
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The role of vanadium substitution in the oxygen sublattice disorder of Ba7Nb4MoO20-based hexagonal perovskite oxide-ion conductors†
Abdulkadir Olatunbosun
Biffo
 a,
Theodosios
Famprikis
*a and
Pedro B.
Groszewicz
*ab
a,
Theodosios
Famprikis
*a and
Pedro B.
Groszewicz
*ab
aDepartment of Radiation Science and Technology, Delft University of Technology, Delft 2629JB, Netherlands. E-mail: t.famprikis@tudelft.nl; p.groszewicz@tudelft.nl
bHelmholtz Zentrum Berlin für Materialien und Energie, Berlin 14109, Germany
First published on 25th September 2024
Abstract
Ba7Nb4MoO20-based hexagonal perovskite derivatives are promising oxygen-ion conductors for solid electrolytes in solid-oxide fuel cells and electrolysers. A thorough understanding of chemical substitution and its impact on structural features conducive to high ionic conductivity is fundamental for decreasing the operation temperature of such devices. Here, a new 7H polytype-based composition, namely Ba7Nb3.9−xVxMo1.1O20.05, is investigated to assess the effect of vanadium substitution. Structural changes upon V incorporation are studied using X-ray and neutron diffraction, as well as 51V and 93Nb solid-state nuclear magnetic resonance spectroscopy. For the undoped composition at room temperature, two distinct oxygen sites (O1 and O5) are found along the palmierite-like layer, corresponding to a mix of four- and six-fold coordination for adjacent M2 cations. At high temperature (527 °C), reorganization of oxygen results in the major occupation of O1 and four-fold (tetrahedral) coordination of the M2 cations. The same rearrangement is observed upon V-substitution, but already at room temperature. From 51V NMR, we identified a tetrahedral coordination for V5+ cations, indicating their preferential occupation of the M2 site. This preferential occupation by V5+ cations is correlated with increasing tetrahedral coordination of Nb5+ cations as observed from 93Nb NMR. Altogether, these observations indicate that V-substitution impacts the oxygen sublattice so as to mimic the high-temperature structure. Additionally, BVSE calculations demonstrate a decreasing energy barrier for O2− migration associated with the presence of vanadium in the structure. This conclusion corroborates the hypothesis that vanadium's propensity for a lower coordination number is beneficial for promoting high O2− mobility in this promising class of oxide-ion conductors.
Introduction
Solid oxide fuel cells (SOFCs) are promising energy conversion devices to propel the transition towards a fossil-fuel-free energy economy.1,2 The widespread use of SOFCs is hampered by their high operation temperatures, in the range of 800 – 1000 °C for current commercial devices based on yttrium-stabilised zirconia as solid-state electrolyte.3 These high operating temperatures pose considerable challenges, such as the degradation of metallic interconnects and slow start/stop times, as well as imposing restrictions on the selection of compatible materials.4,5 Given this context, it is imperative to lower the operation temperature of SOFCs to the so-called intermediate temperature range of less than 600 °C.6 Doing this will require finding a suitable electrolyte, which is the first step towards enabling the efficient operation of SOFCs in the intermediate temperature range. The fine-tuning of the most widely used electrolyte materials, such as fluorite-type oxides, and the use of increasingly thinner membranes are saturated strategies.7–11 Hence, there is a need to explore alternative structures or materials. Several families of oxides have been proposed as alternative structures for oxide-ion conduction such as silicon and germanium apatites (La9.33Si6−xGexO26),12 Bi4V2O11 compounds,13 La2Mo2O9 (ref. 14 and 15) and LaSrGa3O7 materials.16 Recently, a high oxide-ion conduction at the intermediate temperature has been reported in hexagonal perovskite derivatives.17–22An example of a hexagonal perovskite family that has shown a high oxide-ion conduction, comparable to yttrium-stabilised zirconia, is the Ba3M’M”O8.5 series (where M’ = Mo, W and M’’ = V, Nb).21,23–26 This series is a palmierite derivative of the 9R polytype of hexagonal perovskite depicted in Fig. 1(a–d).27 The bulk conductivity of a member of this series Ba3MoNbO8.5 has been reported to be around 2.2 mS cm−1 at 600 °C, predominantly dominated by oxide-ion conduction,26 which has been evidenced using the bond-valence site energy (BVSE) approach to proceed via a 2D migration path involving the partially occupied oxygen sites in the palmierite-like layers.21,28–30 Furthermore, the conductivity of this palmierite derivative has been improved by almost an order of magnitude via an iso-valent substitution of Nb5+ with V5+,21 with a bulk ionic conductivity of 11 mS cm−1 at 600 °C reported for the composition Ba3Nb0.9V0.1MoO8.5. The enhanced conductivity was correlated with an increase of tetrahedral moieties (i.e. (Nb/V/Mo)O4) in the palmierite-like layers of the Ba3Nb0.9V0.1MoO8.5 crystal structure,21 based on changes in the oxygen sublattice upon V-substitution as observed by neutron diffraction.21
 | ||
| Fig. 1 The cb plane projections depicting the relationship between (a) 9R hexagonal perovskite polytype and palmierite-type derivative structures; (b) anion deficiency and partially-occupied face-sharing octahedra; (c) palmierite structure with empty octahedra; (d) the palmierite-perovskite intergrowth in Ba3NbMoO8.5 reported by Fop et al.31 This image is adapted from Mitchell.27 The cb plane projections illustrate the relationship between (e) 7H hexagonal perovskite polytype, (f) oxygen-deficient c1 – BaO3, and (g) unoccupied h1 octahedral units.32 And (h) the structure reported by Fop et al.22 Colored polyhedra denote the different crystallographically equivalent metal-cation sites. | ||
The further attempts on chemical modification of Ba3MoNbO8.5-based oxides via tailored cationic chemical substitution have not yielded any better oxide-ion conductors compared to the aforementioned Ba3Nb0.9V0.1MoO8.5.23–25,28,30,33 However, a high oxide-ion and proton conductivity has been discovered in Ba7Nb4MoO20, an anion-deficient, palmierite-perovskite derivative of a 7H polytype of hexagonal perovskite, depicted in Fig. 1(e–h), which exhibits a bulk ionic conductivity of 4.0 mS cm−1 at 510 °C.22 The compound crystallises in the P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 space group and is described as an intergrowth of (12R) perovskite blocks and (9R) palmierite layers along the crystallographic c-axis (Fig. 1g).22,27,32
m1 space group and is described as an intergrowth of (12R) perovskite blocks and (9R) palmierite layers along the crystallographic c-axis (Fig. 1g).22,27,32
There is a significant oxygen occupational disorder along the palmierite-like layers of Ba7Nb4MoO20, which has been described in terms of partially occupied oxygen sites (O1 and O5, Fig. 1h).22 These disordered sites result in a situation where mixed polyhedral units (Nb/Mo)Ox are evidenced in the palmierite-like layers of Ba7Nb4MoO20-based materials (Fig. 1h). The occupation of the O1 site results in tetrahedral (Nb/Mo)O4 moieties, while occupation of the O5 site yields octahedral (Nb/Mo)O6 units.18,22 The O1 and O5 sites have been reported to reorganise upon increasing temperatures, where the O1 site is more occupied than the O5 site.22 This reorganisation thus leads to a situation where the tetrahedral units are more prevalent in the palmierite-like layers at high temperatures,22 which seems to be a structural trait correlated with increased oxide ion conductivity.
The focus of recent studies on Ba7Nb4MoO20 has been to simultaneously suppress its proton conductivity and increase its oxide-ion conductivity.18–20,34 To achieve this, several doped series have been explored, such as Ba7Nb4−xMo1+xO20+x/2,18 Ba7Ta4−xMo1+xO20+x/2,19 Ba7Nb-CrxMoO20+x/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 20 and Ba7Nb4−xWxMoO20+x/2.34 All of these series have shown enhanced performance with respect to concomitant enhanced oxide-ion conduction and suppressed proton conduction, as well as excellent chemical and electrical stability under reducing atmospheres. The mechanisms of oxide-ion conduction of both the pristine and doped composition have been depicted to proceed via a 2D conduction path along the palmierite-like layers.22
20 and Ba7Nb4−xWxMoO20+x/2.34 All of these series have shown enhanced performance with respect to concomitant enhanced oxide-ion conduction and suppressed proton conduction, as well as excellent chemical and electrical stability under reducing atmospheres. The mechanisms of oxide-ion conduction of both the pristine and doped composition have been depicted to proceed via a 2D conduction path along the palmierite-like layers.22
The conduction properties of Ba7Nb4MoO20-based materials have been hypothesized to improve upon tailored introduction of cations in the palmierite-like layers that preferentially adopt tetrahedral coordination geometry.22 For example, V5+, Cr5+ and Cr6+ would be good chemical substitution candidates as they are known to exhibit a preference for tetrahedral coordination in inorganic oxides.35 Contrastingly, cations such as W6+, Nb5+, Mo6+ and Ti4+ preferentially adopt octahedral coordination geometry.35
Given the improvement in ionic conductivity for oxide ions in Ba3NbMoO8.5 (9R polytype) (Fig. 1d) upon chemical modification with vanadium, it is relevant to determine whether the iso-valent substitution of niobium by vanadium is also feasible for the Ba7Nb4MoO20-based 7H polytype. In case this is feasible, we are interested in the structural modifications brought about by vanadium chemical substitution and whether structural motives conducive to high ionic mobility, such as a propensity for low coordination numbers (i.e. CN = 4 instead of 6) for cation sites, in parallel to structural disorder in the oxide anion sub-lattice, are present or become even more pronounced as a function of vanadium content.
