
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilization of intermediate Mo oxidation states by Nb doping enhancing methane aromatization on Mo/HZSM-5 catalysts†
Stefan
Peters
 a,
Stephan
Bartling
a,
Magdalena
Parlinska-Wojtan
b,
Alexander
Wotzka
a,
Ana Guilherme
Buzanich
c,
Sebastian
Wohlrab
a and
Ali M.
Abdel-Mageed
*a
a,
Stephan
Bartling
a,
Magdalena
Parlinska-Wojtan
b,
Alexander
Wotzka
a,
Ana Guilherme
Buzanich
c,
Sebastian
Wohlrab
a and
Ali M.
Abdel-Mageed
*a
aLeibniz-Institut für Katalyse (LIKAT), 18059, Rostock, Germany. E-mail: ali.abdelmageed@catalysis.de
bInstitute of Nuclear Physics, Polish Academy of Sciences, 31-342 Krakow, Poland
cFederal Institute for Materials Research and Testing (BAM), 12489 Berlin, Germany
First published on 22nd February 2024
Abstract
The dehydroaromatization of the naturally abundant methane is a promising process to produce aromatics and COx-free hydrogen. Low-temperature activity, regenerability and time-on-stream stability are fundamental challenges for the industrial use of the commonly studied benzenoid-selective Mo/HZSM-5 catalysts. We report a promotional effect of Nb doping on the activity and stability of Mo/HZSM-5 catalysts between 600 and 700 °C. Nb addition enhances benzene yields measurably at all investigated temperatures. An increased thermal stability of MoNb/HZSM-5 compared to Mo/HZSM-5 catalysts was found upon exposure to multiple consecutive reaction and oxidative treatment cycles, thus resulting in higher restorability of activity which extends the catalyst lifetime. While high-resolution electron microscopy showed homogeneous mixing of Mo and Nb on the catalyst, in situ time-resolved Mo K-edge X-ray absorption spectroscopy, supported by ex situ and time-resolved in situ near-ambient pressure X-ray photoelectron spectroscopy measurements, revealed a distinct interaction between the transition metals, involving a partial reduction of the otherwise stable Nb and concurrent oxidation of Mo which leads to increased stability of Mo5+/Mo4+ states. These effects were correlated to the enhanced activity and regenerability of the MoNb/HZSM-5 catalyst for methane dehydroaromatization.
1. Introduction
Methane is the most abundant hydrocarbon on Earth, with a large proven world reserve and the possibility of production from renewable resources.1,2 Despite its widespread availability and potential production from renewables (via power-to-gas and biomass3–5) methane is primarily used for power generation or production of synthesis gas (CO + H2) via reforming reactions, which are energy intensive and ecologically unfavorable processes.6–9 Direct conversion of methane to value-added products is strictly limited due to the high activation barrier.9–11 Valorization of methane can often only be accomplished by using oxidizing agents and/or high reaction temperatures, severely impacting product selectivity. Among several approaches, methane dehydroaromatization (MDA) is promising as it is an oxidant-free and greener pathway to directly produce hydrogen, small aliphatics and aromatic hydrocarbons from methane.12–15 Early studies had identified Mo as one of the most active transition metals when supported by zeolites such as HZSM-5.14 These zeolites provide shape selectivity to aid in aromatics formation and stabilization of MoOx species inside their micropores.16,17 All known zeolitic catalysts deactivate, however, during reactions with time on stream, losing more than 50% of their initial activity after a few hours due to the deposition of coke.18–20 Removal of coke is possible either by reductive21,22 or oxidative treatment23–25 of spent catalysts, restoring aromatization activity in the process. Cyclic operations using one of these methods have been demonstrated to extend catalyst lifetime and applicability. Oxidation in air is the simplest method for reactivation of MDA catalysts, though too high regeneration temperatures may lead to irreversible deactivation due to loss of Mo26,27 or damage to the zeolite framework by dealumination.28 On the other hand, reductive catalyst regeneration may be more costly and not easily feasible for larger scale applications.29Other attempts to resolve these problems with Mo/HZSM-5 catalysts were made by modification with promoters. Published results refer to the possible use of other various transition metals such as Fe, Co and Zn (between 6% and 47% enhancement in benzenoid product yield).30–34 However, most of these results cannot be directly compared to one another because of the varying reaction conditions. After probing several different transition metals, we have identified Nb as a promising promoter for the MDA reaction, in particular at lower temperatures down to 600 °C, which can be desirable to limit the high necessary energy consumption.14 A positive effect of Nb has not been deeply investigated so far. In contrast, both Pasupulety et al.35 and Rodrigues et al.36 observed negative effects on the benzene yield after incorporating 1–2 wt% Nb (as oxide or carbide, respectively) into their Mo catalysts. However, the influence of these Nb additives on the activity of molybdenum is not yet fully understood.
In this work, we investigated the effects of Nb doping (as Nb2O5) on the performance of a 6 wt% Mo/HZSM-5 catalyst for the MDA reaction. In the following sections we first show the results from catalytic performance tests including different reaction parameters such as temperature, flow rate and the addition of co-reactants. Additionally, cyclic operation of reactions and oxidative regeneration was employed to probe the stability of the catalysts. Catalytic studies are supplemented by temperature-programmed oxidation of the accumulated coke on the spent catalysts. After that we show detailed analysis of catalyst morphology and the particle size using the results from high resolution scanning transmission electron microscopy (STEM) coupled with elemental mapping of different catalyst constituents. Finally, we discuss the speciation of Mo and Nb and their electronic properties using in situ near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) as well as in situ X-ray absorption spectroscopy (XAS) at the Mo K-edge to elucidate the location and effect of Nb on the Mo active species.
2. Experimental section
2.1 Catalyst preparation
All catalysts have been prepared by wet impregnation of commercially available NH4-ZSM-5 (CBV3024E, Zeolyst International, Si/Al = 15). In a previous study we determined the optimal loading of Mo on HZSM-5 to be 6 wt%,37 achieving the highest methane conversion and benzene yield. In a typical synthesis procedure, 5.057 g of the zeolite material (5 g accounting for NH3 loss) and 612.7 mg of (NH4)6Mo7O24·4H2O precursor were mixed in a round-bottom flask. Subsequently 50 mL of deionized water were added, and the suspension was continuously stirred for 24 h at 25 °C. For the Nb-doped sample, the amount of (NH4)6Mo7O24·4H2O was increased to 622.5 mg and 275.3 mg of NH4NbO(C2O4)2·8.6H2O were added for a Nb loading of 1 wt%. After impregnation the water was carefully removed using a rotary evaporator. The obtained solids were then dried under vacuum. Next the samples were heated up to 550 °C with a temperature ramp of 10 °C min−1 under static air and were calcined at this temperature for 6 h to transform the support material into its acidic form and metal precursors into their respective oxides. The ICP-OES analysis results showed a Mo loading of 6.3 wt% for both samples and a Nb loading of 1.0 wt% in the doped sample (see Table 1). The calcined catalysts were pressed, crushed, and sieved to obtain particle sizes of 400–600 μm. The catalysts are denoted as 6Mo/HZSM-5 and 6Mo1Nb/HZSM-5, respectively. For comparison, other promoters (Fe, Co, and Zn) were introduced by using the corresponding metal nitrates in the procedure described above with a loading of 1 wt%.| Catalyst | Mo loadinga [wt%] | Promoter loadinga [wt%] | S BET [m2 g−1] | V micro [cm3 g−1] | V meso [cm3 g−1] | Adsorbed amount of NH3c [μmol g−1] |
|---|---|---|---|---|---|---|
| a Determined by ICP-OES. b Determined by N2 adsorption at 77 K. c Determined by NH3-TPD. | ||||||
| HZSM-5 | — | — | 407 | 0.155 | 0.165 | 908 |
| 6Mo | 6.3 | — | 328 | 0.118 | 0.145 | 661 |
| 6Mo1Fe | 6.0 | 1.0 | 326 | 0.117 | 0.122 | 669 |
| 6Mo1Co | 6.1 | 1.0 | 308 | 0.106 | 0.145 | 721 |
| 6Mo1Zn | 6.2 | 1.1 | 294 | 0.102 | 0.140 | 706 |
| 6Mo1Nb | 6.3 | 1.0 | 318 | 0.110 | 0.132 | 590 |
2.2 Structural characterization
2.3 Kinetic measurements
The catalytic performance of 6Mo/HZSM-5 and 6Mo1Nb/HZSM-5 catalysts was evaluated using the experimental setup illustrated in Fig. S1 in the ESI.† Gases were fed to the reactor via Bronkhorst mass flow controllers. The reactant gas was made up of 90% CH4 (purity 4.5) and 10% N2 (purity 5.0), the latter being used as an internal standard. The gas mixture was supplied by Linde. For measurements, 300 mg of the catalyst sample were placed inside a quartz reactor (inner diameter 7 mm) and held in place using quartz wool plugs. Under a continuous flow of nitrogen (purity 5.0, supplied by Linde) the catalyst was then heated to reaction temperature (between 600 and 700 °C) with a rate of 33.75 K min−1via an electrical 3-zone furnace. A thermocouple inserted into the reactor via a quartz capillary allowed control of the sample temperature by directly regulating the center zone where the sample was located. When the temperature at the catalyst thermocouple was constant, gas flows were changed from nitrogen to the reactant gas mixture. Unless otherwise specified, catalysts were subjected to methane for 6 h with a gas flow of 16 SCCM (3200 SCC gcat−1 h−1, 1.89 h−1 based on CH4 mass; reference conditions 25 °C, 1 atm) at ambient pressure. Afterwards the setup was continuously flushed with nitrogen and the reactor was allowed to cool down over night. The spent catalysts were obtained from the reactor at room temperature and stored under ambient conditions. For regeneration measurements, the spent catalysts were reheated in situ under a continuous nitrogen flow to 450 °C (rate 33.75 K min−1) and held for 45 min to reach a temperature equilibrium. The gas was then switched to a flow of air (5.5 SCCM, GHSV = 1100 SCC gcat−1 h−1) for 45 min at ambient pressure.Reactants and products were analyzed using a custom Agilent 7890D gas chromatograph with a thermal conductivity detector (TCD), a flame ionization detector (FID), and an Agilent 5977B mass spectrometric detector, as described in a previous publication.37 In short, analysis was performed with two channels and independent sample injection loops. H2, CH4, N2 and CO were separated using a Molsieve 5A column and measured with the TCD. A HayeSep Q column, coupled with a 10-port valve, was used as a backflush system for all C2+ compounds. Hydrocarbons were pre-separated with an HP-5 column. A timed Deans switch allowed for separate analysis of gaseous compounds with the FID (further separated using an Rt-Alumina BOND/MAPD column), and aromatic products with the MSD. Condensation or crystallization of products was prevented by heating all transfer lines after the reactor as well as the bypass valve and GC valve box to 200 °C. The reference between the two detector channels was methane. As reported previously,37 conversions and yields were calculated based on carbon balance according to eqn (1)–(3), with νP being the carbon number of the respective product. Quantification of non-calibrated aromatic compounds was approximated by interpolating the MS responses of benzene, toluene, and naphthalene in dependence of their respective m/z. This interpolation was performed using a second order polynome. Reproducibility experiments have shown only small relative deviations (mostly ≈±1%) for methane conversions and different product yields, underlining the reliability of the obtained data and withdrawn trends (see Fig. S2†).
