
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUncovering the critical function of lanthanum in CH4 production from CO2 using exsolved LaNiO3 perovskite catalysts†
Mathias
Barreau
*a,
Davide
Salusso
 b,
Jinming
Zhang
a,
Michael
Haevecker
cd,
Detre
Teschner
cd,
Anna
Efimenko
ef,
Fabrice
Bournel
gh,
Jean-Jacques
Gallet
gh,
Elisa
Borfecchia
i,
Kamil
Sobczak
j,
Corinne
Petit
a and
Spyridon
Zafeiratos
*a
b,
Jinming
Zhang
a,
Michael
Haevecker
cd,
Detre
Teschner
cd,
Anna
Efimenko
ef,
Fabrice
Bournel
gh,
Jean-Jacques
Gallet
gh,
Elisa
Borfecchia
i,
Kamil
Sobczak
j,
Corinne
Petit
a and
Spyridon
Zafeiratos
*a
aInstitut de Chimie et Procédés pour l’Energie, l’Environnement et la Santé (ICPEES), ECPM, UMR 7515 CNRS – Université de Strasbourg, 25 rue Becquerel, Cedex 02, 67087 Strasbourg, France. E-mail: mathias.barreau@unicaen.fr; spiros.zafeiratos@unistra.fr
bEuropean Synchrotron Radiation Facility, CS 40220, Cedex 9 F-38043 Grenoble, France
cMax-Planck-Institut für Chemische Energiekonversion (MPI-CEC), Stiftstrasse 34-36, D-45470 Mülheim a.d. Ruhr, Germany
dFritz-Haber-Institut der Max-Planck-Gesellschaft, Faradayweg 4-6, D-14195 Berlin, Germany
eInterface Design, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH (HZB), Albert-Einstein-Str. 15, 12489 Berlin, Germany
fEnergy Materials In-situ Laboratory Berlin (EMIL), Helmholtz-Zentrum Berlin für Materialien und Energie GmbH (HZB), Albert-Einstein-Str. 15, 12489 Berlin, Germany
gLaboratoire de Chimie Physique-Matière et Rayonnement, Sorbonne Universite, Campus Curie, CNRS UMR 7614, 4 place Jussieu, 75005 Paris, France
hSynchrotron SOLEIL, L'orme des Merisiers, B.P. 48, Saint Aubin, Gif-sur-Yvette Cedex 91192, France
iDepartment of Chemistry, INSTM Reference Center and NIS Centers, University of Torino, 10125 Torino, Italy
jFaculty of Chemistry, Biological and Chemical Research Centre, University of Warsaw, Zwirki, Wigury 101, 02-089 Warsaw, Poland
First published on 13th February 2024
Abstract
CO2 methanation, also known as the Sabatier reaction, is of great environmental interest as a sustainable process for energy production and storage. Herein, we report the development of a Ni–La2O3 catalyst for CO2 methanation prepared upon reduction of a LaNiO3 perovskite precursor. The perovskite-based catalyst exhibits enhanced activity, high methane selectivity and improved stability when compared to Ni–La2O3 prepared through conventional impregnation methods. The transformation of the LaNiO3 perovskite precursor upon thermal activation in H2 was found to have a profound impact on the catalytic properties of the resulting material. The size and stability of exsolved Ni nanoparticles after prolonged reaction were investigated using ex situ electron microscopy. Synchrotron-based X-ray absorption fine structure (XAFS) spectroscopy, as well as soft and tender X-ray photoelectron spectroscopies (AP-XPS/HAXPES), provides detailed insights into the evolution of bulk and surface phases during the transition of the perovskite to its active catalytic state. Our findings indicate that processes beyond the well-established exsolution of nanoparticles, such as lanthana spillover onto nickel, may occur during H2 activation. More importantly, in situ spectroscopy under CO2 methanation conditions revealed that the surface's affinity for La-hydroxide or La-carbonate formation significantly influences the reactivity. Specifically, we found that La-hydroxide acts as a precursor for the formation of La-oxycarbonate (hexagonal La2O2CO3 phase), a crucial element of the active exsolved catalyst. In contrast, in the absence of La-hydroxide, La-carbonates (La2(CO3)3) formed on the surface, blocking the active sites of the supported catalyst. Our research examines hitherto unrecognized processes affecting the reactivity of exsolved perovskites, highlighting LaNiO3 as a promising catalyst for CO2 methanation.
1. Introduction
Global warming caused by the emission of greenhouse gases has attracted a lot of attention during the last few decades. In this context, CO2, a major component of the anthropogenic emissions (estimated to 35 Gt in 2021 (ref. 1 and 2)), can be captured and used as a carbon source to produce value added chemicals and fuels such as methanol, CO or CH4.3,4 Among the most viable strategies, CO2 methanation (CO2 + 4H2 → CH4 + 2H2O) is gaining increasing interest5,6 as a promising route to recycle CO2 emissions using H2 produced from renewable energies (known as power-to-gas). Highly active CO2 methanation catalysts are required to overcome the chemical inertness of the CO2 molecule. Among them Ni,7–9 Co10–12 or Ru-based13,14 supported catalysts have been reported as active and selective for the production of CH4 from CO or CO2. Notably, Ni-based catalysts are mostly envisaged for the reaction because of their low price and high CH4 selectivity therefore they are more favorable for industrial applications.6 However, Ni exhibits relatively poor activity at low temperature in comparison to Ru-based catalysts and is often subject to deactivation mainly caused by Ni particle sintering and/or poisoning.15,16 Consequently, an improvement in the catalyst formulation is needed to elevate CO2 methanation as one of the major green industrial processes.Typically, nickel-based methanation catalysts are composed of nanosized Ni particles fixed on high surface area oxide supports, such as Al2O3, SiO2 or CeO2.17 The support controls the dispersion, morphology and/or structure of nickel particles via metal–support interactions.18,19 It might also have significant influence on the reactivity by structure-sensitivity effects20 or by directly participating in the reaction offering sites for CO2 adsorption and activation.7,21 Basic elements such as La and Pr have been used as promoters to increase Ni dispersion and help limit deactivation over CeO2- or Al2O3-based materials.22,23 While poorly studied, Ni supported over La2O3 (lanthana) was found to be a promising methanation catalyst mainly due to the stabilization of Ni nanoparticles and limited sintering.24 However, lanthanum-supported catalysts still suffer from relatively poor activity compared for instance to ceria-based materials.25
Stabilization of nickel nanoparticles on a support is not trivial, as nickel undergoes agglomeration easily, which ends up degrading the catalyst performance. In this context, exsolution has received a lot of attention over the last decade as a promising approach to synthesize supported metallic catalysts with specific characteristics.26–28 Exsolution, also known as solid phase crystallization, is a process by which a homogeneous phase is separated into two or more solid phases typically upon annealing at high temperature.29–31 In the case of mixed oxides under reductive conditions, one or more elements will move outwards forming metallic nanoparticles at the oxide surface leading to a morphology resembling that of supported metal catalysts. Exsolution, as opposed to traditional deposition procedures, may offer better control of the metal–support interface.26,27,29 Additionally, exsolved metal nanoparticles usually show strong anchoring to the host oxide (also called socketing) enhancing in this way metal–support interaction and stability.26,27,29,32
Perovskite-based materials (ABO3) containing Ni in the B-site are frequently used as precursors which after a thermal reduction treatment will release Ni from the oxide lattice to the surface in the form of metallic nanoparticles leaving behind the host lattice as the support. So far, numerous combinations of stoichiometric, but mostly A or B-site deficient, perovskite systems have been studied with the main aim of favoring the exsolution of Ni and controlling the nature of the “support” phase. Among all, the stoichiometric LaNiO3 perovskite is the archetype of exsolved materials because it is easy to synthesize, has a relatively simple chemistry upon reduction and above all represents an excellent catalyst for CO2 conversion.31 It is therefore not surprising that LaNiO3 is probably the most studied exsolved catalyst in the literature, both in terms of physicochemical and catalytic properties. However, the great majority of these studies focus on CO2 conversion via dry methane reforming33 and it is hard to find reports on the performance and chemistry of LaNiO3 in the CO2 methanation reaction. This is even more intriguing if one considers the radically different operation conditions between dry reforming and methanation of CO2 dictated by the thermodynamics of the reaction: high temperature (ca. 800 °C) and pressure for reforming, medium temperature (ca. 350 °C) and atmospheric pressure for methanation. Considering that the active catalytic state is dynamic and is formed only under the operation conditions,34 significant differences in the chemistry of the catalysts should be anticipated in the two cases.