Herein, we report the synthesis and characterization of compositions Ba7Nb3.9−xVxMo1.1O20.05 (0 ≤ x ≤ 0.4), by substituting V5+ in place of Nb5+ in Ba7Nb3.9Mo1.1O20.05, a composition that is known for both enhanced oxide-ion conduction and suppressed proton conduction.18 We employ 51V solid-state Nuclear Magnetic Resonance spectroscopy (ss-NMR) to evaluate the local environment of V5+ substituents and 93Nb NMR to keep track of Nb5+ coordination in the series, as well as a combined X-ray and neutron diffraction analysis to study the impact of this chemical modification on O1–O5 oxygen site disorder and occupation of cation sites as a function of temperature. Further insight into the potential conduction pathway and the corresponding energy barriers for oxide-ion migration in the Ba7Nb3.9−xVxMo1.1O20.05 (0 ≤ x ≤ 0.4) series is provided by BVSE calculations, showing how vanadium substitution significantly affects the predominant conduction pathway along the palmierite-like layer.
Experimental
The Ba7Nb3.9−xVxMo1.1O20.05 (0 ≤ x ≤ 0.4) series was synthesized using a solid-state reaction approach. Mixtures of stoichiometric amounts of BaCO3 (99.95%, Chem-Impex), Nb2O5 (99.9985%, Puratronic®), V2O5 (99.99%, Aldrich), and MoO3 (≥99.5%, Aldrich) were ground, pelletised and calcined at 1050 °C for 48 h. The calcined pellets were subsequently re-ground, re-pelletised and re-calcined at 1050 °C for 48 h. This subsequent step was repeated one more time to obtain phase pure materials.Laboratory X-ray powder diffraction (XRD) patterns were collected using a PANanalytical X'Pert Pro X-ray diffractometer equipped with a Cu Kα source (1.54 Å) operated at 45 kV, 40 mA. The XRD patterns were recorded in the range 10° < 2θ < 100°. A LaB6 NIST standard (NIST660C) was used for both the calibration and performance qualification of the diffractometer.
Neutron powder diffraction (NPD) experiments were performed using PEARL diffractometer of the TU Delft Reactor Institute (the Netherlands).36 Approximately 2.0 g of each sample was loaded in a 6 mm diameter can made of a V–Ni null-scattering alloy. The sample can was then placed in a neutron-transparent vacuum box connected to a primary vacuum (∼10−3 mbar). Diffraction patterns were measured over approximately 18 h using a wavelength of 1.667 Å, which was selected using the (533) reflection of a single-crystal Ge[511] monochromator. For high-temperature measurements, the temperature was controlled via a resistive heater consisting of a vanadium heating element wrapped around the sample can (see details in ESI Fig. S20†).
Structural refinements were performed using the GSAS-II program.37 The NPD datasets coupled with the laboratory XRD patterns were used for the refinement of the crystal structure. The instrumental background was fitted with the Chebyshev polynomial, while the unit cell metrics and peak profile parameters were simultaneously refined using the Le Bail intensity extraction.38 The Le Bail refinement was subsequently followed by a Rietveld refinement for the determination of the structural model.39 For Rietveld structural refinements, fractional coordinates that are not fixed using symmetry were refined independently for all atomic sites alongside their atomic displacement parameters. The fractional occupancy for the barium atomic sites was fixed to its nominal value. The fractional occupancy of M1 and M3 transition metal sites was respectively fixed to 1 and shared between Nb and Mo according to their stoichiometry, whereas the fractional occupancies of both M2 and M4 sites (if observed in the structural model) were constrained to 1 i.e. M2 + M4 = 1, and respectively shared between Nb, Mo and V (in the vanadium-doped samples). Likewise, the fractional occupancies of O2, O3 and O4 oxygen sites were respectively fixed to 1 and O1 and O5 were constrained to a nominal value of 2.05 in such a way that [fraction(O(1)) × multiplicity(O(1))] + [fraction(O(5)) × multiplicity(O(5))] = 2.05.
Magic Angle Spinning (MAS) 51V and 93Nb NMR spectra were recorded with a Bruker Ascend 500 spectrometer (B0 = 11.7 T) with a NEO console operating at a resonance frequency of 131.557 MHz (51V) and 122.291 MHz (93Nb), respectively. The samples were spun at desired frequencies in a 3.2 mm rotor. A Bruker two-channel DVT 3.2 mm MAS probe was used for all measurements. The chemical shifts were referenced with respect to V2O5 (99.99%, Aldrich) with an isotropic chemical shift of −613.2 ppm and NaNbO3 (99.997%, Puratronic®) with an isotropic chemical shift of −1130.7 ppm. A solid π/2 pulse length of 0.5 μs was used for 93Nb NMR data acquisition. For 51V measurement, a rotor-synchronised-solid-echo (π/2 – τ – π/2) pulse sequence, with a solid π/2 pulse length of 1.5 μs, was used for data acquisition. The acquired 51V spectra were simulated using the program DMFit.40 We report δ as the reduced anisotropy in the Haeberlen Convention, also referred to as axiality of the CSA tensor (δ33− δiso) in the framework of the DMFit program.
The landscape for oxide-ion dynamics was probed using the softBV41 software. This software utilises a force-field algorithm to compute possible conduction pathways and relative energy barriers for ionic conduction in model crystallographic structures.42,43 The computation method implemented in softBV is based on the bond-valence sum (BVS) approach as described in ref. 41 and 43. The structural models obtained from Rietveld refinement serve as the input file in which the BVSE landscapes for a probe oxide-ion were calculated. A dense grid of 10 points with a resolution of 0.1 Å was used for the calculations. The diffusion pathways were determined from the regions of low BVSE, which were performed by both visualising the connectivity of the iso-surfaces and examining the calculated pathway segments. The BVSE maps were visualised using VESTA,44 and the BVSE model of the migration barrier was plotted from the energy profile of the diffusion pathways.
Results/discussion
X-ray diffraction was employed to assess the crystalline phases that resulted from the synthesis of samples in the Ba7Nb3.9−xVxMo1.1O20.05 series. As shown in Fig. 2a, all five patterns exhibit the intended hexagonal-based perovskite as the main phase. Analysis of these patterns provides information about the evolution of lattice parameters in the Ba7Nb3.9−xVxMo1.1O20.05 series, as depicted in Fig. 2b. The lattice parameters of the unit cell are refined using a Le Bail fit38 adopting the Pm1 space group. Taking into consideration the Shannon ionic radii of both Nb5+ (0.640 Å) and V5+ (0.540 Å) in a six-fold coordination or Nb5+ (0.480 Å) and V5+ (0.355 Å) in four-fold coordination,45 the lattice parameters are expected to change towards lower values upon niobium substitution with vanadium. While this expectation is fulfilled for the a and b lattice parameters, which decrease from 5.858 Å for x = 0 to 5.851 Å for x = 0.4, an opposite trend is observed in the c direction, with an increase of the c lattice parameter from 16.509 Å to 16.530 Å. These trends suggest that vanadium incorporation might affect the oxygen distribution between O1 and O5 sites and possibly the metal (Nb, Mo, and V) distribution in M1, M2, M3 and M4 sites, which could conceivably have a non-trivial effect on the lattice volume and shape (i.e. non-Vegard-law).
 | ||
| Fig. 2 X-ray diffraction analysis of the synthesized Ba7Nb3.9−xVxMo1.1O20.05 series as a function of x. (a) XRD patterns, (b) evolution of a, b, and c lattice parameters as a function of x and (c) percentage of the main phase. Impurity phases are included in refinement tables and figures in the ESI.† | ||
The expansion along the c direction indicates a reorganization within the structure to accommodate the isovalent dopant. Upon inspection of the structural model proposed by Garcia-González et al.32 in Fig. 1h, one can deduce that this expansion could be related to the occupation of additional cation sites (e.g. M4) and/or a re-ordering of the oxygen sites within the palmierite-like layer, leading to the repulsion of like ions and the consequent expansion along this direction. Furthermore, this opposing trend for a and c lattice parameters results in only a minor volume change (∼0.1%) upon vanadium chemical substitution despite the difference in the ionic radii of Nb5+ and V5+ (Fig. S1†).
