
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtrinsic pseudocapacitive ultrathin 2D MoS2 nanoflakes clamped on 1D Sb2S3 nanorods: an advanced heterostructured anode for high-energy ammonium ion hybrid capacitors†
Supriya J.
Marje
a,
Harshitha B.
Tyagaraj
a,
Seung-Kyu
Hwang
b,
Kugalur Shanmugam
Ranjith
a,
Ebrahim
Alhajri
c,
Nilesh R.
Chodankar
 *c,
Yun Suk
Huh
*b and
Young-Kyu
Han
*a
*c,
Yun Suk
Huh
*b and
Young-Kyu
Han
*a
aDepartment of Energy and Material Engineering, Dongguk University-Seoul, Seoul, 04620, South Korea. E-mail: ykenergy@dongguk.edu
bNanoBio High-Tech Materials Research Center, Department of Biological Sciences and Bioengineering, Inha University, Incheon, 22212, South Korea. E-mail: yunsuk.huh@inha.ac.kr
cMechanical Engineering Department, Khalifa University, Abu Dhabi, 127788, United Arab Emirates. E-mail: chodankarnilesh311@gmail.com
First published on 13th February 2024
Abstract
Ammonium-ion (NH4+) charge carriers have recently been considered promising for electrochemical energy storage (EES) systems because of their high safety, low molar mass, and small hydrated radius (3.31 Å). However, finding a kinetically balanced anode and cathode combination for high NH4+-ion storage is challenging. Herein, a new approach for developing a heterostructured electrode was developed by constructing extrinsic pseudocapacitive 2D ultrathin MoS2 nanoflakes clamped on 1D Sb2S3 nanorods (MoS2/Sb2S3) as an anode for high-performance ammonium-ion hybrid capacitors (AIHCs) against the intrinsic pseudocapacitive MnO2 cathode. The engineered MoS2/Sb2S3 heterostructured anode facilitated large interlayer galleries owing to the presence of 2D MoS2 for facial NH4+-ion diffusion and provided a rapid electron pathway through 1D Sb2S3, which promoted a high capacitance of 360 F g−1, low resistance, and stable cycling performance. More importantly, the constructed AIHC delivered a superior energy density of 43.75 W h kg−1 at a power density of 600 W kg−1 and excellent cycling durability over 5000 cycles. These results show that a heterostructured extrinsic pseudocapacitive anode can improve the electrochemical parameters of NH4+ EES systems and replace traditional carbon-based anode materials.
Introduction
The demand for green energy continues to inspire the worldwide research community to research and develop next-generation storage systems.1 Rechargeable batteries play a crucial role in electric vehicles, portable devices, and grid-scale storage applications.2 As current batteries tend to use organic electrolytes, which have safety issues along with toxicity and flammability,3 the use of aqueous electrolytes is desired owing to their inherent merits, such as low cost, high ionic conductivity, environmental friendliness, and non-flammability. Moreover, their flexibility in formation and manufacturing make them attractive in the battery industry.4,5 Therefore, aqueous batteries are the focus of research interest and are considered future substitutes for commercial secondary batteries.6,7 Aqueous batteries usually contain metallic cations, such as Li+, Zn2+, Na+, Mg2+, K+ or Al3+, which act as charge carriers. However, their large hydration volumes and high molar masses hamper the diffusion kinetics of these ions and result in poor rate capability and stability.8 In contrast, non-metallic ions have smaller hydration radii, low molar masses, and high availability. Furthermore, these features result in low costs, high energy densities, and excellent rate capabilities.9,10 In this context, non-metal cation carriers, such as protons,11 hydronium,12 and NH4+ ions, have exceptional features.13 In particular, the highly conductive nature of the NH4+ ion makes it an efficient charge carrier, which promotes ion-diffusion processes, and it also has a lower HER (hydrogen evolution reaction) potential and is less corrosive.8,14 Furthermore, NH4+ ions can migrate rapidly because of their smaller Stokes radius and energy barriers and tetrahedral geometries, which support their superior intercalation in host materials.15,16Relatively few studies have been reported on ammonium-ion storage and those that have been reported mainly focused on cathode materials for ammonium-ion storage. It was found that the exceptional features of cathode materials improve the capacity and cycling stability of devices.16 Several cathode materials have been explored for ammonium-ion storage, such as activated carbon cloth (ACC)@V2O5,15 MnAl LDH,9 MnO2,17–20 and Prussian blue (NiHCF).21 However, little work has been published on high-performance anode materials for ammonium-ion storage, yet the development of such materials is crucial. Generally, a suitable material must meet specific parameters; for example, it should have an appropriate electrode structure and bonding sites. A suitable space and binding sites are required for the accommodation and binding of NH4+ ions during the reaction.22 The primary mechanism underlying rapid NH4+ storage involves the formation and breaking of hydrogen bonds.23,24 However, researchers face challenges with the structural collapse of the host material due to the frequent insertion/extraction of NH4+.21 Therefore, novel electrodes for NH4+-storage systems are still required to enhance the performance and long-term stability of ammonium-ion capacitors.
Traditionally, carbon-based materials have been used as anodes due to their excellent electrochemical cycling stability.25–28 However, the double-layer charge storage limits the capacitance of carbon anodes and causes a large kinetic imbalance between anode and cathode materials. This can negatively affect the capacitance and energy density of cells, as indicated by the equation 1/CT = 1/C1 + 1/C2,29 where CT, C1, and C2 are the capacitances of the whole cell, cathode and anode electrodes, respectively.30 For this reason, it is crucial to find anode materials with energy-storage capacities similar to cathode materials to assemble kinetically balanced ammonium-ion energy-storage systems.31,32 Considering these issues, redox-active anode materials that can simultaneously provide high capacitance and stability at negative operating potentials are needed.31–34 Of the many potential candidates, extrinsic pseudocapacitive metal sulfides are promising for aqueous ion capacitors because of their exceptional electrochemical properties and structural diversities.33,35,36 Specifically, because of its structural stability, Sb2S3 was investigated for electrocatalysis, battery and photovoltaic device applications. In addition, Sb2S3 has the merits of being nontoxic, abundant in nature, and cheap.37 However, the volume expansion of Sb2S3 during the intercalation/deintercalation of ions hampers its stability and rate performance; though this issue can be managed by fabricating sulfide-based heterostructures that can maintain their structures.38 Correspondingly, nanostructures covered with a protective layer provide space for volume changes, stabilize the electrode/electrolyte interfaces, and promote electron transport with shorter ion-diffusion paths.39,40
Thus, we were motivated to develop anode materials that would enhance the electrochemical performance of NH4+-storage systems. Consequently, we investigated heterostructured metal chalcogenide-based anode materials for ammonium-ion hybrid capacitors (AIHCs) using a straightforward solution path. Nanorods of Sb2S3 served as a 1D conductive core, while 2D MoS2 nanosheets offered abundant active sites with large interlayer galleries that could promote facial ion movement during charge–discharge processes. The engineered heterostructured Sb2S3/MoS2 anode delivered a specific capacitance of 360 F g−1 (at 3 A g−1) and excellent cycling performance (5000 cycles at 12 A g−1) within a negative potential window (−0.75 to 0.05 V/SCE) in ammonium-ion electrolyte. Furthermore, an AIHC fabricated by integrating MnO2 as the cathode and Sb2S3/MoS2 as the anode exhibited excellent a high energy density and stability, which represented a substantial improvement over previously reported carbon-anode containing AIHCs.15,20
