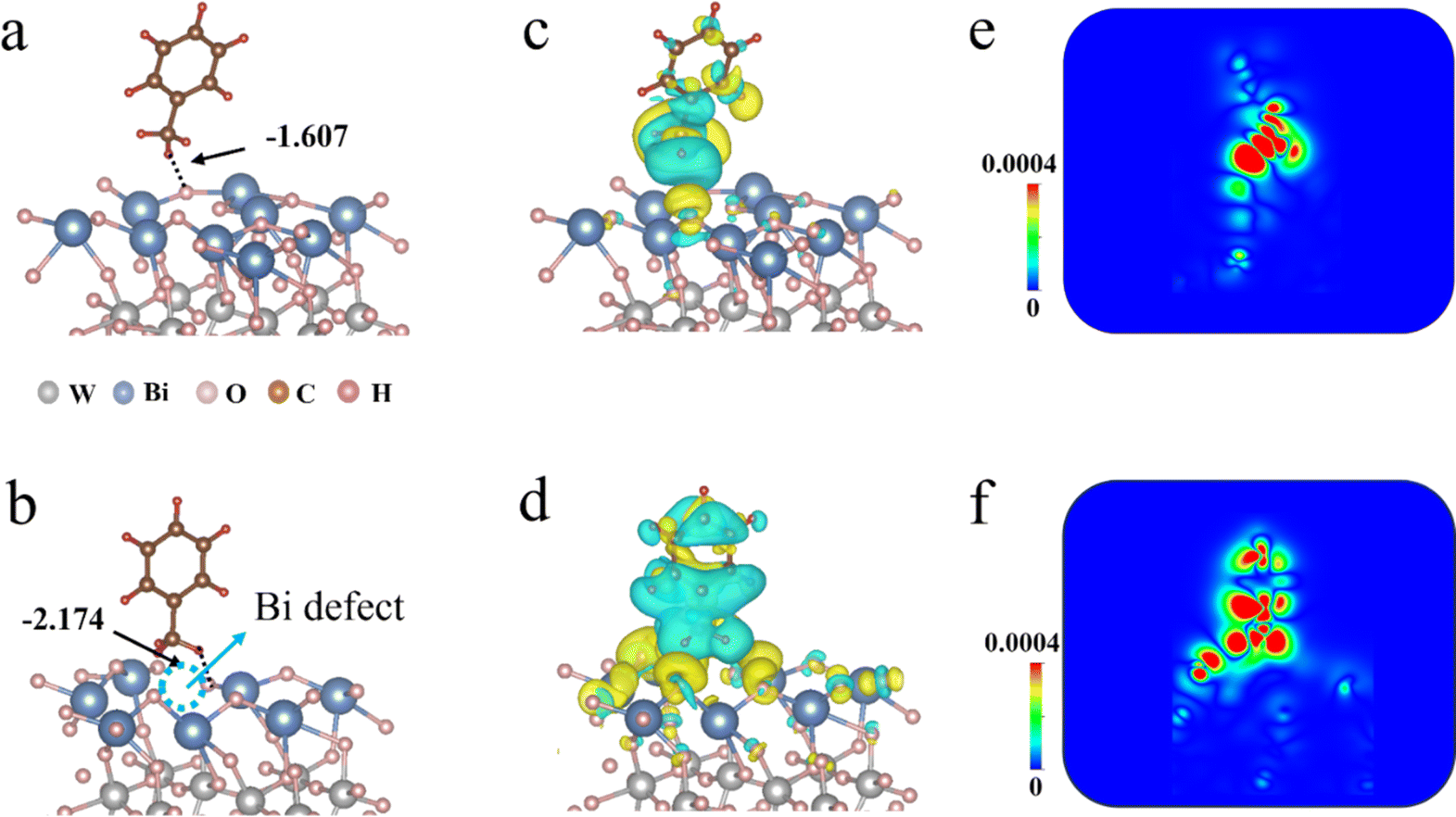 | ||
| Fig. 1 DFT-derived optimized structures and the adsorption energy of toluene on (a) Bulk-BT and (b) BT-48; differential charge density distribution of toluene absorbed on (c) Bulk-BT and (d) BT-48; 2D map of differential charge density maps for toluene absorbed on (e) Bulk-BT and (f) BT-48. | ||
To validate the theoretical predictions, we synthesized a series of Bi defect modified ultrathin Bi2WO6 nanosheet materials using acid treatment for different hours ranging from 6 to 72 hours via a meticulously optimized hydrothermal method (denoted as BT-x, where x represents the acid treatment times, and further details are in the ESI†). We select the optimal material, BT-48, as a representative sample for analysis. The crystal structure was confirmed through powder X-ray diffraction (XRD) analysis (Fig. S3†), confirming the successful synthesis of Bi2WO6.28 The morphologies of these samples were investigated via a scanning electron microscope (SEM). As depicted in Fig. S4,† the morphology of Bulk-BT resembles a flower-like structure composed of nanosheets. Upon subsequent acid treatment, the BT-48 sample maintains its nanosheet morphology, with some thin peeling layers observed on the surface. The transmission electron microscopy (TEM) image in Fig. 2a indicates the nanosheet morphology of the BT-48 sample with a distinct two-dimensional flexible layered structure. EDS (energy dispersive spectroscopy) shows an evenly distributed elemental composition within the BT-48 nanosheets (Fig. S5†). Furthermore, HAADF-STEM (high-angle annular dark-field scanning transmission electron microscopy) in Fig. 2b directly reveals the atomic arrangements of the BT-48 sample with a lattice spacing of 0.27 nm corresponding to the [200] plane of Bi2WO6.29 The relatively heavier Bi makes it appear brighter than W in the dark field.30 Analyzing atom intensities within the yellow box in the STEM image, the leftmost column demonstrates a periodic arrangement of Bi and W atoms, as shown by the purple peaks in Fig. 2c. However, examining the adjacent atom arrays within the same box reveals a conspicuous dip at a Bi site, denoted by the green and blue peaks, which confirms the presence of Bi defects in the BT-48 sample. Additionally, to visually confirm the presence of oxygen vacancies, electron paramagnetic resonance (EPR) spectroscopy was conducted. As shown in