
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTwo new Cd-based metal–organic frameworks for afterglow detection of Fe3+ and NH3†
Wen-Qi
Zhang
 a,
Bo-Lun
Zhang
b,
Ting
Wang
a,
Jun
Chen
b,
Zhong-Yi
Li
*c,
Rui-Hong
Wang
a,
Shuqin
Liu
b and
Jian-Jun
Zhang
*a
a,
Bo-Lun
Zhang
b,
Ting
Wang
a,
Jun
Chen
b,
Zhong-Yi
Li
*c,
Rui-Hong
Wang
a,
Shuqin
Liu
b and
Jian-Jun
Zhang
*a
aSchool of Chemistry, Dalian University of Technology, Dalian 116024, China. E-mail: zhangjj@dlut.edu.cn
bSchool of Chemical Engineering, Dalian University of Technology, Dalian 116024, China
cCollege of Chemistry and Chemical Engineering, Shangqiu Normal University, Shangqiu 476000, China. E-mail: lizhyi84@163.com
First published on 13th February 2024
Abstract
Luminescent metal–organic frameworks (LMOFs) have been extensively employed as detection probes. However, the majority of the reported probes are fluorescence-based, lacking reports on visible afterglow detection. Herein, we present two new afterglow MOFs, compound 1 with a layered structure and 2 with an interpenetrated dia framework. 1 exhibits blue-green room temperature phosphorescence (RTP) (τ485nm = 264.6 ms) and can serve as an afterglow “turn off” probe for selective detection of Fe3+ ions through a framework decomposition mechanism, with a detection limit of 1.63 μM. Displaying blue-green RTP (τ495nm = 83.0 μs), 2 can serve as an afterglow “turn on” probe for the detection of NH3 in both solution and vapor phases. Notably, the afterglow of 2 is visually activated even at concentrations of NH3 vapor as low as 6 ppm. Mechanistic investigations reveal that the “turn on” response is attributed to structural transformation induced by NH3 molecules replacing coordinated solvents. Furthermore, their potential applications were demonstrated through the development of portable test strips for detecting Fe3+ ions and an cryptographic algorithm logic gate for NH3 deciphering. This work opens up new opportunities for the development of LMOF-based afterglow probes.
Introduction
Iron is a vital trace element in living organisms, playing a crucial role in various biological processes including oxygen transportation, cell metabolism,1etc. However, both excessive and insufficient levels of Fe3+ ions in living organisms can contribute to the development of certain diseases.2 Therefore, it is imperative to accurately detect trace amounts of Fe3+ ions. On the other hand, ammonia is a colorless and strongly pungent gas, and is also one of the most widely produced chemicals in the world.3 As per the regulations set by the Occupational Safety and Health Administration (OSHA) in the United States, an acceptable upper limit for human exposure to NH3 within 8 hours is 25 ppm.4 Prolonged exposure to excessive amounts of ammonia can have detrimental effects on the human body. Henceforth, there exists a critical need to develop methods to detect NH3. Currently, a range of analytical techniques including chromatography,5 atomic absorption spectroscopy and electrochemical6 methods are utilized for the detection of Fe3+ and NH3. However, these methodologies entail high costs and operational complexity. In contrast, luminescence detection offers advantages such as rapid detection, convenient sample preparation, and high sensitivity.7–9 In recent years, many luminescent probes have been reported for the detection of Fe3+ and NH3.10–13 However, most of them rely on fluorescence with decay lifetimes on the nanosecond scale or conventional phosphorescence with decay lifetimes on the microsecond scale, which are susceptible to interference from scattered light and background fluorescence.14,15 Conversely, long-lived (>10 ms) probes exhibit a larger Stokes shift and longer lifetime, effectively overcoming this limitation. Moreover, the change in afterglow during detection facilitates naked-eye observation. However, hitherto, there are limited reports on long-lived probes,16,17 and their construction continues to pose a formidable challenge.Long-lived room temperature phosphorescence (LLRTP) materials exhibit persistent visible luminescence even after excitation ceases, thereby offering extensive potential for applications in biological imaging, information encryption, optical devices and sensing.18–20 The primary consideration in the design of such materials lies in promoting the generation and stabilization of triplet excitons. Typically, the introduction of heavy atoms and/or heteroatoms effectively enhances spin–orbit coupling (SOC) and promotes intersystem crossing (ISC), consequently increasing the production of triplet excitons.21,22 Through structural hardening, clustering, cross-linking, and other techniques, a more rigid environment can be created to restrict molecular vibrations and stabilize triplet excitons.23–25 These measures effectively enhance the afterglow performance of the material. Various long-lived RTP materials have been reported thus far, including organic compounds, carbon dots, polymers, and inorganic–organic hybrids.26–29 Among them, metal–organic frameworks (MOFs), as a novel class of afterglow materials, have garnered significant attention recently due to their coordination bond-reinforced rigid structures that effectively suppress molecular vibrations and minimize energy dissipation from excited states.30 Furthermore, the uniform distribution of metal ions within the structure can effectively facilitate the ISC process. These attributes, combined with the inherent features of MOFs such as facile synthesis, tunable and adaptable structures, and high porosity,31–33 confer unique advantages to MOF-based afterglow materials compared to their other counterparts. Some MOF-based afterglow materials have been reported thus far;34,35 however, a significant challenge remains in developing MOFs with larger pore sizes to unlock their full potential for afterglow detection applications.
Luminescent MOFs are considered to be one of the most promising materials in the field of sensing because of their abundant excited state energy levels, and unique pore/channel structures which enable them to selectively capture and preconcentrate analyte molecules.36 However, most of the reported MOF-based probes rely on fluorescence or conventional phosphorescence (generally based on lanthanide or noble metal ions) and thus have short-lived emissions, with very few exceptions of the probes having lifetime levels of 100 ms.37–40 For instance, Xu et al. employed MOF-5 as an afterglow “turn on” probe for detecting Pb2+ ions with an impressive low detection limit of 2 nM; nevertheless, the probe's lifetime post-detection was only 2.45 ms.39 Similarly, Wu and his team utilized a Pb-MOF to detect I−via RTP quenching where the corresponding RTP lifetimes before and after detection were measured to be 3.70 and 2.90 ms respectively.40 Although some MOF-based probes have been reported for Fe3+ and NH3 detection,41–43 they are predominantly short-lived probes; thus far, no long-lived (>10 ms) MOF-based probes capable of detecting the two analytes have been reported. Considering the immense potential offered by combining afterglow with MOF-based probes in sensing applications and recognizing the significance associated with Fe3+ and NH3 detection, it is of great attraction and necessity to construct afterglow MOF-based probes for their detection.