Rare studies over A-deficient perovskites have shown that exsolved Ni catalysts can also be very promising for CO2 hydrogenation. Suk Lim et al. evaluated Ni-exsolved La1−xCaxNiO3 perovskites as methanation catalysts after reduction pretreatment at 600 °C.35 The resulting material showed nearly 100% CH4 selectivity and the yield improved by 45% in comparison to the conventional Ni/Al2O3 catalyst with similar Ni loading. Steiger et al. prepared LaFe0.8Ni0.2O3 perovskite modified in the A-site by Sr doping for CO2 hydrogenation.36 They found by in situ XRD that La substitution with Sr promoted the segregation of Ni after exsolution at 600 °C and therefore the catalytic activity of the material in comparison to undoped LaFe0.8Ni0.2O3. However, the highest yield of CH4 was limited to 20% and could only be achieved at high temperature (around 470 °C). Nevertheless, despite few studies dedicated to dry reforming reaction,37in situ surface-specific characterization of the exsolved perovskite catalysts upon formation of the active phase is missing. In situ studies are of particular importance for this system because La-based perovskites are known to be highly reactive towards water/humidity and CO2, which hinders any attempt for ex situ/post mortem surface characterization.
In light of the above aspects, this study explores the use of the stoichiometric LaNiO3 perovskite as a precursor of highly active and stable Ni-supported catalysts towards CO2 methanation. We used a conventional supported Ni/La2O3 material with similar Ni loading as a benchmark catalyst, to demonstrate the superior CO2 methanation performance of the LaNiO3 exsolved catalyst. We also employed several ex situ and in situ synchrotron-based characterization techniques to examine the exsolution process and the dynamics of surface evolution of the catalyst during H2-activation and reaction conditions. The correlation between the surface state and the catalytic performance of exsolved and supported benchmark catalysts offers valuable insights beyond the commonly observed effect of stabilizing Ni nanoparticles. It also provides crucial information regarding surface transformations that govern the reactivity of CO2 methanation.
2. Experimental part
2.1. Catalyst preparation
The synthesis of Ni/La2O3 was realized by wet impregnation. To this end, La2O3 was first prepared via calcination of La(NO3)3·6H2O at 800 °C for 2 h so as to get a surface area comparable to that of the perovskite.39 Then, Ni(NO3)2·6H2O (8.5 mmol, 2.48 g) was added to 1.5 g of La2O3 dispersed in 30 mL of distilled water, so as to attain similar Ni/La ratio (Ni/La = 1, 25 wt% Ni) to that in the perovskite based-materials. The mixture was stirred for 4 h at 60 °C. The solvent was then evaporated in a rotary evaporator and dried overnight. For comparison purpose, the powder was then calcined at 700 °C for 6 h using a similar protocol to the one described for the perovskite material and finally treated at 600 °C under H2 for 30 minutes prior to the catalytic test.
2.2. Catalytic evaluation at 1 bar
CO2 methanation tests were performed in a fixed-bed flow reactor using a U-shaped quartz micro-reactor (i.d. 6 mm). In a typical catalytic test, 100 mg catalyst was diluted with 100 mg SiC, both sieved between 150 and 250 μm and deposited on a quartz wool bed. The quartz reactor was then introduced in a tubular oven with the catalyst bed located in the isothermal zone. The gases were introduced via calibrated mass flow controllers (Bronkhorst) for a total flow rate of 20 mL min−1 (GHSV = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1). The temperature was controlled by a K-type thermocouple attached to the quartz reactor in intimate contact with the catalytic bed location. Prior to the tests, the samples were first reduced at temperatures ranging between 400 and 600 °C, as already detailed in Section 2.1.2. Then, all catalytic tests were performed at atmospheric pressure in a CO2:H2 gas mixture with a 1:4 molar ratio. Catalytic runs were performed stepwise starting from 150 to 400 °C using a 10 °C min−1 ramp and a dwell time of 30 min at each selected temperature. Long-term stability tests were also conducted at 300 °C for 65 h after H2 pretreatment at 600 °C. The outlet gas was analyzed using a gas chromatograph Agilent 5975C VL MSD equipped with a Molecular Sieve 5A column, a PoraPLOT U GC column and an OV-1 capillary column and TCD for CO, CH4, CO2 and H2O detection. The detection and quantification of H2 was not possible due to the similar retention time to that of the carrier gas (He). The formation of CH4 and CO are due to CO2 methanation and reverse water-gas shift reactions respectively (eqn (1) and (2)):
000 h−1). The temperature was controlled by a K-type thermocouple attached to the quartz reactor in intimate contact with the catalytic bed location. Prior to the tests, the samples were first reduced at temperatures ranging between 400 and 600 °C, as already detailed in Section 2.1.2. Then, all catalytic tests were performed at atmospheric pressure in a CO2:H2 gas mixture with a 1:4 molar ratio. Catalytic runs were performed stepwise starting from 150 to 400 °C using a 10 °C min−1 ramp and a dwell time of 30 min at each selected temperature. Long-term stability tests were also conducted at 300 °C for 65 h after H2 pretreatment at 600 °C. The outlet gas was analyzed using a gas chromatograph Agilent 5975C VL MSD equipped with a Molecular Sieve 5A column, a PoraPLOT U GC column and an OV-1 capillary column and TCD for CO, CH4, CO2 and H2O detection. The detection and quantification of H2 was not possible due to the similar retention time to that of the carrier gas (He). The formation of CH4 and CO are due to CO2 methanation and reverse water-gas shift reactions respectively (eqn (1) and (2)):| CO2 methanation reaction: CO2 + 4H2 → CH4 + 2H2O | (1) |
| Reverse water-gas shift reaction: CO2 + H2 → CO + H2O | (2) |
The CO2 conversion (XCO2) and CH4 selectivity (SCH4) were calculated as follows:
 | (3) |
 | (4) |
2.3. Characterization techniques
N2 physisorption measurements were performed on a Micromeritics Tristar 3000 apparatus. The surface area was determined by a multiple-point Brunauer–Emmett–Teller (BET) approach with relative pressures (p/p0) ranging between 0.05 and 0.35. Prior to the adsorption–desorption of N2 realized at −196 °C, 100 mg sample was first degassed at 200 °C under primary vacuum for a minimum of 6 h.
H2 Temperature Programmed Reduction (H2-TPR) experiments were carried out using a Micromeritics Autochem 2920 device equipped with a thermal conductivity detector (TCD). The profiles were obtained after loading 100 mg of sample in a U-shaped quartz reactor and reduction under 10% H2 in Ar with a flow rate of 20 mL min−1 up to 900 °C, with a heating rate of 10 °C min−1. A cold trap was installed at the outlet of the reactor to remove the moisture produced during reduction.
Transmission Electron Microscopy (TEM) was performed using an FEI Talos F200X microscope operating at 200 kV in scanning transmission electron microscopy (STEM) mode using high-angle annular dark-field (HAADF) imaging. Energy-dispersive X-ray spectroscopy (EDX) using a Super-X system with four silicon drift detectors (SDDs) was applied to the detection of differences in local chemical composition.
In all cases, 20 mg of catalyst was pelletized and fixed on the sample holder, while a 1:1 CO2:H2 mixture was employed. It is important to note that, according to the balanced chemical equation for the Sabatier reaction, a CO2:H2 molar ratio of 1:4 is required, and this ratio was indeed used in the 1 bar catalytic tests. However, under the specific low-pressure conditions of the in situ AP-XPS experiments, this ratio might not be optimal for the reaction. In this context, we investigated the effect of the CO2 to H2 ratio at 5 mbar overall pressure and temperatures between 200 and 300 °C, in a reactor attached to a laboratory XPS setup described previously.43 According to the online gas phase analysis, a CO2:H2 ratio of 1:1 resulted in about 25% higher CH4 and H2O signals compared to 1:4. This increase could be attributed to an extended contact time between CO2 and the catalyst. Therefore, selecting a CO2:H2 ratio of 1:1 is a good compromise to maximize CH4 production in the low-pressure range under AP-XPS testing conditions. Below follows a description of the two AP-XPS instruments used in this study.