Fig. 2c displays the percentage purity of each sample prepared, with respect to the intended phase, as determined by Rietveld refinement (Fig. S2–S10 and Tables S1–S10†). The presence of impurity phases such as BaMoO4, Ba3Nb2O8 (and potentially its solid solution with V: Ba3Nb2−xVxO8) and Ba5Nb4O15 was evidenced (Fig. 2a). While a phase purity above 98% was attained for compositions with x ≤ 0.2, the formation of side phases was more pronounced for x = 0.3 and x = 0.4. We also determined that a phase of Ba7Nb3.9−xVxMo1.1O20.05 cannot be obtained for the V end member of the series (x = 3.9 – ESI Fig. S11†), which results in the stoichiometric formation of Ba3V2O8 and BaMoO4 phases instead. This observation indicates that a high vanadium content destabilizes the 7H polytype and implies a solubility limit for V in the low x range. Given this fact, the following questions can be raised about the distribution of vanadium in the substituted samples: is it located in the main or side phases, and in which local coordination environment can we find it?
These two questions, related to both the location of vanadium and origin of structural changes for the 7H polytype upon vanadium modification, will be addressed in the following sections. For the former, we use 51V MAS NMR as a local-structure characterization method sensitive to the local coordination of V5+. For the latter, we perform an in-depth analysis of combined X-ray and neutron diffraction patterns, leveraging the insight provided by NMR spectroscopy.
51V MAS NMR spectroscopy
Since NMR spectroscopy is sensitive to interactions between the probed nuclei and their immediate surroundings, it offers advantages over diffraction techniques to gauge the local structure of particular ions in materials, especially when structural disorder may be present.46,4751V MAS NMR has been used to derive information about the coordination geometry of vanadium in several studies.48–50 Given the prominent role of chemical shift anisotropy (CSA) in 51V NMR spectra, the evaluation of this parameter plays a key role in the identification of the coordination environment for vanadium.51 In particular, Lapina et al.48 thoroughly compiled CSA tensor parameters of NMR spectra of 51V and related them to the local coordination environment of vanadium. In detail, coordination environments with six neighbouring oxide anions in an octahedral shape (i.e. V2O5, Fig. S12†) are related to a reduced chemical shift anisotropy (δ) in the range of 300 to 1000 ppm, whereas significantly smaller values are observed for tetrahedral environments (δ < 100 ppm).48Fig. 3a shows the 51V MAS NMR spectrum acquired for Ba7Nb3.8V0.1Mo1.1O20.05, which is characteristic of the series. The isotropic line is marked with an arrow situated at around −577 ppm. This line is flanked by a symmetric spinning sidebands envelope of significantly lower intensity, with a span of around 1000 ppm. Considering the hypothesis that 51V NMR spectra are often dominated by the chemical shift anisotropy, the 51V NMR spectrum of Ba7Nb3.8V0.1Mo1.1O20.05 was simulated with only CSA tensor parameters. In this first attempt (Fig. 3b), the distinct intensity between the isotropic peak and spinning sidebands could be reproduced by a small anisotropy, with fitted parameters of δ = 135.50 ppm and η = 0.97. However, these CSA tensor parameters considerably underestimate the span of the spinning sideband (SSB) envelope from experiments. Conversely, increasing the chemical shift anisotropy in order to fit a broader SSB envelope results in a poor fit of the relative intensities of SSB and the isotropic centre band (Fig. 3c). These observations suggest that the CSA interaction is not a dominant trait in the spectra for 51V nuclei in the compounds of interest.
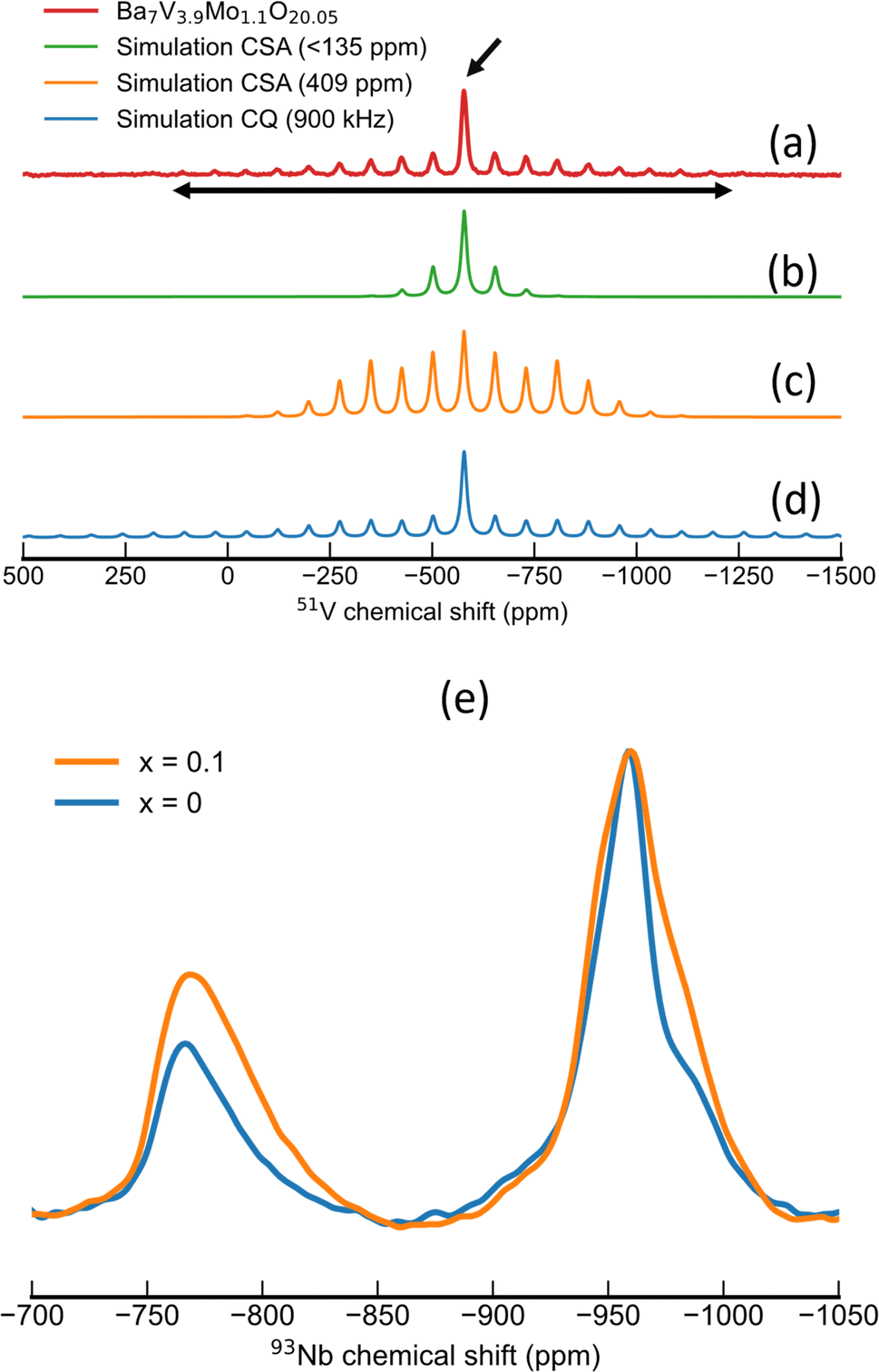 | ||
| Fig. 3 (a–d) 51V MAS NMR spectra of Ba7Nb3.8V0.1Mo1.1O20.05 spun at 10 kHz. Experimental spectrum (a) fitted considering chemical shift anisotropy (b and c) or 1st order quadrupolar interaction (d). The isotropic line is indicated by a slanted, single-headed arrow. The horizontal, double-headed arrow depicts the span of spinning sidebands as a measure for the magnitude of NMR interaction (either chemical shift or quadrupolar coupling). (e) 93Nb MAS NMR spectra recorded with a MAS frequency of 38 kHz, with a noticeable intensity increase for tetrahedral NbO4 environments upon V-substitution. | ||
The 51V MAS NMR spectrum of Ba7Nb3.8V0.1Mo1.1O20.05 could be more appropriately simulated considering only the quadrupolar interaction to first order (Fig. 3d). With this interaction, both the relative intensity of the centre band and spinning sidebands, as well as the span of SSBs, could be well reproduced without the need to include the CSA interaction. From the span and shape of the spinning sideband envelope, a CQ value of 0.9 MHz with an asymmetry parameter (ηQ) of 0.75 could be extracted from the spectrum with a MAS frequency of 10 kHz. Moreover, the fact that CSA was not needed to simulate these spectra indicates a small magnitude for this interaction, in the order of 135 ppm or smaller. A similar pattern was observed across the series Ba7Nb3.9−xVxMo1.1O20.05 with (0.1 ≤ x ≤ 0.4). The corresponding fitted parameters and spectra can be found in the ESI (Table S11 and Fig. S13†).