Experimental section
Synthesis of Sb2S3
Analytical-grade chemicals and reagents were used without further purification. Sb2S3 nanorods were synthesized using a hydrothermal method. For a typical synthesis, SbCl3 (3 mmol), L-cysteine (6 mmol), and Na2S·9H2O (6 mmol) were dissolved in 40 ml of double distilled water (DDW) with stirring. The solution was then transferred to a 50 ml Teflon-lined stainless-steel autoclave and heated for 12 h at 180 °C. A dark-brown precipitate of Sb2S3 was obtained after cooling to room temperature. The product was collected after filtration, washed with DDW several times, and dried at 60 °C overnight.Synthesis of Sb2S3/MoS2
To synthesize Sb2S3/MoS2, 50 mg of Sb2S3 was suspended in 40 ml of DDW containing (NH4)6Mo7O24 (0.01 mol) and thiourea (0.02 mol), stirred, and sonicated for 30 min. The prepared solution was transferred to a 50 ml Teflon-lined stainless-steel autoclave and heated for 24 h at 200 °C. The final product was obtained by suspending it several times sequentially in DDW and ethanol and centrifuging between steps. The obtained Sb2S3/MoS2 was dried overnight at 60 °C.Synthesis of MnO2
MnO2 films were electrodeposited on carbon cloth (CC) using an equimolar MnSO4 (0.1 mol) and Na2SO4 (0.1 mol) solution and an electrochemical workstation. The potential was varied from 0.4–1.5 V at a scan rate of 50 mV s−1 in potentiodynamic mode for 50, 100, 150, or 200 cycles, and the samples produced were denoted as Mn-50, Mn-100, Mn-150, and Mn-200, respectively. In a three-electrode cell, the CC, graphite, and saturated calomel electrodes (SCE) were used as the working, counter, and reference electrodes. The as-deposited MnO2 films were dried overnight at 60 °C.Results and discussion
A two-step hydrothermal process was employed to synthesize heterostructured Sb2S3/MoS2 (Fig. 1a). A simple hydrothermal method was first used to synthesize Sb2S3 nanorods. In this process, SbCl3 and L-cysteine served as the sources of Sb3+ and S2−, respectively. This step was followed by a hydrothermal step to grow MoS2 nanosheets on the surfaces of Sb2S3 nanorods using (NH4)6Mo7O24 and thiourea as the sources of Mo4+ and S2−, respectively. It is known that the self-aggregation and disordered arrangement of a material can restrict exposure of the active sites,41 which can result in a lower redox reaction kinetics and lower specific capacitance. So, it is important to expose an optimum amount of functional material through finely tuning and engineering a suitable architecture to meet definite requirements.42 To load the optimum amount and achieve the required architecture of MoS2 on Sb2S3 nanorods, and also to prevent self-aggregation and a disordered arrangement of MoS2 nanosheets on Sb2S3 nanorods, a second hydrothermal reaction was carried out using different deposition times. Scanning electron microscopy (SEM) analysis showed that the reaction for 24 h obtained good results, and thus, this time was adopted for the further study. The phases and crystallinities of the developed materials were investigated by X-ray diffraction (XRD) analysis. The diffraction peaks of Sb2S3 were well indexed with the orthorhombic phase of Sb2S3 (JCPDS 42-1393), indicating the successful formation of Sb2S3 (Fig. 1b). The low crystallinity and small crystallite size of the as-produced MoS2 were evidenced by the broad diffraction peaks (Fig. S1†). The XRD pattern of Sb2S3/MoS2 was compatible with that of Sb2S3; no extra MoS2 peaks were detected due to its low crystallinity (Fig. 1b).43 The XRD study confirmed that MoS2 nanosheets grew on Sb2S3, and the reduction in the intensities of the Sb2S3/MoS2 diffraction peaks confirmed that MoS2 nanosheets were uniformly decorated on Sb2S3 nanorods. The second hydrothermal step thus produced the required heterostructured Sb2S3/MoS2 material. | ||
| Fig. 1 (a) Schematic of the two-step hydrothermal synthesis process used to produce heterostructured Sb2S3/MoS2. (b) XRD patterns of Sb2S3 and Sb2S3/MoS2. High-resolution XPS spectra of (c) Mo 3d and (d) S 2p. (e) N2 adsorption–desorption isotherms of Sb2S3 and Sb2S3/MoS2. SEM images of (f) Sb2S3 and (g and h) Sb2S3/MoS2. (i–l) TEM images and elemental mapping of Sb2S3/MoS2. | ||
X-Ray photoelectron spectroscopy (XPS) was utilized to characterize the chemical states and composition of the heterostructured Sb2S3/MoS2. The wide-scan XPS survey spectrum confirmed the presence of Mo, Sb, S, and O (Fig. S2a†). In addition, high-resolution spectra was used to gain insights into the valence states. The Sb 3d spectrum (Fig. S2b†) was deconvoluted into two peaks at 530.5 and 539.7 eV, associated with the Sb 3d5/2 and 3d3/2 of metallic Sb, respectively.44 Besides, another peak was observed at 531.6 eV, which belonged to O 1s (Fig. S2c†), representing surface oxidation.38,39 As depicted in Fig. 1d and e, the high-resolution XPS spectra of molybdenum and sulfur elements were investigated to determine the status of Mo 3d and S 2p. The peaks observed at 232.4 and 228.8 eV were ascribed to the Mo 3d3/2 and Mo 3d5/2 of Mo4+ (Fig. 1c).45,46 The peak at 235.7 eV was related to Mo(VI), which reflected a partial reduction of the Mo surface (Mo–O). In this study, (NH4)6Mo7O24 was used as the source of molybdenum to synthesize MoS2. During the 200 °C hydrothermal process, this material thermally decomposed into MoOx, which accelerated the oxidation of Mo.47 Another peak at 229.9 eV was observed corresponding to Mo(V), which may have resulted from the partial reduction of Mo6+.47 Furthermore, the peak that arose at around 226.1 eV was assigned to S 2s.48 The S 2p spectrum displayed a S2− doublet at 161.6 and 162.9 eV, attributed to S 3p3/2 and S 3p1/2 (Fig. 1d).47,49 These XPS results were in accordance with previous studies on Mo, Sb, S, and O, and confirmed the successful formation of the Sb2S3/MoS2 heterostructure with a Mo to S atomic ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.62. The specific surface areas of the prepared samples were determined using N2 adsorption–desorption isotherm measurements. As presented in Fig. 1e, the Sb2S3/MoS2 material displayed a type-III isotherm with an H3 type physisorption hysteresis loop according to the International Union of Pure and Applied Chemistry (IUPAC) classification, which is characteristic of mesoporous materials with a low adsorption energy. According to Brunauer–Emmett–Teller (BET) equation analysis, the surface area of the heterostructured Sb2S3/MoS2 was 40.6 m2 g−1, which was seven times higher than that of Sb2S3 (5.8 m2 g−1). This large specific surface area of the heterostructured Sb2S3/MoS2 suggests that it contains many more active sites than Sb2S3 and mesopores that shorten the diffusion path, which is desirable for rapid charge transfer.
:1.62. The specific surface areas of the prepared samples were determined using N2 adsorption–desorption isotherm measurements. As presented in Fig. 1e, the Sb2S3/MoS2 material displayed a type-III isotherm with an H3 type physisorption hysteresis loop according to the International Union of Pure and Applied Chemistry (IUPAC) classification, which is characteristic of mesoporous materials with a low adsorption energy. According to Brunauer–Emmett–Teller (BET) equation analysis, the surface area of the heterostructured Sb2S3/MoS2 was 40.6 m2 g−1, which was seven times higher than that of Sb2S3 (5.8 m2 g−1). This large specific surface area of the heterostructured Sb2S3/MoS2 suggests that it contains many more active sites than Sb2S3 and mesopores that shorten the diffusion path, which is desirable for rapid charge transfer.