Fig. S6,† no typical signal indicating oxygen vacancies at g = 2.001 was observed,31 revealing that the primary defect type in BT-48 was isolated bismuth vacancies. Meanwhile, no Bi defects were observed in the HAADF-STEM image of Bulk-BT (Fig. S7†). The atomic force microscope (AFM) results in Fig. 2d and e demonstrate the sheet-like structure of BT-48 with a thickness of approximately 2 to 2.5 nm,32 indicating its ultrathin nature. Moreover, Fig. S8† shows that the sizes of the Bulk-BT and BT-48 samples are similar.
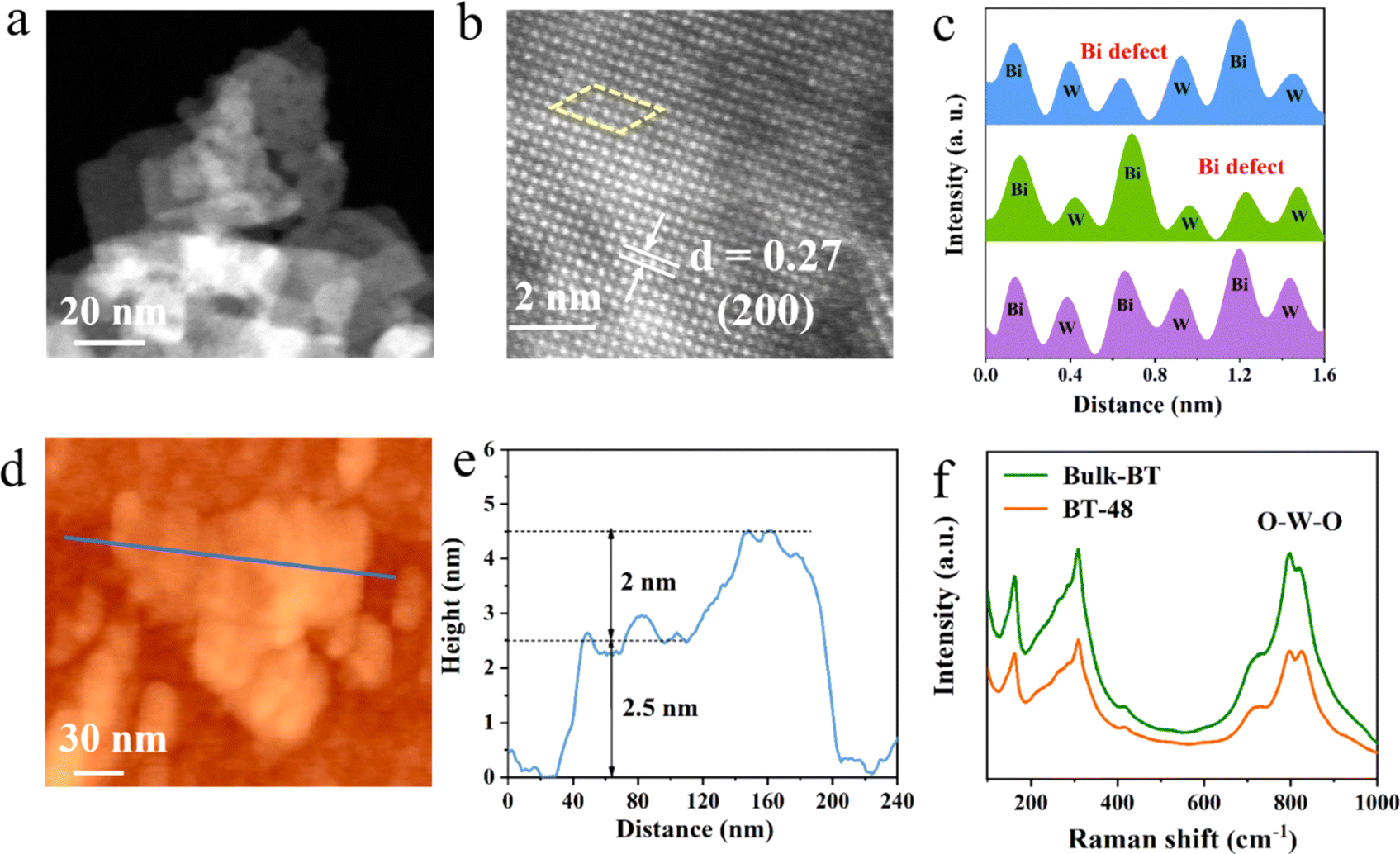 | ||
| Fig. 2 (a) TEM and (b) HAADF-STEM images of the BT-48 sample, (c) intensity along the blue box indicated in the HAADF-STEM image in panel b, (d) AFM image, (e) the cross-section profiles corresponding to the lines shown in the respective AFM image, and (f) Raman spectra of Bulk-BT and BT-48. | ||
The Raman spectra in Fig. 2f show robust peaks at 796 and 829 cm−1, corresponding to the antisymmetric and symmetric Ag stretching modes of terminal O–W–O groups, respectively.33 An enhanced Raman cross-section for the asymmetric stretching vibration of O–W–O is detected in BT-48 compared to Bulk-BT due to alterations induced by bismuth defects.34 X-ray photoelectron spectroscopy (XPS) was used to investigate the valence states of various elements (Bi, W and O) in BT-48 and Bulk-BT (Fig. S9†). As shown in Fig. S10,† the high-resolution Bi 4f spectra of BT-48 shift to higher binding energy compared to Bulk-BT, which is attributed to the reduced electron cloud density around (Bi2O2)2+.24,35 Likewise, a similar shift is also observed for the W 4f peaks of BT-48 (Fig. S11†), which is attributed to the formation of bismuth vacancies.36 The O 1s spectra presented in Fig. S12† indicate a negative shift of the peaks in the BT-48 sample due to the bismuth vacancy-rich configuration.37 Additionally, the peak area of the O–Bi band in BT-48 is observed to decrease by 30% relative to Bulk-BT, further confirming the presence of bismuth defects.29,32 To delve deeper into the Bi defects present in BT-48, we conducted an analysis of the atomic concentration ratio of Bi/W, yielding a calculated value of 1.78, which is lower than the theoretical value of 2 (Table S1†). This finding strongly supports the existence of Bi vacancies in the BT-48 sample, while indicating the absence of Bi defects in the Bulk BT sample.