In light of the empirical correlations from Lapina et al.48 between the magnitude of the CSA and coordination number for oxy-vanadate and from the observation of small, if not negligible CSA parameters in the 51V NMR spectra for the Ba7Nb3.9−xVxMo1.1O20.05 (0.1 ≤ x ≤ 0.4) series, we conclude that all V5+ cations are located in an isolated tetrahedral environment for the 7H polytype samples investigated. We can thus deduce that V5+ is exclusively localised along the palmierite-like layer (M2 site; see Fig. 1h). This conclusion is further corroborated by the observation of a similar pattern of spinning sidebands when analysing the outcome of Ba3V2O8 (Fig. S14†) which features only VO4 units in its structure.52
Given the fact that V5+ exclusively occupies tetrahedral sites, as indicated by the 51V NMR spectra discussed above, this preferential coordination is expected to impact the occupation of equivalent cationic sites populated by other metal atoms throughout the structure. As V5+ substitutes for Nb5+ in the nominal compositions and given the high sensitivity of the latter for local structure investigation via93Nb NMR, we proceed with the analysis of the Nb5+ coordination number.
In the 93Nb MAS NMR spectra in Fig. 3e (also in Fig. S18 and S19†), we observe two broad signals which can be assigned to the central transition of 93Nb in two distinct local environments. One of these signals has a maximum at −765 ppm, which can be assigned to a NbO4 coordination following empirical trends of 93Nb chemical shift.53 This signal exhibits the following: a rather featureless line shape, slight asymmetry and skewness towards low frequencies. These features can be rationalized in terms of the Czjzek model54 and are characteristic of disordered local environments, thus implying a distribution of Nb–O bond lengths or angles for the tetrahedral Nb5+ site. The second 93Nb NMR signal is located at −958 ppm, which is a chemical shift range typical for octahedral coordination.55 Interestingly, this signal presents some finer structures, e.g. a significant shoulder at around −980 ppm. This feature is detected in previous reports on 93Nb NMR of Ba7Nb4MoO20-based composition.56,57 This feature may indicate a well-structured local environment for Nb5+ in octahedral environments, as a feature expected of Nb5+ occupying for example M1, M3 and/or M4 sites.
From the premise that V exclusively occupies tetrahedral sites, one could expect its substitution for Nb to decrease the relative amount of NbO4 moieties. To test this hypothesis, we evaluated the area of 93Nb NMR signals assigned to tetrahedral and octahedral Nb5+ environments, respectively (Fig. 3e). For the pristine composition (x = 0), we observe a tetrahedral-to-octahedral ratio of 25:75 for the area of the respective central transitions. Contrastingly, we observe an increase in the relative intensity of the tetrahedral 93Nb NMR signal and consequently an increase in the tetrahedral-to-octahedral ratio towards 33:67 in the V5+ substituted compositions (Fig. S19†). This trend implies that the presence of vanadium in tetrahedral environments influences the local environment of equivalent sites (i.e. M2); namely, it appears that the preferential occupation of tetrahedral sites by vanadium influences the oxygen sublattice in such a way that it promotes a tetrahedral coordination also in M2 sites occupied by Nb5+. This hypothesis is further evaluated with particular attention to the occupation of O1 and O5 sites along the palmierite layer.
Furthermore, spectral features of the central transition diminished upon vanadium substitution. This is a behaviour often observed58 upon both chemical substitution and the consequent increase in structural disorder in adjacent atomic sites, thus supporting the argument of V5+ substitution having a long-range effect on the oxygen sublattice.
Ba3Nb2−xVxO8 side phase
A vanadium-rich side phase could also be detected. The 51V chemical shifts of Ba3V2O8 and 7H polytype samples are −601.5 and −577.6 ppm, respectively, which can be well resolved in the 51V MAS NMR spectra (Fig. S15†). Despite the similarity in the coordination number, which reflects similar CSA and CQ values, the evaluation of the isotropic chemical shift for the Ba3V2O8 and 7H polytype samples still allows for a clear distinction for the local environment of V5+ between these two oxide structures. Moreover, the observation of a shoulder at approximately −600 ppm for 51V MAS NMR spectra of the 7H polytype samples, whose intensity increases with the substituent concentration (Fig S15b–d†), suggests that Ba3V2O8 (or the isostructural solid-solution Ba3Nb2−xVxO8) is the most probable V-rich side phase—albeit its presence in the sub % concentration as estimated from the spectra in Fig. S15.† We thus further utilize the 51V MAS NMR spectra to quantify the amount of Ba3Nb2−xVxO8 impurities (see Table 1 and Ba3Nb2−xVxO8 side phase discussion in the ESI†).| Nominal composition | Lattice parameters (a, b, and c) of the Ba3Nb2−xVxO8 side phase | x in Ba3Nb2−xVxO8 from Vegard's law | % of Ba3Nb2−xVxO8 from Rietveld refinement | Corrected composition XRD | Corrected composition NMR | |
|---|---|---|---|---|---|---|
| a The Ba3Nb1.9V0.1O8 side phase is not observed for Ba7Nb3.8V0.1Mo1.1O20.05 within the detection limit. | ||||||
| 0 | — | — | — | — | — | — |
| 0.1a | — | — | — | — | — | — |
| 0.2 | 5.9032 | 21.3088 | 1.0369 | 3.50 | 0.16 | 0.18 |
| 0.3 | 5.9012 | 21.2897 | 1.0512 | 4.80 | 0.25 | 0.22 |
| 0.4 | 5.9055 | 21.3055 | 1.0201 | 5.70 | 0.34 | 0.37 |
The resulting actual composition of Ba3Nb2−xVxO8 solid solution is fixed in the structural model of Ba3Nb2−xVxO8 for the further Rietveld refinement of the diffraction patterns. Based on the corrected stoichiometry and knowledge of the local environment of vanadium in the Ba7Nb3.9−xVxMo1.1O20.05 (0 ≤ x ≤ 0.4) series, the combined refinement of both the X-ray and neutron diffraction is performed in a self-consistent way to gauge the impact of vanadium doping on the oxygen sub-lattice of the Ba7Nb3.9−xVxMo1.1O20.05 (0 ≤ x ≤ 0.4) series.