SEM and transmission electron microscopy (TEM) were used to study the morphologies and structures of Sb2S3 and Sb2S3/MoS2. The SEM images indicated that the Sb2S3 material had a rod-like structure and smooth surface (Fig. 1f), and a slightly porous surface was observed at higher magnification (Fig. S3a†). During the hydrothermal reaction, nanosheets of MoS2 grew uniformly on the Sb2S3 nanorods (Fig. 1g and h), and the surface of Sb2S3 became rougher (Fig. S3b†). However, the nanorod-like structure of Sb2S3 was well maintained. The HR-TEM images of Sb2S3 revealed a rod-like structure with a smooth surface (Fig. S4a and b†) and a crystal lattice (d = 0.24 nm) assigned to the (240) plane of orthorhombic Sb2S3 (Fig. S4c and d†). The selected area electron diffraction (SAED) pattern of Sb2S3 revealed a highly regular crystal structure (Fig. S4e†). The uniform coating of MoS2 nanosheets on Sb2S3 nanorods was clearly observed in the HR-TEM images of Sb2S3/MoS2 (Fig. 1i), and the corrugated nanosheets showed a lattice fringe of 0.66 nm (Fig. 1j and k), consistent with the (002) plane of the MoS2 material and sufficient to allow the quick intercalation/deintercalation of NH4+ ions. In addition, the SAED pattern of the corrugated nanosheets on Sb2S3/MoS2 showed that the MoS2 nanosheets were largely amorphous (Fig. 1k). Additionally, energy dispersive X-ray (EDX) elemental mappings of the pristine Sb2S3 and heterostructured Sb2S3/MoS2 material were measured, and disclosed the co-existence of Sb, S, and a small amount of O in the pristine Sb2S3 material (Fig. S4e† and 1l). The EDX mapping of Sb2S3/MoS2 revealed Sb was uniformly distributed in the Sb2S3 core region, whereas Mo was less dense than Sb and was observed in the overall MoS2 shell region. Likewise, uniform S distribution was also observed throughout the heterostructure Sb2S3/MoS2 (Fig. 1l). The small amount of O observed in Sb2S3 and Sb2S3/MoS2 confirmed the partial reduction of the material, which concurred with the XPS results (Fig. S4e, f and S5†). The uniform coverage of Mo and S on the core material confirmed the homogeneous growth of MoS2 nanosheets on the Sb2S3 nanorods, which was consistent with the heterostructure of Sb2S3/MoS2 observed by SEM and TEM analyses. The above investigation verified that MoS2 nanosheets grew on the Sb2S3 nanorods, which aligned with the XRD and XPS results.
A series of tests were performed to evaluate the electrochemical properties of the developed Sb2S3/MoS2 heterostructure for ammonium-ion storage. The ammonium-ion-storage performances of Sb2S3, MoS2, and Sb2S3/MoS2 electrodes were first measured in a half-cell system. Cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), and electrochemical impedance spectroscopy (EIS) analyses of the active materials were performed in 1 M (NH4)2SO4 electrolyte. The CV performances of Sb2S3, MoS2, and Sb2S3/MoS2 electrodes were measured in a similar potential range for comparison purposes in an optimized “−0.75 to 0.05 V (vs. SCE)” potential window (Fig. S6a†). The Sb2S3 electrode displayed oxidation and reduction peaks in the CV curves and delivered minimal capacitive performance due to its small CV area. In contrast, the MoS2 electrode exhibited rectangular CV curves and a higher CV area than the Sb2S3 electrode. Interestingly, the Sb2S3/MoS2 electrode exhibited a much higher current response and almost rectangular shape without any specific redox peaks, indicating the pseudocapacitive nature of the heterostructure due to the MoS2 along with a higher capacity. The GCD profiles of the Sb2S3, MoS2, and Sb2S3/MoS2 electrodes are depicted in Fig. S6b† (at a current density of 10 A g−1) and displayed the significantly distinguished storage behavior. Triangular symmetric curves were noted, without any voltage plateau, for the MoS2 and Sb2S3/MoS2 electrodes, confirming the pseudocapacitive nature of the materials. The discharge time of Sb2S3/MoS2 was much longer than that of Sb2S3 and MoS2. Also, the potential window achieved by the Sb2S3/MoS2 electrode was higher than that of the Sb2S3 electrode at the same current density (10 A g−1). So, the calculated specific capacitance of the heterostructured Sb2S3/MoS2 electrode (258.2 F g−1) was 2.25-fold higher than that of the Sb2S3 (156.8 F g−1) and MoS2 (159.7 F g−1) electrodes. The enhanced electrochemical performance for the Sb2S3/MoS2 electrodes was due to: (1) the rapid electron transfer provided by the conductive core 1D Sb2S3, (2) the greater number of active sites provided by the MoS2 host, and hence, greater specific surface area, (3) the huge number of active sites and increased redox activity provided by the 2D MoS2 layer, and (4) the large MoS2 interlayer distance allowing the rapid intercalation of NH4+ ions.
The comparative EIS spectra of the Sb2S3, MoS2, and Sb2S3/MoS2 electrodes were plotted and simulated based on an equivalent circuit, as shown in Fig. S6c.† Each electrode displayed a semicircle in the high-frequency region and a straight line in the low-frequency region. The semicircle was attributed to charge-transfer resistance (Rct) and the straight line to diffusion resistance (W). The high-frequency interaction with the X-axis corresponded to solution resistance (Rs), which was related to the internal resistance of the electrode, electrolyte conductivity, and electrode/electrolyte contact resistance. In an equivalent circuit, CPE represents a constant phase element and is observed due to electrode/electrolyte interaction inhomogeneities due to the porosity, nature of the electrode, and disorder associated with diffusion.50 It was confirmed that the Rct of the Sb2S3/MoS2 electrode (0.16 Ω cm−2) was significantly lower than that of the Sb2S3 (4.15 Ω cm−2) and MoS2 electrode (0.31 Ω cm−2). The excellent conductivity and large number of contact sites offered by the core Sb2S3 nanorods to clamp MoS2 nanosheets were presumed to be responsible for the high number of uniformly distributed electroactive sites of MoS2 nanosheets and facile ion diffusion. In addition, the straight line existing in the low-frequency region indicated the rapid diffusion of NH4+ ions and supported the idea that the core material could improve the electronic conductivity.
In addition, we investigated the CV performances of the Sb2S3/MoS2 electrode at different scan rates in the range 5–100 mV s−1 (Fig. 2a). The nature of the CV curves indicated the pseudocapacitive nature and the current response increased linearly with the scan rate. The intensities of the curves indicated that the conversion reaction was entirely reversible, which may lead to a high capacity and coulombic efficiency. The kinetic behavior of the Sb2S3/MoS2 electrode was further studied by fitting the power law relationship in the CV profile. Here, ‘b’ is defined as the slope of the plot of the log of the current against the log of the scan rate, and it has specified values like 0.5 and 1, which indicate that the charge-storage mechanism is either by diffusion control (0.5) or surface control (1).51 The ‘b’ value plot for the Sb2S3/MoS2 electrode is shown in Fig. S6d,† and its slope was 0.96, indicating that the electrode stored charges prominently through surface-controlled processes and that it exhibited considerable pseudocapacitive behavior.52 The quantitative distribution of the charge-storage mechanism was further examined to evaluate the exact contribution from the surface and diffusion processes. The capacity contribution with the scan rate was calculated and is shown in Fig. 2b. At a 60 mV s−1 scan rate, the capacitive contribution percentage was 88% and the surface-controlled contribution increased with the increasing scan rate. The kinetic study results indicated that surface-controlled processes were mainly responsible for the charge storage in Sb2S3/MoS2.