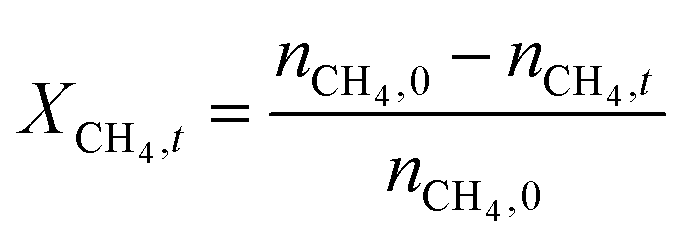 | (1) |
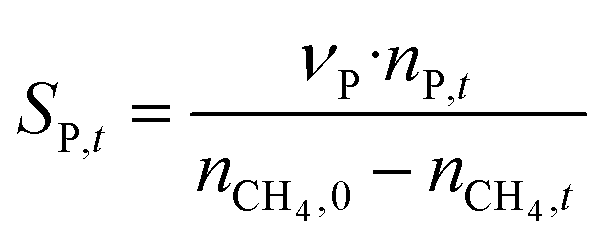 | (2) |
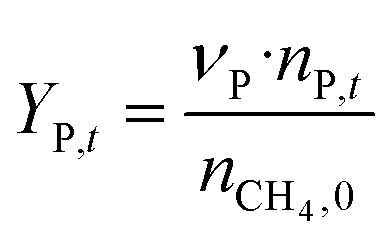 | (3) |
3. Results and discussion
3.1 Physicochemical properties of the catalysts
Chemical composition as well as other structural characteristics of the as-prepared samples are summarized in Table 1. Generally the loading of transition metals by impregnation reduced the specific surface areas, pore volumes and amounts of NH3 that could be adsorbed compared to bare HZSM-5.20 While the addition of Fe did not significantly change the acidity of the catalyst compared to the monometallic sample, modification of the catalyst with Co and Zn increased the amount of adsorbable NH3 of these catalysts, hinting at an increase in the number of accessible acid sites. On the other hand, 1 wt% of Nb resulted in a decrease in the adsorption capacity of NH3 by 11% (see Table 1) and thus lowered the acidity of the catalyst compared to the undoped Mo/HZSM-5 catalyst. The temperature profiles of NH3-TPD show a disappearance of strong Brønsted acid sites (desorption temperature > 400 °C) upon loading with Mo, proving the anchoring of MoOx onto the acidic sites of the zeolites (see Fig. S3†). Measured volumes of meso- and micropores suggest that most Mo is located specifically inside the micropores.40 This is also supported by the results of X-ray powder diffraction of the impregnated samples, which are almost identical to those of the parent zeolite and show neither reflexes for Mo nor for Nb (see Fig. S4†), hinting at very small supported nanoclusters of both metals.3.2 Activity measurements
In a subsequent set of experiments, we studied the impact of the total gas flow (see Fig. 1a). On both catalysts the methane conversion dropped to 36–53% of its initial value after 360 min on stream depending on the space velocity. Higher flow rates (i.e., shorter residence time) resulted in an increased rate of deactivation over time, although the flow-normalized deactivation rate was highest at the lowest space velocity. While the levels of conversion for both catalysts during the reaction remained similar, an increase in benzene yields grew more pronounced at lower residence times for the 6Mo1Nb/HZSM-5 catalyst (see Fig. 1b), hinting at an enhanced catalytic conversion of reaction intermediates to benzene in the presence of niobium. Quantitatively, while the difference from the unmodified catalyst might fall into the margin of uncertainty at 1600 SCC gcat−1 h−1, the 6Mo1Nb catalyst achieved 8.1% relative increased benzene yield compared to the 6Mo/HZSM-5 catalyst at 4800 SCC gcat−1 h−1 over 360 min with only a marginal increase in methane conversion (0.86%), showing a significant enhancement of selectivity toward benzene for the Nb-doped catalyst (see Table S1 and Fig. S6†). We additionally tested a catalyst loaded with 1 wt% of Nb without the addition of Mo and found no detectable product formation or methane conversion at 700 °C and 3200 SCC gcat−1 h−1. From this we can conclude a synergistic effect of Nb on the activity of Mo during the MDA reaction, as Nb is not intrinsically able to activate methane.
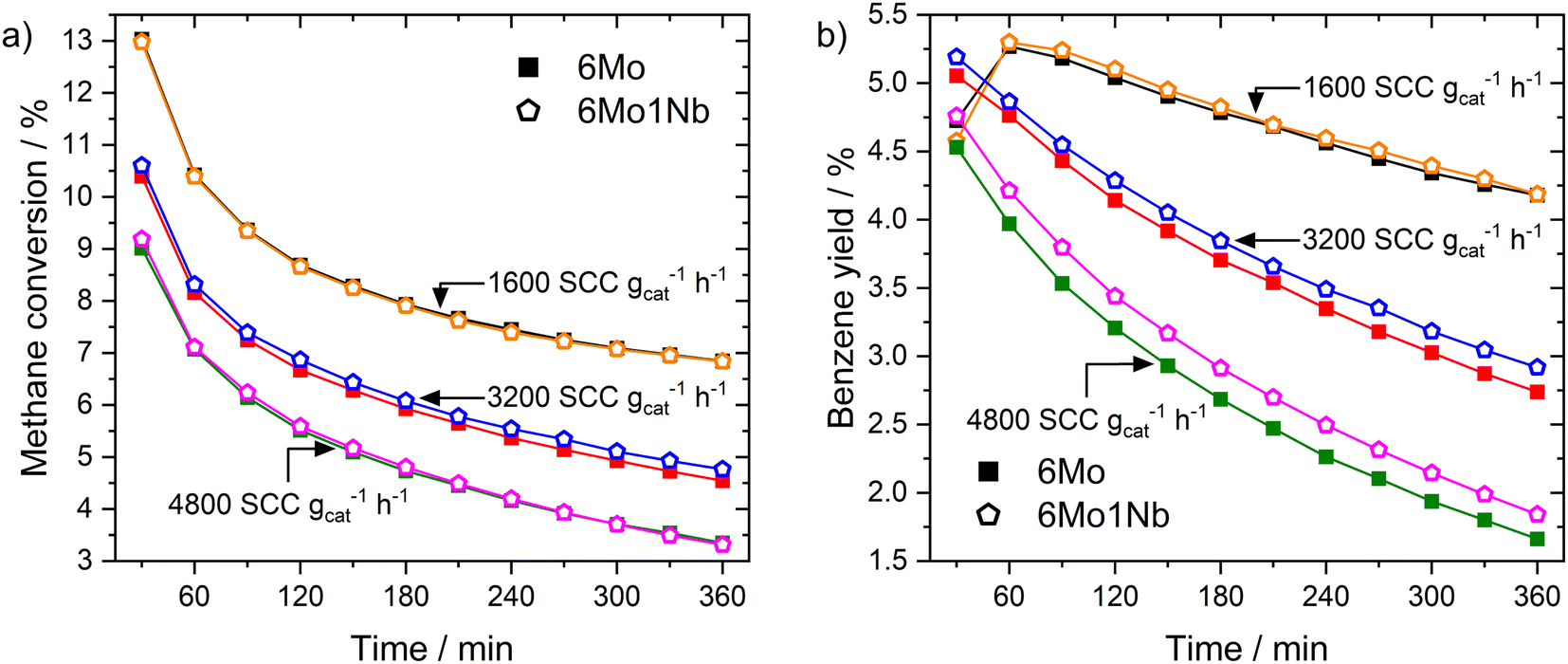 | ||
| Fig. 1 Methane conversions (a) and benzene yields (b) over time for MDA reactions over 6Mo/HZSM-5 and 6Mo1Nb/HZSM-5 catalysts at different reactant flow rates. Reaction conditions: 700 °C, 300 mg catalyst, 90% CH4 + 10% N2, 1 atm. | ||
Next, we examined the activity of both catalysts at lower reaction temperatures, which resulted in overall decreased methane conversions as expected (see Fig. S7†). We could observe, however, a pronounced increase in selectivity to benzene and naphthalene when niobium was present in the sample and the reaction temperature was lowered to 600 °C at 3200 SCC gcat−1 h−1. This resulted in a 10.4% higher relative benzene yield and 22.0% higher relative naphthalene yield on the 6Mo1Nb/HZSM-5 catalyst under these specific reaction conditions compared to the Nb-free sample (see Fig. S8† for the time on stream yields and Fig. S9† for total yields, respectively). Selectivities to coke were also decreased compared to the unmodified 6Mo/HZSM-5 catalyst, while selectivities to C2 and toluene were mostly comparable at 650 °C and 600 °C (see Table S1†). From these results we can surmise that Nb incorporation has no significant effect on methane activation but may improve formation of benzene. Interestingly, an unexpectedly low conversion of methane was found after 30 min on stream for the 6Mo1Nb catalyst at 600 °C, directly followed by 90 min of increased conversion until leveling with the parent catalyst at 150 min. This observation hints towards a side-reaction which slows the formation of the Mo active sites (called the “activation period”) which is only visible at lower methane conversion rates. A concomitant prolonged CO production for 6Mo1Nb/HZSM-5 supports this thesis (see Fig. S10†).
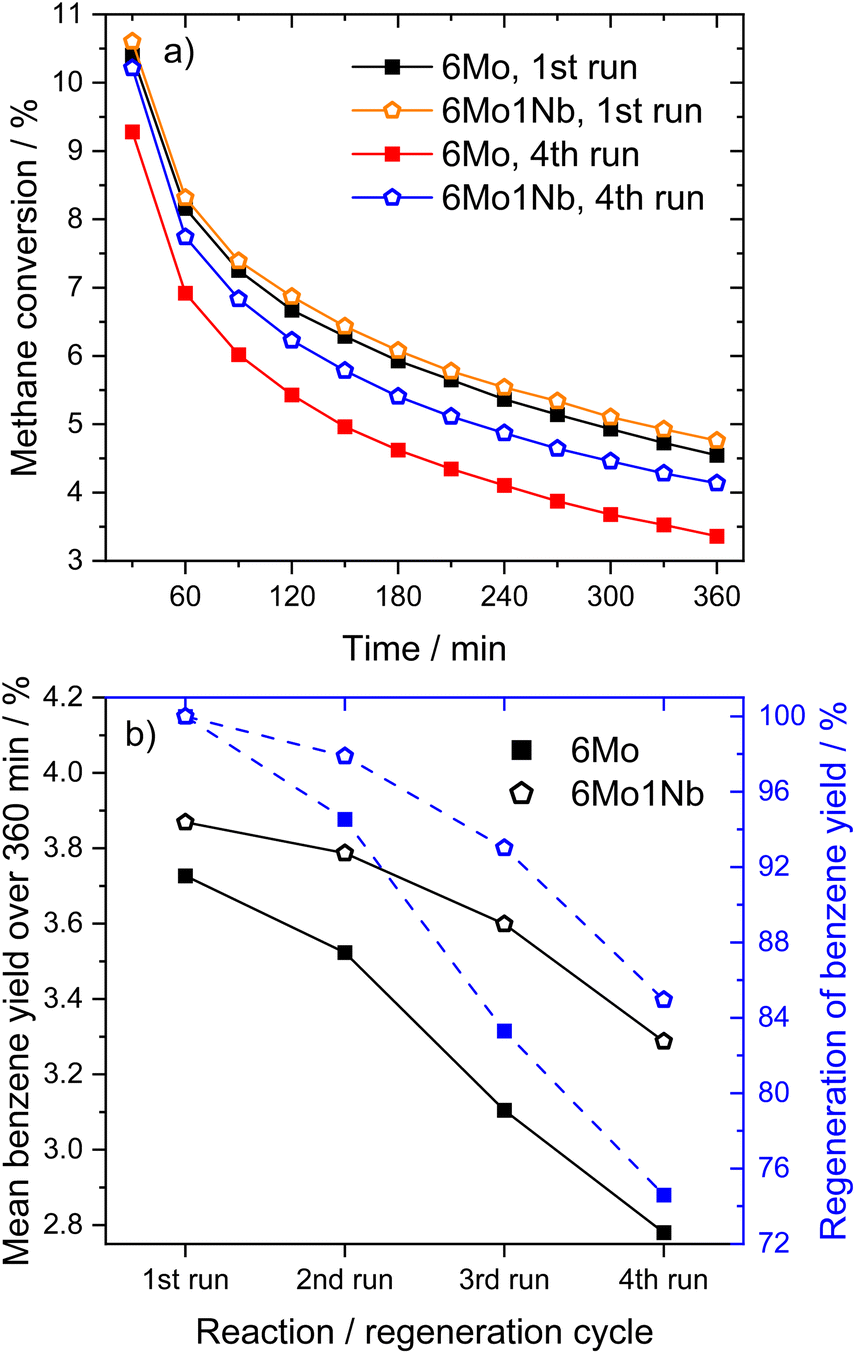 | ||
| Fig. 2 Methane conversions over time (a) and achieved regeneration of benzene yields (b) for MDA reactions over 6Mo/HZSM-5 and 6Mo1Nb/HZSM-5 catalysts in subsequent reaction–regeneration cycles. Reaction conditions: 700 °C, 300 mg catalyst, 90% CH4 + 10% N2, 3200 SCC gcat−1 h−1, 1 atm. Regeneration: 450 °C, air, 1100 SCC gcat−1 h−1, 1 atm, 45 min. For time-resolved changes in benzene yields on stream over the four reaction runs see Fig. S11 in the ESI.† | ||
3.3 Thermal analysis of accumulated carbon
Thermogravimetric analyses (TGA) coupled with differential scanning calorimetry (DSC) have been used to quantify and specify different types of carbon deposited on the catalyst during the reaction. Two significant mass losses were observed for every sample, which correspond to desorption of strongly adsorbed water (see Fig. S12†) and oxidation of carbonaceous species. The second mass loss (i.e., oxidation of carbon deposits) is typically divided into oxidation of proposedly graphitic surface carbon and polyaromatic carbon inside the zeolite micropores.20,51 Additional studies have concluded that both carbon deposits are chemically similar in nature.52 The claimed differentiation by mass-transport limitations of the micropores is, however, dependent on the reaction conditions.53 Furthermore, the two observed mass losses have been ascribed to Mo-adjacent and Brønsted-associated carbon at low and high temperatures, respectively.25,54,55 We have previously found indications of this classification as well.37 In our experiments the acid-related component was only barely visible as a peak shoulder at higher temperatures, whereas the main contribution stemmed from the Mo sites. Carbon mass losses were calculated from sample masses at temperatures between 300 °C and 700 °C to exclude volatile adsorbents and water.56 Based on the quantitative evaluation of the TGA/DSC results (see Table S2†) it is clear that formed coke species are mostly affected by reaction temperature. Different space velocities gave similar DSC peak temperatures, while lowered temperatures resulted in less condensed, more easily oxidizable carbon deposits. The mass loss during oxidation was only slightly reduced when niobium was present in the catalyst sample under regular reaction conditions (700 °C, 3200 SCC gcat−1 h−1). However, the 6Mo1Nb catalyst influenced the coking more severely when employing lower reaction temperatures or reduced GHSV. Interestingly, the Nb-modified catalyst exhibited a third mass change which is generally masked by the simultaneous burn-off of carbon. Due to oxidation of reduced metal species the sample mass increases. This process is visible as a low-temperature shoulder in the DSC curve (see Fig. 3). Considering the reported oxygen transfer from Nb to certain transition metals a direct or catalytic oxidation of MoOxCy by dispersed Nb oxide is possible.57 This is more prominently visible for the spent samples from experiments with methane at 600 °C or 1600 SCC gcat−1 h−1. A mass gain during TG measurements of Nb-containing Mo/MCM-22 was also observed by Rodrigues and coworkers.36 Pasupulety et al.35 reported an increased formation of CO2 at lower temperatures during TPO experiments on Nb–Mo2C/HZSM-5 compared to the undoped catalyst.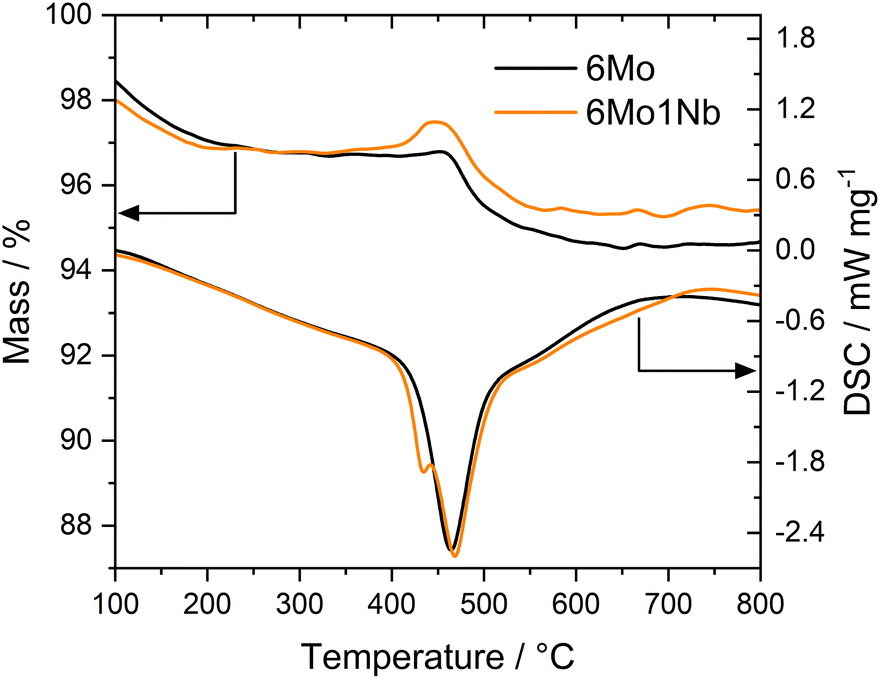 | ||