2.3.2.1 AP-XPS at the tempo beamline. The AP-XPS instrument of Sorbonne University attached to the TEMPO beamline of the SOLEIL Synchrotron radiation facility (France)44 was used to characterize the LaNiO3 catalyst under O2, H2 (sample treated at 600 °C) and CO2
:H2 (1:1) methanation reaction conditions. The gas feeds (O2, H2, CO2) were regulated by leak valves and their purity was monitored using a quadrupole mass spectrometer. The catalyst was fixed on a sample holder with an integrated resistive heater and the temperature was monitored by a K-type thermocouple in contact with the sample surface.
2.3.2.2 AP-XPS and AP-HAXPES at the EMIL beamline. Combined AP-XPS and AP-HAXPES measurements were performed at the new CAT branch of the EMIL beamline at the synchrotron radiation facility BESSY II of the Helmholtz Zentrum Berlin.45,46 The reported measurements of LaNiO3 catalyst in H2 (sample treated at 400 °C) as well as of the supported Ni/La2O3 catalyst under H2 (sample treated at 600 °C) and CO2
:H2 (1:1) methanation reaction conditions were performed using this instrument. The catalyst was mounted on a sample stage heated from the rear with an IR laser irradiating the plate placed under the pellet. Gases (H2, O2, CO2) were introduced into the sample chamber via calibrated mass flow controllers (Bronkhorst). The gas phase composition was monitored using a differentially pumped quadrupole mass spectrometer (QMS, Pfeiffer PrismaPro) connected to the sample chamber through a leak valve. A significant feature of the CAT@EMIL beamline is the possibility to use soft and tender X-ray radiation in one single experiment, allowing depth profiling from the extreme surface to the subsurface. The AP-XPS spectra were measured with a 60 μm exit slit, while for AP-HAXPES a double crystal monochromator (DCM) that works without an exit slit was used. In the AP-HAXPES two fixed photon energies were used (hv = 3005 and 4900 eV) (maximum analysis depth around 21 nm).
:He gas mixture during the temperature ramp protocol reported in Fig. S1.† X-ray Absorption Near Edge Structure (XANES) spectra reported in Fig. 5a in the text concern the regions of the protocol indicated with a red line while the magnitude component of FT-EXAFS spectra reported in Fig. 5d and S9† was collected during steady states indicated by the triangle. Spectra of ex situ samples and steady states of in situ experiment were collected with the step scan mode in the 8.19–9.31 keV energy range. A constant acquisition time of 0.5 s per point was employed throughout the measured energy range. Energy resolution was set to 5 eV per point for the pre-edge (8.19–8.3 keV) and 0.3 eV per point for the XANES (8.31–8.34 keV) while a constant k-spacing of 0.035 k per point was used in the EXAFS region (8.34–9.31 keV). Spectra collected during heating/cooling steps of the in situ experiment were measured in continuous mode in the 8.2–9.2 keV energy range with 0.3 eV per point energy resolution and 0.05 s per point time resolution for a total of 3 minutes per scan. Energy calibration and alignment together with background subtraction and edge jump normalization were conducted with a dedicated Python script based on the Larch library.49 Linear Combination Analysis was done using Larch-based Python scripts. The weight of the single component (wi) was constrained to be 0 < wi < 1 while the sum of the component weights  was not forced to 1. The closeness of
was not forced to 1. The closeness of 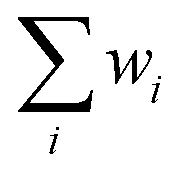 to 1 was used as an additional qualitative indicator of the goodness of the fit. All the presented FT-EXAFS spectra were obtained using a Hanning window in the 2.5–13.3 Å−1k-range. The k-range of FT-EXAFS extraction and R-range of FT-EXAFS fit are reported in Table S1.† FT-EXAFS fit was conducted using the Artemis software from the Demeter package.50 The passive amplitude reduction factor S02 was evaluated from the fit of reference Ni0 spectra (see Fig. S9 and Table S1†) and fixed for the fit of the other spectra. EXAFS Wavelet Transform (WT) was calculated with the Cauchy wavelet function fixing η to 6.4.51
to 1 was used as an additional qualitative indicator of the goodness of the fit. All the presented FT-EXAFS spectra were obtained using a Hanning window in the 2.5–13.3 Å−1k-range. The k-range of FT-EXAFS extraction and R-range of FT-EXAFS fit are reported in Table S1.† FT-EXAFS fit was conducted using the Artemis software from the Demeter package.50 The passive amplitude reduction factor S02 was evaluated from the fit of reference Ni0 spectra (see Fig. S9 and Table S1†) and fixed for the fit of the other spectra. EXAFS Wavelet Transform (WT) was calculated with the Cauchy wavelet function fixing η to 6.4.51
3. Results
3.1. CO2 methanation performance