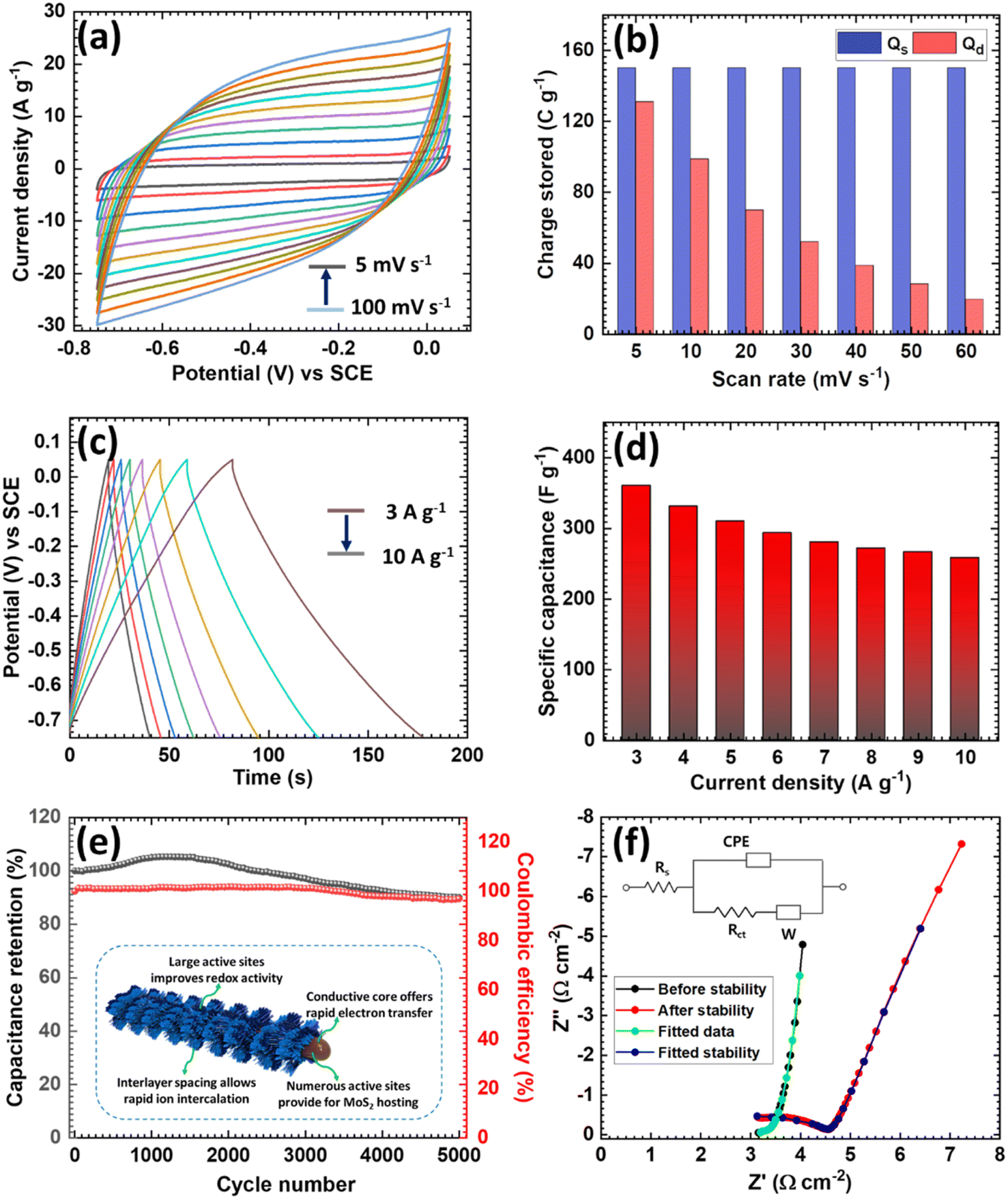 | ||
| Fig. 2 Electrochemical study of heterostructured Sb2S3/MoS2. (a) CV curves of Sb2S3/MoS2, (b) surface and diffusive charges stored at different scan rates for Sb2S3/MoS2, (c) GCD curves of Sb2S3/MoS2, (d) specific capacitance of Sb2S3/MoS2, (e) cycling stability of Sb2S3/MoS2 and (f) Nyquist plots before and after Sb2S3/MoS2 stability tests (inset: fitted circuit). | ||
The GCD curves of the Sb2S3/MoS2 electrode at current densities from 3–10 A g−1 displayed an almost symmetrical curve even at a low current density (3 A g−1) (Fig. 2c) and the potential drop was significantly smaller than for the Sb2S3 electrode, demonstrating the low series resistance of the Sb2S3/MoS2 electrode. Fig. 2d shows the specific capacitance of the Sb2S3/MoS2 electrode, extracted from the discharge profiles at various current densities. The electrode achieved a maximum gravimetric capacitance of 360 F g−1 at a current density of 3 A g−1, demonstrating that the Sb2S3/MoS2 heterostructure significantly improved the charge adsorption and increased NH4+ storage. This high capacitance improvement was mainly due to the enrichment of the MoS2 surface activity. In addition, the long-term cycling stability of the Sb2S3/MoS2 electrode was tested using GCD measurements at a current density of 12 A g−1 (Fig. 2e), revealing 90% of the specific capacitance of the Sb2S3/MoS2 electrodes was retained after 5000 cycles and almost 97% of the corresponding coulombic efficiency during cycling. Moreover, the nature of the initial and final GCD cycles (Fig. S7†) corroborated the stable performance of the Sb2S3/MoS2 electrode.
The excellent cycling stability of the electrode was due to its heterostructure, which prevented dissolution of the core due to the presence of surface oxygen (see the inset of Fig. 2e). It has been well demonstrated that H bonding between NH4+ and lattice O can stabilize lattice structures and suppress material dissolution.53 Next, EIS measurements were performed to investigate the kinetics of the Sb2S3/MoS2 electrode before and after cycling stability tests, and the Nyquist plots and the fitted circuit are provided in Fig. 2f. The small increment in Rct observed after 5000 cycles indicated an increase in internal resistance;54 the other parameters corresponding to the equivalent circuit are provided in Table S1.† The better electrode kinetics and electrochemical properties of the Sb2S3/MoS2 electrode were presumably the result of the synergistic effects between the excellent conductivity of the Sb2S3 nanorods and the abundant electroactive sites provided by MoS2 nanosheets that promote facile ion migration and storage. The above discussion reveals that the Sb2S3/MoS2 is a promising anode candidate for NH4+ storage.
The well-reported MnO2 was selected as a cathode for ammonium-ion storage due to its abundance, high theoretical capacitance, environment friendliness, and cost-effectiveness. A potentiodynamic method was used to synthesize free-standing MnO2 electrodes on carbon cloth (CC), and deposition cycles were adjusted to achieve the optimum mass loading. XRD was used to investigate the structural features of MnO2. The broad diffraction peaks around 2θ = 30° and 43.6° were ascribed to the [002] and [100] planes of the carbon of the CC (Fig. S8a†), but no characteristic peaks of crystalline MnO2 were observed. However, from the XPS study used to investigate the oxide nature of Mn, the survey spectrum confirmed the presence of Mn, O, and C (Fig. S8b†). The high-resolution Mn 2p spectrum is shown in Fig. S8c.† Two peaks could be observed at 654.3 and 642.1 eV, corresponding to the Mn 2p1/2 and Mn 2p3/2 states. After deconvolution, the peaks at 654.3 and 652.8 eV were assigned to Mn3+ and Mn4+ of Mn 2p1/2,55 and the peaks at 642.1 and 644.4 eV to Mn3+ and Mn4+ of Mn 2p3/2.55,56 The high-resolution O 1s spectrum (Fig. S8d†) showed two peaks at binding energies of 529.6 and 531.4 eV, representing lattice oxygen (Mn–O–Mn) and the presence of crystalline water,20,57 which confirmed the formation of MnO2 on the CC.