| Fig. 3 Representative TG (top curves, left y-axis) and DSC (bottom curves, right y-axis) measurements over the temperature range from 100 to 800 °C on spent 6Mo/HZSM-5 and 6Mo1Nb/HZSM-5 catalysts after reaction for 360 min. Reaction conditions: 700 °C, 300 mg catalyst, 90% CH4 + 10% N2, 1600 SCC gcat−1 h−1, 1 atm. | ||
The main DSC peak temperatures of the oxidation process were generally shifted to slightly higher temperatures with a 1 wt% Nb doped catalyst. These temperature shifts can be explained by the deconvolution of Mo oxidation and coke burn-off. As the DSC curve shapes related to coke (>450 °C) remain similar for both catalysts (see Fig. 3 and S13†), we assume that the oxidation process of carbon on Mo sites and the general nature of coke are mostly unaffected by the presence of the Nb dopant. The difference in mass losses between the samples may be partially caused by the oxidation of NbOxCy in addition to MoOxCy. Omitting carbonaceous deposits and water, the theoretical maximum mass gain of this oxidation (NbC → Nb2O5) would be 0.21%, while the formation of MoO3 from Mo2C would result in a mass increase of 1.88%. Based on the stability of Nb2O5 we can infer that Nb is most likely not fully reduced to niobium carbides (NbC and Nb2C) under our reaction conditions.44–47 This will be discussed in more detail in the XPS section.
Finally, TGA investigations of the regenerated catalysts showed a carbon-related mass loss exclusively on 6Mo1Nb/HZSM-5 (see Fig. S14†). Accumulation of high-temperature coke despite oxidative regeneration at 450 °C was reported before. Both results by Kim et al.58 and our own show no detrimental effect of these carbon species on catalyst activity. However, a correlation between this type of coke and the increased catalytic performance of 6Mo1Nb/HZSM-5 could not be confirmed.
3.4 Reaction-induced changes in Mo nanoparticles
To evaluate the structural changes of 6Mo/HZSM-5 and 6Mo1Nb/HZSM-5 during the reaction as well as the differences therein upon doping by niobium, we used high resolution STEM measurements and EDS elemental mapping for the fresh and the spent catalysts. We evaluated the size of Mo nanoparticles from at least 300 particles on each sample. On the undoped catalyst surface (6Mo/HZSM-5) we showed the formation of MoOx particles in the range from 0.5 to 5 nm with an average size of 1.6 ± 0.8 nm (see ref. 37). In addition, isolated monomeric oxo Mo sites as well as sub-nm Mo clusters were visible on the outer surface or inside micropores close to the surface of the zeolite framework. After the reaction bigger particles were observed with an average diameter of 5.8 ± 2.7 nm. At this point we can still observe the tiny nanostructures observed on the fresh catalyst.On the Nb-doped catalyst (6Mo1Nb/HZSM-5) the fresh sample showed a broader size distribution in the range from 0.5 to 15 nm (see Fig. 4a, b and S15†) and an average particle size of around 4.2 ± 2.5 nm. This is significantly larger than that of the undoped sample. It can be clearly seen on the high-resolution micrographs (Fig. S15†) and EDS maps (Fig. 5) of this sample that Nb is well mixed with MoOx particles without visible segregation. This may be a reason for the increase in their average size. Still, a fraction of highly dispersed, possibly mixed MoOx/NbOx species can be traced on the high-resolution micrographs for both the fresh and the spent samples (see Fig. 4c and d). For the spent sample the STEM micrographs indicate at first glance the formation of larger particles compared to the fresh sample (see Fig. 4c and d). Quantitatively the particle size distribution of this sample indicated the presence of a wider range of particle sizes between 1 nm and 19 nm, with the fraction of particles >8 nm being significantly larger than that observed on the fresh sample. This results in an average particle size of 8.0 ± 3.3 nm. Comparable results were found by Pasupulety and coworkers.35
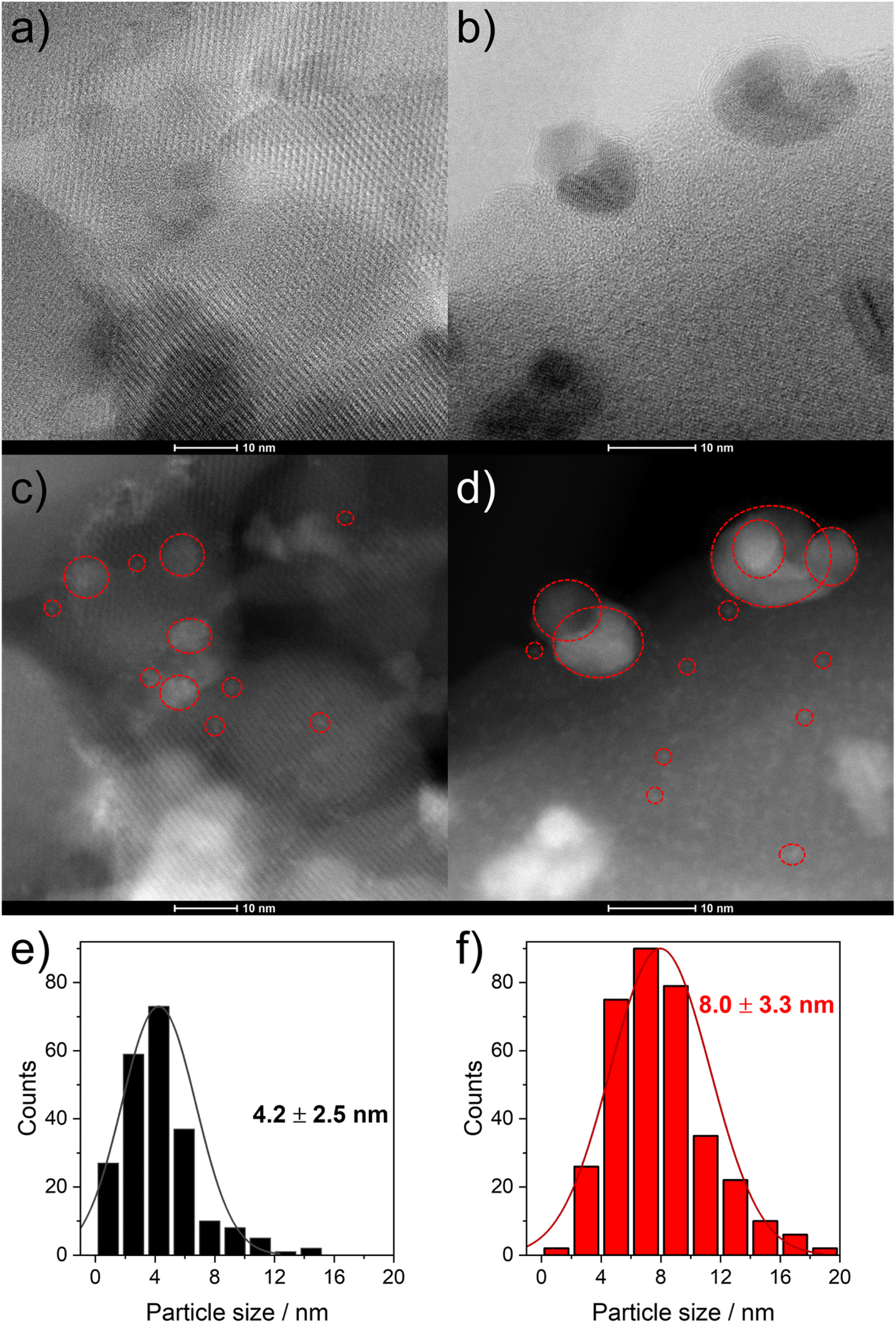 | ||
| Fig. 4 STEM micrographs of the fresh ((a) bright field (BF); (c) dark field (HAADF)) and for the spent 6Mo1Nb/HZSM-5 catalyst ((b) BF; (d) HAADF) as well as the corresponding mixed Mo–Nb representative particle size distributions ((e) fresh catalyst; (f) spent catalyst). Individual particles in agglomerates were distinguished by Z-contrast. Selected particles are highlighted with red circles for visual clarity. The sub-nm particles visible in the HAADF images were not included in these statistics. | ||
 | ||
| Fig. 5 STEM micrograph (a) and the corresponding EDS maps ((b) Mo Lα; (c) Nb Kα; (d) Al K; (e) Si Kα; (f) overlay of O, Si, Al, Mo and Nb) of the fresh 6Mo1Nb/HZSM-5 catalyst. | ||
Next, we coupled STEM with EDS mapping of different elements for the fresh (Fig. 5) and the spent catalysts (Fig. S16†). The comparison of the STEM micrograph in Fig. 5a with Nb Kα and Mo Lα maps (see Fig. 5c and b, respectively) shows that the visible particles are composed of intimately mixed Nb and Mo species. The high-resolution STEM micrographs taken of the Nb-doped catalyst after the reaction suggest similarly pronounced mixing of Nb with Mo species (see Fig. S17†). This is confirmed by detailed EDS maps (see Fig. S16†). The spread of Nb as indicated by the Nb Kα line suggests that it is homogeneously distributed not only on the surface but also inside the zeolite micropores (see Fig. 5c). Larger Mo–Nb agglomerates (visible in Fig. 5b) still contain a measurable amount of Nb. It should also be noted that both the Al and Si signals are homogeneously distributed which indicates that the structure of the zeolite is retained (see Fig. 5d and e as well as Fig. S4 and S18† for XRD analysis). The overlay map indicates a more intense signal for the Mo Kα line which is clearly related to the higher loading of Mo compared to Nb (Fig. 5f). Additionally, the intensity of Si or Al signals on the surface is much lower on the spent sample (see Fig. S16f†) than what can be observed in the overlay map of the fresh sample (see Fig. 5f). We also examined the spent 6Mo1Nb/HZSM-5 (see Fig. S17†) and especially the buildup of carbon using high resolution STEM for relatively larger Mo–Nb particles, about 10 nm (see Fig. S19†). The micrographs showed a thick layer of carbon on the outer surface in a size range of 1–1.5 nm. This observation agrees with our thermal analysis data collected on the doped and undoped catalysts as well as STEM results and reports by others on Mo/HZSM-5.49 Specifically, particles on the zeolite surface were shown to exhibit a higher tendency to form carbon due to the lack of a shape-selective pore environment.37,59,60
Lastly, we investigated the 6Mo/HZSM-5 and 6Mo1Nb/HZSM-5 catalysts after consecutive reaction and regeneration cycles. After a total of 24 h of time on stream (with cycles consisting of 6 h of reaction with methane and 45 min of regeneration in air) only minor average Mo or Mo–Nb particle growth could be observed for both samples compared to the spent samples after 360 min on stream (see Fig. S20 and S21†). Although catalysts retained a significant fraction of the smaller nanoclusters and isolated sites, a notably broadened size distribution was found. Stabilized sub-nm Mo clusters or isolated species were previously proposed as the active centers for this reaction.37 Judging from the regeneration experiments (see Fig. 2), we can infer that the presence of Nb might extend the lifetime of these small active sites. A hint towards higher regeneration stability can also be derived from the N2 sorption results, which showed a higher recovery of surface area and micropore volume for the Nb-doped catalyst compared to 6Mo/HZSM-5 (see Table S3 and Fig. S22†). Finally, we cannot exclude that more complex phenomena such as the dynamic redispersion/agglomeration of the tiny Mo/Mo–Nb clusters or the interconversion of oxy- and carbidic species would occur during the activation and reaction steps, which are essentially difficult to disentangle.