Structure refinement from combined X-ray and neutron diffraction
Next, we perform a combined Rietveld refinement for X-ray and neutron diffraction patterns for all five compositions with 0 < x < 0.4, in order to analyse the impact of the preferential occupation of tetrahedral sites by vanadium on the global structure of these hexagonal-based perovskite oxides of the 7H polytype. This analysis was performed both at room temperature and at an elevated temperature (527 °C, neutrons only), which is representative of the intermediate temperature range for the operation of SOFCs.Base composition
Starting with the pristine sample Ba7Nb3.9Mo1.1O20.05, a combined neutron and X-ray refinement was performed (Fig. 4a) using the structural model (Fig. 4b) described by Garcia-González et al.32 It is worth noting that it is hard to establish the site ordering of each cation (Nb5+ and Mo6+) in Ba7Nb3.9Mo1.1O20.05 because of their similar electron density and coherent neutron scattering cross-section59 meaning that their relative distribution in the metal sites does not affect the quality of the structural refinements. After fitting the structural model by Garcia-González et al.32 to the observed patterns, a large thermal displacement parameter (Uiso = 0.081(8) Å2) was observed for the O1 site (Fig. 4b) which was initially at the Wyckoff position 2d (1/3, 2/3, 0.4862(10)). A similar observation has been reported for 7H polytype Ba7Nb4MoO20.22,32 Thus, a 3-fold split was applied on the O1 site, which assigns it to Wyckoff position 6i (0.31729, 0.63458, 0.98719), depicted in Fig. 4c. Fitting this to the observed data resulted in a more realistic thermal displacement parameter [0.053(9) Å2] for the O1 site. An examination of the neutron difference Fourier map shows two areas of missing scattering density in real space. One of these is at the Wyckoff position 3e (1/2, 0, 0) (Fig. 4d), and it was tagged as O5. The other reveals an additional metal atom, tagged M4, at the Wyckoff position 2d (1/3, 2/3, ∼0.2) (Fig. 4e). The Rietveld refinement of this model resulted in an even better agreement between both the observed and calculated patterns (Fig. 4a). It is worth noting that occupation of O5 and M4 sites has also been reported in Ba7Nb4MoO20 (ref. 22) (Fig. 1h). | ||
| Fig. 4 (a) A plot of combined Rietveld refinement of both the X-ray and neutron diffraction patterns of Ba7Nb3.9Mo1.1O20.05 at 25 °C (x = 0). The crystallographic model (b) proposed by Garcia-González et al.32 and (c) derived in this work (Table S2†). The different Fourier maps observed in the [001] direction: (d) and (e) maps are calculated from the crystallographic model proposed by Garcia-González et al.32 These maps evidenced missing scattering densities of both O5 and M4 from the proposed model. The values of z for (d) and (e) are 0 and ∼0.2. | ||
From the 93Nb ss-NMR observation on the pristine sample, a tetrahedral-to-octahedral ratio of 24.5:75.5 was observed for the local environment of Nb. This implies that 24.5% of the nominal stoichiometry of Nb (0.96/3.9 per formula unit) occupies the tetrahedrally coordinated M2 site alongside Mo and the remaining fraction (2.94)—alongside Mo—is distributed over the octahedrally coordinated M1, M2, M3 and M4 sites. This information about the local environment of Nb5+ thus results in the splitting of the M2 site into M2-tet and M2-oct, respectively. And this splitting leads to the creation of additional refinement constraints such that the occupation of M2-tet is equal to O1 occupation (Fig. 5b).
 | ||
| Fig. 5 Oxygen and metal-ion distribution in Ba7Nb3.9−xVxMo1.1O20.05 (a) ab plane depiction of the thermal reorganisation of oxide-ion site disorder in Ba7Nb3.9Mo1.1O20.05. (b) Bar plots depicting the percentage occupancy of oxide-ion disorder and cations in the mixed coordinated M2 site in the Ba7Nb3.9−xVxMo1.1O20.05 series at 25 °C. | ||
In the obtained crystallographic model (Fig. 4c), the distance between M2 and M4 is approximately 1.659 Å and both cationic sites are partially occupied, leading to face-sharing polyhedral units. Also, the distance between the partially occupied O1 and O5 sites is approximately 1.866 Å. The short distance between both the M2–M4 and O1–O5 indicates that they should not be occupied simultaneously. The presence of O1 and O5 sites will lead to mixed coordination polyhedra for the transition d-block metal cation at the M2 site in the palmierite-like layers of Ba7Nb3.9Mo1.1O20.05. On an average scale, occupation of O1 will lead to a tetrahedral coordination geometry of the M2 site (e.g. M2(O2)3O1). Contrastingly, occupation of the O5 site will result in an octahedral coordination geometry for the M2 site cation (e.g. M2(O2)3(O5)3). This observation is analogous to that reported for the local atomic scale of the palmierite derivative of 9R hexagonal perovskite, Ba3MoNbO8.5, based on data from the neutron pair-distribution function.60 From this comparison between the 9R and the present 7H perovskites, a mix of four- and six-fold local coordination environments is expected for M2 cations in the palmierite-like layers of Ba7Nb3.9−xVxMo1.1O20.05.
After the evaluation of the room temperature structure, we proceed with the refinement of the structural model for the neutron diffraction patterns of Ba7Nb3.9Mo1.1O20.05 recorded at 527 °C using the same approach. However, it is worth noting that the Nb tetrahedral-to-octahedral ratio and splitting of the M2 site into tetrahedral and octahedral were not considered, as we do not have 93Nb NMR data at this temperature. A thermal reorganisation of O1 and O5 sites was observed. This rearrangement led to a situation where O1 is more occupied and O5 is less occupied at 527 °C, when compared to 25 °C (Fig. S16†), resulting effectively in the exclusive tetrahedral coordination for the M2 site at higher temperatures. A similar behaviour has been observed for Ba7Nb4MoO20 by Fop et al.22 Furthermore, the cationic M2 and M4 sites were also discovered to reorganise upon increasing temperature (Fig. S16†), and a similar feature has been proposed by Fop et al. in Ba7Nb4MoO20.22
Vanadium-based compositions
Following the analysis of the base composition, neutron and X-ray diffraction patterns recorded at 25 °C for the vanadium-doped series Ba7Nb3.9−xVxMo1.1O20.05 (0.1 ≤ x ≤ 0.4) were analysed in search of a refined structural model. It is worth noting that it is hard to establish the site ordering of V5+ in the Ba7Nb3.9−xVxMo1.1O20.05 series with diffraction techniques. The first reason is the fact that the percentage of V5+ in each composition of the studied series is so small that refining the fractional occupancies of V5+ in the X-ray diffraction model will be unstable even though the electronic density of V5+ is distinct from that of either Nb5+ or Mo6+. Also, vanadium has a small coherent neutron scattering cross-section with a value of 0.0184 barn,59 thus, making the Bragg intensities in NPD as well as the Rietveld refinement insensitive to the location of the vanadium nucleus compared to either niobium or molybdenum nuclei in the crystal structure; hence, highlighting the importance of insight from 51V MAS NMR for a more in-depth description of the structure.From the 51V NMR parameters, it was observed that vanadium in the Ba7Nb3.9−xVxMo1.1O20.05 series is found in a tetrahedral coordination environment. Based on this observation, the corrected stoichiometry of vanadium (Table 1 and ESI†) in each series was fixed at the M2-tet site of the structural models used in the refinement. The structural models obtained from these refinements are similar to the one derived from pristine Ba7Nb3.9Mo1.1O20.05. However, a correlation can be found between the vanadium content and the relative occupation of O1 and O5 sites for the Ba7Nb3.9−xVxMo1.1O20.05 series at 25 °C (Fig. 5b). For samples with x = 0.25 and 0.34, the percentage of O1 occupation increased by ∼25% in comparison to the pristine sample with x = 0. It is thus apparent that increasing vanadium content increases the tetrahedral M2(O2)3O1 units in the average structure of the 7H polytype samples. This observation of O1–O5 rearrangement is analogous to what was reported in the Ba3Nb1−xVxMoO8.5 series of hexagonal perovskites at room temperature.21
Furthermore, a thermal reorganization of O1 and O5 sites was observed in the Ba7Nb3.9−xVxMo1.1O20.05 (0 ≤ x ≤ 0.4) series at 527 °C (Fig. S16†), where the fractional occupancy of the O5 site is approaching almost zero and O1 site approaching almost 100%. While it is evident that in the pristine Ba7Nb3.9Mo1.1O20.05 composition at 527 °C the tetrahedral M2(O2)3O1 is the prevalent coordination geometry for cations at the M2 site in the palmierite-like layer, it is interesting to note that this behaviour is observed already at much lower temperatures for the V-based compositions. In other words, the combined analysis of neutron and X-ray diffraction data suggests that introduction of vanadium to the structure alters the oxygen sublattice in a way to mimic the high-temperature structure already at room temperature; hence, reproducing relevant structural features only observed in the high-temperature phase and hence potentially related to ionic conductivity.
BVSE
The migration pathways and associated energy barriers for oxide-ion conduction in the average crystal structure of several Ba7Nb4MoO20-based hexagonal perovskites18–20,34 have been estimated using the Bond Valence Site Energy (BVSE) approach on the softBV program.41–43 This program presents the resulting BVSE values of the mobile species (oxide-ions) as an energy landscape, which provides information about the connecting local minima, saddle points (identified using fractional coordinate values), and relative energy barriers along possible conduction pathways.43 The reliability of this method as a probe for ionic conduction pathways in inorganic conductors is ascertained by its consistency with results obtained from techniques such as molecular dynamics simulations and the maximum entropy method.61,62The conduction of oxide ions in Ba7Nb4MoO20-based materials has been predicted, using the BVSE approach, to proceed predominantly in a two-dimensional fashion involving the average crystallographic oxygen sites in the palmierite-like layers.18–20,34 In a bid to correlate the average crystallographic structure of Ba7Nb4MoO20-based hexagonal perovskites with ionic conduction properties, we implemented the BVSE calculation to study the influence of vanadium-doping on the bond-valence site energy landscape for oxide-ion migration in the average crystal structure of the Ba7Nb3.9−xVxMo1.1O20.05 series. Starting with Ba7Nb3.9Mo1.1O20.05, a representative of the series, a BVSE landscape for the interaction of probe oxide-ions was calculated using the refined structural model described in Fig. 4c.