Our group has recently focused on the study of inorganic–organic hybrid afterglow materials.44,45 Herein, H2L (H2L = 4-(5,7-dioxo-5,7-dihydroimidazo[4,5-f]isoindol-6(1H)-yl)benzoic acid) with RTP and abundant coordination sites was selected as the ligand. Through different bindings of ligands to Cd2+ ions, [Cd(L)(DMSO)2] (1) with a two-dimensional (2D) structure and [CdL(H2O)(DMF)0.5 (DMSO)0.5]·DMF (2) featuring an intriguing 3D interpenetrated framework with rhombus channels (∼10 × 10 Å2) were successfully synthesized. The close packing and porous structure of the two compounds lead to their different afterglow properties, which can be employed for different sensing purposes. 1 and 2 exhibit blue-green afterglow (τ = 264.6 ms) with a quantum yield (QY) of 11.94% and relatively weak blue-green RTP (τ = 83.0 μs) with a QY of 5.77%, respectively. Remarkably, 1 can serve as an afterglow “turn off” probe for selective detection of Fe3+ ions with a limit of detection (LOD) of 1.63 μM. On the other hand, 2 can be used as an afterglow “turn on” probe for detecting NH3 both in the solution and vapor state, where its solid-state afterglow is remarkably enhanced even at extremely low NH3 concentrations down to only 6 ppm (τ495nm = 11.9 ms). Mechanistic studies reveal that the quenching behavior of 1 is attributed to framework decomposition. In contrast, the “turn on” response of 2 arises from structural transformation triggered by NH3 molecules replacing the coordinated solvent. Details of preparation, characterization and preliminary application are presented.
Results and discussion
Crystal structures of the compounds
Single crystal X-ray diffraction (SCXRD) shows that 1 crystallizes in the orthorhombic Pbca space group (Fig. 1, Tables S1 and S2†) and has a 2D neutral layered structure. Each Cd2+ ion has a distorted octahedral {N2O4} coordination polyhedron and coordinates with two O atoms from the carboxylate group of one ligand, two O atoms from terminal coordinated DMSO molecules, and two N atoms from the imidazole group of two ligands. Each ligand coordinates with three Cd2+ ions, adopting a coordination mode described as μ3-κO2:κN1:κN1 (Fig. S1†). Both Cd2+ ions and ligands can be considered as 3-connected nodes since each Cd2+ ion is connected to three ligands and each ligand is linked to three metal ions. The arrangement of these nodes results in a honeycomb layered structure extending along the bc plane (Fig. 1(b) and S2(a)†). | ||
| Fig. 1 Structure of compound 1. (a) Coordination environment of Cd2+, symmetry code: A: −x + 1/2, −y+ 2, z− 1/2; B: X, −y+ 3/2, z − 1/2. (b) A 2D layer extending along the bc plane. (c) The packing of the layers viewed along the c direction. H atoms and S/C atoms of DMSO molecules are omitted for clarity. | ||
Compound 2 possesses a 3D neutral framework with two independent Cd2+ ions in its asymmetric unit (Fig. 2). The six-coordinated Cd1 exhibits a distorted octahedral geometry completed by four carboxylate O atoms from two ligands and two imidazole N atoms from other ligands. Each Cd2 ion also adopts a distorted octahedral {N2O4} donor set formed by coordinating with two O atoms from coordinated H2O molecules, two O atoms from coordinated organic solvent molecules (DMSO and DMF occupy positions statistically with occupancy factors of 0.5 each), and two N atoms from the ligands. The ligand also adopts a μ3-κO2:κN1:κN1 coordination mode and coordinates to three metal ions (Fig. S1†).
 | ||
| Fig. 2 Structure of compound 2. (a) Coordination environments of Cd2+ ions. Symmetry code: A: −x + 1, y, −z+ 1/2. B: −x + 1/2, y− 1/2, −z + 1/2. C: X + 1/2, y − 1/2, z. D: −x+ 1, y, −z − 1/2. A view of the single (b) and 2-fold interpenetrated (c) framework along the c direction. The two types of C–H⋯π interactions are represented as pink and green dotted lines, respectively. Some H atoms and S/C atoms of DMSO molecules are omitted for clarity. | ||
Each Cd2 connects with two L2− to form a {CdL2} supramolecule building block (SBB), which can be regarded as a 4-connected tetrahedral node. Additionally, Cd1 serves as another tetrahedral node connecting four {CdL2} SBBs together at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to construct the overall 3D neutral framework. Topological analysis reveals that the framework has a uninodal 4-connected dia network with a point symbol of 66 (Fig. S2b†).46 Along the c direction, the framework exhibits rhombus channels with dimensions of ∼15 × 15 Å2, providing ample space for accommodating another set of frameworks. Interestingly, the ligands from neighboring frameworks are closely stacked, resulting in strong quadruple edge-to-face π–π interactions. The corresponding H⋯Ph separations range from 2.92 to 2.98 Å. Even with the presence of 2-fold interpenetration, the resulting framework still has channels of ∼10 × 10 Å2 along the c direction, and the solvent accessible volume without guest molecules accounts for ∼22% of the unit cell volume as calculated by PLATON.47 Free DMF solvent molecules are captured in the channels.
:1 to construct the overall 3D neutral framework. Topological analysis reveals that the framework has a uninodal 4-connected dia network with a point symbol of 66 (Fig. S2b†).46 Along the c direction, the framework exhibits rhombus channels with dimensions of ∼15 × 15 Å2, providing ample space for accommodating another set of frameworks. Interestingly, the ligands from neighboring frameworks are closely stacked, resulting in strong quadruple edge-to-face π–π interactions. The corresponding H⋯Ph separations range from 2.92 to 2.98 Å. Even with the presence of 2-fold interpenetration, the resulting framework still has channels of ∼10 × 10 Å2 along the c direction, and the solvent accessible volume without guest molecules accounts for ∼22% of the unit cell volume as calculated by PLATON.47 Free DMF solvent molecules are captured in the channels.