3.5 Insights into Mo and Nb speciation and the dominant oxidation state
We investigated the fresh, spent, and regenerated 6Mo/HZSM-5 and 6Mo1Nb/HZSM-5 catalysts by employing XPS measurements to elucidate the effect of Nb on the Mo active phase and their possible interplay during the reaction. The XPS spectra of the Mo 3d core level were deconvoluted according to procedures described by Murugappan and coworkers,61 which were also applied in our previous work.37 The fresh catalysts contained mostly Mo6+ with small amounts of Mo5+ (see Fig. 6a). The surface concentration of Mo was higher for 6Mo1Nb/HZSM-5 than that observed for the undoped sample, mirroring the determined differences in surface particle size visible by STEM. It is important to note that Al concentration was reduced in turn (see Table 2). Furthermore, the ratio of Mo5+/Mo6+ increased in the Nb-doped sample. Our analysis revealed the presence of several different oxidation states of Mo in the spent catalysts, ranging from Mo6+ to Mo2+, showing incomplete reduction of Mo and formation of carbidic and oxycarbidic species (see Fig. 6b). Similar surface concentrations for Mo2+ and Mo4+ could be found for both catalysts after the reaction at 700 °C for 360 min. The chosen deconvolution procedure assigns two doublets to Mo4+ cations, based on research proposing species with either more carbidic or oxidic characteristics.62,63 Overall, surface concentrations of Mo and Al were also almost equal in both samples. When compared to 6Mo/HZSM-5, the doped 6Mo1Nb/HZSM-5 catalyst exhibited a higher amount of Mo5+ species at all investigated reaction steps (see Table 2).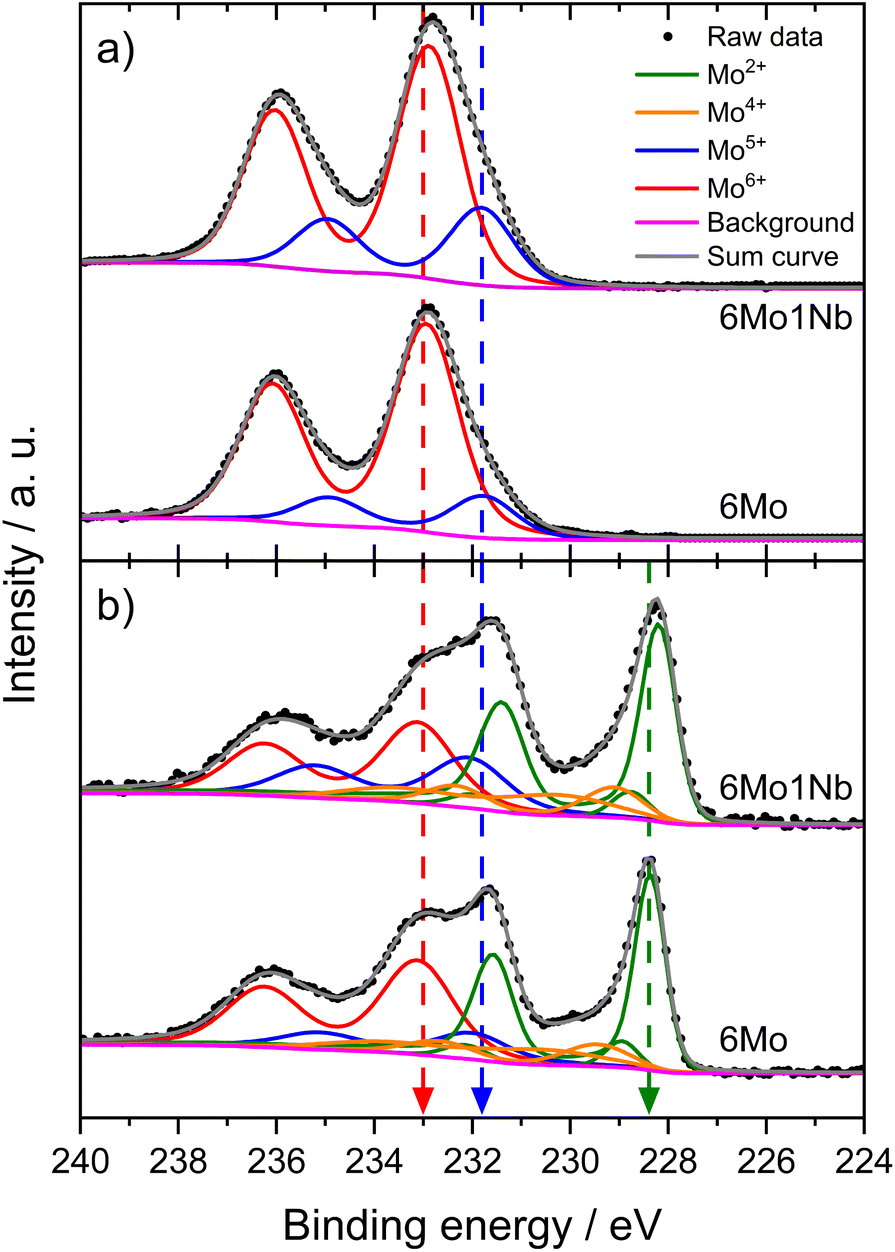 | ||
| Fig. 6 Intensity-normalized XP spectra for the Mo 3d core level of fresh (a) and spent (b) 6Mo/HZSM-5 and 6Mo1Nb/HZSM-5 catalysts. Reaction conditions: 700 °C, 300 mg catalyst, 90% CH4 + 10% N2, 3200 SCC gcat−1 h−1, 1 atm, 6 h. Dashed lines were added as a visual guide. | ||
| Sample | Surface concentration [at%] | Si/Al | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Mo6+ | Mo5+ | Mo4+ | Mo2+ | Nb | Al | Si | C | ||
| 6Mo fresh | 2.3 | 0.46 | 0 | 0 | 0 | 1.4 | 27.6 | 9.6 | 19.7 |
| 6Mo1Nb fresh | 2.8 | 0.95 | 0 | 0 | 0.54 | 1.2 | 26.4 | 10.6 | 22.0 |
| 6Mo spent | 0.49 | 0.15 | 0.23 | 0.57 | 0 | 1.4 | 23.6 | 31.3 | 16.9 |
| 6Mo1Nb spent | 0.36 | 0.24 | 0.24 | 0.55 | 0.35 | 1.4 | 24.5 | 28.2 | 17.5 |
| 6Mo reg. | 3.1 | 0.33 | 0 | 0 | 0 | 1.8 | 27.8 | 7.3 | 15.4 |
| 6Mo1Nb reg. | 3.6 | 0.86 | 0 | 0 | 0.56 | 1.6 | 26.0 | 8.7 | 16.3 |
| 1Nb fresh | 0 | 0 | 0 | 0 | 0.37 | 1.6 | 32.1 | 4.6 | 20.1 |
| 1Nb spent | 0 | 0 | 0 | 0 | 0.20 | 1.2 | 23.9 | 5.8 | 19.9 |
This suggests an improved reducibility of Mo in the presence of Nb. In this context, Jentoft et al.64 reported a beneficial effect of Nb on the reduction and carburization temperatures of Mo. While not directly comparable due to the absence of methane, a similar effect was observed in our own H2-TPR investigations where the high temperature reduction step is significantly diminished in favor of low-temperature processes (see Fig. S23†). This might indicate that while Mo6+ reduction occurs more thoroughly, further reduction is hindered by stabilization of intermediate Mo oxidation states by Nb. An interaction between Mo and Nb (electronic or coordinative in nature) might cause an increased concentration of Mo5+ as well, since this effect is also observed in the fresh and regenerated samples. Both cases may be correlated to the slightly increased conversion of methane on 6Mo1Nb/HZSM-5. It should be noted at this point that XPS is a surface-sensitive method. Most active sites should be situated deep inside the micropores of the zeolite. However, it has been shown before that surface (XPS) and bulk (XAS) speciation of Mo in MDA catalysts are comparable.60 Metallic Mo0 was not detected in contrast to reports from Pasupulety and coworkers.17 They performed in situ carburization in 20% CH4/H2 for 20 min and employed lower flow rates for a longer reaction time on stream (1800 mL gcat−1 h−1 for 10 h), which might be a cause for their observation of fully reduced Mo. The lower surface concentration of C suggests a decreased tendency for coke formation on 6Mo1Nb/HZSM-5 during the MDA reaction, which is consistent with our findings from thermogravimetric analyses (see Table S2† and Fig. 3).
The Mo 3d XPS spectra of both regenerated catalysts are comparable to those of the fresh samples, showing the reoxidation of MoOxCy to the original Mo oxide species (see Fig. S24†). After four reaction/regeneration cycles the surface concentrations of Mo6+ were significantly increased for both catalysts compared to the fresh samples (see Table 2). The Mo5+ concentration was similar to that before regeneration, though the Mo5+/Mo6+ ratio decreased. Combined with the higher surface concentrations of Al, this observation suggests the occurrence of either Mo agglomeration or a partial dealumination of the zeolite framework and concomitant diffusion of Al and Mo species from the micropore system to the surface of the support. Damage to the zeolite support could not be detected through XRD measurements (see Fig. S18†). Thus, agglomeration of Mo or Mo–Nb into larger particles, which was also clearly visible by STEM, is more likely and would result in the exposure of Al sites.37 Still, the higher Mo5+/Mo6+ ratio for 6Mo1Nb persists, further underlining a possible electronic or coordinative interaction between both transition metals.
For evaluation of Nb speciation in the doped catalyst before and after the reaction with methane, we first analyzed fresh and spent 1Nb/HZSM-5 samples. No change in the oxidation state for niobium was visible in this case (see Fig. S25†). This was expected since no product formation was observed over 360 min on stream at 700 °C. Niobium oxide is known for its high reduction stability under a methane atmosphere.45,46 Hence, the observed decrease in the surface concentrations of Al, Si and Nb goes along with an increased concentration of oxygen.