The light absorption properties of Bulk-BT and BT-48 were investigated by diffuse reflectance spectroscopy (DRS), as depicted in Fig. S13.† The absorption edge of BT-48 shows a slight redshift as compared to Bulk-BT, hinting at a possible improvement in visible light absorption capability and a consequent reduction in the band gap.38 Calculations based on the formula: αhν = A(hν − Eg)1/2 (ref. 32) yield band gap energies of 3.26 eV and 3.21 eV for Bulk-BT and BT-48 samples, respectively. Mott–Schottky analysis in Fig. S14† shows flat band energies of −0.92 eV for Bulk-BT and −1.04 eV for BT-48 samples, which indicates that the conduction band energy values for Bulk-BT and BT-48 are −1.02 and −1.14 eV (vs. NHE), respectively. Therefore, the valence band energies for Bulk-BT and BT-48 are determined to be 2.24 eV and 2.07 eV, respectively, as depicted in Fig. S15.† The photocatalytic oxidation of C(sp3)-H in toluene to generate benzyl alcohol (BA) and benzaldehyde (BAD) was conducted to evaluate the performance of different samples under simulated solar light irradiation (AM 1.5 G). As illustrated in Fig. 3a, Bulk-BT demonstrates production rates of 1109 μmol g−1 h−1 for BAD and 85 μmol g−1 h−1 for BA. BT-6 h exhibits significantly increased production rates for BAD (3081 μmol g−1 h−1) and BA (239 μmol g−1 h−1), indicating that acid treatment enhances the photocatalytic performance of the BT materials. Further prolonging the acid treatment time to 48 h, the optimal BT-48 sample displays the highest photocatalytic activity in toluene oxidation among these materials, boasting production rates of 6781 μmol g−1 h−1 for BAD and 275 μmol g−1 h−1 for BA. This represents a 6-fold increase compared to the Bulk-BT sample. The catalyst's performance surpasses that of previously reported Bi2WO6-based catalysts (Fig. 3c and Table S2†).19,23,24,28,31,32,39–42 Extending the acid treatment to 72 hours leads to reduced BAD and BA production, which is potentially attributed to the damage to the Bi2WO6 structure. Furthermore, the photostability of the BT-48 sample was evaluated by a recycling experiment, as shown in Fig. 3b. No obvious activity loss is observed during five consecutive reaction cycles, demonstrating the excellent photostability of BT-48. The XRD analysis of the BT-48 sample after reactions as shown in Fig. S16† confirms the high stability and its crystal structure is well-maintained during the photocatalytic reaction process.