Fig. 6 depicts the BVSE landscape for oxide-ions and associated conduction pathways obtained for Ba7Nb3.9Mo1.1O20.05. In the framework of the BVSE calculation, the disordered O1 and O5 sites in the diffraction-refined model, which are responsible for the mixed coordination of the M2 site (Fig. 4c), are both combined into a partially occupied site tagged O5’ (Fig. 6a). This O5′ site is approximately at the same position as the O5 site from the refined model, only slightly displaced, resulting in double multiplicity (O5′ at (0.486, 0.514, 0.018), Wyckoff 6i; O5 at (1/2, 0, 0), Wyckoff 3e). The O5′ site can be considered an ellipsoidal minimum in the oxygen energy landscape, bridged by a negligible (0.012 eV – Fig. 6c) barrier, denoted as s1 and located at the coordinates of the refined O5.
 | ||
| Fig. 6 Bond-valence site energy map calculated for Ba7Nb3.9Mo1.1O20.05 as seen along the (a) c-axis and (b) a-axis. (c) Bond-valence energy landscape of Ba7Nb3.9Mo1.1O20.05 depicting both two- and three-dimensional conduction pathways. | ||
Interestingly, the BVSE calculation does not identify a local minimum in energy around the coordinates of O1, as would be expected for an appreciably occupied crystallographic position. The reasons for this discrepancy between the diffraction-derived model and the BVSE-derived oxygen positions are not obvious, but the same results have been consistently observed for analogous Ba7Nb4MoO20-based 7H materials in the literature.18–20,34 In an attempt, we hypothesize that the BVSE approach is unable to capture the electronic rearrangement of the transition metal in M2 involved in changing its bonding from octahedral (O5) to tetrahedral (O1) coordination, integral to the oxide-ion conduction process; as such, the predominant octahedrally coordinating site (O5) is identified as most stable. The remaining oxygen sites in the structure along with their respective energies and occupancies are listed in Table S13† and exhibit close correspondence between the diffraction- and BVSE-derived structure models.
Furthermore, two-dimensional conduction pathways that involve the diffusion of oxide-ions via the partially occupied O5′ sites along the ab plane in the palmierite-like layer were evidenced in the computed BVSE map and landscape, consistent with similar studies on Ba7Nb4MoO20-related oxide-ion conductors.18–20,34 This 2D-migration pathway has a significantly lower energy barrier than any of the 3D-pathways identified in Fig. 6c, confirming the view of Ba7Nb4MoO20-based 7H materials as 2D conductors. The two-dimensional conduction pathway identified involves two saddle points, tagged s1 and s2 (Fig. 6a and c). The s1 saddle point can be correlated with the local oscillation of oxide-ions in an ellipsoidal minimum, as mentioned above, while the s2 saddle point corresponds to the migration of oxide-ions between different O5′ sites along the O1 position and as such is our main descriptor for oxide-ion mobility in the structure.
Fig. 7 depicts the bond-valence energy landscape for the Ba7Nb3.9−xVxMo1.1O20.05 series and the respective energy barriers for s1 and s2 saddle points from the refined structure models as a function of vanadium content. It can be observed that the relative energy barrier of s1 is lower than that of s2 for every member of the series (Fig. 7a and b). Whereas the energy barrier for the s1 saddle point increases linearly with increasing vanadium content in the models, the energy barrier of the s2 saddle point decreases linearly (Fig. 7b). The linear decrease of the energy barrier of the s2 saddle point (Fig. 7b) upon increasing vanadium content suggests that the overall barrier for migration of oxide-ions is lowered by the presence of V5+. This observation strongly suggests enhanced oxide-ion mobility in the palmierite-like layer of the Ba7Nb3.9−xVxMo1.1O20.05 series with increasing vanadium content.
 | ||
| Fig. 7 (a) The bond-valence energy landscape for the Ba7Nb3.9−xVxMo1.1O20.05 series illustrating the two saddle points and their respective energy barriers for two-dimensional conduction pathways, and (b) a plot showing the linear variation of the relative bond valence energy of s1 and s2 saddle points as a function of vanadium content in the Ba7Nb3.9−xVxMo1.1O20.05 series. | ||
Given the fact that a similar bond-valence energy landscape is observed for the structural model reported by Yashima et al. for the base composition (x = 0) of this 7H polytype,18 we expand our BVSE analysis by also considering the potential effect of vanadium occupation on the various metal cation sites of their structural model. In resonance with the trend seen for the relative energy barrier of the s2 saddle in the present structure model, the relative energy barrier of the s2 saddle point for the former model also decreases upon increasing the nominal composition of vanadium at the average M2 crystallographic site (Fig. S17†).
Conclusion
In this work, we assessed the feasibility of vanadium substitution into the hexagonal-based perovskite oxide of the 7H polytype. We successfully synthesized the Ba7Nb3.9−xVxMo1.1O20.05 series in the 0 ≤ x ≤ 0.4 range and thoroughly characterized its average and local structure by a combination of solid-state NMR, X-ray and neutron diffraction.Based on parameters from 51V MAS NMR, we identified a tetrahedral coordination for V5+ cations as a chemical substituent for Nb5+, indicating its presence in the palmierite-like layers of the structure. Also, it was observed that the preferential occupation of vanadium influences the local environment of adjacent cations in the palmierite layers such that a tetrahedral coordination geometry is enforced on the Nb cation in the M2 site, with 93Nb NMR data reinforcing this trend. Combining insight from NMR and XRD, we recognized a solubility limit for vanadium in the low x range and identified and quantified V-rich side phases.
Information on the local structures of both vanadium and niobium was essential for the accurate description of the average structure of the main phase and its evolution as a function of V content. A combined X-ray and neutron Rietveld refinement for the undoped sample demonstrated the presence of two distinct oxygen sites along the palmierite-like layer (O1 and O5), whose occupation leads to a mix of four- and six-fold coordination, respectively, for the adjacent M2 cation site. At high temperature (527 °C), a reorganization of oxygen disorder is observed and O1 is found to be more occupied, resulting effectively in a tetrahedral coordination of the M2 site. The same behaviour is observed upon V-substitution, but already at room-temperature, implying that the presence of vanadium impacts the oxygen sub-lattice so as to mimic the high-temperature structure, thereby reproducing at lower temperature structural features conducive to high ionic conductivity.
Our analysis is extended using BVSE calculations to shed light on potential conduction pathways for oxide ions. We found a decreasing energy barrier for O2− migration when vanadium is present in the structure, namely at the M2 site next to the palmierite-like layers, which is the only site able to accommodate a tetrahedral coordination for this substituent. This observation corroborates the hypothesis that vanadium's propensity for a lower coordination number is beneficial for promoting high O2− mobility in this promising class of oxide ion conductors and prompts the study of its effect on the ionic conductivity of this and analogous oxide-ion conductors.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. G. and A. O. B. acknowledge financial support from the Dutch Research Council (NWO) for the ECCM Tenure Track funding under project number ECCM.TT.ECCM.006. T. F. acknowledges the funding provided by the European Union's HORIZON EUROPE programme in the form of a Marie Skłodowska-Curie individual postdoctoral fellowship (project no. 101066486). The authors would like to thank Michel Thijs and Robert Dankelman for support with the neutron powder diffraction measurements.Notes and references
- R. Mark Ormerod, Solid oxide fuel cells, Chem. Soc. Rev., 2003, 32, 17–28, 10.1039/B105764M
.