SEM and TEM were used to investigate the morphological and chemical compositions of the MnO2 material. The microstructures of the synthesized Mn materials are presented in Fig. 3a, b and S9.† A single CC thread was used in the SEM study to investigate the morphological changes that occurred during cycling. The Mn-50 sample displayed fewer, more unevenly deposited particles on the CC (Fig. S9a†), and at higher magnification, a bundle of uniformly sized nanoparticles could be observed (Fig. S9b†) the other samples (Mn-100, Mn-150, and Mn-200) produced similar morphologies (Fig. S9c–f†). Upon increasing the number of deposition cycles, the MnO2 loading on CC increased, and for the Mn-150 sample, a uniform coating of nanoparticles over the surface of CC was observed (Fig. 3a and b). The overgrowth of nanoparticles was observed for the Mn-200 sample. The TEM analysis (Fig. 3c and d) showed a similar morphology for the Mn-150 sample. Dense particles could be observed in Fig. 3c and at higher magnification, the crystalline lattice fringes (d = 0.27 nm) were clearly marked (Fig. 3d and e), corresponding to the (400) plane of MnO2 (JCPDS card no. 44-0141). The SAED pattern also verified the crystalline nature of MnO2. Furthermore, EDX mapping of Mn-150 (Fig. 3f and S10†) showed the uniform distribution of Mn and O. These findings concurred with the XPS results and confirmed the successful deposition of MnO2 on the CC.
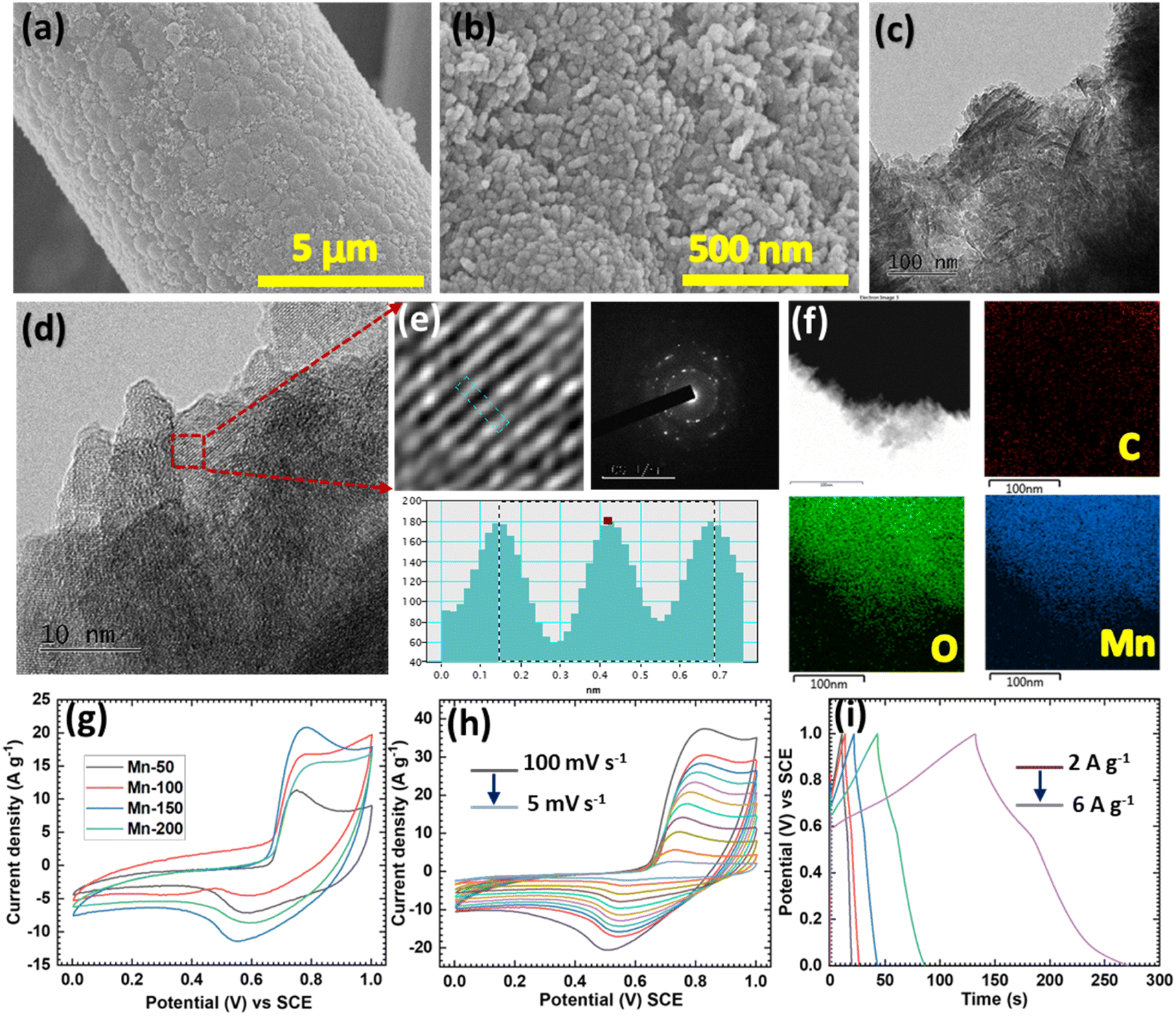 | ||
| Fig. 3 (a and b) SEM images of MnO2 and (c–e) TEM images and (f) elemental mapping of MnO2 (Mn-150 sample). (g) Comparative CV curves for the Mn series of electrodes. (h) CV curves at different scan rates and (i) GCD curves at different current densities for the Mn-150 electrode. | ||
The ammonium-ion-storage performances of a series of Mn electrodes were tested using half-cells in 1 M (NH4)2SO4 electrolyte. Comparative CV curves at a constant scan rate (50 mV s−1) and GCD curves at a current density of 3 A g−1 for the Mn series of electrodes measured in an optimized potential window of 0–1 V are shown in Fig. 3g and S11a.† The uniformly coated Mn-150 electrode delivered the highest current response in the CV study and the longest discharge time in the GCD study. A comparative study showed that the uniformly coated nanoparticles had higher NH4+-storage capacities than the uneven or overgrown nanoparticle structures (Fig. S10b†). Also, we investigated the scan rate and current density dependent analysis of the Mn-150 electrode (Fig. 3h and i). Upon increasing the scan rate, the current response of an electrode increased, and a minor shift occurred in the redox peaks due to the surface polarization of the charges.58 In addition, the nature of the CV curves was well maintained over the scan rate study, suggesting the good reversibility of the Mn-150 electrode. Furthermore, at a lower current density of 2 A g−1, the Mn-150 electrode delivered the highest specific capacitance of 278.6 F g−1 (Fig. S11c†). Nyquist plots of the Mn series of electrodes fitted with the equivalent circuit (Fig. S11d†) revealed that the Mn-150 electrodes had lower Rct (98 Ω cm−2) values than the other electrodes. Other parameters corresponding to the fitted circuit of the Mn series of electrodes are summarized in Table S2.† The above study shows that the electrodeposited MnO2 cathodes are strong candidates for ammonium-ion capacitors.