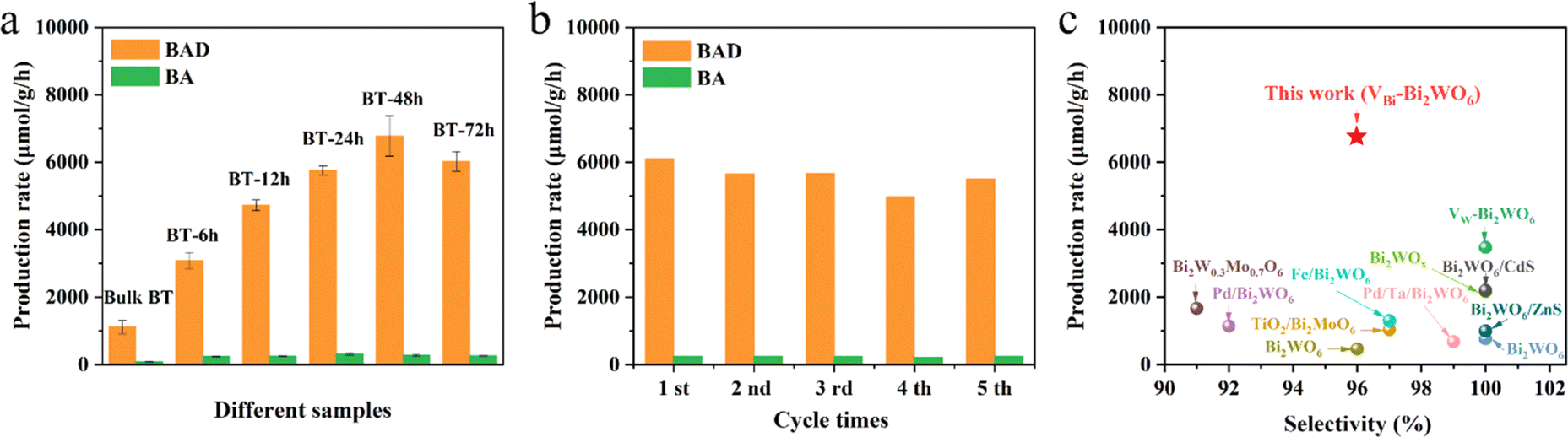 | ||
| Fig. 3 (a) Photocatalytic oxidation of toluene over samples, (b) recycling tests of toluene oxidation over BT-48, and (c) comparison of the catalytic activity for the benzaldehyde formulation rate over various Bi2WO6-based photocatalysts. Reaction conditions: photocatalysts (15 mg), Tol (2.5 mL) saturated with O2, simulated light irradiation using a 150 W xenon lamp (AM 1.5 G), and irradiation time (4 h). | ||
To reveal the origin of the photoactivity enhancement in the BT-48 samples compared to Bulk-BT, a series of characterization studies was performed. Nitrogen physisorption (Fig. S17†) revealed that the BT-48 sample exhibits a higher surface area (38 m2 g−1) compared to Bulk-BT (29 m2 g−1). This expanded surface area could provide more active sites,43,44 which potentially enhances the overall photocatalytic activity. Moreover, the photo-electrochemical (PEC) and photoluminescence (PL) spectra of BT-48 and Bulk-BT were recorded to elucidate the charge transfer and charge separation efficiency in these samples. The cyclic voltammetry curves (Fig. S18†), photocurrent (Fig. S19†) and linear sweep voltage (Fig. 4a) indicate faster charge transfer of photogenerated carriers in BT-48 compared to Bulk-BT.45–47 Electrochemical impedance spectroscopy spectra in Fig. S20† exhibit more efficient charge transfer between the electrode and electrolyte solution in the BT-48 sample than in the Bulk-BT sample.48,49 Moreover, as shown in Fig. S21,† the PL spectra of these samples exhibit a similar PL emission peak position but with lower intensity for the BT-48 sample, indicating reduced radiative recombination.50 The density of states (DOS) for BT-48 and Bulk-BT was obtained via DFT to investigate the change in the Bi2WO6 electronic band structure induced by Bi defects. Fig. 4c illustrates that introducing Bi defects in BT-48 creates a new impurity state in the gray area, reducing the band gap compared to Bulk-BT (Fig. 4b),32 consistent with the DRS findings. A new defect level in the forbidden band can be observed relative to Bulk-BT, favoring the electron transition into the conduction band under light irradiation, enabling a higher carrier density and improving electron conductivity.51 The collective analysis of PEC, PL, and DFT affirms that the Bi defects in BT-48 facilitate the separation and transfer process of photogenerated electron–hole pairs while suppressing their recombination. These factors collectively contribute to promoting the selective oxidation of toluene and enhancing photocatalytic performance.