Fig. 1a displays the CO2 conversion (XCO2) and CH4 selectivity (SCH4) profiles of the methanation light-off tests over perovskite LaNiO3 catalyst activated by reduction in H2 at 400, 500 and 600 °C, hereafter abbreviated as LaNiO3-400, LaNiO3-500, LaNiO3-600. The XCO2 and SCH4 curves of supported Ni/La2O3 pre-reduced at 600 °C are included for comparison. The XCO2 increases rapidly above 200 °C, while at 400 °C it attains CO2 conversion values close to thermodynamic equilibrium indicated by the dashed-brown line in Fig. 1a. The CO2 conversion curves of LaNiO3 are shifted towards lower temperatures compared to Ni/La2O3. The shift becomes more pronounced as the activation temperature of LaNiO3 increases from 400 °C to 600 °C, indicating improvement of the catalytic activity with the increase of the activation temperature. This can be quantified by the light-off temperatures corresponding to 50% conversion (T50) determined from Fig. 1a and listed in Table 1. It is evident that in the case of LaNiO3 the T50 shifts up to 100 °C compared to Ni/La2O3, which represents an enormous difference in the catalytic performance. Note that the XCO2 and SCH4 of Ni/La2O3 is comparable to those of similar La2O3 supported Ni catalysts.25 In addition, the LaNiO3 is more selective than Ni/La2O3 in the production of CH4 (top part of Fig. 1a), while SCH4 further improves with H2-activation temperature. For example, SCH4 of LaNiO3-600 remains above 97% throughout the experiment and reaches 99.7% at 350 °C while that of Ni/La2O3 rests around 92%. The apparent activation energies (Eapp) determined from Arrhenius plots (Fig. S2†) are listed in Table 1. In all cases the Eapp is between 87 and 99 kJ mol−1, which is in the range typically reported for CO2 methanation over Ni-based catalysts.52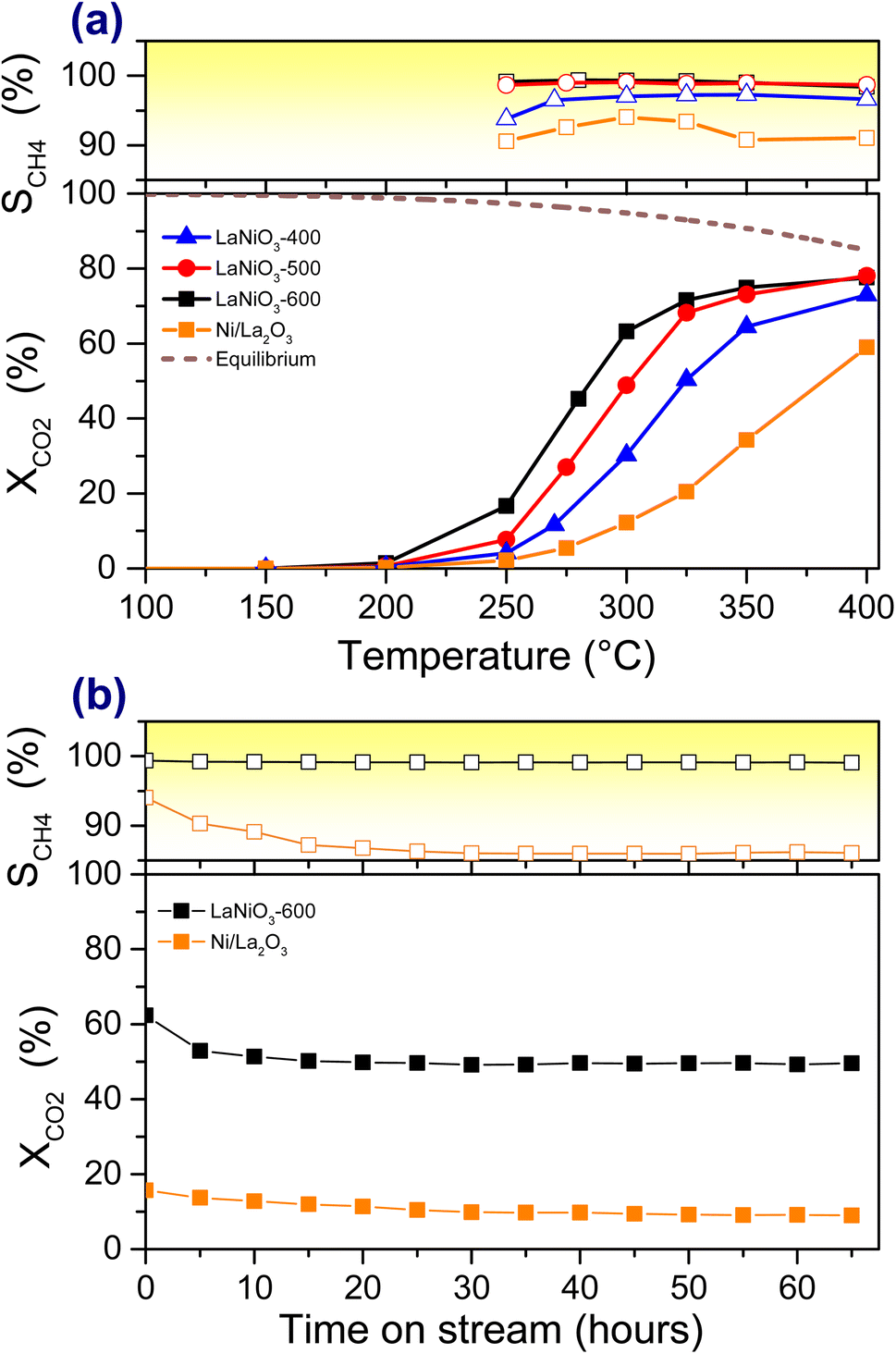 | ||
| Fig. 1 CO2 methanation performance of LaNiO3 and Ni/La2O3 catalysts: (a) CO2 conversion and CH4 selectivity as a function of the reaction temperature. The thermodynamic limit of CO2 conversion is included as a dashed-line. (b) Long-term stability tests at 300 °C for catalysts pre-reduced at 600 °C. All catalytic tests were performed in 20% CO2/H2 at 1 bar (100 mg mixed in 100 mg of SiC, GHSV ∼12000 h−1). | ||
| Catalyst | T 50 °C | E app kJ mol−1 |
|---|---|---|
| LaNiO3-400 | 325 | 99 |
| LaNiO3-500 | 300 | 96 |
| LaNiO3-600 | 285 | 87 |
| Ni/La2O3 | 381 | 93 |
The durability tests of LaNiO3-600 and Ni/La2O3 catalysts (Fig. 1b) reveal that the perovskite catalyst is not only more active and selective, but also far more stable than the supported one. In particular, after a decrease in the relative CO2 conversion of about 20% observed within the first 10 h, LaNiO3 does not present any sign of further deactivation up to 65 h. In addition, throughout these tests, the CH4 selectivity remains always above 99%. This performance clearly outperforms that of the Ni/La2O3 catalyst, which has lower CO2 conversion and CH4 selectivity also being less stable.
Overall, the catalytic tests show that the LaNiO3 perovskite catalyst outperforms the conventional supported Ni/La2O3, while higher H2 activation temperatures seem to promote both CO2 conversion and CH4 selectivity of LaNiO3.
3.2. Ex situ structural and morphological characterization
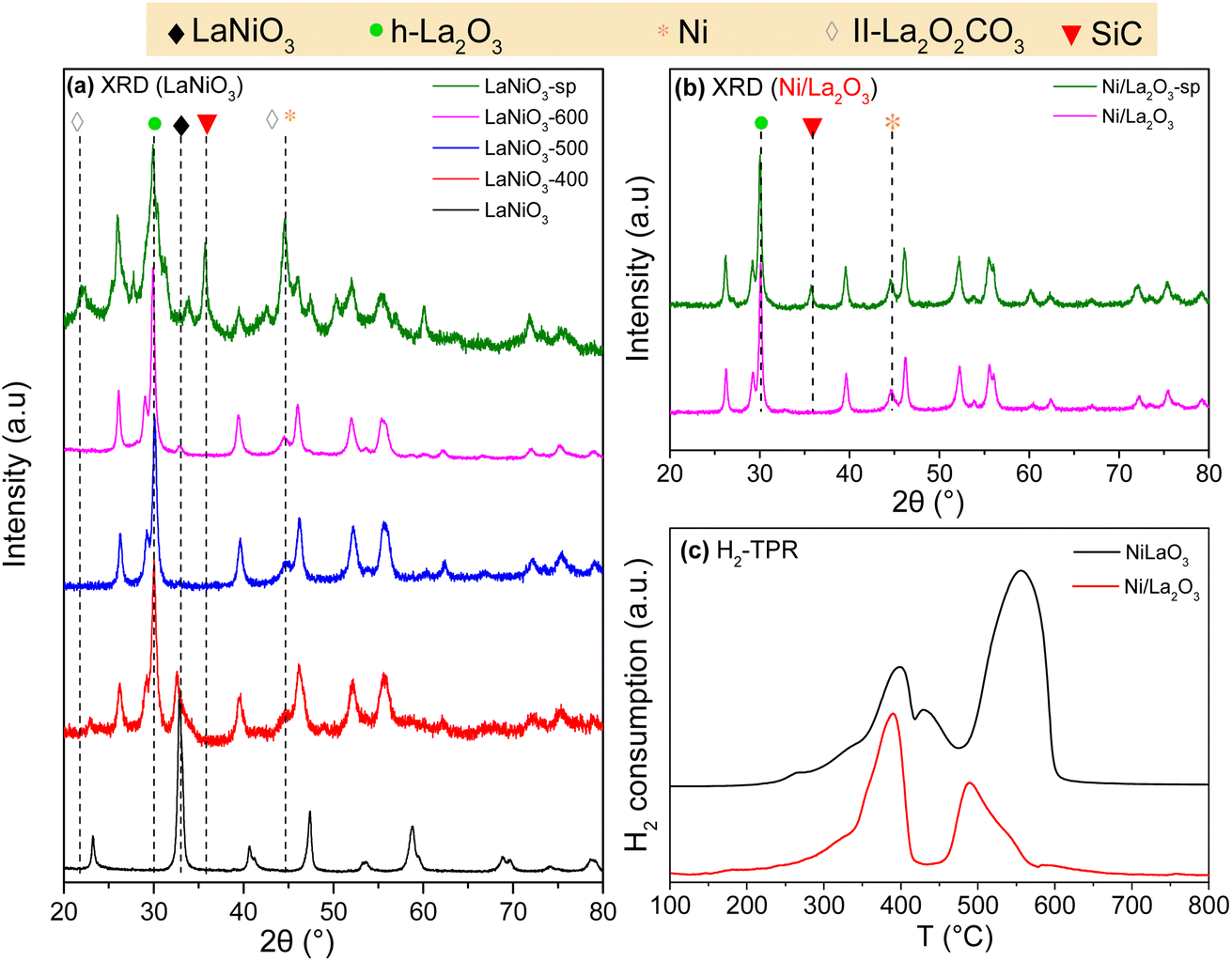 | ||
| Fig. 2 XRD patterns of (a) LaNiO3 catalysts measured after calcination, H2-activation at 400, 500 and 600 °C (LaNiO3-400, LaNiO3-500 and LaNiO3-600) and after long-term stability methanation test (LaNiO3-sp), (b) Ni/La2O3 catalysts measured after H2-activation at 600 °C (Ni/La2O3) and after long-term stability methanation test (Ni/La2O3-sp). The SiC-related peaks appear in the diffractograms of the spent catalysts, (c) H2-TPR profile of LaNiO3 and Ni/La2O3 catalysts. | ||
| Sample | S BET (m2.g−1) | D Ni (nm) | La-based crystalline phases detected | D LaOx (nm) | dNib (nm) |
|---|---|---|---|---|---|
| a Average crystallite size estimated by XRD using the Scherrer equation. b Average particle size determined by TEM. | |||||
| LaNiO3 | 3 | — | LaNiO3 | — | — |
| LaNiO3-400 | 2 | 6 | LaNiO3, h-La2O3 | 12.9 (h-La2O3) | 4.2 ± 0.5 |
| LaNiO3-500 | <1 | 8 | h-La2O3 | 14.5 (h-La2O3) | N/A |
| LaNiO3-600 | <1 | 8 | h-La2O3 | 17.5 (h-La2O3) | 6.4 ± 0.2 |
| LaNiO3-sp | N/A | N/A | h-La2O2CO3, h-La2O3 | 17.3 (h-La2O3) | 6.4 ± 0.2 |
| Ni/La2O3-600 | <1 | 11 | h-La2O3 | 18.9 (h-La2O3) | N/A |
| Ni/La2O3-sp | N/A | 11 | h-La2O3 | 19.5 (h-La2O3) | N/A |
The H2-TPR profile of LaNiO3 calcined at 700 °C (Fig. 2c) is dominated by two main and several smaller H2 consumption peaks. This shows that the reduction of LaNiO3 occurs in two steps at approximately 400 and 550 °C. The H2 consumption ratio between the two main reduction peaks is about 1.5. The H2-TPR profile of Fig. 2c is in fair agreement with previous reports.37,55,56 Usually, the low temperature peak is attributed to the Ni3+ → Ni2+ reduction and the one at higher temperature to Ni2+ → Ni0.37 Although formation of Ni0 in the second reduction step is related to the transformation of the perovskite to La2O3, both La2Ni2O5 (ref. 37) and La2NiO4 (ref. 57 and 58) have been proposed as the intermediate lanthanum phase of the first reduction step. The in situ XANES spectra (see infra) provide more insight on LaNiO3 reduction. The profile of Ni/La2O3 shows two main peaks around 400 and 500 °C. According to previous reports59 the one at 400 °C is due to the reduction of Ni2+ species with possible contribution of Ni3+. The smaller peak around 500 °C has been ascribed to the reduction of Ni–La2O3 solid solution probably formed during calcination at the interface of the two elements.
The specific surface areas deduced from N2 physisorption measurements are included in Table 2. The surface area of the calcined catalyst is low and, as might be expected, decreases more with the increase of the reduction temperature and remains below the detection limit of conventional N2-BET measurements. Nevertheless, the surface areas of the perovskite and supported catalyst after reduction are similar, which essentially rules out the possibility that the observed differences in the reactivity are related to the surface area of the catalysts.
 | ||