The practical application of the developed materials was investigated by fabricating a full cell (AIHCs) by combining Sb2S3/MoS2 as the anode and MnO2 as the cathode. Here, the charge balance theory was employed to balance the mass of the cathode and anode (1:0.96) and the obtained electrochemical performances are presented. A schematic presentation of the AIHC is illustrated in Fig. 4a. As shown in the figure, during the first charge, NH4+ ions are inserted in the Sb2S3/MoS2 anode and extracted from the Sb2S3/MoS2 during discharge. During the charge–discharge process, NH4+ ions are continuously shuttled between the anode and cathode.16 The CV curves of the anode and cathode in different working potential windows at a constant scan rate are shown in Fig. 4b. A series of CV curves in different potential windows at a constant scan rate were used to define the optimum potential window for the AIHC, indicating 0–1.6 V was the optimum potential window for the AIHC (Fig. 4c). In addition, CV measurements of the AIHC at different scan rates (5–100 mV s−1) obtained using the 0–1.6 V window (Fig. 4d) revealed a pair of reduction/oxidation peaks. At high scan rates, polarization shifted the positions of the oxidation and reduction peaks with the scan rates.21 The symmetric nature of the CV curves suggested the excellent capacitor reversibility. The kinetics of the AIHC were investigated using the Power law, the relationship between the log of the current density and the log of the scan rate (Fig. S12a†).59 Here, the ‘b’ values of the cathodic and anodic peaks were accordingly 0.92 and 0.90, suggesting a combination of surface- and diffusion-controlled processes. These values indicate that the majority of the charges were contributed from the rapid reaction kinetics of a surface-controlled process.60 The exact contributions from the surface- and diffusion-controlled processes were also calculated. Battery-type materials follow a slow charge-storage process due to the diffusion process while capacitive-type materials follow a fast charge-storage process due to the double-layer/surface charge-transfer process.61,62 Here, the contribution from diffusive-(Qd) and surface-controlled (Qs) processes was calculated at different scan rates and estimated in Fig. S12b,† showing the contribution from the diffusion processes decreased with increasing the scan rate. Conversely, the charge contribution from the surface processes increased with increasing the scan rate from 53% (5 mV s−1) to 89% (60 mV s−1). Even at lower scan rates, the surface-controlled process still dominated the charge-transfer kinetics, which was favorable for high-rate performance and indicated that the Sb2S3/MoS2 anodes are promising candidates for ammonium-ion capacitors.
 | ||
| Fig. 4 Electrochemical study of AIHC. (a) Schematic presentation of ammonium-ion storage. (b) CV curves of MnO2 and Sb2S3/MoS2 in different voltage windows, (c) CV curves at different potential windows, (d) CV curves at various scan rates, (e) GCD curves at different current densities, and (f) Ragone plot comparison with materials reported in the available literature. | ||
GCD curves were also measured in different potential windows, and a symmetrical curve potential window was selected for further study (Fig. S12c†). The GCD performance was measured at various current densities, and symmetric curves were obtained without an IR drop (Fig. 4e). Specific capacitances were also extracted at different current densities from 0.8 to 4 A g−1. The specific capacitances of the AIHC were 140, 133.4, 124.3, 117.6, and 114.5 F g−1 at 0.8, 1.2, 2.4, 3.2, and 4 A g−1, respectively, with an excellent capacitive retention of 82% (Fig. S12d†). This suggests a high reversibility of NH4+ ion insertion/extraction and rapid kinetics at the cathode and anode, which favor long-term cycle stability. The energy and power density of the AIHC were also determined to investigate the electrochemical features of the cell from the charge–discharge data (Fig. 4f). As can be seen in the Ragone plot, the AIHC delivered a maximum energy density of 43.75 W h kg−1 at a power density of 600 W kg−1, which is comparable with those previously reported for AIHCs and supercapacitors.9,15,20,63–67 In addition, a 5-fold increase in power density was observed; the AIHC achieved a power density of 3000 W kg−1 at an energy density of 35.77 W h kg−1, indicating high reversibility and a rapid transmission of the charge carriers. Also, the long-term cycling performance of the AIHC was determined, because this is a crucial feature of energy-storage devices and is essential for commercial consideration. The cycling stability of the as-fabricated AIHC was examined at a 5 A g−1 current density (Fig. 5a). It was notable that the fabricated AIHC exhibited excellent cycle stability and retained almost 98.6% of its initial capacitance after 4000 cycles with 100% coulombic efficiency. The initial and final GCD cycles of the study are presented in the inset of Fig. 5a, which demonstrates the excellent retention and stability of the AIHC. In addition to its excellent cycle stability, the AIHC also showed an excellent rate capability (Fig. 5b). The specific capacitance of the AIHC was measured when the current density returned to a low current density of 0.8 A g−1 (140.8 F g−1), which was almost identical to its initial value (140 F g−1) at the same current density. Moreover, the AIHC maintained 100% coulombic efficiency at different current densities.
 | ||
| Fig. 5 (a) Cycling stability and (b) rate performance of the AIHC. (c) Nyquist plots before and after the cycling stability test of the AIHC with the fitted data (inset: fitted circuit). (d) Different charging/discharging state points on the GCD cycle. (e) Ex situ XPS survey spectra of the Sb2S3/MoS2 electrode. Ex situ study of the high-resolution (f) N 1s and (g) S 2p spectra of the Sb2S3/MoS2 electrode. | ||
The reaction kinetics of the AIHC were investigated using the EIS technique. Nyquist plots of the AIHC before and after the cycling stability test are shown in Fig. 5c. The semicircles observed at high and intermediate frequencies corresponded to the solution resistance (Rs) and charge-transfer resistance (Rct). The straight line in the low-frequency region signified Warburg impedance related to the NH4+-diffusion kinetics in the active material. The AIHC showed a smaller Rct (0.5 Ω), revealing its easy charge-transfer kinetics. In addition, after continuous cycling, the Rct value increased slightly (5.6 Ω) due to negligible deterioration of the electrode structure.68 Other parameters corresponding to the circuit are provided in Table S3.† The superior charge transfer and ion-diffusion kinetics of the AIHC were attributed to the highly conductive and porous structure of the electroactive material, which provides rapid electron pathways and allows easy electrolyte penetration and more easily accessible active sites for ammonium-ion storage.68 These results suggest a new route toward the development of high-performance, stable ammonium-ion-storage systems for grid-scale applications.
Next, ex situ XPS characterizations were employed to study the structural evolution of the Sb2S3/MoS2 material during NH4+ charging–discharging at different stages of the charge–discharge cycle, as shown in Fig. 5d. The full survey spectrum of the anode at various stages of charge–discharge is presented in Fig. 5e and confirmed the evolution in the S 2p and N 1s spectra. The high-resolution N 1s spectra of the anode at different states are shown in Fig. 5f. In the pristine state, the N 1s spectrum of Sb2S3/MoS2 was deconvoluted into two peaks at 394.9 and 397.8 eV, associated with Mo 3p and –N–H–, respectively.69–72 At fully charged, a new peak arose at 401.8 eV, ascribed to –N + H–segments, indicating the successful insertion of NH4+.73 Furthermore, the –N–H– content was reduced and assigned to the protonation state (–N + H–) caused by the adsorption of NH4+ on the surface of the electrode.19 After full discharge, the –N + H– segment-associated peak intensity (401.7 eV) sharply decreased, indicating NH4+ extraction during discharge.74 Furthermore, a new peak could be observed in Fig. 5g in the S 2p spectrum at 168.87 eV, corresponding to the oxidized sulfur remaining after discharge. This suggested that the sulfur was not totally returned to its original state and the inserted ions had not been completely extracted. In the high-resolution Mo 3d spectrum (Fig. S13a†), only a very slight shift in binding energy was observed in the fully charged state with a decreased intensity. In the fully discharged state, the peaks shifted toward the original state, confirming the insertion and extraction of NH4+ ions during the charge/discharge process. Similar results were observed in the high-resolution Sb 3d spectrum (Fig. S13b†), where the Sb 3d peaks were shifted toward higher binding energy on charging and returned to their original positions after discharging. In addition, the O 1s spectra were also examined (Fig. S13c†), and two deconvoluted peaks were observed, representing O from the core of the material (530.5 eV) and absorbed water (532.2 eV).75 In the fully charged state, the intensity of the hydroxyl group increased, and the slight shift to higher energy indicated the insertion of NH4+ ions. After full discharge, the NH4+ ions were extracted, and the peaks re-emerged at their original energies. The existence of a peak at 401.7 eV after full discharge in the N 1s spectrum confirmed the insertion and presence of NH4+ ions in the electrode. The shifting and diminished intensities of the peaks in the elemental spectra that occurred upon charging also supported the insertion of NH4+ ions. These results disclosed that NH4+ ions were inserted and extracted from the Sb2S3/MoS2 anode during charging and discharging.