Characterization of the compounds
Powder XRD (PXRD) was employed to verify the phase purity of the synthesized samples (Fig. S3†). The patterns of the as-synthesized samples match well with theoretical patterns derived from SCXRD data, indicating that the products have high purity. Subsequently, thermogravimetric analysis (TGA) was conducted to investigate the thermal stability of 1 and 2. As depicted in Fig. S4,†1 remains stable up to 180 °C before losing two coordinated DMSO molecules within a temperature range of 180 to 250 °C (obs. 25.6% and cal. 27.1%). After reaching a plateau in the range of 250–480 °C, further weight loss is observed signifying framework collapse. 2 loses 1 coordinated H2O molecule and 0.5 free DMF molecules within a temperature range of 60–100 °C (obs. 8.5% and cal. 9.3%). Then all remaining solvent molecules are removed in the range of 100–280 °C (obs. 8.5% and cal. 9.3%). Beyond 450 °C, the framework rapidly decomposes.Under the irradiation of a 365 nm UV lamp, the ligand and the two compounds emit blue-green, blue and blue light, respectively (Fig. 3(a)). After removing the UV lamp, the ligand and 1 emit visible blue-green afterglow lasting for ∼1.0 and ∼2.5 s respectively. In contrast, 2 can only emit a very short blue-green RTP. Then their solid-state PL spectra were measured (Fig. 3(b)). Strong emission in the prompt and delayed spectra peaked at 482 and 505 nm for H2L, 420 and 485 nm for 1, and 445 and 495 nm for 2 can be observed respectively. Note that 1 also has a shoulder delayed peak located at 525 nm. Compared with that of H2L, both 1 and 2 exhibit different degrees of blue-shift for the prompt and delayed emission. This can be attributed to the different coordination patterns of metal ions in compounds. Neither compound demonstrates excitation-dependent properties (Fig. S7†). The lifetimes of the delayed emission of H2L, 1 and 2 are 139.2 ms (505 nm), 264.6 ms (485 nm), and 83.0 μs (495 nm), respectively (Fig. 3(c) and S8†). Obviously, the unique binding of ligands to metal ions can result in varying afterglow lifetimes of the MOFs compared to that of the initial ligand, highlighting the remarkable ability of MOFs to control afterglow characteristics. The QYs of 1 and 2 are 11.94% and 5.77%, respectively (Fig. S9†).
 | ||
| Fig. 3 Luminescence properties of H2L, 1 and 2. (a) Photographs of the solid samples before and after turning off a 365 nm UV lamp under ambient conditions. (b) The prompt (solid line) and delayed (dotted line) spectra (λex = 365 nm); (c) the lifetime decay curves at 505 nm for H2L and 485 nm for 1. | ||
Although 1 and 2 were synthesized using the same ligand and metal ion, their luminescent properties differ significantly due to variations in stacking structures and intermolecular interactions between them.48,49 In the structure of 1, no significant hydrogen bond or π–π interaction can be observed. However, there is strong edge-to-face interaction between interpenetrated frameworks in the structure of 2. Such an interaction helps to stabilize the triplet state exciton and promote phosphorescence emission,50–52 but the presence of a large number of coordinated solvent molecules in the structure, especially H2O molecules, is not conducive to the emission, as O–H vibration can effectively consume the energy of the excited state. In addition, the framework of 2 also contains channels capable of trapping free solvent molecules whose vibrations further consume excited state energy. As a result, both the luminescence efficiency and RTP lifetime of 2 are not as good as those of 1.
The effects of heating on the luminescence properties of 1 and 2 were also investigated. First, 1 was heated at 150 °C for 20 min, and the resulting sample 1-150 °C shows the same PXRD pattern and green afterglow as that of the initial sample, although its prompt/delayed emission has a certain red shift (Fig. S10 and S13†). Subsequent soaking of 1-150 °C in DMSO can restore the initial luminescent behavior. Such results indicate that the structure and RTP properties of 1 exhibit good thermal stability. In contrast, the afterglow intensity and lifetime of the sample prepared by heating 1 at 250 °C (1-250 °C) are significantly reduced, accompanied by a noticeable red shift in prompt/delayed emission spectra. PXRD analysis confirms the destruction of the initial framework structure, highlighting the decisive role of coordinated solvent molecules in shaping both the structure and RTP behavior of 1. Although the structure can be restored after soaking 1-250 °C in DMSO, the afterglow properties cannot be restored (Fig. S11 and S13†). Solvent molecules also play a crucial role in the structure and luminescence behavior of 2, as it exhibits irreversible luminescence changes upon heating at 105 °C (Fig. S12 and S13†).
The influence of solvent on the luminescence and structure stability of 1 and 2 was studied. First, the PXRD patterns of the samples soaked in various solvents (DMSO, DMF, DMAc, MeOH, EtOH and CH3CN) for 24 h were obtained (Fig. S3†). The results show that the pattern of soaked samples of 1 closely resembles that of the initial sample, indicating its excellent stability. For 2, although several new peaks can be observed in the soaked sample due to the solvation effect, the main diffraction peaks are still well maintained.
Subsequently, suspensions of both compounds in several organic solvents were prepared and their corresponding prompt/delayed spectra were recorded (Fig. S14†). Due to the solvent effect, the prompt and delayed emission peak of 1 in different solvents is located in the range of 420–440 nm and 480–510 nm, respectively. In contrast, 2 displays prompt emission peaks spanning from 428 nm to 478 nm in various solvents; however, only DMF and DMAc suspensions show detectable delayed emissions with peak wavelengths at ∼455 nm and ∼465 nm, respectively. Therefore, considering the stability, dispersibility, and low toxicity of 1 and 2 in the solvents, EtOH and DMAc were ultimately selected as dispersing solvents for subsequent luminescence sensing experiments, respectively. Furthermore, the delayed emission lifetimes of 1's EtOH suspensions and 2's DMAc suspensions were measured to be 175.4 ms (485 nm) and 434.6 μs (465 nm), respectively (Fig. S15†).
Afterglow detection of Fe3+ ions by compound 1
The sensing behaviors of 1 toward various metal ions (Ni2+, Zn2+, Cd2+, Co2+, Mn2+, Mg2+, Cu2+, K+, Li+, Na+, Ca2+, Al3+, Cr3+, Pb2+ and Fe3+) were studied. Under excitation with a UV lamp at 365 nm, only Fe3+ ions effectively quench the fluorescence of 1's suspension (Fig. S16†).Upon removal of the lamp, only Fe3+ ions can almost completely quench the afterglow of 1's EtOH suspension (Fig. 4(a)). In contrast, other metal ions have negligible influence on the fluorescence and afterglow of 1's suspensions. Then the prompt/delayed emission spectra of the above mixtures were tested. The impact efficiency of metal ions on 1's suspension was evaluated using the formula [I/I0 − 1] × 100% (I0 and I are the prompt/delayed emission intensity of 1's suspension before and after the addition of 1 mM metal ions). As depicted in Fig. 4, S17 and S18,† only Fe3+ ions exhibit a significant quenching effect on prompt/delayed emission from 1's suspension, while other metal ions show no discernible impact on such emissions as visually observed. The quenching efficiency of Fe3+ on 1's suspension is remarkably high, reaching 95.5% and 97.2% for prompt and delayed emissions, respectively. In contrast, Cu2+, Cr3+ and Ni2+ can only moderately quench the emission of the suspensions. The corresponding quenching efficiency is 20.2%, 13.3% and 10.7% (prompt) and 10.6%, 6.7% and 10.8% (delayed) for the three ions, respectively. Other metal ions only have a weak impact on the luminescence of the suspension. Notably, compared to the prompt spectrum, delayed luminescence demonstrates superior selectivity and higher quenching efficiency in detecting Fe3+ ions. Moreover, delayed emission detection effectively eliminates interference from scattered light and background luminescence, resulting in excellent sensitivity and signal-to-noise ratio performance. The above findings indicate that 1 can serve as an effective afterglow probe for Fe3+.