Next, we investigated the binding energies of Nb in 6Mo1Nb/HZSM-5 before and after the reaction. In the fresh catalyst only Nb5+ could be detected (see Fig. 7). Contrary to the observations on 1Nb/HZSM-5, a significant reduction of the Nb took place over the course of the reaction in the presence of Mo, resulting in a multitude of different binding energies for the Nb 3d level. These can be attributed to Nb2+ species (NbO)35 or NbC at 204.0 eV and oxycarbidic species at 205.1 eV (presumed as Nb4+), which agrees with assignments reported by Gupta and coworkers.65 This finding shows the activation of otherwise inert Nb2O5 by MoOx or MoOxCy species under our chosen reaction conditions. A partial reduction of Nb on 6Mo1Nb/HZSM-5 during the MDA reaction was also reported by Pasupulety et al.,35 though it is important to note that pre-carburization was not performed in our experiments. Interestingly, a completely new species of Nb was found at even higher binding energy (208.8 eV) than that of fully oxidized Nb2O5. Since the binding energy of Nb5+ species in the 1Nb/HZSM-5 catalyst is shifted slightly higher than that in the bimetallic 6Mo1Nb sample (208.0 eV and 207.1 eV, respectively), an electronic interaction with Mo species is likely, although a more thorough investigation is needed. After oxidative regeneration, Nb2O5 was fully restored and exhibited no differences compared to the fresh catalyst.
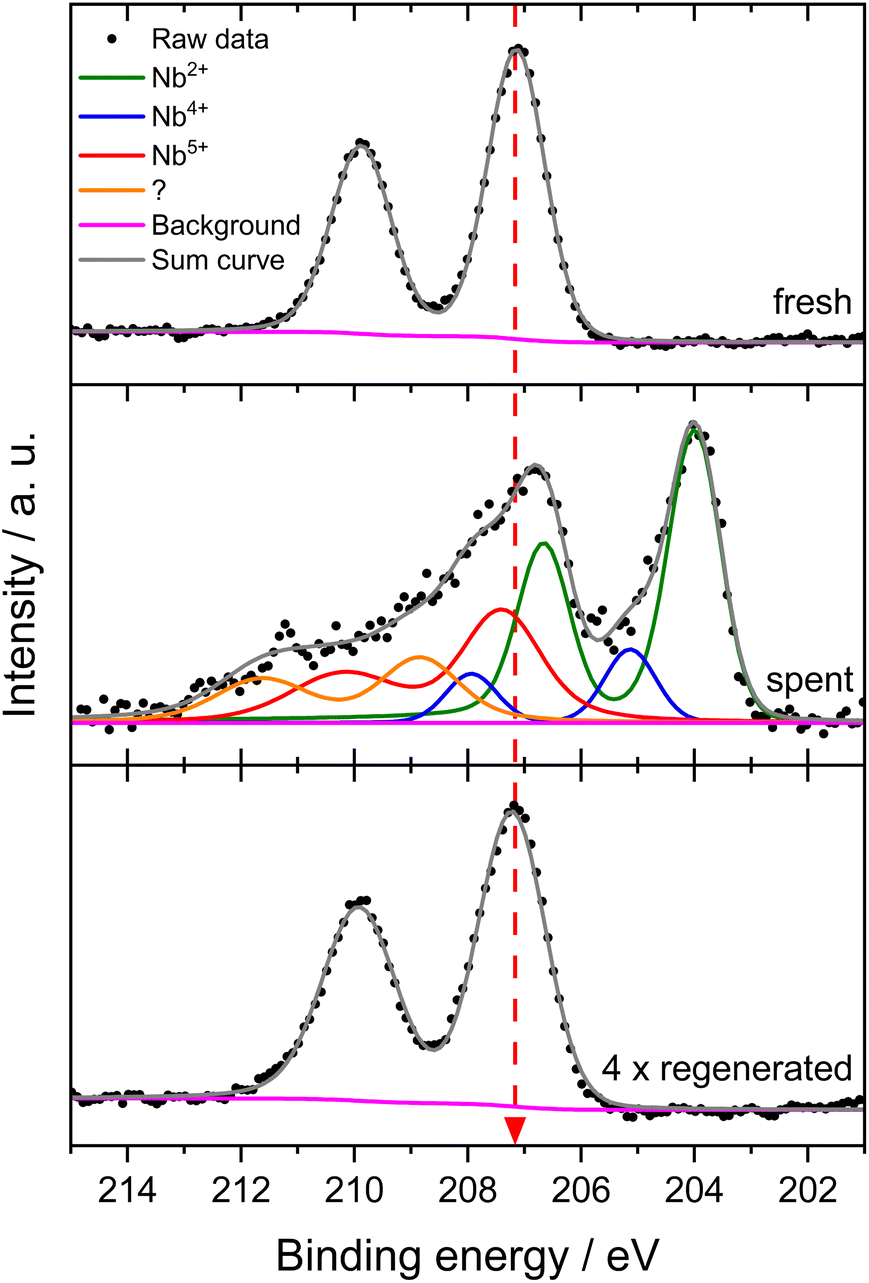 | ||
| Fig. 7 Intensity-normalized XP spectra for the Nb 3d level of the 6Mo1Nb/HZSM-5 catalyst before the reaction (bottom), after the reaction with methane at 700 °C for 360 min (middle) and after four reaction/regeneration cycles. Reaction conditions: 700 °C, 300 mg catalyst, 90% CH4 + 10% N2, 3200 SCC gcat−1 h−1, 1 atm, 6 h. Regeneration: 450 °C, air, 1100 SCC gcat−1 h−1, 1 atm, 45 min. | ||
3.6 In situ investigations on reduction/carburization effects of Nb
To further elucidate the possible interactions of Mo and Nb during MDA we expanded our analytical approaches with two different in situ methods. First, we investigated the dynamic change in Mo and Nb oxidation states via in situ NAP-XPS analogous to our previous investigations performed on 6Mo/HZSM-5.37,39 During the initial measurement in N2 at 127 °C the surface composition of 6Mo1Nb/HZSM-5 was slightly different from that observed in the ex situ results, with the major fraction of Mo present as Mo6+ together with only a minor amount of Mo5+ (see Fig. 8d). Nb5+ was the only niobium species observed (see Fig. S26a†). Changing from inert gas to the CH4 mixture resulted in an increased occurrence of Mo5+ without an apparent change in total Mo concentration (see Fig. 8a and Table S4†) and composition more similar to that of the ex situ sample. Upon heating to 627 °C the Nb-modified sample showed reduction of Mo6+ to Mo4+ (see Fig. 8b) with a simultaneous appearance of two additional Nb species already observed ex situ (see Fig. S26b†). While the Nb signal at lower binding energy can be attributed to Nb4+, the species at higher binding energy (relative to Nb2O5) is likely a result of a direct electronic interaction with Mo. Interestingly, the concentration of Mo4+ reached a maximum after 70 min at 627 °C (relative time 2.92 h) and gradually decreased thereafter in favor of the formation of carbidic Mo species (labeled simply as Mo2+, although it was reported to include both MoC and Mo2C66). During the next 60 min (relative time 3–4 h) the concentration of Nb5+ species dropped significantly while contributions of Mo6+ and Mo4+ were mostly stable, supporting the proposed redox interaction of Mo and Nb.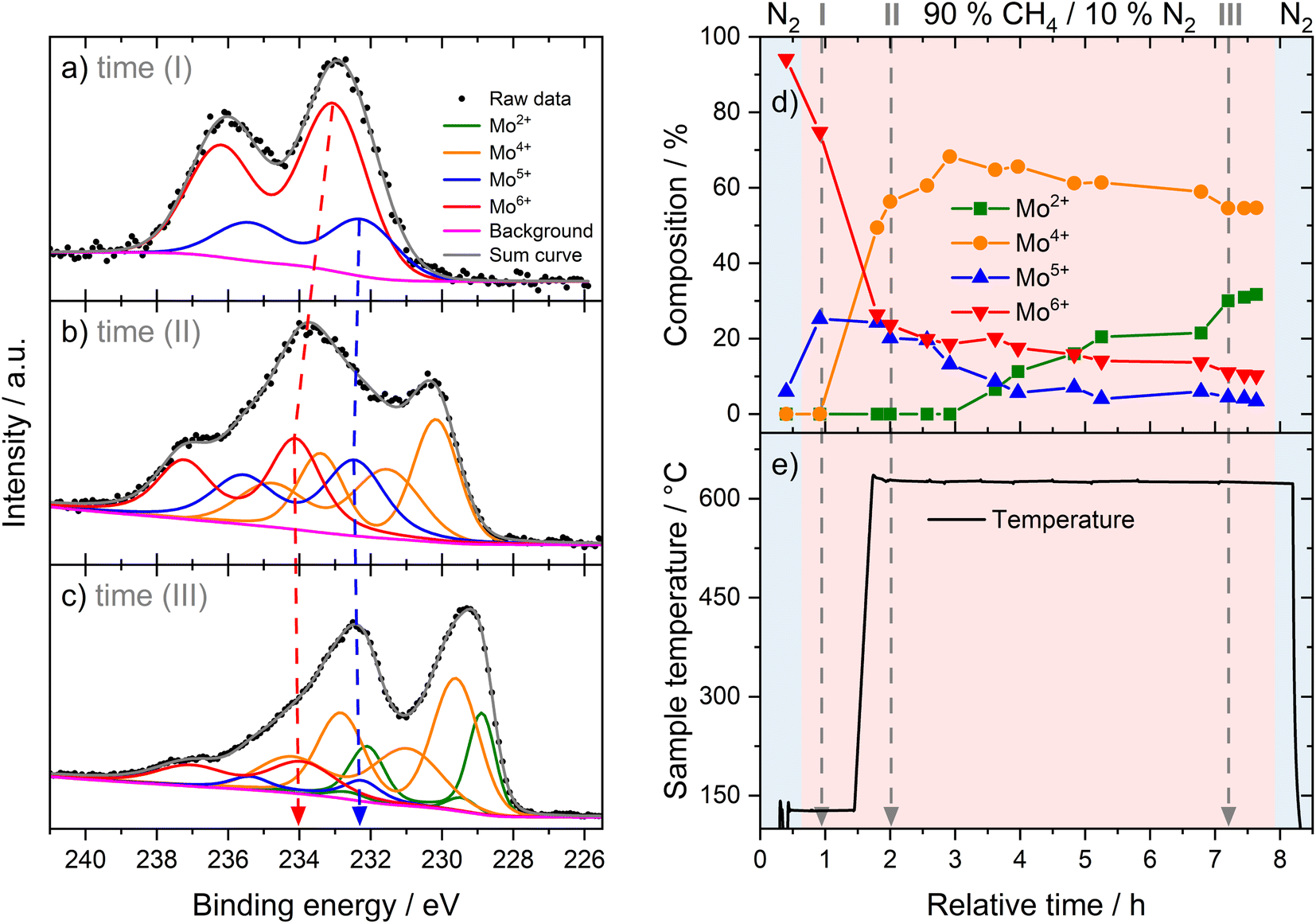 | ||
| Fig. 8 In situ collected Mo 3d spectra at different times ((a) time (I) – 55 min@127 °C; (b) time (II) – 2.0 h@627 °C; (c) time (III) – 7.2 h@627 °C) showing the reduction of Mo during the NAP-XPS study of 6Mo1Nb/HZSM-5 at a total pressure of 2 mbar in the reaction atmosphere 90% CH4/10% N2. For higher temperatures a shift of about 1 eV to higher binding energies can be observed for the Mo6+ component. (d) Contribution of different oxidation states of Mo during in situ NAP-XPS study of the 6Mo1Nb/HZSM-5 catalyst during heating up and the subsequent reaction in 90% CH4/10% N2 (highlighted in light pink); (e) temperature program as a function of reaction time. | ||
By the end of the experiment around 30% of the observed Mo was carburized (see Fig. 8c and Table S4†) compared to 8% for the 6Mo/HZSM-5 catalyst under similar reaction conditions.39 Additionally, binding energies for reduced Mo (Mo5+ to Mo2+) were shifted slightly lower in comparison, further supporting an interaction between Mo and Nb. The formation of carbidic Mo species was accompanied by a significant relative concentration increase in the Nb species at higher binding energy (up to 34% of detected Nb, see Fig. S26c†). Meanwhile, a higher concentration of carbidic C together with a lower amount of oxygen was found. We have observed a strong increase in the Mo total surface concentration for the undoped 6Mo/HZSM-5 catalyst under NAP reaction conditions before,39 which was also apparent for the Nb-doped sample in this study. Despite its higher initial surface Mo concentration before the reaction compared to 6Mo/HZSM-5 (see Table 2), the 6Mo1Nb/HZSM-5 sample showed a ≈22% lower total concentration of Mo after comparable reaction times (see Table S4† and ref. 39). This suggests a higher retention of Mo inside the pores under the employed reaction conditions. From our ex situ XPS investigations a similar effect can be concluded, as the ≈36% higher surface Mo concentration on the fresh 6Mo1Nb/HZSM-5 sample resulted in an almost identical total concentration after 6 h of MDA reaction compared to undoped 6Mo/HZSM-5 (see Table 2). According to Jentoft et al.,64 who noticed a more facile carburization of Mo mixed with Nb, a stable MoC/NbC mixed carbide phase can be formed. Since deeply carburized Mo2C was reported to detach from zeolite acid sites,67 we propose an increased stability of molybdenum(IV) carbide in MDA by doping with niobium.