The above results corroborate that the Sb2S3/MoS2 anode is a promising candidate for ammonium-ion storage. The highly conductive Sb2S3 core helps to improve the conductivity of the heterostructured electrode material and effectively optimize the charge transport. The interlayer spacing of the MoS2 shell material enables the easy insertion and extraction of ions, and improves the storage capacity of NH4+ ions. In addition, the heterostructure increases the specific surface area and provides highly active sites to interact with NH4+ ions. Furthermore, the presence of O increases the surface interactions, forms hydrogen bonds, and thus, increases the charge storage. This formation and breaking of bonds enables excellent NH4+ storage in the Sb2S3/MoS2 electrode, while the interlayer spacing of the MoS2 nanoshell increases the device storage capacity.
Conclusions
The use of non-metal charge carriers, especially ammonium ions, in electrochemical energy-storage devices has garnered attention because of their cost-effectiveness, safety, and environmentally friendly characteristics. We successfully developed a heterostructured Sb2S3/MoS2 anode material using a two-step hydrothermal method. The heterostructure of the electrode improved the electrochemical performance due to the synergy between the nanorods and nanoflakes, facilitated easy ion transfer, and demonstrated a high specific capacitance of 360 F g−1. In addition, an ammonium-ion hybrid capacitor constructed using a MnO2 cathode and an Sb2S3/MoS2 anode delivered superior energy and power density. Also, it exhibited excellent cycling performance, with a capacitive retention of 98.6% after 4000 cycles and a coulombic efficiency of 100%. In addition, ex situ XPS analysis was used to investigate the reversible insertion/extraction of NH4+ ions in the Sb2S3/MoS2 anode during the charging and discharging process. This work demonstrates the potential of ammonium-ion hybrid capacitors and provides perspectives for the future anode materials required for ammonium-ion-storage systems.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Research Foundation (NRF) of South Korea grant funded by the Korean government (MSIT) (2022R1A2C2008968 and 2022M3J7A1062940). NRC and EA appreciate the financial support from the Khalifa University of Science and Technology under Award No. 8474000620.References
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- M. T. F. Rodrigues, G. Babu, H. Gullapalli, K. Kalaga, F. N. Sayed, K. Kato, J. Joyner and P. M. Ajayan, Nat. Energy, 2017, 2, 17108 CrossRef CAS.
- M. R. Lukatskaya, J. I. Feldblyum, D. G. Mackanic, F. Lissel, D. L. Michels, Y. Cui and Z. Bao, Energy Environ. Sci., 2018, 11, 2876–2883 RSC.
- X. Wen, J. Luo, K. Xiang, W. Zhou, C. Zhang and H. Chen, Chem. Eng. J., 2023, 458, 141381 CrossRef CAS.
- F. Wang, O. Borodin, M. S. Ding, M. Gobet, J. Vatamanu, X. Fan, T. Gao, N. Edison, Y. Liang, W. Sun, S. Greenbaum, K. Xu and C. Wang, Joule, 2018, 2, 927–937 CrossRef CAS.
- D. Chao and H. J. Fan, Chem, 2019, 5, 1359–1361 CAS.
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S. Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS PubMed.
- Y. Ma, T. Sun, Q. Nian, S. Zheng, T. Ma, Q. Wang, H. Du and Z. Tao, Nano Res., 2022, 15, 2047–2051 CrossRef CAS.
- Q. Liu, F. Ye, K. Guan, Y. Yang, H. Dong, Y. Wu, Z. Tang and L. Hu, Adv. Energy Mater., 2023, 13, 1–11 Search PubMed.
- G. Liang, F. Mo, X. Ji and C. Zhi, Nat. Rev. Mater., 2021, 6, 109–123 CrossRef CAS.
- C. Lv, Y. Li, Y. Zhu, Y. Zhang, J. Kuang, Q. Zhao, Y. Tang and H. Wang, Adv. Sci., 2023, 2304214, 1–9 Search PubMed.
- Z. Guo, J. Huang, X. Dong, Y. Xia, L. Yan, Z. Wang and Y. Wang, Nat. Commun., 2020, 11, 959 CrossRef CAS PubMed.
- X. Zhang, G. Sun, S. Jia, H. Xie, Z. Kang, W. Chen, M. Cui, B. Wang, B. Wang, X. Chen and D. P. Yang, Chem. Eng. J., 2022, 438, 135624 CrossRef CAS.
- D. Yu, Z. Wei, X. Zhang, Y. Zeng, C. Wang, G. Chen, Z. X. Shen and F. Du, Adv. Funct. Mater., 2021, 31, 2008743 CrossRef CAS.
- X. Chen, P. Wang, Z. Feng, C. Meng and Y. Zhang, Chem. Eng. J., 2022, 445, 136747 CrossRef CAS.
- J. Han, A. Varzi and S. Passerini, Angew. Chem., 2022, 134, e202115046 CrossRef.
- R. Slade and A. Roberts, ECS Meet. Abstr., 2011, MA2011-02, 530 CrossRef.
- Q. Chen, J. Jin, M. Song, X. Zhang, H. Li, J. Zhang, G. Hou, Y. Tang, L. Mai and L. Zhou, Adv. Mater., 2022, 34, 1–8 Search PubMed.
- T. H. Lu, C. Zeng, H. Zhang, X. Shi, Y. Yu and X. Lu, Small, 2023, 19, 2206727 CrossRef CAS PubMed.
- S. G. Krishnan, H. D. Pham, C. Padwal, H. Weerathunga, X. Wang, K. Mahale and D. Dubal, J. Power Sources, 2023, 570, 232994 CrossRef CAS.
- L. Yan, Y. e. Qi, X. Dong, Y. Wang and Y. Xia, eScience, 2021, 1, 212–218 CrossRef.
- Q. Chen, H. Li, X. Lou, J. Zhang, G. Hou, J. Lu and Y. Tang, Adv. Funct. Mater., 2023, 33, 2214920 CrossRef CAS.
- R. Zheng, Y. Li, H. Yu, X. Zhang, D. Yang, L. Yan, Y. Li, J. Shu and B. Su, Angew. Chem., 2023, 135, e202301629 CrossRef.
- S. F. Kuchena and Y. Wang, ACS Appl. Energy Mater., 2020, 3, 11690–11698 CrossRef CAS.
- N. Murugan, S. Thangarasu, S. Bin Seo, Y. R. Choi, S. S. Magdum, T. H. Oh and Y. A. Kim, Carbon Lett., 2023, 33, 791–802 CrossRef.
- Q. Li, Y. Zhang, Y. Song, H. Yang, L. Yang, L. Bai, D. Wei, W. Wang, Y. Liang and H. Chen, Carbon Lett., 2023, 33, 549–560 CrossRef.
- X. Qin, J. Wan, Q. Zhang, Y. Zhang, H. Yu and S. Shi, Carbon Lett., 2023, 33, 781–790 CrossRef.
- C. Wu, W. Xiong and H. Li, Carbon Lett., 2023, 33, 2327–2334 CrossRef.
- N. W. Duffy, W. Baldsing and A. G. Pandolfo, Electrochim. Acta, 2008, 54, 535–539 CrossRef CAS.
- K. Wang, Z. Wang, J. Liu, C. Li, F. Mao, H. Wu and Q. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 47482–47489 CrossRef CAS PubMed.
- C. H. An, Y. Q. Li, S. Wu, L. X. Gao, L. Y. Lin, Q. B. Deng and N. Hu, Rare Met., 2023, 42, 1959–1968 CrossRef CAS.
- L. Wang, X. Zhang, C. Li, Y. Xu, Y. An, W. Liu, T. Hu, S. Yi, K. Wang, X. Sun, Y. Gong, Z. S. Wu and Y. Ma, Chem. Eng. J., 2023, 468, 143507 CrossRef CAS.
- T. Yan, F. Wen, J. Duan, C. Zhu, J. Wen, Y. Wang, J. Tong and Z. Chen, Chem. Eng. J., 2023, 474, 145839 CrossRef CAS.
- A. Panagiotopoulos, G. Nagaraju, S. Tagliaferri, C. Grotta, P. C. Sherrell, M. Sokolikova, G. Cheng, F. Iacoviello, K. Sharda and C. Mattevi, J. Mater. Chem. A, 2023, 11, 16190–16200 RSC.
- Y. Xiao, S. H. Lee and Y. K. Sun, Adv. Energy Mater., 2017, 7, 1601329 CrossRef.
- K. Prakash, S. Harish, S. Kamalakannan, T. Logu, M. Shimomura, J. Archana and M. Navaneethan, J. Energy Chem., 2023, 80, 335–349 CrossRef CAS.
- W. Li, T. H. Ma, Y. Y. Dang, X. Y. Liu, J. Y. Li and C. Y. Wang, Adv. Mater. Interfaces, 2022, 9, 2–9 Search PubMed.
- R. Jia, L. Li, G. Shen and D. Chen, Sci. China Mater., 2022, 65, 1443–1452 CrossRef CAS.
- C. Li, H. Song, C. Mao, H. Peng and G. Li, J. Alloys Compd., 2019, 786, 169–176 CrossRef CAS.
- Q. An, F. Lv, Q. Liu, C. Han, K. Zhao, J. Sheng, Q. Wei, M. Yan and L. Mai, Nano Lett., 2014, 14, 6250–6256 CrossRef CAS PubMed.
- G. Zhang, C. Zhou, J. Long, Y. Li, L. Lv, K. Yan, X. Chen, C. Dong, X. Xu and L. Mai, Nano Res., 2023, 16, 8488–8496 CrossRef CAS.
- W. He, C. Wang, H. Li, X. Deng, X. Xu and T. Zhai, Adv. Energy Mater., 2017, 7, 1700983 CrossRef.
- X. Mu, Y. Song, Z. Qin, J. Meng, Z. Wang and X. X. Liu, Chem. Eng. J., 2023, 453, 139575 CrossRef CAS.
- Z. Zhang, J. Zhao, M. Xu, H. Wang, Y. Gong and J. Xu, Nanotechnology, 2018, 29, 335401 CrossRef PubMed.
- Z. Li, Y. Xu, X. Ren and W. Wang, J. Mater. Sci., 2020, 55, 13892–13904 CrossRef CAS.
- Y. Wang, M. Zhen and H. Liu, J. Solid State Electrochem., 2018, 22, 3069–3076 CrossRef CAS.
- L. Zou, R. Qu, H. Gao, X. Guan, X. Qi, C. Liu, Z. Zhang and X. Lei, Results Phys., 2019, 14, 102458 CrossRef.
- Y. Yao, K. Ao, P. Lv and Q. Wei, Nanomaterials, 2019, 9, 844 CrossRef CAS PubMed.
- B. Li, L. Jiang, X. Li, P. Ran, P. Zuo, A. Wang, L. Qu, Y. Zhao, Z. Cheng and Y. Lu, Sci. Rep., 2017, 7, 1–12 CrossRef PubMed.
- J. Theerthagiri, K. Thiagarajan, B. Senthilkumar, Z. Khan, R. A. Senthil, P. Arunachalam, J. Madhavan and M. Ashokkumar, ChemistrySelect, 2017, 2, 201–210 CrossRef CAS.
- C. Costentin, T. R. Porter and J. M. Savéant, ACS Appl. Mater. Interfaces, 2017, 9, 8649–8658 CrossRef CAS PubMed.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CrossRef CAS.
- H. Zhang, Y. Tian, W. Wang, Z. Jian and W. Chen, Angew. Chem., 2022, 134, e202204351 CrossRef.
- J. Dai, X. Qi, L. Xia, Q. Xue, L. Luo, X. Wang, C. Yang, D. Li, H. Xie, A. Cabot, L. Dai and Y. Xu, Adv. Funct. Mater., 2023, 33, 2212440 CrossRef CAS.
- Y. Z. Wu, Y. Ding, T. Hayat, A. Alsaedi and S. Y. Dai, Appl. Surf. Sci., 2018, 459, 430–437 CrossRef CAS.
- L. Yang, W. Zheng, J. Halim, J. Rosen, Z. M. Sun and M. W. Barsoum, Batteries Supercaps, 2023, 6, e202200432 CrossRef CAS.
- M. Zhang, Y. Chen, D. Yang and J. Li, J. Energy Storage, 2020, 29, 101363 CrossRef.
- S. G. Krishnan, M. Harilal, B. Pal, I. I. Misnon, C. Karuppiah, C. C. Yang and R. Jose, J. Electroanal. Chem., 2017, 805, 126–132 CrossRef CAS.
- Y. An, Z. Li, Y. Sun, S. Li, Y. Xu, H. Dou and X. Zhang, Batteries Supercaps, 2022, 5, e202200036 CrossRef CAS.
- S. Li, D. Yu, J. Liu, N. Chen, Z. Shen, G. Chen, S. Yao and F. Du, Adv. Sci., 2023, 10, 2206836 CrossRef CAS PubMed.
- M. Huang, X. Wang, X. Liu and L. Mai, Adv. Mater., 2022, 34, 1–33 Search PubMed.
- D. Li, S. Zhao, J. Li, Z. Yuan, J. Cao, X. Fu, Y. Zhang, L. Wang and W. Han, Electrochim. Acta, 2022, 411, 140067 CrossRef CAS.
- C. Zhao, Y. Zhou, Z. Ge, C. Zhao and X. Qian, Carbon, 2018, 127, 699–706 CrossRef CAS.
- Y. Li, H. Wang, T. Shu, J. Yuan, G. Lu, B. Lin, Z. Gao, F. Wei, C. Ma, J. Qi and Y. Sui, J. Energy Storage, 2022, 51, 1–9 CrossRef.
- R. Govindan, X. J. Hong, P. Sathishkumar, Y. P. Cai and F. L. Gu, Electrochim. Acta, 2020, 353, 136502 CrossRef CAS.
- J. Hao, H. Liu, S. Han and J. Lian, ACS Appl. Nano Mater., 2021, 4, 1330–1339 CrossRef CAS.
- Q. Qin, L. Chen, T. Wei and X. Liu, Small, 2019, 15, 1803639 CrossRef PubMed.
- J. Yang, T. Wang, X. Guo and G. Wang, Nano Res., 2023, 16, 5592–5600 CrossRef CAS.
- Z. Yin, Y. Sun, C. Zhu, C. Li, X. Zhang and Y. Chen, J. Mater. Chem. A, 2017, 5, 13648–13658 RSC.
- J. Jia, M. Zhai, J. Lv, B. Zhao, H. Du and J. Zhu, ACS Appl. Mater. Interfaces, 2018, 10, 30400–30408 CrossRef CAS PubMed.
- B. Chang, J. Yang, Y. Shao, L. Zhang, W. Fan, B. Huang, Y. Wu and X. Hao, ChemSusChem, 2018, 11, 3198–3207 CrossRef CAS PubMed.
- S. H. Park, S. H. Kang and D. H. Youn, Materials, 2021, 14, 1–10 CrossRef.
- L. Xing, H. Chen, X. Wen, W. Zhou and K. Xiang, J. Alloys Compd., 2022, 925, 166652 CrossRef CAS.
- N. Lv, R. Ren, Y. Wu, Z. Xu, D. Wu, X. You, G. Zhu, Y. Zhang and S. Dong, Electrochim. Acta, 2022, 431, 141097 CrossRef CAS.
- H. Idriss, Surf. Sci., 2021, 712, 121894 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of materials synthesis, characterization, and electrochemical measurements, additional XRD, XPS, SEM, TEM, and electrochemical performance. See DOI: https://doi.org/10.1039/d4ta00262h. |
| This journal is © The Royal Society of Chemistry 2024 |