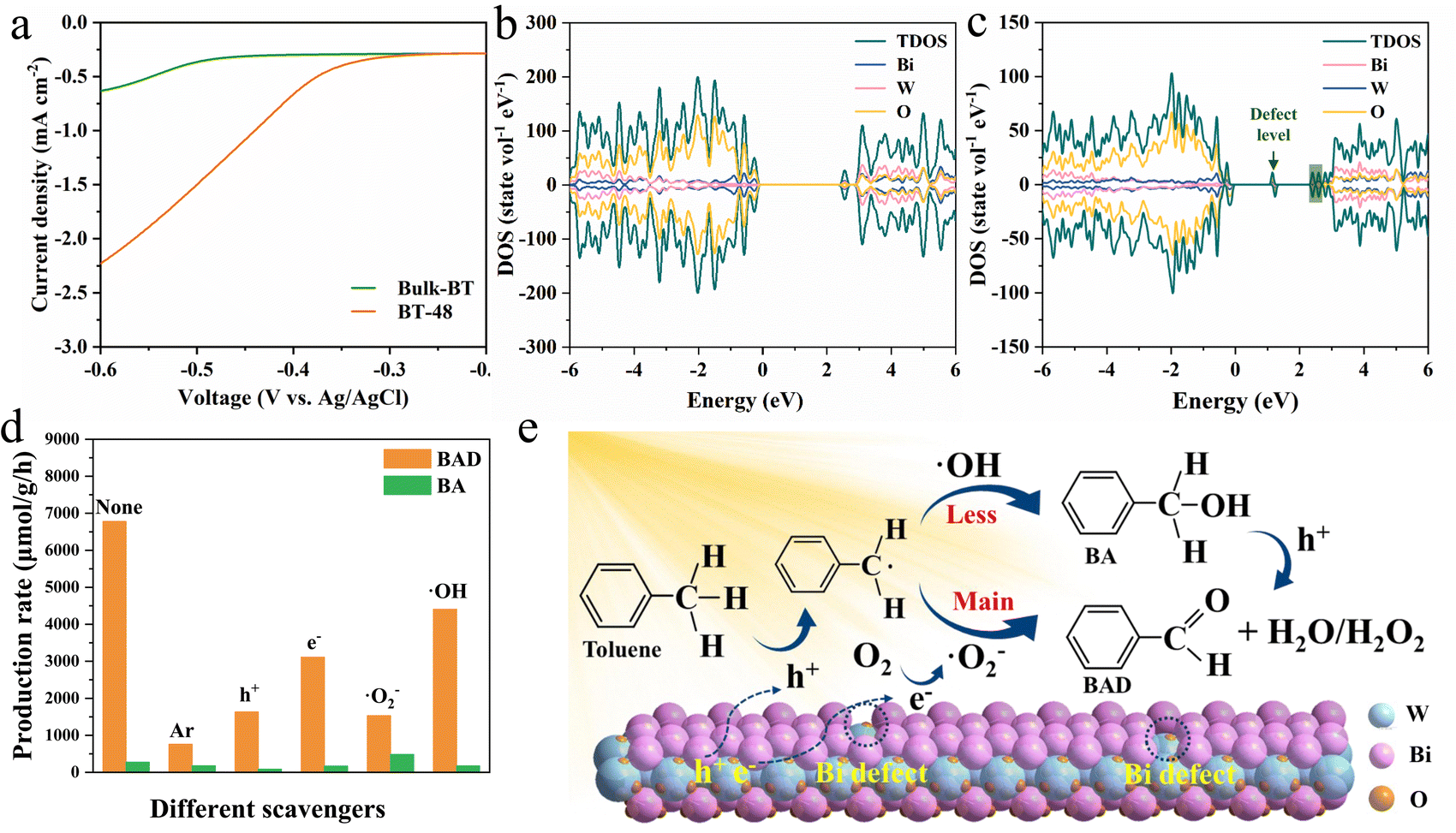 | ||
| Fig. 4 (a) LSV curves of Bulk-BT and BT-48 samples. The calculated density of states (DOS) of (b) Bulk-BT and (c) BT-48 samples. (d) Toluene oxidation in the absence or presence of various radical scavengers of BT-48. (e) Schematic illustration of the microscopic structure and the mechanism of the photocatalytic reaction and photochromism of BT-48. | ||
To further unravel the detailed photocatalytic reaction mechanism, control experiments were conducted with specific radical scavengers, such as ammonium oxalate for holes, potassium persulfate for electrons, 1,4-benzoquinone for ˙O2− and t-butanol for OH−.52Fig. 4d demonstrates that replacing oxygen with argon in the reaction process significantly inhibits toluene oxidation, indicating the crucial role of oxygen in this oxidation process. Moreover, the introduction of hole scavengers into the reaction system reduces BAD formation, highlighting the significant involvement of holes in toluene oxidation. Toluene oxidation is also hindered by the introduction of electron scavengers, but their inhibitory impact on the reaction is less significant compared to that of the hole. Importantly, the yield of BA increases while that of BAD significantly diminishes with the inclusion of ˙O2− scavengers. This could be due to an alternative pathway for BA formation, where ˙O2− radicals are not involved. In contrast, the hindrance to BAD production is lower with the addition of ˙OH scavengers, indicating that the secondary ˙OH-involved pathway is not the primary route for BAD formation. Notably, both the ˙O2− and ˙OH radicals are confirmed by EPR analysis in Fig. S22.†23,53,54
Based on the aforementioned data, we propose a photocatalytic mechanism for toluene oxidation over the BT-48 sample. As shown in Fig. 4e, under simulated sunlight illumination, the BT-48 catalyst efficiently generates electron–hole pairs (eqn S1, see the ESI† for details), with the Bi defects in BT-48 facilitating charge carrier separation.22 Photoexcited holes activate the C–H bond of toluene (eqn S2†), forming toluene radicals and protons.17 Simultaneously, molecular oxygen adsorbed on the BT-48 surface can be reduced to ˙O2− by electrons (eqn S3†).55 The produced toluene radicals can react with ˙O2− to yield BAD (eqn S4†), constituting the primary pathway for converting toluene into BAD.56 Additionally, holes may oxidize with surface-bound H2O or OH− to generate ˙OH radicals (eqn S5†), further reacting with toluene radicals to produce BA (eqn S6†). Generated BA can then oxidize into BAD facilitated by holes and ˙OH radicals (eqn S7†).57 Meanwhile the adsorbed benzylalcohol molecule is activated by a hole to generate the active intermediate of carbon-centered radical, along with the release of a proton (eqn S8†). The ˙O2− radical combines with a proton to form an ˙OOH radical (eqn S9†). Finally, the ˙OOH radical acquires a proton from the carbon centered radical to produce H2O2 and benzaldehyde (eqn S10†).58 To verify the presence of H2O2, we probed the production of H2O2 after the photocatalytic oxidation reaction.59 The findings in Fig. S23† reveal that the H2O2 signal at 325 nm was detected within the reaction system, indicating the coexistence of both mechanisms for aldehyde production from toluene under light irradiation.