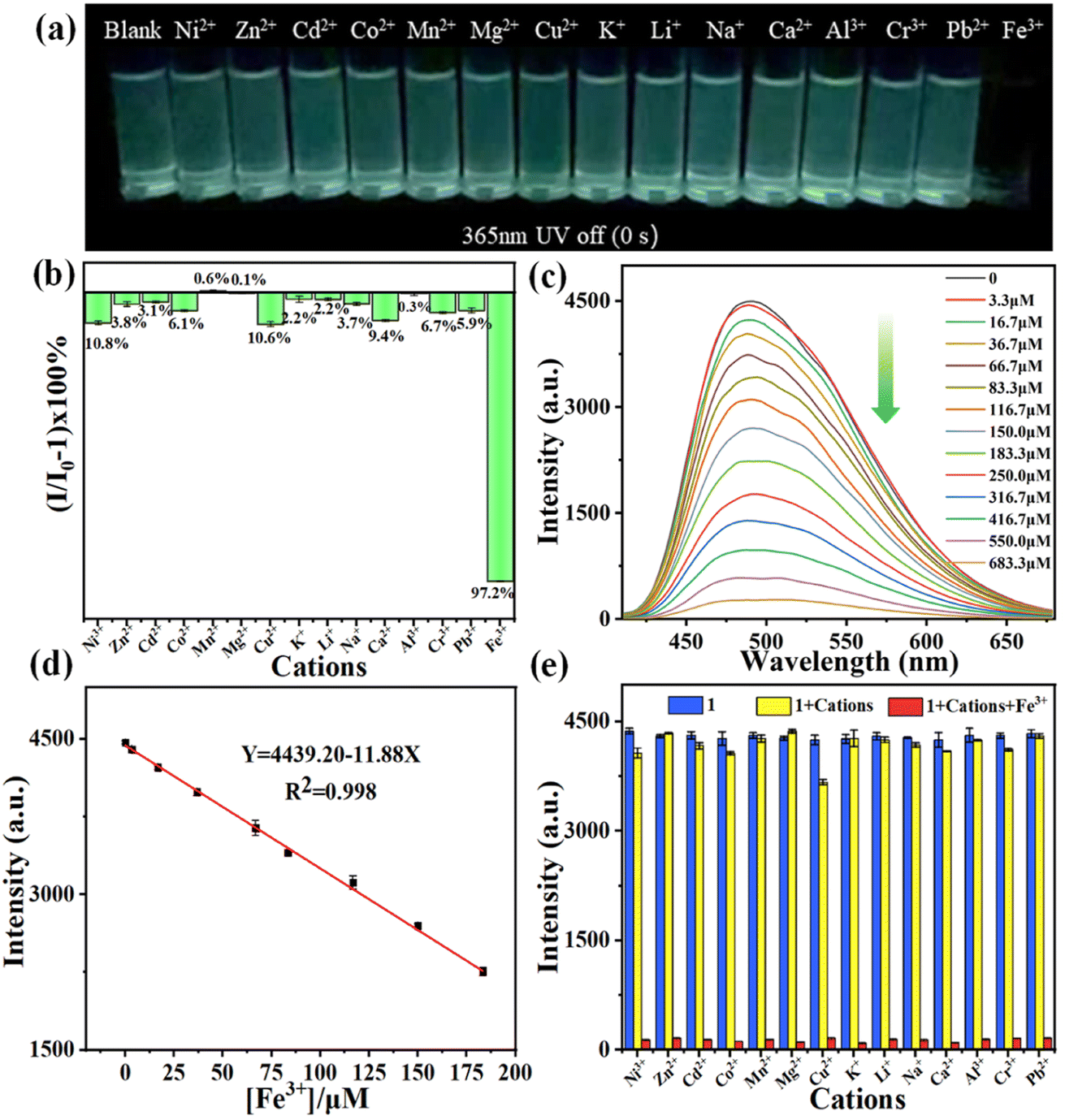 | ||
| Fig. 4 Detection of Fe3+ ions by 1. (a) Afterglow photos of 1's EtOH suspension in the presence of 1 mM various metal ions. (b) A comparison of delayed emission impact efficiency of 1's suspensions after the addition of various metal ions. (c) Delayed emission spectra of 1's suspension upon an incremental addition of Fe3+ solution (0–683.3 μM). (d) Linear relationships between the delayed emission intensity (I485) and concentration of Fe3+ (0–183 μM). (e) A comparison of the delayed emission intensities (I485) of 1's suspension containing different interferents (1 mM) before and after the addition of 1 mM Fe3+. λex = 365 nm. | ||
The kinetic curve of 1's response to Fe3+ ions was further measured (Fig. S19(a)†). After the addition of 1 mM Fe3+ ions, the delayed emission of the suspension immediately decreases and reaches an equilibrium within ∼30 s, and remains stable in the following 30 min. Such a result indicates that 1 has a fast response for detection of Fe3+. To gain further insights into the delayed luminescence response of Fe3+ to 1, a luminescence titration experiment was conducted. The delayed emission intensity of 1's EtOH suspension was measured after adding different concentrations of Fe3+ solutions.
Obvious changes in luminescence intensity can be observed after the addition of 3.3 μM Fe3+ solution. As the concentration of Fe3+ gradually increases, the emission intensity at 485 nm weakens progressively, and the suspension gradually becomes clear. When the concentration increases to 683 μM, the emission intensity is almost completely quenched (Fig. 4(c)). Within the concentration range of 0–183 μM of Fe3+, there exists a linear correlation between concentration and delayed emission intensity in the suspension. The linear equation obtained from the fitting is I485 = 4439.20 − 11.88 × C[Fe3+], with a correlation coefficient of R2 = 0.99802 (Fig. 4(d)). According to LOD = 3σ/slope (where σ represents the relative standard deviation of delayed emission intensity of the blank sample in 10 tests), the LOD is calculated to be ∼1.63 μM. Then 1's anti-interference ability in detecting Fe3+ ions was studied (Fig. 4(e)). The quenching ability of Fe3+ on the delayed emission is almost unaffected by other metal ions, demonstrating its exceptional selectivity as an afterglow probe for Fe3+ ions.