| Fig. 3 HR-TEM and TEM/EDX elemental mapping images of (a) LaNiO3-400, (b) LaNiO3-600, and (c) LaNiO3-sp catalysts. The Ni particle-size distribution histogram for each catalyst is included at the bottom of the figure. Between 120 and 200 particles were analyzed depending on the sample. Isolated O, Ni and La elemental mapping of the selected regions are depicted in Fig. S4.† | ||
The La overlayer and the mixed Ni–La areas disappear in LaNiO3-600 but the Ni particles still look cast into lanthanum (see areas marked with arrows). This morphology resembles the classical exsolution process involving socking of metal nanoparticles into the support.60 The HR-TEM and TEM/EDX images after the long-term stability tests (LaNiO3-sp) show that the morphology generated during the activation process, comprising homogeneously distributed nano-sized Ni particles, is largely preserved. In addition, there is no evidence of filamentous carbon deposition after the reaction.
The Ni particle size distribution is included in the form of histograms at the bottom of Fig. 3. It is estimated by measuring the size of at least 120 Ni particles identified in the TEM images. Accordingly, the average Ni particle size increases slightly from around 4 to 6 nm when the reduction temperature rises from 400 to 600 °C, in fair agreement with the crystallite sizes estimated from XRD measurements (Table 2). The histograms of the spent catalyst show that the long-term reaction does not affect the Ni particle size and distribution. The stability of highly dispersed metal nanoparticles formed through exsolution from the perovskite matrix is consistent with previous reports.26 What is of particular interest here is that XRD and TEM results confirm that there is practically no sintering of Ni particles even after 65 hours under the reaction conditions which is a key feature that explains the long-term stability found in Fig. 1b.
 | ||
| Fig. 4 (a) Ex situ Ni K-edge XANES spectra collected on LaNiO3 and Ni/La2O3 catalysts and reference materials. Details of pre-edge transitions is reported in the inset. (b) Results of LCA analysis conducted on the Ni K-edge of LaNiO3 and Ni/La2O3. The experimental spectra and the best fit curves are reported in Fig. S6.† | ||
Aiming to confirm LCA results and to eventually estimate NiO and Ni0 particle dimensions, FT-EXAFS spectra were fitted considering the components evaluated by the LCA procedure (Fig. S7a and b†). The results (Table S1†) show that for both LaNiO3 and Ni/La2O3 catalysts metallic and oxidized Ni particles possess bulk-like coordination numbers (CNs) obtained for average particle dimension larger than 3 nm. This finding is complementary to the TEM/EDX analysis since particles smaller than 3 nm fall within the detection limit of the TEM/EDX.
3.3. In situ spectroscopy of LaNiO3 during H2-activation treatment
The results of Fig. 1 showed a critical effect of the H2-activation temperature on the catalytic performance, while ex situ analysis reveals major transformations of the perovskite catalyst after the activation and methanation reaction. For a better assessment, the fresh LaNiO3 catalyst was studied during H2-activation by in situ XAFS and AP-XPS. Hard, tender and soft X-rays were employed in these measurements providing complementary information in a broad analysis depth range. | ||
| Fig. 5 (a) Ni K edge evolution from RT to 600 °C, (b) separate graph of references and spectra recorded under steady state conditions, (c) magnitude component of phase uncorrected FT-EXAFS and (d) phase evolution as a function of temperature according to LCF. Experimental EXAFS spectra together with LCA results on the whole thermal treatment and LCA best/worst fit are reported in Fig. S10.† | ||
LCA of the Ni K-edge by four reference line shapes was performed to quantify the evolution of the different Ni phases during LaNiO3 reduction. As displayed in Fig. 5d the fresh/calcined catalyst is a mixture of 82% LaNiO3, 12% La2NiO4 phases and 3% NiO. At 400 °C the LaNiO3 phase is converted to 33% Ni0–66% La2NiO4 mixture following reaction (1). Above this temperature La2NiO4 reduces fast to Ni0, most likely leading to the production of La2O3 by reaction (2). At 500 °C the LaNiO3 reduction is completed and at higher temperatures, the Ni K-edge shows no further changes.
| 2LaNiO3 + 2H2 → La2NiO4 + Ni + 2H2O (100–400 °C) | (1) |
| La2NiO4 + H2 → Ni + La2O3 + H2O (400–500 °C) | (2) |
The gradual decomposition of LaNiO3 into La2NiO4 and presumably to La2O3 in the final reduction step, along with the appearance of metallic Ni deduced by our XAFS results agrees with the findings of a recent TEM and XRD study of LaNiO3 treated under vacuum and inner gas (i.e. He).60 However, in our case the addition of H2 changes the process suggested in ref. 60 in two ways. The first is related to the NiO formation clearly evident under inert or vacuum conditions due to oxygen diffusion from the perovskite lattice towards exsolved Ni. In our case, NiO does not appear in the absorption spectra because, if it forms, it is quickly reduced by gas phase H2 to Ni0. The second difference is the temperature of the perovskite transformation. We found that the LaNiO3-phase in H2 decomposes between 200 and 500 °C, while under an inert atmosphere this window shifts to considerably higher temperatures (ca. 550–800 °C). Interestingly, a small quantity of NiO was observed above 500 °C which would imply La2NiO4 decomposition to La2O3 and NiO. Since at T > 500 °C under H2 the latter should be directly reduced to Ni, this spectral feature should be more reasonably ascribed to a not fully reduced Ni-oxidic phase.