Due to the possible limitations of NAP-XPS (i.e., the pressure gap and surface sensitivity), we decided to expand our investigations to a bulk characterization method applicable under realistic reaction conditions. Therefore, we carried out in situ dispersive XAS studies at the Mo K-edge on both the 6Mo/HZSM-5 and the 6Mo1Nb/HZSM-5 catalysts. We followed the change of the Mo K-edge region (XANES) during heating from RT to 600 °C in 20% CH4/N2 to achieve a reasonable time resolution of the reduction/carburization step. Below 470 °C no reaction with methane occurred and no changes in the X-ray absorption were visible. Beginning at 490 °C we observed the onset of the reduction of Mo6+ ions to lower oxidation states (Mo5+/Mo4+) on the 6Mo1Nb sample, while this change only happened at temperatures >520 °C for the Nb-free catalyst (see Fig. 9). This is in line with findings by Jentoft et al.64 who reported a significant decrease in the carburization temperature of MoO3 by addition of Nb2O5 in a similar ratio to our catalyst. They confirmed the formation of a single-phase mixed metal carbide made up of MoC and NbC with cubic structure which we assume is a possible explanation for the observed intimate mixing of Mo and Nb in our EDS investigations (see Fig. 5). As temperature increases, the XANES edge of 6Mo/HZSM-5 shifted only slightly to lower energies (Fig. 9, upper layer), while the change for 6Mo1Nb/HZSM-5 is more severe (Fig. 9, bottom layer). Further heating and isothermal treatment at 600 °C did not lead to any more visible changes in the X-ray absorption of both catalysts.
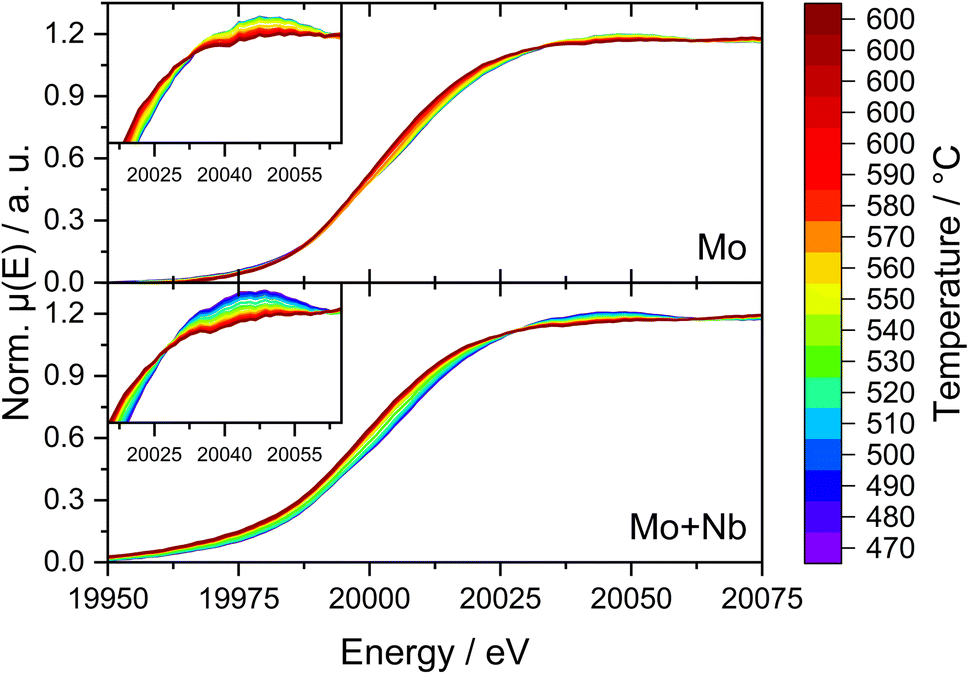 | ||
| Fig. 9 Normalized XANES at the Mo K-edge recorded in situ of 6Mo/HZSM-5 (top) and 6Mo1Nb/HZSM-5 (bottom) during continuous heating. Curves depict the normalized sum of 10 measurements (60 s) in the range of T ± 5 °C. Insets show zoom-ins on the edge features. Reaction conditions: 50 mL min−1 20% CH4/N2, 1 atm, heating rate 10 °C min−1, heating from RT to 600 °C. | ||
An explanation for this behavior can be discovered when looking at the first derivative of the XANES spectra (see Fig. 10). Both samples exhibited a maximum at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 007 eV (corresponding to MoO3 species), whereas only the unmodified 6Mo/HZSM-5 showed a second maximum at 19994 eV. The latter can be assigned to a pre-peak transition associated with tetrahedrally coordinated Mo species.68 These species are hardly visible in the Nb-doped catalyst, suggesting the presence of Mo in a mainly octahedral environment. Following the reduction/carburization these maxima converged, leading to a singular broad maximum centered at ≈20001 eV (width ≈ 20 eV) for 6Mo/HZSM-5. This suggests the presence of a multitude of different Mo species ranging from Mo6+ to Mo2+ in the working catalyst, comparable to our XPS results (see Fig. 6 and 8). However, the reaction of 6Mo1Nb/HZSM-5 with methane yields a denser maximum (width ≈ 13 eV) centered at ≈19999 eV. Since the observed absorption edge as well as the derivative maximum shifted more significantly for the niobium-doped sample, it can be argued that the reduction of Mo6+ species happens more easily and uniformly compared to the undoped catalyst. The reduced broadness of the derivative maximum after reduction/carburization infers a more narrow distribution of Mo oxidation states in addition to fewer Mo–O species in the presence of Nb. Together with the observed reduction of Nb2O5 only with Mo (see Fig. 7) a significant interaction between both elements is likely. Due to their similar derivative XANES maxima an exact differentiation between MoC and Mo2C is not possible from this data (see Fig. S27†). However, the observed mixing of Mo with Nb inside the micropores might create sufficient local Mo/Nb ratios to enable the stabilization of a MoC/NbC mixed carbide.64
007 eV (corresponding to MoO3 species), whereas only the unmodified 6Mo/HZSM-5 showed a second maximum at 19994 eV. The latter can be assigned to a pre-peak transition associated with tetrahedrally coordinated Mo species.68 These species are hardly visible in the Nb-doped catalyst, suggesting the presence of Mo in a mainly octahedral environment. Following the reduction/carburization these maxima converged, leading to a singular broad maximum centered at ≈20001 eV (width ≈ 20 eV) for 6Mo/HZSM-5. This suggests the presence of a multitude of different Mo species ranging from Mo6+ to Mo2+ in the working catalyst, comparable to our XPS results (see Fig. 6 and 8). However, the reaction of 6Mo1Nb/HZSM-5 with methane yields a denser maximum (width ≈ 13 eV) centered at ≈19999 eV. Since the observed absorption edge as well as the derivative maximum shifted more significantly for the niobium-doped sample, it can be argued that the reduction of Mo6+ species happens more easily and uniformly compared to the undoped catalyst. The reduced broadness of the derivative maximum after reduction/carburization infers a more narrow distribution of Mo oxidation states in addition to fewer Mo–O species in the presence of Nb. Together with the observed reduction of Nb2O5 only with Mo (see Fig. 7) a significant interaction between both elements is likely. Due to their similar derivative XANES maxima an exact differentiation between MoC and Mo2C is not possible from this data (see Fig. S27†). However, the observed mixing of Mo with Nb inside the micropores might create sufficient local Mo/Nb ratios to enable the stabilization of a MoC/NbC mixed carbide.64
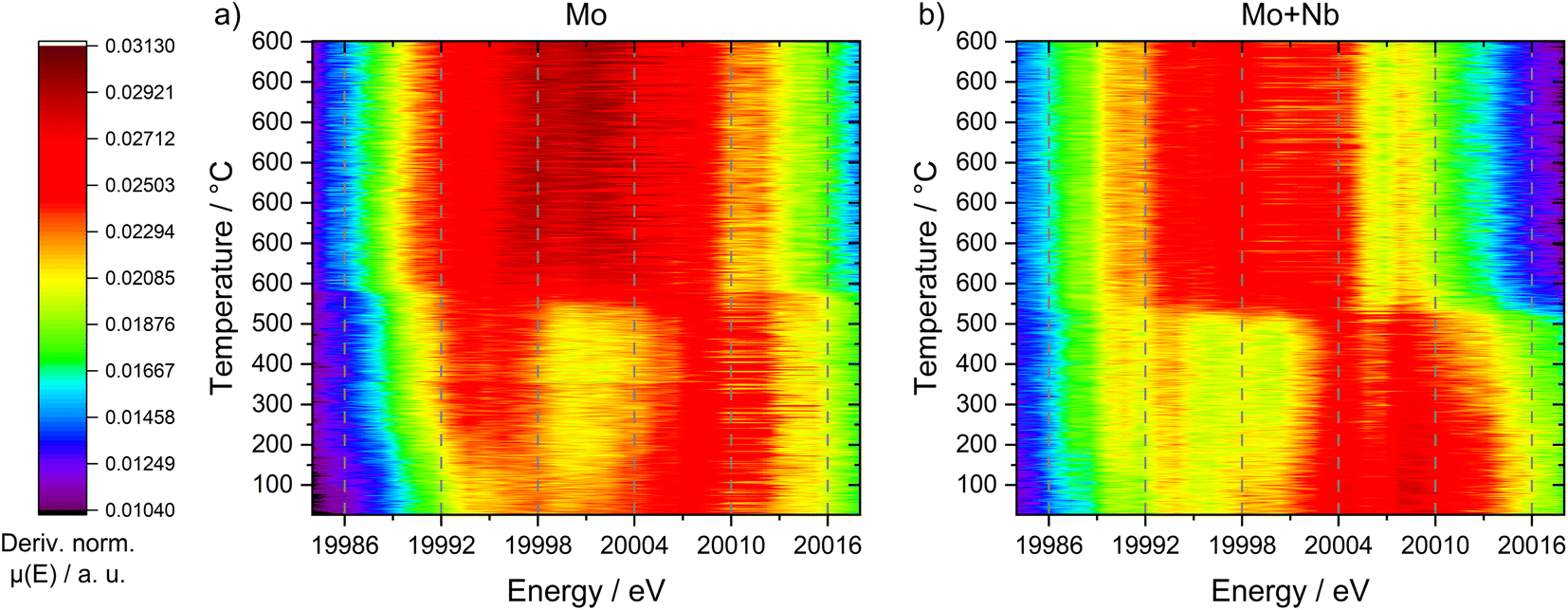 | ||
| Fig. 10 Heatmap depictions of the first derivative of all recorded XANES measurements during the temperature-programmed reaction of 6Mo/HZSM-5 (a) and 6Mo1Nb/HZSM-5 (b) with 50 mL min−1 20% CH4/N2 at 1 atm and a heating rate of 10 °C min−1. | ||
4. Discussion
In this section we would like to first summarize and highlight the most important findings obtained from different measurements and connect them to draw a more comprehensive mechanistic overview of the Nb-doping effect on the activity of the Mo/HZSM-5 catalysts for the dehydroaromatization of methane. These can be listed as follows:(i) The catalytic activity of the Mo/HZSM-5 catalysts for the MDA can be improved by doping with Nb especially under reaction conditions with lower conversions (low temperature down to 600 °C or a high space velocity of 4800 SCC gcat−1 h−1). This is associated with a pronounced enhancement in benzene productivity for the Nb-doped Mo/HZSM-5 catalysts.
(ii) Based on thermal analysis measurements (TGA/DSC) the Nb-doped catalyst exhibits a slight decrease in the tendency for the buildup of coke species (carbon) but at the same time it shows enhanced oxidation of MoCx species.
(iii) Electron microscopy reveals intimate mixing of Mo and Nb oxides on the doped catalyst, as well as the presence of subnanometer particles at all stages of reaction similar to the undoped sample; the average particle size of MoNbOx in 6Mo1Nb/HZSM-5 before and after reaction with methane is larger than that of MoOx in 6Mo/HZSM-5, while the relative particle growth under reaction conditions is less pronounced (4.2 → 8.0 nm for 6Mo1Nb, 1.6 → 5.8 nm for 6Mo).