To date, some reports have been published on MOF-based Fe3+ probes; however, to the best of our knowledge, almost all of them are either fluorescent or Ln-based phosphorescent probes with lifetimes in the range of several hundred microseconds (Table S5†). For instance, Chand et al. reported two 3D Cd-MOFs that serve as fluorescent “turn off” probes for detecting Al3+ and Fe3+.14 Kang and his team synthesized an Eu-MOF which can be utilized for Fe3+ detection with a LOD of 45 μM.10 However, such a detection based on short lifetime luminescence is susceptible to interference from scattered light and the fluorescence background. Additionally, there have been several reports on RTP-based Fe3+ detection. Wu et al. synthesized carbon dot RTP materials and employed them for Fe3+ detection through RTP quenching.11 Nevertheless, the specific structure of carbon dots cannot be determined, limiting their further development to some extent. In comparison with these probes, 1 possesses certain advantages as it is a MOF-based afterglow probe with a periodic structure, and can show more effective avoidance of interference from background luminescence. Moreover, its LOD is comparable to those of reported probes (Table S5†).
The mechanism of detecting Fe3+
To elucidate the mechanism behind Fe3+ detection, the following control experiments were conducted. The absorption spectrum of Fe3+ ions only slightly overlaps with the delayed emission spectrum of 1's suspension (Fig. S20†), indicating that there is only a weak resonance energy transfer (RET) between the probe and Fe3+. The significant overlap between the UV-vis absorption spectra of Fe3+ ions and 1 in the range of 240–500 nm, indicates that there may be a competitive absorption between them. In the aforementioned experiments, the rapid response of 1 to Fe3+ (∼30 s) indicates that it is almost impossible for a metal metathesis reaction to explain the RTP quenching. Most importantly, during the detection process, 1's suspension gradually becomes clear as the concentration of Fe3+ ions gradually increases, until reaching up to 4 mM at which concentration it transforms into a clear solution. Subsequently, a mixture of 1's suspension containing 683 μM Fe3+ was stirred for 20 minutes, followed by centrifugation. The resulting supernatant was subjected to mass spectrum measurement (Fig. S21†). The dominant peak observed at 306.05 can be attributed to [HL−]. Therefore, it can be reasonably inferred that the primary cause of RTP quenching is framework collapse; however, the contribution of competitive absorption cannot be disregarded. A similar structural collapse has also been reported in some MOF-based probes for metal ion detection.53,54Afterglow detection of NH3 by compound 2
The sensing behaviors of 2 towards NH3 and eight different amines (methylamine (MA), dimethylamine (DMA), trimethylamine (TMA), triethylamine (TEA), diisopropylamine (DIPA), di-n-propylamine(DPA), n-butylamine (BUA) and aniline (PhA)) were investigated. After the addition of 44.3 mM of NH3·H2O or various amines (Fig. S22†), the prompt emission of 2's suspensions exhibits varying degrees of indistinguishable enhancement. In contrast, only NH3·H2O can induce a ∼37 times increase of the delayed emission, while amine molecules only cause different degrees of “turn off” response (Fig. S23†). Such results indicate that 2 shows excellent selectivity towards NH3via delayed emission. Furthermore, the response kinetic test also shows a rapid response (<2 min) of 2 to NH3 (Fig. S19(b)†). Then the titration experiment was conducted to further evaluate the delayed luminescence sensing ability of 2 to NH3. As the concentration of NH3 gradually increases, the delayed emission intensity of the suspension also increases until reaching its maximum value at a concentration of 44.3 mM with a measured lifetime of 1.63 ms (Fig. S11(b)†). A linear correlation between the emission intensity and concentration of NH3 was observed within the range of concentration from 0 to 35.4 mM with a fitted linear equation I465 = 47.83 × C[NH3] + 42.36, and a correlation coefficient of R2 = 0.990 (Fig. 5(c)). The LOD was calculated to be approximately 0.40 mM. An anti-interference experiment was also performed. MA, DMA and BUA can significantly interfere with the detection of NH3, whereas other tested amines show no interference with the detection (Fig. 5(d)). | ||
| Fig. 5 Detection of NH3 by 2's DMAc suspension. (a) A comparison of delayed emission impact efficiency after the addition of 44.3 mM NH3·H2O or amine. (b) Delayed emission spectra of 2's suspension upon an incremental addition of NH3·H2O solution (0–53.2 mM). (c) Linear relationships between the delayed emission intensity (I465) and concentration of NH3·H2O (0–35.4 mM). (d) A comparison of the delayed emission intensities (I465) of 2's suspension containing different interferents (44.3 mM) before and after the addition of 44.3 mM NH3·H2O. λex = 365 nm. | ||
Additionally, the response of solid sample 2 to NH3 vapor was investigated. A sample holder loaded with 8 mg of 2 was used to measure its kinetic curve of the delayed luminescence response towards NH3 vapor (25% NH3·H2O) (Fig. S24†). Initially, there is a slight decrease in luminescence intensity which then gradually increases over time, reaching a maximum within 30 min, and then remains equilibrate. Subsequently, the response to trace amounts of NH3 vapor (3–27 ppm) was studied using the same amount of 2 with a contact time of 35 min (Fig. 6(a)).
 | ||
| Fig. 6 The sensing of NH3 vapor by a solid sample of 2. (a) Delayed emission spectra of 2 after 35 min of exposure to different concentrations of NH3 vapor (0–27 ppm). (b) The corresponding photos of 2 before and after turning off a 365 nm UV lamp. | ||
The results demonstrate that significant changes in luminescence intensity can be detected even when the concentration of NH3 vapor is as low as 3 ppm. At a concentration level reaching up to 6 ppm, afterglow changes can be clearly observed by the naked eye along with a corresponding lifetime of 11.9 ms. The luminescence intensity and lifetime increase as the concentration of NH3 vapor increases. The lifetime of the solid is up to 72.4 ms after detecting 24 ppm NH3 vapor (Fig. S8†). Remarkably, the NH3 vapor detection of 2 has a lower response concentration compared to the suspension detection, which may be due to the strong competition interference from DMAc and water molecules in suspension detection. In addition, molecular vibration in the suspension is stronger than that in the solid, resulting in a greater dissipation of excited state energy and less conduciveness to delayed emission.
So far, there have been some reports on NH3 probes based on MOFs (Table S6†). However, the majority of these studies focus on fluorescence or conventional phosphorescence. For example, Shustova et al. demonstrated the effectiveness of two MOFs as fluorescent probes for NH3 at high temperatures.15 Ibarra et al. also reported a luminescent Tb-MOF that can serve as a “turn off” probe for detecting NH3.12 In addition, the number of non-MOF RTP probes capable of NH3 detection is extremely limited. For instance, Wang et al. developed a bimetallic cluster compound consisting of a Ag10Cu6 core, which serves as an effective RTP quenching probe for the detection of NH3 in both vapor and solution states.13 The corresponding LODs are 53 ppm and 21 μM respectively. Compared with these probes, compound 2 in this paper is a rare example of a MOF-based probe with an afterglow “turn on” detection behavior for NH3, which is more conducive to naked eye observation.