Finally, comparison of the Ni K-edge LCA results between ex situ (Fig. 4b) and in situ (Fig. 5d) treated LaNiO3 catalyst shows many similarities but also some differences. In particular, both set of measurements agree on the decomposition of the LaNiO3-phase to La2NiO4 up to 400 °C and its full conversion to Ni0 at 600 °C. However, in situ measurements show relatively more La2NiO4 at 400 °C and considerably less NiO as compared to the ex situ results. In addition, ex situ FT-EXAFS analysis suggested Ni clusters with a bulk-like electronic structure while in situ results anticipated the CN to be lower than the bulk value. These differences can be rationalized by the milder reduction conditions applied in the in situ XAFS measurements (diluted H2 in the Microtomo cell instead of pure H2 in the quartz tube for the ex situ treatment). It is therefore possible that in situ spectra depict the formation of atomically dispersed Ni at the early stages of the exsolution process, before their agglomeration into nanoparticles with a bulk-like electronic structure shown in the ex situ results. Besides, the small amount of NiO during in situ measurements suggests that the NiO observed during ex situ analysis was the result of sample oxidation in air. Based on that, one can also speculate that the reason behind the differences in the relative NiO amount among the catalysts shown in Fig. 4b is due to differences in Ni particle size. The NiO upon exposure in air forms a passivation layer around Ni0 particles. The bigger the particles the lower the NiO contribution in the Ni K-edge (bulk sensitive method) since they have a lower surface to volume ratio. If this assumption is valid, one can conclude that LCA results of Fig. 4b indirectly show that the Ni particle size on our perovskite catalyst is in the order LaNiO3-400 < LaNiO3-600 < LaNiO3-sp. In addition, the size of Ni particles for Ni/La2O3 catalysts is in similar order to that of LaNiO3 after reduction, but probably becomes much bigger after the reaction.
 | ||
| Fig. 6 (a) In situ La 3d and Ni 2p AP-XPS spectra of the fresh/oxidized LaNiO3 catalyst (2 mbar O2 at 400 °C), H2-activated catalyst (2 mbar H2 at 400 °C and 600 °C) and Ni/La2O3 catalyst (2 mbar H2 at 600 °C), (b) depth-dependent La 3d and Ni 2p spectra of LaNiO3-600 (left panel) Ni/La2O3 catalysts (left panel) measured at 400 °C in 2 mbar H2 at three different photon energies. For the La 3d & Ni 2p curve fitting peak line-shapes derived from reference materials and synthetic line-shapes derived by a mathematical formula were used. For clarity only the La 3d5/2 and Ni 2p fitting components are shown. (c) Bar graph showing the %Ni0 atomic concentration of different samples calculated by the Ni 2p and La 3d peak areas (spectra measured at 400 °C in 2 mbar H2 with photons of hv = 1320 eV giving an estimated analysis depth of approx. 3.9 nm). (d) The evolution of %Ni0 as a function of the excitation energy for the two catalysts measured at 400 °C in 2 mbar H2. The information depth in each photon energy is estimated as 3 times the photoelectron inelastic mean free path. | ||
To quantify the evident differences in the curve shape, the Ni 2p and La 3d spectra in Fig. 6a were fitted by a linear combination of synthetic peaks and spectra of reference compounds (details are presented in Fig. S11†). The spectrum of the reduced Ni/La2O3 catalyst was fitted using the same approach and is included at the bottom of Fig. 6 for comparison. The curve fitting reveals the coexistence of 2 Ni and 2 La components. In particular, ionic Ni (likely Ni3+ of LaNiO3) dominates the calcined sample and upon H2 annealing it is gradually replaced by Ni0. Likewise, La from the perovskite lattice (La3+ of LaNiO3) is stepwise transformed into La3+ of La2O3 (vide infra). The %Ni, determined by the La 3d and Ni 2p areas (Fig. 6c), reveals a net enhancement of nickel surface concentration for the LaNiO3-600 sample. This ratio is about 4 times higher than that of Ni/La2O3, despite the fact that both catalysts have the same nominal Ni loading and were treated under identical conditions. The enhancement of Ni concentration at the surface of the perovskite catalyst is a direct consequence of nickel exsolution upon H2-activation. The greater surface Ni0 content of LaNiO3-600 can also be linked to its better CO2 conversion (Fig. 1).
To further elaborate these findings, photoemission spectra with varied photon energies, thus different analysis depths, were collected in order to distinguish surface from subsurface species. As shown in Fig. 6b and d, the Ni0 fraction relative to La3+ (La2O3) decreases in deeper analysis depths for LaNiO3. As it is clearly evident in the HAXPES spectrum for deeper analysis depths (ca. 12 nm), Ni3+ and La3+ components of LaNiO3 are required to properly fit the Ni 2p and La 3d regions. This shows that under the employed conditions, reduction is limited to the outer 2–3 nm and the unreduced perovskite structure is still preserved in the subsurface.
The inverse Ni and La arrangement is noticed for the supported Ni/La2O3 catalyst (Fig. 6b and d) with Ni0 fraction being low at the outer surface and increasing in deeper layers (up to 20 nm). This trend suggests total or partial Ni encapsulation by lanthana alluding strong metal support interaction (SMSI) phenomena.64,65 Assuming that the reactivity is proportional to the amount of accessible Ni sites on the surface, increase of Ni surface concentration in the case of LaNiO3 might explain its greater CO2 methanation activity when compared to Ni/La2O3.
3.4. In situ AP-XPS/HAXPES investigation under CO2 hydrogenation conditions
Ex situ TEM/EDX and XAFS analyses of the spent LaNiO3 catalyst do not show any considerable difference with the earlier activated state (Fig. 3 and 4), while XRD indicates that La2O2CO3 oxycarbonates are built after long term CO2 methanation tests (Fig. 2). The surface chemical state, under conditions relevant to CO2 methanation, was analyzed by AP-XPS. Fig. 7a compares the Ni 2p and La 3d spectra of LaNiO3-600 and Ni/La2O3 catalysts measured at 400 °C in 2 mbar CO2:H2 (1:1). The sharp feature at 855.6 eV suggests that Ni remains metallic in both catalysts similar to the finding in H2. In contrast, the La 3d5/2 peak under the reaction conditions is modified as compared to the prior state in H2 (Fig. S12†). A fitting attempt by the linear combination of La3+(LaNiO3) and La3+(La2O3) reference peak profiles failed to reproduce La 3d5/2, excluding the possibility that the La3+(LaNiO3) phase is partially restored during the reaction. The quality of the fitting (i.e., lower STD value) is considerably improved when a synthetic La 3d curve combining 2 pairs of main and satellite peaks is used. More details about this peak, hereafter abbreviated as s.La 3d, are given in Table S2.† The fraction of s.La 3d is more pronounced in LaNiO3 than Ni/La2O3 sample. The fit's optimal s.La 3d peak profile exhibited a somewhat different main to satellite peak BE splitting (abbreviated as ΔLa 3d) for the two catalysts. Specifically, ΔLa 3d for LaNiO3 (hereafter s.La 3d(LaNiO3)) was 3.6 eV while for Ni/La2O3 (hereafter s.La 3d(Ni/La2O3)) was 3.4 eV. The ΔLa 3d is very sensitive to the lanthanum chemical state and has been used to distinguish between various lanthanum compounds (Table 3). Accordingly, the ΔLa 3d of s.La 3d(LaNiO3) corresponds to hydroxide or carbonate lanthanum species, while s.La 3d(Ni/La2O3) primarily to carbonate (vide infra). Depth-dependent La 3d measurements (Fig. S13†) reveal possible differences in the location of syn.La 3d between the two catalysts. In particular, for the LaNiO3 catalyst the s.La 3d(LaNiO3) fraction is independent of the analysis depth suggesting homogeneous distribution within the outer 4 nm. In contrast, the s.La 3d(Ni/La2O3) fraction increases at the surface indicating its preferential location at the outermost surface.