In summary, ultrathin Bi2WO6 nanosheets with bismuth defects were engineered to functionalize sp3 C–H bonds in toluene under simulated solar light irradiation. The optimal BT-48 sample demonstrated a notably higher benzaldehyde production rate (6781 μmol g−1 h−1) than Bulk-BT (1109 μmol g−1 h−1), surpassing the highest performance among reported Bi2WO6-based photocatalysts for toluene oxidation. Various characterization studies suggest that introducing bismuth defects enhances the activation of C–H bonds in toluene, improves reactant absorption, and facilitates the separation and transfer of photogenerated charge carriers, thus achieving improved photocatalytic performance towards toluene oxidation under light irradiation. These findings are anticipated to deepen our insight into crafting efficient photocatalysts for C–H bond activation.
Author contributions
X. Li and B. Weng designed the idea. B. Weng, L. Zeng and Q. Qian supervised this study. X. Li, L. Luo, H. Guo, S. Li, G. Velpulaand and I. Aslam collected and analyzed the data. L. Luo built the structure models. X. Li and L. Luo visualized the results. X. Li, B. Weng, L. Zeng and Q. Qian wrote the draft. M. B. J. Roeffaers, Q. Chen and M. Q. Yang revised the manuscript. All the authors contributed to writing and finalizing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2023YFC3906300), the Research Foundation – Flanders (FWO grants G098319N and 1280021N), the KU Leuven Research Fund (C14/19/079 and iBOF-21-085 PERSIST), the KU Leuven Industrial Research Fund (C3/19/046), the Flemish government through long term structural funding Methusalem (CASAS2 and Meth/15/04), the Young Top Talent of Fujian Young Eagle Program, and the Natural Science Foundation of Fujian Province (2023J02013 and 2023YZ038001).References
- L. J. Sun, H. W. Su, Q. Q. Liu, J. Hu, L. L. Wang and H. Tang, Rare Met., 2022, 41, 2387–2404 CrossRef CAS.
- Á. Velasco-Rubio, P. Martínez-Balart, A. M. Álvarez-Constantino and M. Fañanás-Mastral, Chem. Commun., 2023, 59, 9424–9444 RSC.
- Y. Wang, J. Wu, G. Wang, D. Yang, T. Ishihara and L. Guo, Appl. Catal., B, 2021, 285, 119873 CrossRef CAS.
- H. H. Zhang, Z. C. Zhou, Y. J. Dong, L. Zhang, H. Y. Chen and D. B. Kuang, Sol. RRL, 2021, 5, 2100559 CrossRef CAS.
- B. K. Singh, S. Lee and K. Na, Rare Met., 2019, 39, 751–766 CrossRef.
- Z. J. Bai, X. P. Tan, L. Chen, B. Hu, Y. X. Tan, Y. Mao, S. Shen, J. K. Guo, C. T. Au, Z. W. Liang and S. F. Yin, Chem. Eng. Sci., 2022, 247, 120393 CrossRef.
- H. Huang, D. Verhaeghe, B. Weng, B. Ghosh, H. Zhang, J. Hofkens, J. A. Steele and M. B. J. Roeffaers, Angew. Chem., Int. Ed., 2022, 61, e202203261 CrossRef CAS PubMed.
- S. Kim, K. H. Kim, C. Oh, K. Zhang and J. H. Park, Carbon Energy, 2022, 4, 21–44 CrossRef CAS.
- T. Chen, L. Liu, C. Hu and H. Huang, Chin. J. Catal., 2021, 42, 1413–1438 CrossRef CAS.
- T. Wang, C. Feng, J. Liu, D. Wang, H. Hu, J. Hu, Z. Chen and G. Xue, Chem. Eng. J., 2021, 414, 128827 CrossRef CAS.
- Y. Wang, W. Jiang, W. Luo, X. Chen and Y. Zhu, Appl. Catal., B, 2018, 237, 633–640 CrossRef CAS.
- X. Cao, T. Han, Q. Peng, C. Chen and Y. Li, Chem. Commun., 2020, 56, 13918–13932 RSC.