- O. Yamamoto, Solid oxide fuel cells: fundamental aspects and prospects, Electrochim. Acta, 2000, 45, 2423–2435, DOI:10.1016/S0013-4686(00)00330-3
- E. Ivers-Tiffée, A. Weber and D. Herbstritt, Materials and technologies for SOFC-components, J. Eur. Ceram. Soc., 2001, 21, 1805–1811, DOI:10.1016/S0955-2219(01)00120-0
- B. C. H. Steele and A. Heinzel, Materials for fuel-cell technologies, Nature, 2001, 414, 345–352, DOI:10.1038/35104620
- A. J. Jacobson, Materials for Solid Oxide Fuel Cells, Chem. Mater., 2010, 22, 660–674, DOI:10.1021/cm902640j
- D. J. L. Brett, A. Atkinson, N. P. Brandon and S. J. Skinner, Intermediate temperature solid oxide fuel cells, Chem. Soc. Rev., 2008, 37, 1568–1578, 10.1039/B612060C
- L. Malavasi, C. A. J. Fisher and M. S. Islam, Oxide-ion and proton conducting electrolyte materials for clean energy applications: structural and mechanistic features, Chem. Soc. Rev., 2010, 39, 4370–4387, 10.1039/B915141A
- F. Liang, J. Yang, Y. Zhao, Y. Zhou, Z. Yan, J. He, Q. Yuan, J. Wu, P. Liu, Z. Zhong and M. Han, A review of thin film electrolytes fabricated by physical vapor deposition for solid oxide fuel cells, Int. J. Hydrogen Energy, 2022, 47, 36926–36952, DOI:10.1016/j.ijhydene.2022.08.237
- Y. Yang, Y. Zhang and M. Yan, A review on the preparation of thin-film YSZ electrolyte of SOFCs by magnetron sputtering technology, Sep. Purif. Technol., 2022, 298, 121627, DOI:10.1016/j.seppur.2022.121627
- J. Zhang, S. Ricote, P. V. Hendriksen and Y. Chen, Advanced Materials for Thin-Film Solid Oxide Fuel Cells: Recent Progress and Challenges in Boosting the Device Performance at Low Temperatures, Adv. Funct. Mater., 2022, 32, 2111205, DOI:10.1002/adfm.202111205
- H. Shi, C. Su, R. Ran, J. Cao and Z. Shao, Electrolyte materials for intermediate-temperature solid oxide fuel cells, Prog. Nat. Sci.: Mater. Int., 2020, 30, 764–774, DOI:10.1016/j.pnsc.2020.09.003
- P. r. Slater, J. e. h. Sansom and J. r. Tolchard, Development of apatite-type oxide ion conductors, Chem. Rec., 2004, 4, 373–384, DOI:10.1002/tcr.20028
- F. Abraham, J. C. Boivin, G. Mairesse and G. Nowogrocki, The bimevox series: a new family of high performances oxide ion conductors, Solid State Ionics, 1990, 40–41, 934–937, DOI:10.1016/0167-2738(90)90157-M
- S. Georges, F. Goutenoire, O. Bohnke, M. C. Steil, S. J. Skinner, H.-D. Wiemhoefer and P. Lacorre, The LAMOX Family of Fast Oxide-Ion Conductors: Overview and Recent Results, J. New Mater. Electrochem. Syst., 2004, 7(1), 51–57 CAS
- F. Goutenoire, O. Isnard, E. Suard, O. Bohnke, Y. Laligant, R. Retoux and P. Lacorre, Structural and transport characteristics of the LAMOX family of fast oxide-ion conductors, based on lanthanum molybdenum oxide La2Mo2O9 basis of a presentation given at Materials Discussion No. 3, 26–29 September, 2000, University of Cambridge, UK., J. Mater. Chem., 2001, 11, 119–124, 10.1039/B002962I
- M. Morales, J. J. Roa, J. Tartaj and M. Segarra, A review of doped lanthanum gallates as electrolytes for intermediate temperature solid oxides fuel cells: from materials processing to electrical and thermo-mechanical properties, J. Eur. Ceram. Soc., 2016, 36, 1–16, DOI:10.1016/j.jeurceramsoc.2015.09.025
- S. Fop, K. S. McCombie, E. J. Wildman, J. M. S. Skakle and A. C. Mclaughlin, Hexagonal perovskite derivatives: a new direction in the design of oxide ion conducting materials, Chem. Commun., 2019, 55, 2127–2137, 10.1039/C8CC09534E
- M. Yashima, T. Tsujiguchi, Y. Sakuda, Y. Yasui, Y. Zhou, K. Fujii, S. Torii, T. Kamiyama and S. J. Skinner, High oxide-ion conductivity through the interstitial oxygen site in Ba7Nb4MoO20-based hexagonal perovskite related oxides, Nat. Commun., 2021, 12, 556, DOI:10.1038/s41467-020-20859-w
- T. Murakami, T. Shibata, Y. Yasui, K. Fujii, J. R. Hester and M. Yashima, High Oxide-Ion Conductivity in a Hexagonal Perovskite-Related Oxide Ba7Ta3.7Mo1.3O20.15 with Cation Site Preference and Interstitial Oxide Ions, Small, 2022, 18, 2106785, DOI:10.1002/smll.202106785
- Y. Sakuda, J. R. Hester and M. Yashima, Improved oxide-ion and lower proton conduction of hexagonal perovskite-related oxides based on Ba7Nb4MoO20 by Cr6+ doping, J. Ceram. Soc. Jpn., 2022, 130, 442–447, DOI:10.2109/jcersj2.21192
- S. Fop, K. McCombie, R. I. Smith and A. C. Mclaughlin, Enhanced Oxygen Ion Conductivity and Mechanistic Understanding in Ba3Nb1−xVxMoO8.5, Chem. Mater., 2020, 32, 4724–4733, DOI:10.1021/acs.chemmater.0c01322
- S. Fop, K. S. McCombie, E. J. Wildman, J. M. S. Skakle, J. T. S. Irvine, P. A. Connor, C. Savaniu, C. Ritter and A. C. Mclaughlin, High oxide ion and proton conductivity in a disordered hexagonal perovskite, Nat. Mater., 2020, 19, 752–757, DOI:10.1038/s41563-020-0629-4
- K. S. McCombie, E. J. Wildman, S. Fop, R. I. Smith, J. M. S. Skakle and A. C. Mclaughlin, The crystal structure and electrical properties of the oxide ion conductor Ba3WNbO8.5, J. Mater. Chem. A, 2018, 6, 5290–5295, 10.1039/C7TA08989A
- K. S. McCombie, E. J. Wildman, C. Ritter, R. I. Smith, J. M. S. Skakle and A. C. Mclaughlin, Relationship between the Crystal Structure and Electrical Properties of Oxide Ion Conducting Ba3W1.2Nb0.8O8.6, Inorg. Chem., 2018, 57, 11942–11947, DOI:10.1021/acs.inorgchem.8b01366
- A. Bernasconi, C. Tealdi, M. Mühlbauer and L. Malavasi, Synthesis, crystal structure and ionic conductivity of the Ba3Mo1−xWxNbO8.5 solid solution, J. Solid State Chem., 2018, 258, 628–633, DOI:10.1016/j.jssc.2017.11.030
- S. Fop, E. J. Wildman, J. T. S. Irvine, P. A. Connor, J. M. S. Skakle, C. Ritter and A. C. Mclaughlin, Investigation of the Relationship between the Structure and Conductivity of the Novel Oxide Ionic Conductor Ba3MoNbO8.5, Chem. Mater., 2017, 29, 4146–4152, DOI:10.1021/acs.chemmater.7b01298
-
R. H. Mitchell, Perovskites: Modern and Ancient, Almaz Press, 2002 Search PubMed
- A. Gilane, S. Fop, F. Sher, R. I. Smith and A. C. Mclaughlin, The relationship between oxide-ion conductivity and cation vacancy order in the hybrid hexagonal perovskite Ba3VWO8.5, J. Mater. Chem. A, 2020, 8, 16506–16514, 10.1039/D0TA05581F
- D. N. Tawse, A. Gilane, S. Fop, A. Martinez-Felipe, F. Sher, R. I. Smith and A. C. Mclaughlin, Investigation of the Crystal Structure and Ionic Pathways of the Hexagonal Perovskite Derivative Ba3–xVMoO8.5−x, Inorg. Chem., 2021, 60, 13550–13556, DOI:10.1021/acs.inorgchem.1c01840
- B. Sherwood, E. J. Wildman, R. I. Smith and A. C. Mclaughlin, Enhanced Oxide Ion Conductivity by Ta Doping of Ba3Nb1–xTaxMoO8.5, Inorg. Chem., 2023, 62(4), 1628–1635, DOI:10.1021/acs.inorgchem.2c03943
- S. Fop, J. M. S. Skakle, A. C. McLaughlin, P. A. Connor, J. T. S. Irvine, R. I. Smith and E. J. Wildman, Oxide Ion Conductivity in the Hexagonal Perovskite Derivative Ba3MoNbO8.5, J. Am. Chem. Soc., 2016, 138, 16764–16769, DOI:10.1021/jacs.6b10730