The mechanism of detecting NH3
To elucidate the NH3 detection mechanism, a series of control experiments were conducted. (1) The ligand H2L was exposed to NH3 vapor under identical conditions, and its delayed spectrum was measured (Fig. S25†). In contrast to that of the initial H2L sample, no enhancement of RTP or change in the emission peak position can be observed in the fumigated samples. Thus, it can be inferred that the interaction between NH3 and the ligand does not affect the aforementioned sensing behavior. (2) Compared with that of the original sample, the PXRD pattern of the fumigated sample changes obviously (Fig. S3†). Even after washing with DMAc solvent, the initial pattern cannot be recovered (Fig. S26†). These results suggest an irreversible framework transformation induced by NH3 fumigation. (3) IR spectra were obtained for both pre- and post-fumigation samples with NH3 vapor (Fig. S27†). Notably, a substantial decrease in intensity at 1650 cm−1 associated with amide group stretching vibration can be observed in the fumigated sample compared to the original spectrum, which is attributed to the reduction of the DMF solvent molecule. Simultaneously, a new peak emerges at ∼586 cm−1 attributed to Cd–N bond stretching vibration, indicating that the NH3 molecule enters the pores of 2 and coordinates with Cd2+ ions. Furthermore, peaks at ∼1000 and ∼1540 cm−1 corresponding to the vibration (δ(HNH)) of the NH3 molecule demonstrate a significant increase in intensity.55 Additionally, UV-vis absorption spectra of the samples before and after contact with NH3 display notable differences (Fig. S28†). The results indicate that NH3 molecules enter the pore of 2, replace the solvent molecules in the original structure, and successfully coordinate with Cd2+ ions. (4) X-ray photoelectron spectroscopy (XPS) characterization reveals an increase in Cd 3d5/2 binding energy from 404.75 eV to 404.95 eV upon exposure to NH3 vapor indicating an altered coordination environment surrounding Cd2+ ions after fumigation (Fig. S29†). Furthermore, the O1s core level of 2 exhibits three components at 530.6 (Cd–O), 531.3 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, SO), and 532.5 (H2O) eV, respectively.56–58 After NH3 fumigation, a decrease in intensity is observed for the components at 530.6 and 532.5 eV, indicating a reduction in Cd–O bonds and coordinated water molecules.
O, C–O, SO), and 532.5 (H2O) eV, respectively.56–58 After NH3 fumigation, a decrease in intensity is observed for the components at 530.6 and 532.5 eV, indicating a reduction in Cd–O bonds and coordinated water molecules.
Based on these findings, it is reasonable to hypothesize that NH3 molecules permeate into the pores of 2, displacing coordinated solvents and forming coordination bonds with Cd2+ ions during the detection process, which triggers framework transformation and ultimately induces a “turn on” response in RTP.
Among all tested amines/NH3, only ammonia can induce an enhanced RTP response in 2 due to its smaller size compared to other amines tested herein; this facilitates easier entry into the channels of 2. Additionally, NH3 may exhibit stronger coordination ability with Cd2+ ions possessing potential coordination vacancies. Finally, trapped NH3 molecules are more likely to form hydrogen bonds within the framework compared to other amines tested herein, which can weaken molecular motion and minimize energy dissipation from excited states, thereby enhancing RTP accordingly. Similar beneficial effects of hydrogen bond networks on RTP have been reported in the literature.59,60
Application—test paper detection
The interesting afterglow detection properties of compound 1 for iron ions drove us to further investigate the possibility of using it to prepare test strips for rapid detection. A long strip of filter paper was immersed in 1's EtOH suspension (1 mg mL−1) for 1 hour, and then taken out to dry. The dried paper strips were used for subsequent sensing experiments. Different concentrations of Fe3+ solutions ranging from 1.0 × 10−3 to 1.0 × 10−2 M were applied onto the paper strips, resulting in a spot area of approximately 2 cm2 (Fig. 7(a)). Upon deactivating the 365 nm UV lamp, it can be visually observed that the test paper exhibits noticeable quenching for Fe3+ concentrations above 5.0 × 10−3 M, indicating a visible detection limit of around 4.2 × 10−6 g cm−2. Repeated experiments consistently yielded similar outcomes, thus highlighting the significant potential of 1 for on-site identification and measurement of Fe3+ ions. | ||
| Fig. 7 Photograph of the test papers after adding 30 μL different concentrations of (1.0 × 10−2–1.0 × 10−3 M) Fe3+ ions. (a) Under daylight (the red dashed box is a spot area of ∼2 cm2). (b) With and without 365 nm light irradiation. | ||
Encryption application triggered by NH3 fumigation
The different afterglow stimulation responses of compounds 1 and 2 to NH3 can be used to design cryptographic applications that are decrypted by NH3. As shown in Fig. 8, the painting “60” is generated using only 1 that has no afterglow response to NH3 stimulation. In contrast, paintings “80” and “88” are obtained by combining 1 and 2, wherein 1 fills the portion corresponding to number “00” and 2 fills the center horizontal line (the part within the orange dotted box). Upon illumination with a 365 nm UV lamp, blue numbers “60”, “80” and “88” representing A, B and C can be observed, respectively. Due to 1 having a longer blue-green afterglow and 2 having a very short RTP lifetime, the blue-green messages “60”, “00” and “00” will be displayed once the UV lamp is turned off. By applying an algorithm that calculates their sum, “A + B + C” equals “60” (this is denoted as encrypted message X1, X1 = 60). When decryption was performed with NH3 fumigation (>6 ppm, ∼30 min of exposure time), the RTP of 2 is “turned on” and a green afterglow lasting for ∼1.5 s can be observed by the naked eye. Therefore, in the time range of 0–1.5 s, the values of A, B and C are “60”, “80” and “88” respectively, resulting in the total output value being equal to “228” (denoted as encrypted message X2, X2 = 228). | ||
| Fig. 8 A schematic diagram for the process of information encryption to decryption. | ||
Once the turn off time exceeds 1.5 s, the observed information reverts back again displaying values as “60”, “00” and “00”, and a corresponding total output value equal to “60” (denoted as encrypted message X3, X3 = 60). Finally, by using the encryption algorithm “X2 − (X1 + X3)”, the true encrypted information “108” can be obtained.