- J. Zhong, Y. Zeng, D. Chen, S. Mo, M. Zhang, M. Fu, J. Wu, Z. Su, P. Chen and D. Ye, J. Hazard. Mater., 2020, 386, 121957 CrossRef CAS PubMed.
- D. Tantraviwat, A. Nattestad, J. Chen and B. Inceesungvorn, J. Colloid Interface Sci., 2023, 629, 854–863 CrossRef CAS PubMed.
- S. Lei, C. Yang, H. Liao, J. Chen, J. Zhong and J. Li, Mater. Res. Bull., 2021, 133, 111075 CrossRef CAS.
- Y. Dai, C. Poidevin, C. Ochoa-Hernandez, A. A. Auer and H. Tuysuz, Angew. Chem., Int. Ed., 2020, 59, 5788–5796 CrossRef CAS PubMed.
- Z. J. Bai, Y. Mao, B. H. Wang, L. Chen, S. Tian, B. Hu, Y. J. Li, C. T. Au and S. F. Yin, Nano Res., 2023, 16, 6104–6112 CrossRef CAS.
- L. Xiong and J. Tang, Adv. Energy Mater., 2021, 11, 2003216 CrossRef CAS.
- X. Wei, K. Li, X. Zhang, Q. Tong, J. Ji, Y. Cai, B. Gao, W. Zou and L. Dong, Appl. Catal., B, 2022, 317, 121694 CrossRef CAS.
- Y. Liu, L. Chen, Q. Yuan, J. He, C. T. Au and S. F. Yin, Chem. Commun., 2016, 52, 1274–1277 RSC.
- H. Zhang, S. Zhou, D. Li, W. Zhan, H. Liu, L. Sun, M. Li, L. Yang, J. Zhao, X. He, J. Xu and X. Han, ACS Catal., 2023, 13, 14657–14669 CrossRef CAS.
- Z. Guan, X. Li, Y. Wu, Z. Chen, X. Huang, D. Wang, Q. Yang, J. Liu, S. Tian, X. Chen and H. Zhao, Chem. Eng. J., 2021, 41, 128283 CrossRef.
- X. Cao, Z. Chen, R. Lin, W. C. Cheong, S. Liu, J. Zhang, Q. Peng, C. Chen, T. Han, X. Tong, Y. Wang, R. Shen, W. Zhu, D. Wang and Y. Li, Nat. Catal., 2018, 1, 704–710 CrossRef CAS.
- J. Di, C. Chen, C. Zhu, M. Ji, J. Xia, C. Yan, W. Hao, S. Li, H. Li and Z. Liu, Appl. Catal., B, 2018, 238, 119–125 CrossRef CAS.
- Y. C. Zhang, N. Afzal, L. Pan, X. Zhang and J. J. Zou, Adv. Sci., 2019, 6, 1900053 CrossRef PubMed.
- X. Zhang, F. Bi, Z. Zhu, Y. Yang, S. Zhao, J. Chen, X. Lv, Y. Wang, J. Xu and N. Liu, Appl. Catal., B, 2021, 297, 120393 CrossRef CAS.
- Z. Shen, Y. Hu, Q. Pan, C. Huang, B. Zhu, W. Xia, H. Wang, J. Yue, M. Muhler, G. Zhao, X. Wang and X. Huang, Appl. Surf. Sci., 2022, 571, 151370 CrossRef CAS.
- V. Sharma, A. Kumar and V. Krishnan, Chemosphere, 2022, 287, 132119 CrossRef CAS PubMed.
- H. Huang, J. Zhao, B. Weng, F. Lai, M. Zhang, J. Hofkens, M. B. J. Roeffaers, J. A. Steele and J. Long, Angew. Chem., Int. Ed., 2022, 61, e202204563 CrossRef CAS PubMed.
- J. Bai, B. Zhang, T. Xiong, D. Jiang, X. Ren, P. Lu and M. Fu, J. Alloys Compd., 2021, 887, 161297 CrossRef CAS.
- W. C. Huo, X. Dong, J. Y. Li, M. Liu, X. Y. Liu, Y. X. Zhang and F. Dong, Chem. Eng. J., 2019, 361, 129–138 CrossRef CAS.
- H. Huang, C. Zhou, X. Jiao, H. Yuan, J. Zhao, C. He, J. Hofkens, M. B. J. Roeffaers, J. Long and J. A. Steele, ACS Catal., 2019, 10, 1439–1443 CrossRef.
- J. Wang, H. Liang, C. Zhang, B. Jin and Y. Men, Appl. Catal., B, 2019, 256, 117874 CrossRef CAS.
- T. Xie, Y. Zhang, W. Yao, Y. Liu, H. Wang and Z. Wu, Catal. Sci. Technol., 2019, 9, 1178–1188 RSC.
- Q. Gu, K. Zhang, P. Jiang, Y. Shen, Y. Leng, P. Zhang and P. T. Wai, Mol. Catal., 2021, 515, 111932 CrossRef CAS.
- G. Zhang, Z. Hu, M. Sun, Y. Liu, L. Liu, H. Liu, C. P. Huang, J. Qu and J. Li, Adv. Funct. Mater., 2015, 25, 3726–3734 CrossRef CAS.