(iv) Oxidative regeneration restores activity for both 6Mo/HZSM-5 and 6Mo1Nb/HZSM-5; the presence of Nb increases the stability of the catalyst against irreversible deactivation.
(v) Surface analysis with XPS indicated that Mo is mainly present as Mo6+ (with a small fraction of Mo5+) for both fresh 6Mo/HZSM-5 and 6Mo1Nb/HZSM-5; larger surface particles observed by STEM led to an increased Mo surface concentration.
(vi) After the reaction, both the doped and undoped catalysts exhibit similar overall Mo surface concentrations but differ in the distribution of oxidation states; the observable increase in the Mo5+/Mo6+ ratio for the Nb-doped sample refers to interaction of Nb with MoOx species.
(vii) Oxidative regeneration restores the originally present MoOx species; the Nb-doped catalyst retains its higher relative Mo5+ content.
(viii) In situ NAP-XPS experiments show an increased degree of Mo carburization on the Nb-doped catalyst, while a relatively decreased Mo surface concentration compared to the undoped catalyst suggests a lower likelihood of deep reduction during the reaction.
(ix) In situ Mo K-edge XANES measurements show more facile reduction of Mo6+ as indicated by changes at lower temperature in the presence of Nb; formation of a more uniform carbidic phase is suggested by the width of the derivative feature compared to the undoped 6Mo/HZSM-5.
According to the detailed analysis of the undoped and the Nb doped catalysts, we propose that electron transfer occurs between Nb and Mo species. An electronic interaction between Mo and Nb can be deduced based on the binding energy shifts of both Mo 3d and Nb 3d in the bimetallic catalyst compared to the respective monometallic samples. The interaction between both metals is made possible due to the intimate mixing of Mo and Nb on the 6Mo1Nb/HZSM-5 catalyst as observed in EDS measurements (see Fig. 5). In this redox process, Nb is reduced and Mo is concurrently oxidized. Nb was proven to not be reducible by hydrocarbons and barely reducible by molecular hydrogen below 900 °C, so reduction by Mo-activated CHx species is unlikely.45,46 This interaction was especially visible during in situ NAP-XPS measurements (see Fig. 8 and S26†), as reduction and noticeable binding energy shifts of both Mo and Nb occurred simultaneously. Due to the existence of the activation period (reduction of Mo6+ by CH4) at the beginning of the MDA reaction, it is generally assumed that Mo6+ species are not active for C–C coupling reactions and at least partial reduction to lower oxidation states (Mo5+/Mo4+/Mo2+) is necessary for C2+ hydrocarbon formation. Pre-reduction with H2 or CO was reported to shorten or eliminate the activation period for the catalyst.69 The subsequent occurrence of aromatic products and their increase in selectivity over time is the so-called induction period, which was related to the formation of (oxy-)carbidic active sites.70,71 In recent studies larger carbonaceous species were proposed as a key intermediate for benzene production via hydrogenolysis and radical reactions.53,60,72,73 In this case, the induction period was referred to as the buildup of a hydrocarbon pool. The observed redox interaction between Nb and Mo would explain the slightly delayed formation of aromatics and an prolonged formation of CO with 6Mo1Nb/HZSM-5 catalysts clearly visible at 600 °C (see Fig. S10†), as the generation of active sites and aromatic precursors is slowed by reoxidation of MoOxCy and MoCx.67 As an example, Lai et al. found reoxidation of Mo to be the cause of quenched aromatic yields when co-feeding CO2 to the MDA reaction.74
While oxidation of active sites might appear counterproductive, it can be concluded from the presented results that the redox interaction of Mo and Nb increases the stabilization and lifetime of intermediate Mo oxidation states which seem to have a pronounced impact on catalytic performance of the Mo/HZSM-5 catalysts. It was proposed that completely carburized Mo species (Mo2C) are not active towards aromatics formation, but rather produce aliphatics instead. This was correlated by Lezcano-González et al. to a detachment of deeply reduced Mo carbides from the Brønsted acid sites of the zeolite.67 This leads to the loss of both electronic stabilization and pore-induced shape selectivity due to diffusion and surface agglomeration. The assumed critical role of Brønsted acid sites in C2+ oligomerization during the MDA reaction75–77 has been disputed in the recent literature. Kosinov et al.16 found that aromatization of methane is possible without any acid sites present in the catalyst. The acid sites on HZSM-5 are rather proposed to disperse and stabilize Mo species finely inside the zeolite micropores. We have recently demonstrated further dispersion of Mo upon first contact with methane via27Al MAS NMR, so the initial state of Mo is not easily relatable to the catalyst performance under reaction conditions.37
The intimate mixing and interaction of Mo and Nb species confirmed by STEM/EDS and XPS investigations, together with the observation of homogeneous carburization of Mo in the presence of Nb at lower temperatures by XANES measurements, suggest that a Mo–Nb mixed carbide is formed during MDA. This is supported by the simultaneous reduction and change in speciation of both Mo and Nb visible in NAP-XPS. As seen from our experimental results, methane conversion is thermodynamically controlled in the temperature range studies. In this case Nb did not affect the time-on-stream behaviour. Formation of aliphatic products such as ethylene or ethane is also mostly unchanged. Production of aromatics, however, was generally higher with Nb, suggesting improved kinetics especially visible at low methane conversions where subsequent product condensation is less likely. Jentoft et al.64 found that mixed Mo–Nb carbide allows for more facile creation of carbon vacancies. Isotopic labeling experiments by Vollmer et al.78 have shown the direct incorporation of carbidic carbon into the MDA products, suggesting a Mars–van-Krevelen-like interaction with a formed hydrocarbon pool. With this in mind, we tend to propose an improved carbon exchange from transition metal carbides into aromatic products as a possible explanation for the increased benzene productivities observed for 6Mo1Nb/HZSM-5. While autonomous aromatization activity of reduced Nb species cannot be completely excluded, reactivity of Mo and Nb cannot be clearly separated in the doped catalyst.
The proposed redox interaction between Nb and Mo might also be responsible for the measurably increased catalyst stability against long-term irreversible deactivation mechanisms. Judging from the surface concentrations determined by XPS and the particle sizes observed in STEM analyses, the abundance of surface Mo is not a direct indicator for catalyst activity. It should be noted that the MoNb average particle size (for the doped 6Mo1Nb/HZSM-5) was, at all investigated reaction states, higher than that of the reference sample. Despite this observation 6Mo1Nb/HZSM-5 was proven to be almost universally more active for benzene formation. It can thus be concluded that surface Mo–Nb particles do not contribute significantly towards the desired reaction, but rather subnanometer-sized centers inside the micropores are responsible. The significantly higher concentration of Mo5+ (commonly associated with Al-stabilized Mo) in the fresh and regenerated 6Mo1Nb/HZSM-5 catalysts might indicate that Nb improves the anchoring of Mo onto the zeolitic acid sites or by itself presents an anchoring site. Through stabilization of intermediate Mo oxidation states by Nb (i.e., formation of mixed Mo–Nb carbide) during the reaction, deep reduction to Mo2+ could be partially avoided, which would otherwise lead to detachment of the active site from the framework aluminium.60,67 Based on our in situ experiments, which suggest a lower occurrence of Mo-bound oxygen and stronger retention of Mo species inside the zeolite pores in the presence of Nb, it can be concluded that the abundance of stabilized carbidic Mo is increased, in turn reducing the likelihood of active site loss by surface diffusion and agglomeration. The more facile oxidation of mixed Mo–Nb carbide in 6Mo1Nb/HZSM-5 compared to the undoped sample (see Fig. 3) might also assist in an increased regenerability. Therefore, we conclude that despite the observably increased surface particle size of 6Mo1Nb/HZSM-5, the actual concentration of active sites inside the zeolite pores after each regeneration step is higher than that for the unmodified 6Mo/HZSM-5.
One other possible explanation for the beneficial effect of Nb would be a decreased rate of dealumination of the zeolite framework during the reaction or regeneration. This loss of tetrahedral Al species is most likely caused by the oxidative regeneration step,28 although the Si/Al ratios measured by XPS are reduced in the spent samples as well. This can be explained by surface coverage of Si with carbon deposits. Due to the Nb-catalyzed oxidation of Mo at lower temperatures (visible in TGA, see Fig. 3) the formation of Al2(MoO4)3 could possibly be reduced in favor of a Mo–Nb mixed oxide. The initial absence of tetrahedrally coordinated Mo during our XAS investigations on the Nb-doped catalyst might be associated with a Mo–Nb mixed oxide. However, additional XRD measurements (see Fig. S18†) did not show any significant loss of crystallinity, making dealumination less likely. Furthermore, minor changes to framework aluminium are difficult to analyze and might not be conclusive in regard to a structure–reactivity relationship in MDA.18,71,79
5. Conclusions
In total we conclude that the doping of a 6.3 wt% Mo/HZSM-5 catalyst with a small amount of Nb (1 wt%) enhances the activity and stability of these catalysts for the dehydroaromatization of methane, with a measurable impact on the yield of aromatics especially at low reaction temperatures and increased space velocities. In addition, doping with Nb enhanced the regenerability of the catalyst via oxidative treatment. The improved catalytic performance was associated with a more facile conversion of MoOx to active Mo–Nb carbide species and their stabilization under reaction and regeneration conditions which extends the lifetime productivity of the catalyst. While a complete solution to the deactivation of MDA catalysts is still a highly sought-after target, these results will help broaden the scope of applicable dopants and further understand and control the catalytic properties of Mo-based catalysts for this reaction and similar chemical conversions based on catalysts using zeolitic frameworks. This gives a positive outlook for further optimizations in regard to productivity and regenerability, as well as potential industrial applications.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was financed by the Deutsche Forschungsgemeinschaft (DFG) (Grants: 351914377 and 442613239). XAS measurements were performed at the BAMline at the BESSY-II storage ring (Helmholtz-Zentrum Berlin für Materialien und Energie, HZB). We thank Dr Kirill Yusenko, Ralf Britzke, Michael Sintschuk, and Sven Schlau (BAM) for technical support at the beamline. We thank Reinhard Eckelt for N2 adsorption experiments, Anja Simmula for ICP-OES analyses, Dr Hanan Atia for NH3-TPD measurements, as well as Dr Henrik Lund and Kathleen Schubert for XRD experiments.References
- K. Kaygusuz, J. Eng. Res. Appl. Sci., 2015, 4, 317–327 Search PubMed.
- E. V. Kondratenko, T. Peppel, D. Seeburg, V. A. Kondratenko, N. Kalevaru, A. Martin and S. Wohlrab, Catal. Sci. Technol., 2017, 7, 366–381 RSC.
- A. M. Abdel-Mageed and S. Wohlrab, Catalysts, 2022, 12, 16 CrossRef CAS.
- S. Chen and A. M. Abdel-Mageed, Int. J. Hydrogen Energy, 2023, 48, 24915–24935 CrossRef CAS.
- P. Weiland, Appl. Microbiol. Biotechnol., 2010, 85, 849–860 CrossRef CAS PubMed.
- U. Dingerdissen, A. Martin, D. Herein and H. J. Wernicke, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH, Weinheim, 2nd edn, 2008, vol. 1, pp. 37–56 Search PubMed.
- A. Farsi and S. S. Mansouri, Arabian J. Chem., 2016, 9, S28–S34 CrossRef CAS.
- J. H. Lunsford, Catal. Today, 2000, 63, 165–174 CrossRef CAS.
- R. Horn and R. Schloegl, Catal. Lett., 2015, 145, 23–39 CrossRef CAS.
- M. Ahlquist, R. J. Nielsen, R. A. Periana and W. A. Goddard III, J. Am. Chem. Soc., 2009, 131, 17110–17115 CrossRef CAS PubMed.
- P. M. Bijani, M. Sohrabi and S. Sahebdelfar, Ind. Eng. Chem. Res., 2014, 53, 572–581 CrossRef CAS.
- S. Majhi, P. Mohanty, H. Wang and K. K. Pant, J. Energy Chem., 2013, 22, 543–554 CrossRef CAS.
- J. J. Spivey and G. Hutchings, Chem. Soc. Rev., 2014, 43, 792–803 RSC.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- K. Sun, D. M. Ginosar, T. He, Y. Zhang, M. Fan and R. Chen, Ind. Eng. Chem. Res., 2018, 57, 1768–1789 CrossRef CAS.
- N. Kosinov, F. J. A. G. Coumans, E. A. Uslamin, A. S. G. Wijpkema, B. Mezari and E. J. M. Hensen, ACS Catal., 2017, 7, 520–529 CrossRef CAS.