Conclusions
In summary, two new Cd-MOFs, [Cd(L)(DMSO)2] (1) and [CdL(H2O)(DMF)0.5(DMSO)0.5]·DMF (2) based on a ligand featuring imidazole/carboxylate functional groups have been successfully prepared. 1 has a honeycomb layered structure, whereas 2 possess a 2-fold interpenetrated framework with dia topology. Photoluminescence investigations have demonstrated that the formation of distinct MOFs can effectively modulate the afterglow behavior of the ligand. Compared with that of the ligand, 1 has a blue-green RTP with an extended lifetime of 264.6 ms. Notably, this RTP emission selectively responds to Fe3+ ions and thus can serve as an afterglow “turn off” probe for detecting Fe3+ ions with a LOD of 1.63 μM. Conversely, after the formation of compound 2, the RTP lifetime of the ligand is reduced to 83.0 μs. This RTP shows a “turn on” response towards NH3, and can be employed as a probe to detect NH3 both in solution and vapor states. Interestingly, the afterglow shows a remarkable enhancement which can be visually observed when the NH3 concentration is as low as 6 ppm. Mechanistic studies reveal that the quenching behavior of 1 and the enhancement behavior of 2 in detection are attributed to framework decomposition and structural transformation, respectively. This work provides new insights into the development of MOF-based probes with exceptional afterglow detection capabilities.Author contributions
Wen-Qi Zhang: formal analysis, investigation, writing, original-draft, data curation, visualization. Bo-Lun Zhang: investigation. Ting Wang: investigation. Jun Chen: validation. Zhong-Yi Li: supervision. Shuqin Liu: validation. Rui-Hong Wang: investigation. Jian-Jun Zhang: conceptualization, supervision, writing – review & editing, project administration, resources, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Natural Science Foundation of China (21871038). Special thanks are due to the instrumental/data analysis from Instrumental Analysis Center of Dalian University of Technology.References
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- H. N. Abdelhamid, A. Bermejo-Gómez, B. Martín-Matute and X. Zou, Microchim. Acta, 2017, 184, 3363–3371 CrossRef CAS PubMed.
- J. Zhang, J. Ouyang, Y. Ye, Z. Li, Q. Lin, T. Chen, Z. Zhang and S. Xiang, ACS Appl. Mater. Interfaces, 2018, 10, 27465–27471 CrossRef CAS PubMed.
- S. Abdulla, T. L. Mathew and B. Pullithadathil, Sens. Actuators, B, 2015, 221, 1523–1534 CrossRef CAS.
- H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC.
- J. Gong, Y. Li, Z. Hu, Z. Zhou and Y. Deng, J. Phys. Chem. C, 2010, 114, 9970–9974 CrossRef CAS.
- L. Huang, B. Chen, X. Zhang, C. O. Trindle, F. Liao, Y. Wang, H. Miao, Y. Luo and G. Zhang, Angew. Chem., Int. Ed., 2018, 57, 16046–16050 CrossRef CAS PubMed.
- Y. Zhao, M.-Y. Wan, J.-P. Bai, H. Zeng, W. Lu and D. Li, J. Mater. Chem. A, 2019, 7, 11127–11133 RSC.
- Y.-S. Wong, M. Ng, M. C.-L. Yeung and V. W.-W. Yam, J. Am. Chem. Soc., 2021, 143, 973–982 CrossRef CAS PubMed.
- Y. Kang, X.-J. Zheng and L.-P. Jin, J. Colloid Interface Sci., 2016, 471, 1–6 CrossRef CAS PubMed.
- X. Wu, C. Ma, J. Liu, Y. Liu, S. Luo, M. Xu, P. Wu, W. Li and S. Liu, ACS Sustainable Chem. Eng., 2019, 7, 18801–18809 CrossRef CAS.
- I. A. Ibarra, T. W. Hesterberg, J.-S. Chang, J. W. Yoon, B. J. Holliday and S. M. Humphrey, Chem. Commun., 2013, 49, 7156 RSC.
- Y. Wang, J. J. Yan, S. Hu, D. James Young, H. X. Li and Z. G. Ren, Chem. - Asian J., 2021, 16, 1–7 CrossRef.
- S. Chand, M. Mondal, S. C. Pal, A. Pal, S. Maji, D. Mandal and M. C. Das, New J. Chem., 2018, 42, 12865–12871 RSC.
- N. B. Shustova, A. F. Cozzolino, S. Reineke, M. Baldo and M. Dincă, J. Am. Chem. Soc., 2013, 135, 13326–13329 CrossRef CAS PubMed.
- Q. Wu, H. Ma, K. Ling, N. Gan, Z. Cheng, L. Gu, S. Cai, Z. An, H. Shi and W. Huang, ACS Appl. Mater. Interfaces, 2018, 10, 33730–33736 CrossRef CAS PubMed.
- Y. Zheng, Z. Wang, J. Liu, Y. Zhang, L. Gao, C. Wang, X. Zheng, Q. Zhou, Y. Yang, Y. Li, H. Tang, L. Qu, Y. Zhao and C. Yang, ACS Appl. Mater. Interfaces, 2022, 14, 15706–15715 CrossRef CAS PubMed.
- Y. Wang, H. Gao, J. Yang, M. Fang, D. Ding, B. Z. Tang and Z. Li, Adv. Mater., 2021, 33, 2007811 CrossRef CAS PubMed.
- T. K. Pal, Mater. Chem. Front., 2023, 7, 405–441 RSC.
- L. J. Small, S. E. Henkelis, D. X. Rademacher, M. E. Schindelholz, J. L. Krumhansl, D. J. Vogel and T. M. Nenoff, Adv. Funct. Mater., 2020, 30, 2006598 CrossRef CAS.
- J. H. Wei, W. T. Ou, J. B. Luo and D. B. Kuang, Angew. Chem., Int. Ed., 2022, 61, e202207985 CrossRef CAS PubMed.
- Y. He, J. Wang, Q. Li, S. Qu, C. Zhou, C. Yin, H. Ma, H. Shi, Z. Meng and Z. An, Highly Efficient Room-Temperature Phosphorescence Promoted via Intramolecular-Space Heavy-Atom Effect, Adv. Opt. Mater., 2023, 22201641 Search PubMed.
- S. Jena, J. Eyyathiyil, S. K. Behera, M. Kitahara, Y. Imai and P. Thilagar, Chem. Sci., 2022, 13, 5893–5901 RSC.
- Y. Gong, G. Chen, Q. Peng, W. Z. Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 6195–6201 CrossRef CAS PubMed.
- Y. Zhang, X. Chen, J. Xu, Q. Zhang, L. Gao, Z. Wang, L. Qu, K. Wang, Y. Li, Z. Cai, Y. Zhao and C. Yang, J. Am. Chem. Soc., 2022, 144, 6107–6117 CrossRef CAS PubMed.