 | ||
| Fig. 7 (a) In situ La 3d and Ni 2p AP-XPS spectra of LaNiO3 and Ni/La2O3 catalysts under CO2 methanation conditions (2 mbar CO2:H2 (1:1) at 400 °C). Prior to the AP-XPS measurements both catalysts were pre-activated in H2 at 600 °C. Peak line-shapes derived from reference materials and synthetic line-shapes derived by a mathematical formula are used to fit the La 3d & Ni 2p curves. For clarity only the La 3d5/2 and Ni 2p fitting components are shown. The corresponding (b) O 1s spectra fitted by three O 1s symmetric peaks and (c) C 1s spectra fitted with asymmetric and symmetric peaks. The estimated information depth (three times the photoelectron inelastic mean free path) is about 4 for La 3d & Ni 2p and O 1s spectra, and 2 nm for C 1s. | ||
| Name | ΔLa 3d (STD) (eV) | Reference |
|---|---|---|
| Lanthanum oxide (lanthana) La2O3 | 4.6(±0.3) | 63 and 66–68 |
| Lanthanum oxycarbonate La2O2CO3 | 4.3(±0.2) | 66, 68 and 69 |
| Lanthanum hydroxide La(OH)3 | 3.7(±0.3) | 63, 66–68, 70 and 71 |
| Lanthanum carbonate La2(CO3)3 | 3.5(±0.1) | 70–72 |
| LaNiO3 | 3.8 | This work |
| La2O3 | 4.7 | This work |
| s.La 3d(LaNiO3) | 3.6 | This work |
| s.La 3d(Ni/La2O3) | 3.4 | This work |
The O 1s and C 1s peaks corroborate the proposed arrangement and provide further details on the chemical nature of the species behind the s.La 3d peaks. The O 1s spectra shown in Fig. 7b are fitted with three O 1s components with BE at 529.2, 530.7 and 532.2 eV (±0.2 eV). The peak at 529.2 eV is typical of La2O3 lattice oxygen (abbreviated as Olat),63,68 while the broad peak at 532.2 eV is probably the convolution of OH− groups (OH) and carbonates (CO32−) both appearing within this energy region.68 The one at 530.7 eV is more difficult to assign since its BE is relatively low for OH and high for Olat species. Its presence is also clearly manifested in the spectrum measured in an H2 atmosphere (Fig. S14†). Based on the evolution of the three oxygen species observed in the depth-dependent O 1s measurements (Fig. S15†) their distribution within the outer layers of the catalyst can be determined. Accordingly, the peak at 529.2 eV is located subsurface, while the peak at 532.2 eV is predominantly on the surface. Additionally, the peak at 530.7 eV seems to be distributed rather homogeneously with the analysis depth. The analysis of the Ni/La2O3 catalyst gives very similar results, the only difference is that the fraction of Olat on this sample is relatively less.
The C 1s spectrum of LaNiO3 (Fig. 7c) is composed by a main C 1s peak at 284.4 eV typical of C–C and C–H bonds (for simplicity noted as C–C hereafter) and two smaller peaks at 287.3 eV and 289.8 eV assigned to C–O or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (C–O hereafter) and carbonate (CO32−) species, respectively.37,63,73 The lack of peaks around 283 eV excludes the presence of Ni-carbides.37 The C–C peak was present during the H2 reduction step (Fig. S14†), therefore it is attributed to residual carbon preexisting under the reaction conditions. Depth-dependent C 1s spectra (Fig. S16a†) suggest that CO32− is in contact with lanthana beneath a C–C layer. The CO32−/s.La 3d(LaNiO3) stoichiometry calculated from the corresponding photoemission peaks was 0.1, far below any known La carbonate species. Since extensive La carbonate formation is excluded, La(OH)3 species formation remains the most credible scenario for the origin of s.La 3d(LaNiO3). The s.La 3d(LaNiO3) value of 3.6 eV discussed above further supports this argument. The evident reason for the formation of La(OH)3 under the reaction conditions and not in H2, is the presence of H2O which is produced by the methanation reaction.
O (C–O hereafter) and carbonate (CO32−) species, respectively.37,63,73 The lack of peaks around 283 eV excludes the presence of Ni-carbides.37 The C–C peak was present during the H2 reduction step (Fig. S14†), therefore it is attributed to residual carbon preexisting under the reaction conditions. Depth-dependent C 1s spectra (Fig. S16a†) suggest that CO32− is in contact with lanthana beneath a C–C layer. The CO32−/s.La 3d(LaNiO3) stoichiometry calculated from the corresponding photoemission peaks was 0.1, far below any known La carbonate species. Since extensive La carbonate formation is excluded, La(OH)3 species formation remains the most credible scenario for the origin of s.La 3d(LaNiO3). The s.La 3d(LaNiO3) value of 3.6 eV discussed above further supports this argument. The evident reason for the formation of La(OH)3 under the reaction conditions and not in H2, is the presence of H2O which is produced by the methanation reaction.
The C 1s of Ni/La2O3 catalyst is dominated by the CO32− peak at 290.1 eV, while the signal from other components falls to the background level. Depth-dependent C 1s measurements (Fig. S16b†) suggest that CO32− is mainly located at the outer surface, which differs from LaNiO3. The CO32−/s.La 3d(Ni/La2O3) stoichiometry calculated by XPS is around 1.6, in fair agreement with the La2(CO3)3 stoichiometry. The above two experimental findings, in addition to ΔLa 3d = 3.4, are sound evidence for La2(CO3)3 formation on Ni/La2O3 in sharp contrast with La(OH)3 found for LaNiO3.
To summarize, operando NAP-XPS showed that under the reaction conditions Ni is maintained in the metallic state, while new types of lanthanum species are formed. Analysis of the surface sensitive spectroscopic results suggest that the perovskite LaNiO3 catalyst turns into La-hydroxide (probably La(OH)3) while La2(CO3)3 is the main species on supported Ni/La2O3. The presence of oxycarbonates (i.e., La2O2CO3) on Ni/La2O3 is unlikely since it is incompatible with both the s.La 3d(Ni/La2O3) and the expected compound stoichiometry (0.5 rather than 1.6).
4. Discussion
Catalytic tests of perovskite and supported Ni–La2O3 catalysts show that the CO2 methanation performance is critically affected by the starting material. This might seem surprising since both catalysts have similar Ni loading and surface area, and after activation in H2 they also show largely similar XRD patterns. One explanation of the superior perovskite-derived Ni–La2O3 catalyst performance frequently suggested in the literature26,31,33 is the creation of highly dispersed nanoparticles socketing into the oxide matrix. Actually, this morphology was also identified in our exsolved perovskite catalyst by TEM/EDX showing the formation of 6 nm Ni particles that resist sintering under the reaction conditions. Although particle size fixation may be a crucial factor in explaining the exsolved LaNiO3 catalyst's long-term stability (Fig. 1b), thorough in situ characterization points to significant differences in the surface chemistry of perovskite and supported catalysts that may be responsible for their distinct performances.In particular, AP-XPS shows that the amount of Ni exposed to the surface might be up to 4 times higher on LaNiO3 than Ni/La2O3 (Fig. 6c). Although this can be partly explained by the dispersion of Ni in the two catalysts, depth-dependent AP-XPS and HAXPES measurements revealed an additional clue, the encapsulation of Ni by lanthana, on the line of the well-known SMSI effect. Indeed, the two catalysts have a very different profile of Ni distribution in the outer 10–20 nm (Fig. 6d). This was found during the H2-activation step but it is also conserved under the reaction conditions as illustrated in Fig. 8. A similar phenomenon has already been observed when Ni/CeO2 is exposed under reducing conditions and attributed to Ce2O3 migration of the reduced oxide to the metal surface.74 In another example, closer to the materials investigated in this study, a different mechanism was given to explain the covering of Rh particles by La2O3.75,76 As lanthana is much less prone to reduction, a combination of sintering and phase transformation of the support was proposed to explain the encapsulation of Rh during sample preparation, clearly observed by HRTEM measurements. Although we cannot exclude partial lanthana reduction at the extreme surface of Ni/La2O3, if it exists it should be very local according to La 3d peak analysis.