- E. García-González, M. Parras and J. M. González-Calbet, Crystal Structure of an Unusual Polytype: 7H-Ba7Nb4MoO20, Chem. Mater., 1999, 11, 433–437, DOI:10.1021/cm981011i
- A. Bernasconi, C. Tealdi and L. Malavasi, High-Temperature Structural Evolution in the Ba3Mo(1–x)WxNbO8.5 System and Correlation with Ionic Transport Properties, Inorg. Chem., 2018, 57, 6746–6752, DOI:10.1021/acs.inorgchem.8b01093
- Y. Suzuki, T. Murakami, K. Fujii, J. R. Hester, Y. Yasui and M. Yashima, Simultaneous Reduction of Proton Conductivity and Enhancement of Oxide-Ion Conductivity by Aliovalent Doping in Ba7Nb4MoO20, Inorg. Chem., 2022, 61, 7537–7545, DOI:10.1021/acs.inorgchem.2c00671
- D. Waroquiers, X. Gonze, G.-M. Rignanese, C. Welker-Nieuwoudt, F. Rosowski, M. Göbel, S. Schenk, P. Degelmann, R. André, R. Glaum and G. Hautier, Statistical Analysis of Coordination Environments in Oxides, Chem. Mater., 2017, 29, 8346–8360, DOI:10.1021/acs.chemmater.7b02766
- L. van Eijck, L. D. Cussen, G. J. Sykora, E. M. Schooneveld, N. J. Rhodes, A. A. van Well and C. Pappas, Design and performance of a novel neutron powder diffractometer: PEARL at TU Delft, J. Appl. Crystallogr., 2016, 49, 1398–1401, DOI:10.1107/S160057671601089X
- B. H. Toby and R. B. Von Dreele, GSAS-II: the genesis of a modern open-source all purpose crystallography software package, J. Appl. Crystallogr., 2013, 46, 544–549, DOI:10.1107/S0021889813003531
- A. Le Bail, H. Duroy and J. L. Fourquet, Ab-initio structure determination of LiSbWO6 by X-ray powder diffraction, Mater. Res. Bull., 1988, 23, 447–452, DOI:10.1016/0025-5408(88)90019-0
- H. M. Rietveld, A profile refinement method for nuclear and magnetic structures, J. Appl. Crystallogr., 1969, 2, 65–71, DOI:10.1107/S0021889869006558
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Modelling one- and two-dimensional solid-state NMR spectra, Magn. Reson. Chem., 2002, 40, 70–76, DOI:10.1002/mrc.984
- H. Chen, L. L. Wong and S. Adams, SoftBV – a software tool for screening the materials genome of inorganic fast ion conductors, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 18–33, DOI:10.1107/S2052520618015718
-
S. Adams and R. P. Rao, Understanding Ionic Conduction and Energy Storage Materials with Bond-Valence-Based Methods, in: I. D. Brown and K. R. Poeppelmeier ed. Bond Valences, Springer, Berlin, Heidelberg, 2014: pp. 129–159. doi: DOI:10.1007/430_2013_137
- S. Adams, From bond valence maps to energy landscapes for mobile ions in ion-conducting solids, Solid State Ionics, 2006, 177, 1625–1630, DOI:10.1016/j.ssi.2006.03.054
- K. Momma and F. Izumi,
VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276, DOI:10.1107/S0021889811038970
- R. D. Shannon and C. T. Prewitt, Effective ionic radii in oxides and fluorides, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 925–946, DOI:10.1107/S0567740869003220
-
A. Abragam, The Principles of Nuclear Magnetism, Clarendon Press, 1961 Search PubMed
-
C. P. Slichter, Principles of Magnetic Resonance, Springer Science & Business Media, 2013 Search PubMed
- O. B. Lapina, D. F. Khabibulin and A. A. Shubin, Modern solid-state NMR of quadrupolar nuclei, J. Struct. Chem., 2010, 51, 28–46, DOI:10.1007/s10947-010-0187-8
- O. B. Lapina, D. F. Khabibulin, A. A. Shubin and V. V. Terskikh, Practical aspects of 51V
and 93Nb solid-state NMR spectroscopy and applications to oxide materials, Prog. Nucl. Magn. Reson. Spectrosc., 2008, 53, 128–191, DOI:10.1016/j.pnmrs.2007.12.001
- O. B. Lapina, V. M. Mastikhin, A. A. Shubin, V. N. Krasilnikov and K. I. Zamaraev, 51V Solid state NMR studies of vanadia based catalysts, Prog. Nucl. Magn. Reson. Spectrosc., 1992, 24, 457–525, DOI:10.1016/0079-6565(92)80008-4
-
R. E. Wasylishen, S. E. Ashbrook and S. Wimperis, NMR of Quadrupolar Nuclei in Solid Materials, Wiley, Hoboken, NJ, 2012 Search PubMed
- S. Fop, J. A. Dawson, D. N. Tawse, M. G. Skellern, J. M. S. Skakle and A. C. Mclaughlin, Proton and Oxide Ion Conductivity in Palmierite Oxides, Chem. Mater., 2022, 34, 8190–8197, DOI:10.1021/acs.chemmater.2c01218
- O. B. Lapina, D. F. Khabibulin, K. V. Romanenko, Z. Gan, M. G. Zuev, V. N. Krasil’nikov and V. E. Fedorov, 93Nb NMR chemical shift scale for niobia systems, Solid State Nucl. Magn. Reson., 2005, 28, 204–224, DOI:10.1016/j.ssnmr.2005.09.003
- J.-B. d'Espinose de Lacaillerie, C. Fretigny and D. Massiot, MAS NMR spectra of quadrupolar nuclei in disordered solids: the Czjzek model, J. Magn. Reson., 2008, 192, 244–251, DOI:10.1016/j.jmr.2008.03.001
- O. B. Lapina, D. F. Khabibulin, K. V. Romanenko, Z. Gan, M. G. Zuev, V. N. Krasil’nikov and V. E. Fedorov, 93Nb NMR chemical shift scale for niobia systems, Solid State Nucl. Magn. Reson., 2005, 28, 204–224, DOI:10.1016/j.ssnmr.2005.09.003
- I. Oikawa, T. Nakajima, A. Ishii and H. Takamura, Acceptor Doping and Local Structure in Ba7Nb4MoO20 Investigated by 93Nb NMR, Spectroscopy, 2024, DOI:10.2139/ssrn.4683603.
- Y. Yasui, M. Tansho, K. Fujii, Y. Sakuda, A. Goto, S. Ohki, Y. Mogami, T. Iijima, S. Kobayashi, S. Kawaguchi, K. Osaka, K. Ikeda, T. Otomo and M. Yashima, Hidden chemical order in disordered Ba7Nb4MoO20 revealed by resonant X-ray diffraction and solid-state NMR, Nat. Commun., 2023, 14, 2337, DOI:10.1038/s41467-023-37802-4
- V. Veerapandiyan, M. N. Popov, F. Mayer, J. Spitaler, S. Svirskas, V. Kalendra, J. Lins, G. Canu, M. T. Buscaglia, M. Pasciak, J. Banys, P. B. Groszewicz, V. Buscaglia, J. Hlinka and M. Deluca, Origin of Relaxor Behavior in Barium-Titanate-Based Lead-Free Perovskites, Adv. Electron. Mater., 2022, 8, 2100812, DOI:10.1002/aelm.202100812
- V. F. Sears, Neutron scattering lengths and cross sections, J. Neutron Res., 1992, 3, 26–37, DOI:10.1080/10448639208218770
- M. S. Chambers, K. S. McCombie, J. E. Auckett, A. C. McLaughlin, J. T. S. Irvine, P. A. Chater, J. S. O. Evans and I. R. Evans, Hexagonal perovskite related oxide ion conductor Ba3NbMoO8.5: phase transition, temperature evolution of the local structure and properties, J. Mater. Chem. A, 2019, 7, 25503–25510, 10.1039/C9TA08378B
- W. Zhang, K. Fujii, E. Niwa, M. Hagihala, T. Kamiyama and M. Yashima, Oxide-ion conduction in the Dion–Jacobson phase CsBi2Ti2NbO10−δ, Nat. Commun., 2020, 11, 1224, DOI:10.1038/s41467-020-15043-z
- W. Uno, K. Fujii, E. Niwa, S. Torii, P. Miao, T. Kamiyama and M. Yashima, Experimental visualization of oxide-ion diffusion paths in pyrochlore-type Yb2Ti2O7, J. Ceram. Soc. Jpn., 2018, 126, 341–345, DOI:10.2109/jcersj2.17253
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01540a |
| This journal is © The Royal Society of Chemistry 2024 |