- X. Chen, W. Dai, X. Wu, H. Su, C. Chao, Y. Lei, J. Shi, B. Tong, Z. Cai and Y. Dong, Chem. Eng. J., 2021, 426, 131607 CrossRef CAS.
- J. Liu, Y. Luo, Z. Ran, F. Wang, M. Sun, Y. Luo, J. Zhuang, X. Zhang, B. Lei, Y. Liu and C. Hu, Chem. Eng. J., 2023, 474, 145597 CrossRef CAS.
- A. Cheng, H. Su, X. Gu, W. Zhang, B. Zhang, M. Zhou, J. Jiang, X. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202312627 CrossRef CAS PubMed.
- Y. J. Ma, X. Fang, G. Xiao and D. Yan, Angew. Chem., Int. Ed., 2022, 61, e202114100 CrossRef CAS PubMed.
- J. Yuan, J. Dong, S. Lei and W. Hu, Mater. Chem. Front., 2021, 5, 6824–6849 RSC.
- M. G. Campbell, D. Sheberla, S. F. Liu, T. M. Swager and M. Dincă, Angew. Chem., Int. Ed., 2015, 54, 4349–4352 CrossRef CAS PubMed.
- J. Yang, D. He, W. Chen, W. Zhu, H. Zhang, S. Ren, X. Wang, Q. Yang, Y. Wu and Y. Li, ACS Appl. Mater. Interfaces, 2017, 9, 39450–39455 CrossRef CAS PubMed.
- A. H. Assen, O. Yassine, O. Shekhah, M. Eddaoudi and K. N. Salama, ACS Sens., 2017, 2, 1294–1301 CrossRef CAS PubMed.
- H. Liu, W. Ye, Y. Mu, H. Ma, A. Lv, S. Han, H. Shi, J. Li, Z. An, G. Wang and W. Huang, Adv. Mater., 2022, 34, 2107612 CrossRef CAS PubMed.
- R. Feng, Z.-A. Guo, Y.-N. Zhang, H.-X. Sun, X.-H. Bu, Z.-Y. Li, Z.-Q. Yao and W. Li, Sci. China: Chem., 2022, 65(1), 128–134 CrossRef CAS.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- C.-Y. Zhu, Z. Wang, J.-T. Mo, Y.-N. Fana and M. Pan, J. Mater. Chem. C, 2020, 8, 9916–9922 RSC.
- X. Yang and D. Yan, J. Mater. Chem. C, 2017, 5, 7898–7903 RSC.
- S. Xu, L. Zhan, C. Hong, X. Chen, X. Chen and M. Oyama, Sens. Actuators, B, 2020, 308, 127733 CrossRef CAS.
- Z. Wu, J. Wei, T. Jiao, Q. Chen, M. Oyama, Q. Chen and X. Chen, Food Chem., 2022, 385, 132710 CrossRef CAS PubMed.
- E. P. Asiwal, D. S. Shelar, C. S. Gujja, S. T. Manjare and S. D. Pawar, New J. Chem., 2022, 46, 12679–12685 RSC.
- N. B. Shustova, A. F. Cozzolino, S. Reineke, M. Baldo and M. Dincă, J. Am. Chem. Soc., 2013, 135, 13326–13329 CrossRef CAS PubMed.
- L. Sun, A. Rotaru and Y. Garcia, J. Hazard. Mater., 2022, 437, 129364 CrossRef CAS PubMed.
- A.-Y. Ni, H. Zhao, P.-P. Zhang, B.-L. Zhang, J.-J. Zhang, S. Liu, J. Chen and C. Duan, Inorg. Chem. Front., 2023, 10, 4065–4075 RSC.
- B.-L. Zhang, P.-P. Zhang, A.-Y. Ni, J.-J. Zhang, H.-Y. Wang, K.-X. Feng, S. Liu, Z. Zhao and C. Duan, Adv. Opt. Mater., 2023, 2300717 CrossRef CAS.
- X.-X. Wang, M.-X. Zhang, B. Yu, K. V. Hecke and G.-h. Cui, Spectrochim. Acta, Part A, 2015, 139, 442–448 CrossRef CAS PubMed.
- L.-L. Zhu, Y.-E. Huang, L.-K. Gong, X.-Y. Huang, X.-H. Qi, X.-H. Wu and K.-Z. Du, Chem. Mater., 2020, 32, 1454–1460 CrossRef CAS.
- Q. Dang, L. Hu, J. Wang, Q. Zhang, M. Han, S. Luo, Y. Gong, C. Wang, Q. Li and Z. Li, Chem. - Eur. J., 2019, 25, 7031–7037 CrossRef CAS PubMed.
- T. Seki, T. Ozaki, T. Okura, K. Asakura, A. Sakon, H. Uekusa and H. Ito, Chem. Sci., 2015, 6, 2187–2195 RSC.
- X. Song, G. Lu, Y. Man, J. Zhang, S. Chen, C. Han and H. Xu, Angew. Chem., Int. Ed., 2023, 62, e202300980 CrossRef CAS PubMed.
- J. Ren, Y. Tian, Y. Wang, J. Yang, M. Fang and Z. Li, J. Mater. Chem. C, 2022, 10, 13741–13746 RSC.
- S. Cai, H. Shi, Z. Zhang, X. Wang, H. Ma, N. Gan, Q. Wu, Z. Cheng, K. Ling, M. Gu, C. Ma, L. Gu, Z. An and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 4005–4009 CrossRef CAS PubMed.
- M. Yan, T. Gu, S. Yang and K.-L. Zhang, J. Mol. Struct., 2022, 1260, 132802 CrossRef CAS.
- G. Zhang, Y. Ma, H. Chai, K. Yu, Y. Li, S. Wang, J. Ma, L. Qu, W. Tan and X. Zhang, Sens. Actuators, B, 2022, 370, 132395 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, 1986 Search PubMed.
- M. S. Setty and A. P. B. Sinha, Thin Solid Films, 1986, 144(1), 7–19 CrossRef CAS.
- V. I. Nefedov, N. P. Sergushin, Y. V. Salyn, I. M. Band and M. B. Trzhaskovskaya, J. Electron Spectrosc. Relat. Phenom., 1975, 7(2), 175–185 CrossRef CAS.
- E. Agostinelli, C. Battistoni, D. Fiorani, G. Mattogno and M. Nogues, J. Phys. Chem. Solids, 1989, 50, 269 CrossRef CAS.
- B. Zhou and D. Yan, Adv. Funct. Mater., 2019, 29, 1807599 CrossRef.
- N. Liu, Y. Pan, Y. Lei, M. Liu, C. Peng, Z. Cai, G. Shen, H. Wu, X. Huang and Y. Dong, Chem. Eng. J., 2022, 433, 133530 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2321760–2321761. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ta00356j |
| This journal is © The Royal Society of Chemistry 2024 |