- N. Pasupulety, A. A. Al-Zahrani, M. A. Daous, H. Driss and L. A. Petrov, Arabian J. Chem., 2020, 13, 5199–5207 CrossRef CAS.
- C. H. L. Tempelman and E. J. M. Hensen, Appl. Catal., B, 2015, 176–177, 731–739 CrossRef CAS.
- V. I. Zaikovskii, A. V. Vosmerikov, V. F. Anufrienko, L. L. Korobitsyna, E. G. Kodenev, G. V. Echevskii, N. T. Vasenin, S. P. Zhuravkov, E. V. Matus, Z. R. Ismagilov and V. N. Parmon, Kinet. Catal., 2006, 47, 389–394 CrossRef CAS.
- Y. Song, Q. Zhang, Y. Xu, Y. Zhang, K. Matsuoka and Z.-G. Zhang, Appl. Catal., A, 2017, 530, 12–20 CrossRef CAS.
- Y. Xu, Y. Song and Z.-G. Zhang, Catal. Today, 2017, 279, 115–123 CrossRef CAS.
- H. Liu, S. Yang, J. Hu, F. Shang, Z. Li, C. Xu, J. Guan and Q. Kan, Fuel Process. Technol., 2012, 96, 195–202 CrossRef CAS.
- E. V. Matus, O. B. Sukhova, I. Z. Ismagilov, V. I. Zaikovskii, M. A. Kerzhentsev, Z. R. Ismagilov, K. D. Dosumov and A. G. Mustafin, Eurasian Chem.-Technol. J., 2010, 12, 1–8 CAS.
- M. T. Portilla, F. J. Llopis and C. Martinez, Catal. Sci. Technol., 2015, 5, 3806–3821 RSC.
- N. Kosinov, F. J. A. G. Coumans, E. Uslamin, F. Kapteijn and E. J. M. Hensen, Angew. Chem., Int. Ed., 2016, 55, 15086–15090 CrossRef CAS PubMed.
- S. Herzog, K. Gustav and J. Strähle, in Handbuch der Präparativen Anorganischen Chemie, ed. G. Brauer, Ferdinand Enke Verlag, Stuttgart, 3rd edn, 1981, vol. 3, pp. 1543–1544 Search PubMed.
- H. C. Zeng, J. Cryst. Growth, 1998, 186, 393–402 CrossRef CAS.
- N. Kosinov, F. J. A. G. Coumans, G. Li, E. Uslamin, B. Mezari, A. S. G. Wijpkema, E. A. Pidko and E. J. M. Hensen, J. Catal., 2017, 346, 125–133 CrossRef CAS.
- P. Tan, Appl. Catal., A, 2019, 580, 111–120 CrossRef CAS.
- V. Abdelsayed, D. Shekhawat and M. W. Smith, Fuel, 2015, 139, 401–410 CrossRef CAS.
- A. K. Aboul-Gheit, A. E. Awadallah, A. A. Aboul-Enein and A.-L. H. Mahmoud, Fuel, 2011, 90, 3040–3046 CrossRef CAS.
- S. Burns, J. S. J. Hargreaves, P. Pal, K. M. Parida and S. Parija, Catal. Today, 2006, 114, 383–387 CrossRef CAS.
- S. Liu, Q. Dong, R. Ohnishi and M. Ichikawa, Chem. Commun., 1997, 1455–1456, 10.1039/A702731A.
- S. Ma, X. Guo, L. Zhao, S. Scott and X. Bao, J. Energy Chem., 2013, 22, 1–20 CrossRef CAS.
- N. Pasupulety, A. A. Al-Zahrani, M. A. Daous, H. Driss and L. A. Petrov, J. Mater. Res. Technol., 2021, 14, 363–373 CrossRef CAS.
- A. C. C. Rodrigues and J. L. F. Monteiro, React. Kinet., Mech. Catal., 2012, 105, 441–450 CrossRef CAS.
- S. Peters, C. Rieg, S. Bartling, M. Parlinska-Wojtan, M. Dyballa, S. Wohlrab and A. M. Abdel-Mageed, ACS Catal., 2023, 13056–13070, DOI:10.1021/acscatal.3c02385.
- A. Guilherme Buzanich, M. Radtke, K. V. Yusenko, T. M. Stawski, A. Kulow, C. T. Cakir, B. Röder, C. Naese, R. Britzke, M. Sintschuk and F. Emmerling, J. Chem. Phys., 2023, 158, 244202 CrossRef CAS PubMed.
- S. Peters, B. Kunkel, C. T. Cakir, A. Kabelitz, S. Witte, T. Bernstein, S. Bartling, M. Radtke, F. Emmerling and A. M. Abdel-Mageed, Chem. Commun., 2023, 59, 12120–12123 RSC.
- T. Kubota, N. Oshima, Y. Nakahara, M. Yanagimoto and Y. Okamoto, J. Jpn. Pet. Inst., 2006, 49, 127–133 CrossRef CAS.
- V. Abdelsayed, M. W. Smith and D. Shekhawat, Appl. Catal., A, 2015, 505, 365–374 CrossRef CAS.
- Y. Lai and G. Veser, Environ. Prog. Sustainable Energy, 2016, 35, 334–344 CrossRef CAS.
- C. Karakaya and R. J. Kee, Prog. Energy Combust. Sci., 2016, 55, 60–97 CrossRef.
- G. Brauer and A. Simon, in Handbuch der Präparativen Anorganischen Chemie, ed. G. Brauer, Ferdinand Enke Verlag, Stuttgart, 3rd edn, 1981, vol. 3, pp. 1462–1476 Search PubMed.
- V. L. S. Teixeira da Silva, E. I. Ko, M. Schmal and S. T. Oyama, Chem. Mater., 1995, 7, 179–184 CrossRef CAS.
- V. L. S. Teixeira da Silva, M. Schmal and S. T. Oyama, J. Solid State Chem., 1996, 123, 168–182 CrossRef CAS.
- I. E. Wachs, L. E. Briand, J.-M. Jehng, L. Burcham and X. Gao, Catal. Today, 2000, 57, 323–330 CrossRef CAS.
- D. Ma, D. Wang, L. Su, Y. Shu, Y. Xu and X. Bao, J. Catal., 2002, 208, 260–269 CrossRef CAS.
- E. V. Matus, I. Z. Ismagilov, O. B. Sukhova, V. I. Zaikovskii, L. T. Tsikoza, Z. R. Ismagilov and J. A. Moulijn, Ind. Eng. Chem. Res., 2007, 46, 4063–4074 CrossRef CAS.
- B. M. Weckhuysen, M. P. Rosynek and J. H. Lunsford, Catal. Lett., 1998, 52, 31–36 CrossRef CAS.
- Y. Song, Y. Xu, Y. Suzuki, H. Nakagome and Z.-G. Zhang, Appl. Catal., A, 2014, 482, 387–396 CrossRef CAS.
- N. Kosinov, E. A. Uslamin, F. J. A. G. Coumans, A. S. G. Wijpkema, R. Y. Rohling and E. J. M. Hensen, ACS Catal., 2018, 8, 8459–8467 CrossRef CAS PubMed.
- A. Beuque, H. Hao, E. Berrier, N. Batalha, A. Sachse, J.-F. Paul and L. Pinard, Appl. Catal., B, 2022, 309, 121274 CrossRef CAS.
- B. S. Liu, L. Jiang, H. Sun and C. T. Au, Appl. Surf. Sci., 2007, 253, 5092–5100 CrossRef CAS.
- N. Kosinov, E. A. Uslamin, L. Meng, A. Parastaev, Y. Liu and E. J. M. Hensen, Angew. Chem., Int. Ed., 2019, 58, 7068–7072 CrossRef CAS PubMed.
- C. Bornes, J. A. Amelse, M. Peacock, C. L. Marshall, M. M. Schwartz, C. F. G. C. Geraldes, J. Rocha and L. Mafra, Eur. J. Inorg. Chem., 2020, 2020, 1860–1866 CrossRef CAS.
- M. Ziolek and I. Sobczak, Catal. Today, 2017, 285, 211–225 CrossRef CAS.
- S. J. Han, S. K. Kim, A. Hwang, S. Kim, D.-Y. Hong, G. Kwak, K.-W. Jun and Y. T. Kim, Appl. Catal., B, 2019, 241, 305–318 CrossRef CAS.
- N. Wang, X. Dong, L. Liu, D. Cai, Q. Cheng, J. Wang, Y. Hou, A.-H. Emwas, J. Gascon and Y. Han, Cell Rep. Phys. Sci., 2021, 2, 100309 CrossRef CAS.
- N. Kosinov, A. S. Wijpkema, E. Uslamin, R. Rohling, F. J. Coumans, B. Mezari, A. Parastaev, A. S. Poryvaev, M. V. Fedin and E. A. Pidko, Angew. Chem., Int. Ed., 2018, 57, 1016–1020 CrossRef CAS PubMed.
- K. Murugappan, E. M. Anderson, D. Teschner, T. E. Jones, K. Skorupska and Y. Román-Leshkov, Nat. Catal., 2018, 1, 960–967 CrossRef CAS.
- B. Frank, T. P. Cotter, M. E. Schuster, R. Schlögl and A. Trunschke, Chem.–Eur. J., 2013, 19, 16938–16945 CrossRef CAS PubMed.
- J. Baltrusaitis, B. Mendoza-Sanchez, V. Fernandez, R. Veenstra, N. Dukstiene, A. Roberts and N. Fairley, Appl. Surf. Sci., 2015, 326, 151–161 CrossRef CAS.
- A. Mehdad, R. E. Jentoft and F. C. Jentoft, J. Catal., 2017, 351, 161–173 CrossRef CAS.
- A. Gupta, M. Mittal, M. K. Singh, S. L. Suib and O. P. Pandey, Sci. Rep., 2018, 8, 13597 CrossRef PubMed.
- C. Wan, Y. N. Regmi and B. M. Leonard, Angew. Chem., Int. Ed., 2014, 53, 6407–6410 CrossRef CAS PubMed.
- I. Lezcano-González, R. Oord, M. Rovezzi, P. Glatzel, S. W. Botchway, B. M. Weckhuysen and A. M. Beale, Angew. Chem., Int. Ed., 2016, 55, 5215–5219 CrossRef PubMed.
- A. Gaur, M. Stehle, K. V. Raun, J. Thrane, A. D. Jensen, J.-D. Grunwaldt and M. Høj, Phys. Chem. Chem. Phys., 2020, 22, 11713–11723 RSC.
- B. M. Weckhuysen, D. Wang, M. P. Rosynek and J. H. Lunsford, J. Catal., 1998, 175, 338–346 CrossRef CAS.
- D. Wang, J. H. Lunsford and M. P. Rosynek, J. Catal., 1997, 169, 347–358 CrossRef CAS.
- M. Rahman, A. Sridhar and S. J. Khatib, Appl. Catal., A, 2018, 558, 67–80 CrossRef CAS.
- I. Vollmer, N. Kosinov, Á. Szécsényi, G. Li, I. Yarulina, E. Abou-Hamad, A. Gurinov, S. Ould-Chikh, A. Aguilar-Tapia and J.-L. Hazemann, J. Catal., 2019, 370, 321–331 CrossRef CAS.
- G. Li, I. Vollmer, C. Liu, J. Gascon and E. A. Pidko, ACS Catal., 2019, 9, 8731–8737 CrossRef CAS.
- P. Tan, K. W. Wong, C. T. Au and S. Y. Lai, Appl. Catal., A, 2003, 253, 305–316 CrossRef CAS.
- C. Karakaya, S. H. Morejudo, H. Zhu and R. J. Kee, Ind. Eng. Chem. Res., 2016, 55, 9895–9906 CrossRef CAS.
- S. Liu, L. Wang, R. Ohnishi and M. Ichikawa, J. Catal., 1999, 181, 175–188 CrossRef CAS.
- K. S. Wong, J. W. Thybaut, E. Tangstad, M. W. Stöcker and G. B. Marin, Microporous Mesoporous Mater., 2012, 164, 302–312 CrossRef CAS.
- I. Vollmer, B. van der Linden, S. Ould-Chikh, A. Aguilar-Tapia, I. Yarulina, E. Abou-Hamad, Y. G. Sneider, A. I. Olivos Suarez, J.-L. Hazemann, F. Kapteijn and J. Gascon, Chem. Sci., 2018, 9, 4801–4807 RSC.
- C. Rieg, D. Dittmann, Z. Li, A. Kurtz, E. Kaya, S. Peters, B. Kunkel, M. Parlinska-Wojtan, S. Wohlrab, A. M. Abdel-Mageed and M. Dyballa, J. Phys. Chem. C, 2022, 126, 13213–13223 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta07532j |
| This journal is © The Royal Society of Chemistry 2024 |