- Y. Zhao, Y. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2019, 31, e1806482 CrossRef PubMed.
- S. Liu, W. Qi, S. Adimi, H. Guo, B. Weng, J. P. Attfield and M. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 7238–7247 CrossRef CAS PubMed.
- L. N. Song, F. Ding, Y. K. Yang, D. Ding, L. Chen, C. T. Au and S. F. Yin, ACS Sustain. Chem. Eng., 2018, 6, 17044–17050 CrossRef CAS.
- X. X. Deng, S. Tian, Z. M. Chai, Z. J. Bai, Y. X. Tan, L. Chen, J. K. Guo, S. Shen, M. Q. Cai, C. T. Au and S. F. Yin, Ind. Eng. Chem. Res., 2020, 59, 13528–13538 CrossRef CAS.
- E. Safaei and S. Mohebbi, J. Photochem. Photobiol., A, 2019, 371, 173–181 CrossRef CAS.
- K. Zhang, H. Chen, Y. Liu, J. Deng, L. Jing, A. Rastegarpanah, W. Pei, Z. Han and H. Dai, Appl. Catal., B, 2022, 315, 121545 CrossRef CAS.
- H. Wang, P. Hu, J. Zhou, M. B. J. Roeffaers, B. Weng, Y. Wang and H. Ji, J. Mater. Chem. A, 2021, 9, 19984–19993 RSC.
- D. Wei, J. Wu, Y. Wang, J. Zhong, D. Li, X. Jin, Y. Wu, P. Chen, H. Liu, W. Lv and G. Liu, Chem. Eng. J., 2023, 462, 142291 CrossRef CAS.
- S. Zhang, Y. Zhao, R. Shi, C. Zhou, G. I. N. Waterhouse, L. Z. Wu and C. H. Adv, Energy Mater., 2020, 10, 1901973 CrossRef CAS.
- P. Song, J. Du, X. Ma, Y. Shi, X. Fang, D. Liu, S. Wei, Z. Liu, Y. Cao, B. Lin, J. Di, Y. Wang, J. Cui, T. Kong, C. Gao and Y. Xiong, EcoEnergy, 2023, 1, 197–206 CrossRef.
- H. Huang, J. Zhao, H. Guo, B. Weng, H. Zhang, R. A. Saha, M. Zhang, F. Lai, Y. Zhou, R.-Z. Juan, P.-C. Chen, S. Wang, J. A. Steele, F. Zhong, T. Liu, J. Hofkens, Y.-M. Zheng, J. Long and M. B. J. Roeffaers, Adv. Mater., 2024, 2313209 CrossRef PubMed.
- W. Feng, Y. Lei, X. Wu, J. Yuan, J. Chen, D. Xu, X. Zhang, S. Zhang, P. Liu, L. Zhang and B. Weng, J. Mater. Chem. A, 2021, 9, 1759–1769 RSC.
- H. Gao, L. Sun, M. Li, W. Zhan, X. Wang and X. Han, Nano Res., 2022, 15(5), 4638–4645 CrossRef CAS.
- S. Wan, J. Xu, S. Cao and J. Yu, Interdiscip. Mater., 2022, 1, 294–308 CrossRef CAS.
- Y. W. Teh, C. C. Er, X. Y. Kong, B. J. Ng, S. T. Yong and S. P. Chai, ChemSusChem, 2022, 15, e202200471 CrossRef CAS PubMed.
- H. Huang, H. Yuan, J. Zhao, G. Solís-Fernández, C. Zhou, J. W. Seo, J. Hendrix, E. Debroye and J. A. Steele, ACS Energy Lett., 2018, 4, 203–208 CrossRef.
- G. Qiu, T. Wang, X. Li, X. Tao and B. Li, Ind. Eng. Chem. Res., 2020, 59, 11517–11526 CrossRef CAS.
- X. Li, T. Wang, X. Tao, G. Qiu, C. Li and B. Li, J. Mater. Chem. A, 2020, 8, 17657 RSC.
- F. Liu, C. X. Xiao, L. H. Meng, L. Chen, Q. Zhang, J. B. Liu, S. Shen, J. K. Guo, C. T. Au and S. F. Yin, ACS Sustain. Chem. Eng., 2020, 8, 1302–1310 CrossRef CAS.
- G. Han, X. Liu, Z. Cao and Y. Sun, ACS Catal., 2020, 10, 9346–9355 CrossRef CAS.
- Q. Cheng, Y. J. Yuan, R. Tang, Q. Y. Liu, L. Bao, P. Wang, J. Zhong, Z. Y. Zhao, Z. T. Yu and Z. Zou, ACS Catal., 2022, 12, 2118–2125 CrossRef CAS.
- C. Chen, G. Qiu, T. Wang, Z. Zheng, M. Huang and B. Li, J. Colloid Interface Sci., 2021, 592, 1–12 CrossRef CAS PubMed.
- Y. Chen, X. Yan, J. Xu and L. Wang, J. Mater. Chem. A, 2021, 9, 24056 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed description including the materials, preparation of the catalysts, physical characterization studies, electrochemical measurements, computational details, and supporting figures. See DOI: https://doi.org/10.1039/d4ta01573h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |