
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular engineering with CuanCl for effective optimization of a defective interface for wide-bandgap perovskite solar cells†
Maoxia
Xu
a,
Rui
Liu
a,
Haoran
Ye
a,
Haorong
Ren
a,
Jinyu
Li
a,
Chen
Deng
a,
Zetan
Zhang
a,
Chengbin
Yang
a,
Kexin
Hu
a,
Xiaoran
Sun
*a and
Hua
Yu
 *b
*b
aSchool of New Energy and Materials, Southwest Petroleum University, Chengdu, 610500, China. E-mail: xiaoran.sun@swpu.edu.cn
bGreat Bay University School of Physical Sciences Dongguan, Guangdong, China. E-mail: yuhua@gbu.edu.cn
First published on 5th March 2024
Abstract
In wide-bandgap (WBG) perovskite solar cells (PSCs), the energy level misalignment between the hole transport layer (HTL) and the perovskite layer, coupled with the high-density defects at their buried interface, causes severe non-radiative recombination within PSCs. Herein, CuanCl (carbamoyl-guanidine amidino urea salt, hydrochloride salt) with multifunctional molecular groups is introduced to optimize the WBG perovskite/HTL interface. This strategic introduction aims to suppress non-radiative recombination, consequently mitigating open-circuit voltage loss (Vloss). The findings demonstrate the bifunctional chemical passivation effect of the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and imine cations (NH+–) within CuanCl molecules on surface defects of perovskite, effectively suppressing diverse defect-assisted non-radiative recombination. Furthermore, the surface-bound CuanCl on the perovskite provides supplementary electronic states at the valence band maximum, achieving a more harmonized energy level alignment and effectively inhibiting charge recombination at the interface. The resultant CuanCl-treated WBG PSCs produce a high open-circuit voltage of 1.27 V, and a decent fill factor of 77.28%, which leads to a power conversion efficiency of 19.36%. Furthermore, the devices exhibit superior stability, maintaining 84% of their initial efficiency after 1000 hours in air with a humidity of 40%. This work provides new insight for optimizing a defective interface with the molecular engineering approach for fabricating efficient and stable WBG PSCs.
O) and imine cations (NH+–) within CuanCl molecules on surface defects of perovskite, effectively suppressing diverse defect-assisted non-radiative recombination. Furthermore, the surface-bound CuanCl on the perovskite provides supplementary electronic states at the valence band maximum, achieving a more harmonized energy level alignment and effectively inhibiting charge recombination at the interface. The resultant CuanCl-treated WBG PSCs produce a high open-circuit voltage of 1.27 V, and a decent fill factor of 77.28%, which leads to a power conversion efficiency of 19.36%. Furthermore, the devices exhibit superior stability, maintaining 84% of their initial efficiency after 1000 hours in air with a humidity of 40%. This work provides new insight for optimizing a defective interface with the molecular engineering approach for fabricating efficient and stable WBG PSCs.
Introduction
The promising applications of wide-bandgap (WBG) perovskite solar cells (PSCs), in areas such as tandem cells,1 semi-transparent cells,2 and building-integrated photovoltaics3 have garnered great attention and made tremendous advances in efficiency and stability. Despite the impressive achievements of the WBG PSCs, the open-circuit voltage loss (Vloss) caused by non-radiative recombination remains a critical factor that hampers their efficiency and stability.4 Previous studies have shown that the main reason for non-radiative recombination includes two aspects: high-density defects of the perovskite film and energy level mismatch between different function layers.5,6 Minimizing non-radiative recombination within the PSCs and optimizing charge extraction at adjacent transport layers are pivotal for realizing high performance photovoltaic devices.Due to the ionic nature of halide perovskite materials, various charged defect states tend to accumulate at grain boundaries and interfaces within the perovskite films.7 To eliminate these charged defects within perovskite films, an extensive exploration into the role of the functional passivator has been undertaken. Generally, passivating agent molecules encompass structural moieties with non-bonding electron pairs (Lewis base groups), such as phosphate (PO),8 carbonyl (CO),9 cyano (–CN),10 and sulfoxide (SO),11 capable of passivating electron-deficient defects within the perovskite, including undercoordinated Pb2+ ions, I−/Br− vacancies, and Pb clusters. In contrast, passivating agents bearing electron-deficient groups (Lewis acids), such as C60, PCBM, ICBA, and iodopentafluorobenzene, possess the capability to accept a non-bonding electron pair.12–15 These Lewis acid passivating agents can coordinate with electron-rich perovskite defects (including undercoordinated I−/Br− ions, antisite halide lead anions, and MA+/FA+ vacancies) to form coordination bonds, consequently reducing defects. Considering the charged nature of perovskite defects, ionic bonding stands as an alternative passivation technique for eliminating positively and negatively charged defects. The introduction of cations (metallic and organic cations) and anions (Cl−, I−, SCN−, and COO−) has been demonstrated to selectively passivate charge-complementary perovskite defects.16 To circumvent the constraints posed by single-functional passivating agents, the integration of zwitterionic molecules exhibiting spatially segregated negative and positive domains has emerged as a promising strategy. These composite molecules demonstrate an adaptive capacity to selectively target and passivate defects contingent upon their relative charges. Zwitterionic compounds like choline chloride and choline iodide have shown prowess in concurrently passivating both antisite PbI3− anions and cationic Pb clusters within the perovskite structure.17 The quaternary ammonium moiety of the 3-(decyldimethylammonio)-propane-sulfonate inner salt passivates the I− vacancy and MA+ vacancy, while the sulfonate group passivates the undercoordinated Pb2+.18 However, only a limited number of zwitterions have been reported to optimize the energy level alignment between the perovskite and the transport layers. This may be attributed to the electron cloud distribution and the highest occupied molecular orbital (HOMO) of the zwitterions. Wang et al. introduced squaraine molecules with a zwitterionic structure to passivate undercoordinated Pb2+ and antisite PbI3− defects while facilitating hole extraction at the perovskite/HTL interface, leading to a notable increase in the power conversion efficiency (PCE) to 18.83%.19
In this work, we introduced a multifunctional passivating agent, carbamoyl-guanidine amidino urea salt hydrochloride salt (CuanCl), at the perovskite/spiro-OMeTAD interface. The pair of non-bonding electrons in CO of CuanCl forms a coordinate covalent bond with undercoordinated Pb2+ in the perovskite, effectively mitigating cationic traps. Additionally, the imine cation (NH+–) within CuanCl passivates I− and FA+ vacancies in the perovskite through electrostatic interactions. Furthermore, the CuanCl molecule provides an additional electronic state at the valence band maximum (VBM) of the perovskite surface, thereby altering the surface potential to optimize the energy level alignment. With this multifunctional interface passivation strategy, the Vloss of the prepared WBG PSCs was significantly reduced. The addition of CuanCl resulted in an improvement in the open-circuit voltage (VOC) of the champion device from 1.19 V to 1.27 V, the PCE increased from 16.77% to 19.36%, and the fill factor (FF) increased from 71.56% to 77.28%. Moreover, the CuanCl-treated perovskite exhibits improved crystallinity, larger grain sizes, and reduced traps, thus enhancing the structural stability of the perovskite crystals. The fabricated WBG PSCs maintained 84% of their initial PCE after 1000 hours of aging under air conditions with a relative humidity of 40%.
Results and discussion
Initially, a WBG perovskite film with a composition of Cs0.2FA0.8Pb(I0.7Br0.3)3 was deposited via a one-step spin-coating method. Then, the perovskite surface was coated with a precise volume of CuanCl/IPA solution using a spin-coating method. The schematic representation of the perovskite film deposition method and the CuanCl post-treatment are depicted in Fig. 1. The multifaceted passivation mechanism of CuanCl is observed to be achieved through defect binding and the modulation of the VBM on the perovskite surface, as elucidated by the subsequent studies. | ||
| Fig. 1 The deposition method of WBG perovskite films and the impact of CuanCl on the perovskite film. | ||
To investigate the impact of CuanCl modification on defects within WBG perovskite films; X-ray photoelectron spectroscopy (XPS) of the control and the CuanCl-treated WBG perovskite was performed. Fig. 2a depicts the C 1s spectra of the control and the CuanCl-treated samples. As can be seen from the C 1s spectrum, in contrast to the control sample, the CuanCl-treated perovskite reveals an additional peak (CN), which indicates the presence of CuanCl at WBG perovskite interfaces. Additionally, in Fig. 2b and c, a shift in the core levels of the elements in WBG perovskite (Pb and I spectra) is observed after CuanCl treatment, attributed to electrostatic interaction.20 Analysis reveals a shift to higher binding energy positions for both the Pb 4f and I 3d peaks. In specific, the shift of the Pb 4f peak is due to the Lewis acid–base interaction between the CO bond in CuanCl and Pb2+.21 CuanCl forms a covalent coordination bond with the under-coordinated Pb2+ ions, reducing the Pb2+ defects of the perovskite structure. The shift observed in the I 3d peak can be attributed to the ionic bonding occurring between the imine cation (NH+–) of CuanCl and the I− ions. The observed shift in the I 3d peak suggests a strong interaction between CuanCl and the perovskite, attributed to the formation of ionic bonds between the imine cation (NH+–) and the halide ions. The bonding of NH+– with I− suppresses the migration behavior of halides. Therefore, CuanCl significantly inhibits the generation of halide vacancies, enhancing the stability of WBG PSCs. Additionally, it is well-known that Cl−, upon protonation of CuanCl, tends to enter the perovskite lattice and occupy halide vacancies, thereby reducing halide vacancy defects in the perovskite bulk phase.22 The impact of CuanCl on the crystallization of the WBG perovskite film was evaluated using X-ray diffraction (XRD). Fig. 2d reveals that the CuanCl-treated film (1.5 mg mL−1 CuanCl treatment) has a strong intensity of the (100) peak in comparison with the control film, implying that CuanCl post-treatment facilitates the formation of a (100) plane crystal.23 Furthermore, as shown in Fig. 2e and f, the (100), (200), and (210) peak positions are slightly shifted to lower angles, indicating that the lattices of the perovskite are expanded, which is caused by the cations of large radius embedded in the perovskite lattice.24 We consider this as strong evidence that CuanCl passivates the FA+ vacancies of the WBG perovskite. Based on the XPS and XRD results, CuanCl can be anchored on the perovskite surface due to its interaction with surface defects, thereby enhancing the stability of the perovskite.
 | ||
| Fig. 2 XPS spectra of (a) C 1s, (b) Pb 4f, and (c) I 3d. (d) XRD patterns. (e) The zoomed-in view of the (100) peak. (f) Amplification of (200) and (210) peaks. | ||
To analyze the impact of CuanCl anchored on the WBG perovskite surface on the band structures, ultraviolet photoelectron spectroscopy (UPS) measurements were conducted for control and CuanCl-treated samples. Fig. 3a illustrates the secondary electron cutoff spectrum, with calculated work functions of 4.19 eV for the control and 3.81 eV for the CuanCl-treated film. Additionally, as shown in Fig. 3b, the VBM for the control is −5.98 eV, whereas for the CuanCl-treated film, it is calculated to be −5.65 eV. According to previous work, the VBM of spiro-OMeTAD is −5.22 eV.25Fig. 3c shows the energy level arrangement of WBG PSCs. After CuanCl treatment, the work function of WBG perovskite films decreased, resulting in a weaker surface p-type doping.26 The change in the work function reduces the band offset between perovskite and spiro-OMeTAD by 0.33 eV. The VBM of the CuanCl-treated film exhibits closer alignment with that of spiro-OMeTAD, which facilitates hole transport at the WBG perovskite interface providing strong support for improved VOC and FF. Furthermore, Kelvin probe force microscopy (KPFM) was employed to assess the difference in surface potential between the perovskite films before and after CuanCl treatment. As illustrated in Fig. 3d and e, the surface potential distribution of the perovskite film becomes more uniform after CuanCl treatment, possibly due to the passivation of sub-bandgap defects.27 KPFM extracts the contact potential difference (CPD) between the tip and the sample by probing electrostatic forces. CPD enables the derivation of the sample's work function (Φsample), with the relationship expressed as follows:28,29
 | (1) |
 | ||
| Fig. 3 UPS spectra: (a) secondary electron cutoff spectrum. (b) Valence band edge region intensity. (c) Band alignment of the PSCs after CuanCl post-treatment. KPFM images of perovskite films: (d) control and (e) CuanCl. (f) Histogram of the contact potential difference. | ||
To explore disparities in the surface morphology between the modified and unmodified perovskite, top-view scanning electron microscopy (SEM) was conducted (Fig. 4a–d). In comparison with the control film, the films treated with CuanCl exhibit larger grains, implying that the crystal growth of CuanCl-treated perovskite films has been improved.31 This result can be further confirmed by XRD results. The enhanced grain size diminishes the grain boundary, which suppresses carrier accumulation and improves the WBG perovskite film quality. Hence, CuanCl treatment is advantageous for improving the stability of the WBG PSCs. Nevertheless, upon reaching a concentration of 3 mg mL−1, an excessive buildup of CuanCl on the film was evident, potentially obstructing carrier transport at the WBG perovskite interface. Notably, upon reaching a concentration of 1.5 mg mL−1, CuanCl accumulates moderately at the grain boundaries, consequently promoting film surface smoothing and facilitating the reduction of defects. A CuanCl-treated WBG perovskite film displaying a flatter morphology was confirmed by atomic force microscopy (AFM) measurements. Fig. 4e and f show that the CuanCl-treated perovskite film reveals a lower surface root-mean-square (RMS) value of 19.5 nm than that of the control one of 31.5 nm. A lower RMS suggests a smoother surface confirming that the non-electronic defects of the WBG perovskite surface are reduced after CuanCl treatment.32 The smooth morphology of the perovskite layer promotes the formation of excellent interface contacts with the charge transport layer, thus suppressing carrier interface transport losses.
 | ||
| Fig. 4 SEM images of the perovskite films: (a) control, (b) 0.75 mg mL−1 CuanCl-treated, (c) 1.5 mg mL−1 CuanCl-treated and (d) 3 mg mL−1 CuanCl-treated. AFM images of perovskite films: (e) control and (f) CuanCl treated. | ||
To further explore the effect of CuanCl accumulation at grain boundaries on the passivation of defects, photoluminescence (PL) and time-resolved photoluminescence (TRPL) measurements were conducted for the WBG perovskite film. As depicted in Fig. 5a, the 1.5 mg mL−1 CuanCl-treated perovskite film demonstrates the strongest PL intensity, markedly higher than that of the control and other concentration CuanCl-treated films (1 mg mL−1 and 3 mg mL−1). The increase in PL intensity is attributed to the filling of defects at the grain boundaries of the WBG perovskite film by CuanCl, resulting in a reduction in defect-assisted recombination. Following CuanCl treatment, a slight blue shift is observed in the emission peak of the WBG perovskite film, indicating a reduction in shallow defects within the perovskite film.33,34 As illustrated in Fig. 5b and Table S1 (ESI†), the TRPL fitting results demonstrate that the CuanCl-treated films exhibit a longer average decay lifetime than the control films, which further indicates a lower concentration of defects in the CuanCl-treated WBG perovskite film. The reduced defect concentration inhibits the non-radiative recombination of carriers, leading to reduced Vloss. In addition, to confirm the decrease in non-radiative recombination, we conducted dark current characterization for both the control device and the CuanCl-treated device. As shown in Fig. 5c, the CuanCl-treated device exhibits a dramatically lower dark current value of 5.59 × 10−3 mA cm2 than that of the control device of 2.12 × 10−2 mA cm2. This further confirms the inhibitory effect of CuanCl treatment on carrier recombination in the film, consistent with the results of TRPL and PL. Additionally, space-charge-limited current (SCLC) was employed to quantitatively assess the effect of CuanCl treatment on the trap density of WBG PSCs. The following formula was used to calculate the trap state density (Nt) (2):35
 | (2) |
 | (3) |
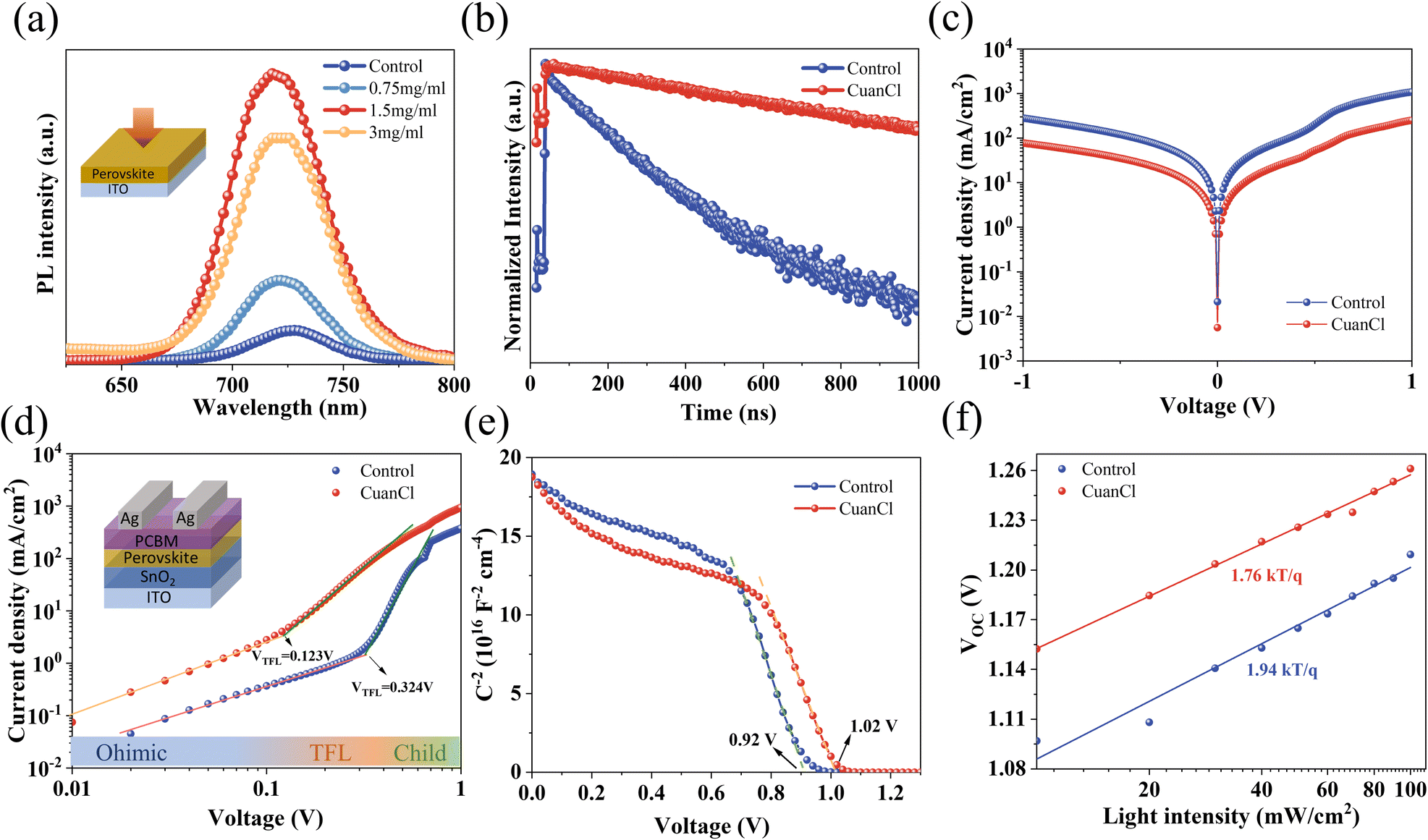 | ||
| Fig. 5 Passivation of defects. (a) PL spectra of perovskite films. (b) TRPL spectra. (c) Dark J–V characteristics recorded with and without CuanCl treatment. (d) Electron-only SCLC curves of control and CuanCl. (e) Mott–Schottky curve. (f) Illumination intensity dependence plot. | ||
A stands for the active device area, N is the carrier density, and V is the applied bias voltage. As depicted in Fig. 5e, the CuanCl-treated device exhibits a higher Vbi value of 1.01 V in comparison with that of the control one of 0.91 V. The increased Vbi indicates that the synergistic strategy of defect reduction and energy level arrangement optimization facilitates carrier extraction at the perovskite/spiro-OMeTAD interface, which helps to improve VOC. We assessed the potential of CuanCl treatment to suppress non-radiative recombination by analyzing the VOC of the devices at varying light intensities. As depicted in Fig. 5f, the slope of the line fitted using the ln-scale of VOC and light intensity is nkT/q, in which n, k, and T denote the ideality factor, Boltzmann constant, and absolute temperature, respectively.37 When n > 1, the main recombination mechanism is defect-assisted Shockley–Read–Hall recombination, and the lower n value indicates that nonradiative recombination is suppressed.38 The untreated samples exhibit an ideality factor n of 1.94, whereas the ideality factor n of WBG PSCs treated with CuanCl decreases to 1.76. This serves as compelling evidence that CuanCl treatment significantly reduces defects and suppresses carrier recombination. The characterization analyses above anticipate the high VOC and FF of WBG PSCs treated with CuanCl.
We fabricated n-i-p structured solar cells using the ITO/SnO2/perovskite/CuanCl/spiro-OMeTAD/Ag stack configuration to assess the impact of CuanCl post-treatment on the performance of WBG PSCs (Fig. 6a). After CuanCl post-treatment, the champion PCE of WBG PSCs increased from 16.77% to 19.36%, the VOC improved from 1.19 V to 1.27 V, and the FF increased from 71.56% to 77.28% (Fig. 6b). The change in short-circuit current density (JSC) was not pronounced, as corroborated by UV-vis spectroscopy (Fig. S2, ESI†) and external quantum efficiency (EQE) measurements (Fig. 6c). The EQE data indicate an integrated JSC of 18.98 mA cm−2 and 18.67 mA cm−2 for WBG PSCs before and after CuanCl treatment, respectively (Table S2†). The perovskites before and after CuanCl treatment have similar band gaps (∼1.73 eV) (Fig. S3, ESI†). The passivation effect of CuanCl significantly enhances the photovoltaic performance of wide-bandgap perovskite solar cells, particularly in reducing Vloss (Table S3†). Fig. S4a and b (ESI†) display the photovoltaic performance parameters of the WBG PSCs manufactured with different contents of CuanCl (0 mg mL−1, 1 mg mL−1, 1.5 mg mL−1, and 3 mg mL−1). Notably, all passivation routes exhibit significant reproducibility for different CuanCl concentrations. The average PCE of 1.5 mg mL−1 CuanCl-treated devices shows an improvement of 2.39% (Fig. 6d), which is caused by the synchronous increase in VOC and FF. The CuanCl post-treatment, with its optimal concentration, tunes the band-edge state of perovskite and reducing defects, thereby improving energy level alignment and mitigating defect-related recombination. The improvement of VOC and FF was attributed to an improved carrier transport channel, which promoted photogenerated carrier transport within the WBG film and device. In addition, the number of defects on the grain boundary and surface and the quality of the perovskite film are important factors that affect the stability of WGB PSCs. The effect of CuanCl passivation on the stability of WGB PSCs was investigated through PCE testing (under air conditions with a relative humidity of 40% ± 10%) (Fig. 6e). The champion devices treated with CuanCl maintained 84% of their initial PCE after 1000 hours. In contrast, the champion devices untreated with CuanCl only retained 71% of their initial PCE. With the extension of time, the decrease in PCE gradually slows down for the CuanCl-treated device, indirectly indicating that CuanCl post-treatment effectively prevented water and oxygen invasion owing to improved crystallization and reduced defects. In addition, the modified device also shows good thermal stability. Subjecting both the CuanCl-treated and untreated devices to 85 °C on a hotplate in a N2 atmosphere for 250 hours revealed that the CuanCl-treated devices maintained 70% of their initial efficiency, a notable enhancement compared to the control devices, which retained just 56% of their initial efficiency (Fig. 6f).
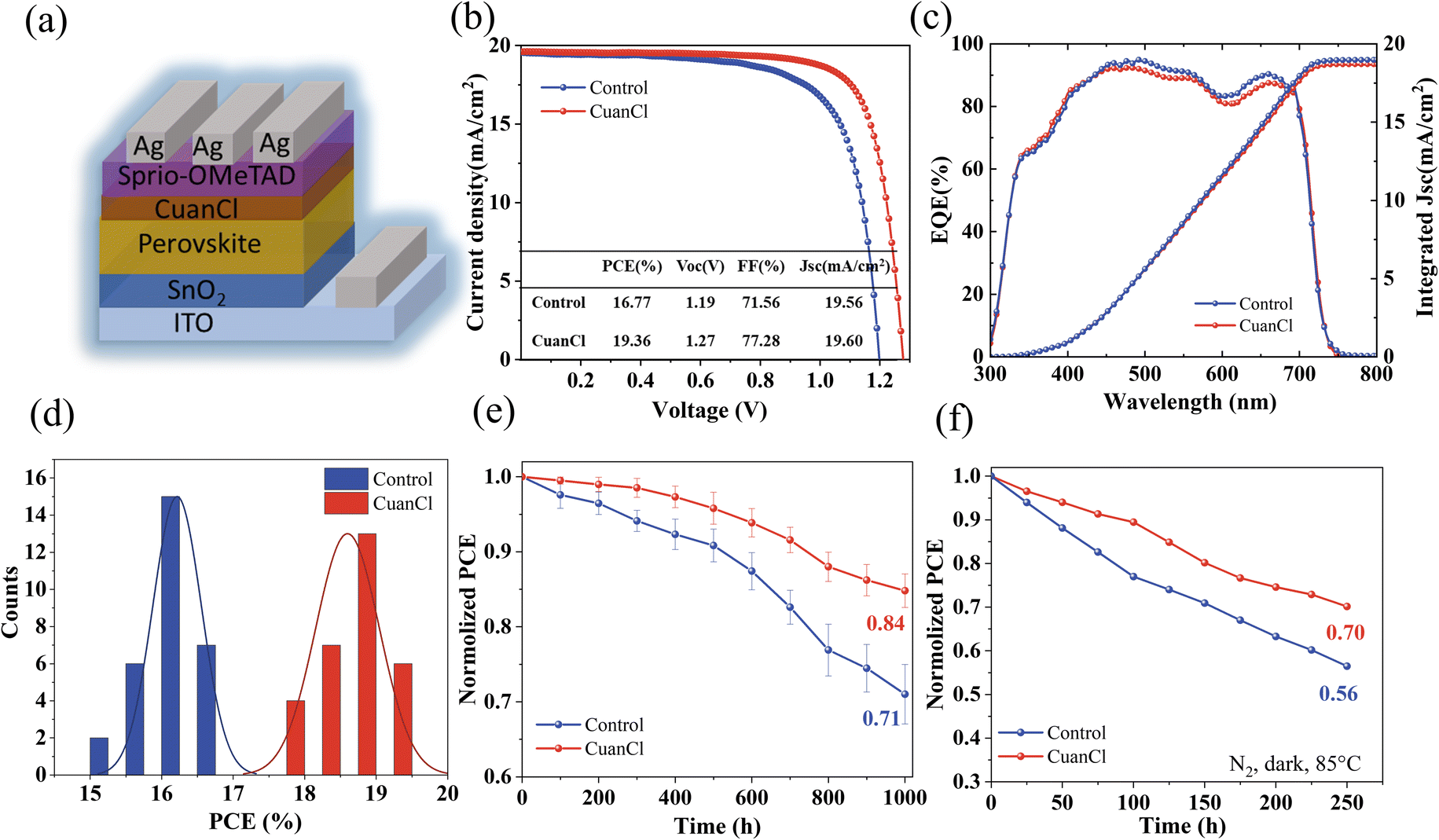 | ||
| Fig. 6 Performance of PSCs. (a) Device architecture of WBG PSCs. (b) J–V curves of the optimal device. (c) EQE curves. (d) PCE histogram of WBG PSCs prepared with and without CuanCl treatment. (e) Long-term stability of the two devices under non-encapsulated conditions with a relative humidity of 40% ± 10%. (f) The change in PCE for unencapsulated devices under exposure to an 85 °C N2 atmosphere. | ||
Conclusion
We introduced a multifunctional CuanCl molecule with a zwitterionic structure capable of passivating under-coordinated Pb2+, halide vacancies, and FA+ vacancyieson the perovskite surface, resulting in a significant reduction in the defect density of the perovskite. The robust interaction between CuanCl molecules and the perovskite surface facilitates crystal growth. Meanwhile, CuanCl molecules induce a reduction of 0.33 eV in the energy band offset between spiro-OMeTAD and the perovskite by altering the perovskite's surface potential. The multifunctional passivation strategy significantly reduces carrier recombination, thus improving the VOC and PCE of WBG PSCs. Furthermore, with the CuanCl binding to the perovskite surface, the reduction in defect states, along with an increase in perovskite grain size, results in improved perovskite film quality. This improvement is advantageous for enhancing the stability of WBG PSCs. Finally, after treatment with CuanCl, the WBG PSCs exhibited an increase in VOC from 1.19 V to 1.27 V and an improvement in PCE from 16.77% to 19.36%. The unpackaged CuanCl-treated device maintained 84% of its initial efficiency in an air environment with a relative humidity of 40% ± 10% over 1000 hours. This study proposes a straightforward and cost-effective strategy that effectively facilitates charge transfer, significantly reducing Vloss and enabling the realization of highly efficient and stable WBG PSCs.Experimental section
Materials
Dimethylformamide (DMF), bis(trifluoromethane)sulfonamide lithium salt (LiTFSI, >99%), isopropanol (IPA, 99.9%), chlorobenzene (CB), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich. SnO2 (15% colloidal dispersion in H2O) was ordered from Alfa Aesar. Spiro-OMeTAD (99.8%) was ordered from Lumtec. Carbamoyl-guanidine amidino urea salt hydrochloride salt (CuanCl, ≥99.5%) has been purchased from Aladdin. Formamidinium iodide (FAI, ≥99.5%), 4-tert-butylpyridine (tBP, 96%), PbBr2 (99.99%), cesium iodide (CsI, 99.99%), and PbI2 (99.99%) was purchased from Xi'an Polymeric Optics Technology Corp. All materials were used directly without further purification.Preparation and characterization
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52376210) and Chengdu Science and Technology Program (2021GH0200032HZ).References
- R. Lin, Y. Wang, Q. Lu, B. Tang, J. Li, H. Gao, Y. Gao, H. Li, C. Ding, J. Wen, P. Wu, C. Liu, S. Zhao, K. Xiao, Z. Liu, C. Ma, Y. Deng, L. Li, F. Fan and H. Tan, Nature, 2023, 620, 994–1000 CrossRef CAS PubMed.
- L. Yang, Y. Jin, Z. Fang, J. Zhang, Z. Nan, L. Zheng, H. Zhuang, Q. Zeng, K. Liu, B. Deng, H. Feng, Y. Luo, C. Tian, C. Cui, L. Xie, X. Xu and Z. Wei, Nano-Micro Lett., 2023, 15, 111 CrossRef CAS PubMed.
- M. Batmunkh, Y. L. Zhong and H. Zhao, Adv. Mater., 2020, 32, 2000631 CrossRef CAS PubMed.
- Y. Yu, R. Liu, C. Liu, X. Shi, H. Yu and Z. Chen, Adv. Energy Mater., 2022, 12, 2201509 CrossRef CAS.
- L. Canil, T. Cramer, B. Fraboni, D. Ricciarelli, D. Meggiolaro, A. Singh, M. Liu, M. Rusu, C. M. Wolff, N. Phung, Q. Wang, D. Neher, T. Unold, P. Vivo, A. Gagliardi, F. De Angelis and A. Abate, Energy Environ. Sci., 2021, 14, 1429–1438 RSC.
- S. Mahesh, J. M. Ball, R. D. J. Oliver, D. P. McMeekin, P. K. Nayak, M. B. Johnston and H. J. Snaith, Energy Environ. Sci., 2020, 13, 258–267 RSC.
- Z. Ni, C. Bao, Y. Liu, Q. Jiang, W.-Q. Wu, S. Chen, X. Dai, B. Chen, B. Hartweg, Z. Yu, Z. Holman and J. Huang, Science, 2020, 367, 1352–1358 CrossRef CAS PubMed.
- S. Yang, S. Chen, E. Mosconi, Y. Fang, X. Xiao, C. Wang, Y. Zhou, Z. Yu, J. Zhao, Y. Gao, F. De Angelis and J. Huang, Science, 2019, 365, 473–478 CrossRef CAS PubMed.
- Y. Li, J. Shi, J. Zheng, J. Bing, J. Yuan, Y. Cho, S. Tang, M. Zhang, Y. Yao, C. F. J. Lau, D. S. Lee, C. Liao, M. A. Green, S. Huang, W. Ma and A. W. Y. Ho-Baillie, Advanced Science, 2020, 7, 1903368 CrossRef CAS PubMed.
- L. Yang, Q. Xiong, Y. Li, P. Gao, B. Xu, H. Lin, X. Li and T. Miyasaka, J. Mater. Chem. A, 2021, 9, 1574–1582 RSC.
- R. Chen, Y. Wang, S. Nie, H. Shen, Y. Hui, J. Peng, B. Wu, J. Yin, J. Li and N. Zheng, J. Am. Chem. Soc., 2021, 143, 10624–10632 CrossRef CAS PubMed.
- Y. Lin, B. Chen, F. Zhao, X. Zheng, Y. Deng, Y. Shao, Y. Fang, Y. Bai, C. Wang and J. Huang, Adv. Mater., 2017, 29, 1700607 CrossRef PubMed.
- Y. Shao, Z. Xiao, C. Bi, Y. Yuan and J. Huang, Nat. Commun., 2014, 5, 5784 CrossRef CAS PubMed.
- A. Abate, M. Saliba, D. J. Hollman, S. D. Stranks, K. Wojciechowski, R. Avolio, G. Grancini, A. Petrozza and H. J. Snaith, Nano Lett., 2014, 14, 3247–3254 CrossRef CAS PubMed.
- R. C. Haddon, R. E. Palmer, H. W. Kroto, P. A. Sermon, H. W. Kroto, A. L. Mackay, G. Turner and D. R. M. Walton, Philos. Trans. R. Soc. London, Ser. A, 1997, 343, 53–62 Search PubMed.
- B. Chen, P. N. Rudd, S. Yang, Y. Yuan and J. Huang, Chem. Soc. Rev., 2019, 48, 3842–3867 RSC.
- X. Zheng, B. Chen, J. Dai, Y. Fang, Y. Bai, Y. Lin, H. Wei, X. C. Zeng and J. Huang, Nat. Energy, 2017, 2, 1–9 Search PubMed.
- X. Zheng, Y. Deng, B. Chen, H. Wei, X. Xiao, Y. Fang, Y. Lin, Z. Yu, Y. Liu, Q. Wang and J. Huang, Adv. Mater., 2018, 30, 1803428 CrossRef PubMed.
- Z. Wang, A. Pradhan, M. A. Kamarudin, M. Pandey, S. S. Pandey, P. Zhang, C. Huey Ng, A. S. M. Tripathi, T. Ma and S. Hayase, ACS Appl. Mater. Interfaces, 2019, 11, 10012–10020 CrossRef CAS PubMed.
- C. Li, X. Wang, E. Bi, F. Jiang, S. M. Park, Y. Li, L. Chen, Z. Wang, L. Zeng, H. Chen, Y. Liu, C. R. Grice, A. Abudulimu, J. Chung, Y. Xian, T. Zhu, H. Lai, B. Chen, R. J. Ellingson, F. Fu, D. S. Ginger, Z. Song, E. H. Sargent and Y. Yan, Science, 2023, 379, 690–694 CrossRef CAS PubMed.
- Z. Zhang, Y. Gao, Z. Li, L. Qiao, Q. Xiong, L. Deng, Z. Zhang, R. Long, Q. Zhou, Y. Du, Z. Lan, Y. Zhao, C. Li, K. Müllen and P. Gao, Adv. Mater., 2021, 33, 2008405 CrossRef CAS PubMed.
- H. Si, C. Xu, Y. Ou, G. Zhang, W. Fan, Z. Xiong, A. Kausar, Q. Liao, Z. Zhang, A. Sattar, Z. Kang and Y. Zhang, Nano Energy, 2020, 68, 104320 CrossRef CAS.
- X. Zheng, Y. Hou, C. Bao, J. Yin, F. Yuan, Z. Huang, K. Song, J. Liu, J. Troughton, N. Gasparini, C. Zhou, Y. Lin, D.-J. Xue, B. Chen, A. K. Johnston, N. Wei, M. N. Hedhili, M. Wei, A. Y. Alsalloum, P. Maity, B. Turedi, C. Yang, D. Baran, T. D. Anthopoulos, Y. Han, Z.-H. Lu, O. F. Mohammed, F. Gao, E. H. Sargent and O. M. Bakr, Nat. Energy, 2020, 5, 131–140 CrossRef CAS.
- Z. Zhang, J. Jiang, X. X. Liu, X. Wang, L. Wang, Y. Qiu, Z. Zhang, Y. Zheng, X. Wu, J. Liang, C. Tian and C. Chen, Small, 2022, 18, 2105184 CrossRef CAS PubMed.
- Y. Cai, J. Cui, M. Chen, M. Zhang, Y. Han, F. Qian, H. Zhao, S. Yang, Z. Yang, H. Bian, T. Wang, K. Guo, M. Cai, S. Dai, Z. Liu and S. F. Liu, Adv. Funct. Mater., 2021, 31, 2005776 CrossRef CAS.
- H. Chen, A. Maxwell, C. Li, S. Teale, B. Chen, T. Zhu, E. Ugur, G. Harrison, L. Grater, J. Wang, Z. Wang, L. Zeng, S. M. Park, L. Chen, P. Serles, R. A. Awni, B. Subedi, X. Zheng, C. Xiao, N. J. Podraza, T. Filleter, C. Liu, Y. Yang, J. M. Luther, S. De Wolf, M. G. Kanatzidis, Y. Yan and E. H. Sargent, Nature, 2023, 613, 676–681 CrossRef CAS PubMed.
- S. You, F. T. Eickemeyer, J. Gao, J.-H. Yum, X. Zheng, D. Ren, M. Xia, R. Guo, Y. Rong, S. M. Zakeeruddin, K. Sivula, J. Tang, Z. Shen, X. Li and M. Grätzel, Nat. Energy, 2023, 8, 515–525 CrossRef CAS.
- Y. Yu, R. Liu, M. Xu and H. Yu, EcoMat, 2023, 5, e12272 CrossRef CAS.
- R. Liu, C. Liu, Y. Yu, H. Yu and X. Xu, J. Phys. Chem. Lett., 2022, 13, 8945–8952 CrossRef CAS PubMed.
- H. Chen, A. Maxwell, C. Li, S. Teale, B. Chen, T. Zhu, E. Ugur, G. Harrison, L. Grater, J. Wang, Z. Wang, L. Zeng, S. M. Park, L. Chen, P. Serles, R. A. Awni, B. Subedi, X. Zheng, C. Xiao, N. J. Podraza, T. Filleter, C. Liu, Y. Yang, J. M. Luther, S. De Wolf, M. G. Kanatzidis, Y. Yan and E. H. Sargent, Nature, 2023, 613, 676–681 CrossRef CAS PubMed.
- Y. Li, Z. Chen, B. Yu, S. Tan, Y. Cui, H. Wu, Y. Luo, J. Shi, D. Li and Q. Meng, Joule, 2022, 6, 676–689 CrossRef CAS.
- S. Chen, Y. Liu, X. Xiao, Z. Yu, Y. Deng, X. Dai, Z. Ni and J. Huang, Joule, 2020, 4, 2661–2674 CrossRef CAS.
- D. W. de Quilettes, S. M. Vorpahl, S. D. Stranks, H. Nagaoka, G. E. Eperon, M. E. Ziffer, H. J. Snaith and D. S. Ginger, Science, 2015, 348, 683–686 CrossRef CAS PubMed.
- Z. Li, J. Zhang, S. Wu, X. Deng, F. Li, D. Liu, C. Lee, F. Lin, D. Lei, C.-C. Chueh, Z. Zhu and A. K.-Y. Jen, Nano Energy, 2020, 78, 105377 CrossRef CAS.
- Y. Huang, L. Li, Z. Liu, H. Jiao, Y. He, X. Wang, R. Zhu, D. Wang, J. Sun, Q. Chen and H. Zhou, J. Mater. Chem. A, 2017, 5, 8537–8544 RSC.
- O. Almora, C. Aranda, E. Mas-Marzá and G. Garcia-Belmonte, Appl. Phys. Lett., 2016, 109, 173903 CrossRef.
- D. Yang, X. Zhou, R. Yang, Z. Yang, W. Yu, X. Wang, C. Li, S. F. Liu and R. P. H. Chang, Energy Environ. Sci., 2016, 9, 3071–3078 RSC.
- P. Caprioglio, C. M. Wolff, O. J. Sandberg, A. Armin, B. Rech, S. Albrecht, D. Neher and M. Stolterfoht, Adv. Energy Mater., 2020, 10, 2000502 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta07690c |
| This journal is © The Royal Society of Chemistry 2024 |