 | ||
| Fig. 8 Illustration of the proposed surface arrangement of LaNiO3 and Ni/La2O3 catalysts under calcination, activation and CO2 methanation (initial and steady state reaction stages) conditions. The proposed surface reactions when the catalysts pass from one condition to the next are shown in boxes. | ||
However, the configuration of Ni and lanthana at the outer surface is most probably not the only difference between the two catalysts that contributes to their distinct catalytic performance. By careful analysis of photoemission spectra we identify also crucial differences in their surface chemistry. As displayed in Fig. 8, La(OH)3 species can be easily formed at the early reaction stage on the surface of the exsolved LaNiO3 catalyst. In contrast, on supported Ni/La2O3 a thin La2(CO3)3 layer is evidenced. The latter, being quite stable and inert towards CO2, can block surface sites with a detrimental effect on the catalytic performance.
It now remains to correlate AP-XPS finding with ex situ XRD of the spent catalysts which at a first glance seems quite contradictory. According to AP-XPS, La(OH)3 species are formed on the LaNiO3 catalyst under the reaction conditions, but XRD of the spent LaNiO3 show crystalline II-La2O2CO3 and not La(OH)3 at all. This might seem even more confusing when one considers results on Ni/La2O3 where XRD did not show any additional species after reaction (neither carbonates nor oxycarbonates) despite the fact that the presence of La2(CO3)3 was clearly evident in AP-XPS. To explain this, we have to recall first the difference in catalytic reaction conditions between the two experiments. XRD is recorded after 65 hours of reaction at 1 bar, while AP-XPS during 1 hour reaction at 2 mbar total pressure. Evidently, one can anticipate that due to considerably “milder” reaction conditions AP-XPS data represent the initial stages of the catalyst transformation while ex situ XRD gives the steady state picture of the catalyst (see Fig. 8). Nevertheless, despite its limitation, AP-XPS remains the best available option to understand the surface chemistry since any ex situ XPS analysis will be dubious considering the affinity of lanthana to form carbonates and hydroxides upon exposure to air.
To explain the XRD finding we should extrapolate the AP-XPS data toward the long-term steady state. Indeed, the La(OH)3 species might function as a precursor for La2O2CO3. It has been shown that La(OH)3 can be converted to La2O2CO3 even after 2 h annealing at 400 °C in air (CO2 < 0.04% or 0.4 mbar).77,78 The initial stage of La2O2CO3 formation is most probably already observed in the C 1s spectra of LaNiO3 catalyst under the reaction conditions, showing a C 1s peak around 290 eV (Fig. 7c). One can assume that at significantly higher CO2 concentrations, like in the 1 bar reaction mixture, the La2O2CO3 formation rate is enhanced giving rise to the formation of the bulk crystalline La2O2CO3 observed in the XRD patterns of Fig. 2a.
The above analysis can also be used to explain the poor catalytic activity of Ni/La2O3. In this case La(OH)3, the La2O2CO3 precursor, is not readily formed therefore even after hours of reaction oxycarbonates are not observed by XRD. Of course, carbonates may be generated directly by carbonizing La2O3 in CO2 gas, but this requires significantly higher temperatures (e.g. 850 °C) than the methanation reaction conditions (350–400 °C).53 This might suggest that the active state of the exsolved LaNiO3 perovskite catalyst under reforming and methanation conditions could be quite different.
At this stage it is unclear whether the lower CO2 conversion of Ni/La2O3 and the resulting lower production of H2O is due to the absence of La(OH)3, or if it is an intrinsic characteristic related to the La2O3 surface microstructure. As shown earlier, the surface termination plays an important role in lanthana's reactivity with H2O,79 suggesting that the surface structure of La2O3 is critical in determining whether La(OH)3 forms or not.
The oxycarbonate phases seen in the spent NiLaO3 catalyst have already been observed before on lanthana-based catalysts but were correlated with catalyst deactivation.80–82 Recently, the presence of La2O2CO3 has been claimed to benefit methanation acting as a CO2 reservoir available for hydrogenation.83 Our results agree with the latter assessment since the most active and stable sample contained oxycarbonate crystalline phases whereas Ni/La2O3 did not. However, the influence of morphological parameters (crystallite size, surface area, Ni dispersion) that could possibly have a role in the reactivity should not be overlooked.84
5. Conclusions
LaNiO3 perovskite was synthesized by the Pechini sol gel method and tested in CO2 methanation after activation by a reductive treatment. The catalytic tests show a net enhancement of the CO2 conversion with reduction temperature, and superior catalytic performance and stability compared to a Ni/La2O3 benchmark catalyst synthesized by conventional wet impregnation. The transformation of the LaNiO3 precursor into the active phase during the thermal reduction treatment was monitored by multiple ex situ and in situ techniques. Electron microscopy displays the formation of 6 nm Ni particles with narrow size distribution anchored into the La2O3 matrix, while in situ XAFS detects the intermediate phases of this transition. Although the XRD crystal structures of reduced, exsolved and supported Ni/La2O3 seem identical, combined surface/subsurface analysis by in situ AP-XPS/HAXPES identified critical differences in the arrangement between La and Ni. Furthermore, it is shown that the active phase is formed during the catalytic cycle and it is quite different from the H2-activated state. Specifically, although the Ni particle size and oxidation state are maintained, lanthana reacts with the gas phase during the reaction to form new compounds. This information was used to rationalize the differences in the catalytic performance. Notably, encapsulation of Ni by lanthana and the affinity of the latter towards hydroxide or carbonate species formation are proposed to critically affect the catalytic performance. We present strong evidence that the nature of carbon species formed under the reaction conditions is a crucial element of the reactivity, which is promoted by oxycarbonates but suppressed by carbonates. This work highlights the distinct surface chemistry of metal oxide catalysts derived from perovskite precursors in comparison to conventional supported catalysts and extends our understanding on their unique catalytic properties. Differences in the surface chemistry are usually disregarded in the relevant literature, where the high activity and stability of exsolved perovskite catalysts in thermo- or electrochemical reactions are primarily attributed to the socketing of Ni nanoparticles.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. B. and S. Z. acknowledge the support from Strasbourg University and the French Agence Nationale de la Recherche (ANR), under the projects IdEX-2018 (Post-doctorants), DuCaCO2 (No. 236294). J. Z. would like to thank the China Scholarship Council (CSC) for supporting his stay at ICPEES. ESRF is acknowledged for the allocation of synchrotron radiation beamtime and BM23 beamline staff for the collaboration during the experiments. We acknowledge HZB for the allocation of synchrotron radiation beamtime (proposal no 222-11356-ST) and BESSY Synchrotron staff for the collaboration during the experiments. The authors acknowledge ESRF for beamtime allocation together with Dr K. Lomachenko for support during XAS measurements. A. Molokova is acknowledged for support with development of XAS analysis using Python scripts. Dr A. Martini is acknowledged for providing the EXAFS Wavelet Transform analysis code employed in this work. EB acknowledges support from Project no. 2017KKP5Z PRIN-2017 MOSCATo and Project CH4.0 under the MUR program “Dipartimenti di Eccellenza 2023-2027” (CUP: D13C22003520001). Finally, we thank X. Thanh Nguyen for his considerable contribution to the experiments during his master thesis at ICPEES and Dr S. Labidi for the support with the catalytic measurements.References
- Z. Liu, Z. Deng, S. J. Davis, C. Giron and P. Ciais, Nat. Rev. Earth Environ., 2022, 3, 217–219 CrossRef PubMed.
- International Energy and Agency, 2022, https://iea.blob.core.windows.net/assets/c3086240-732b-4f6a-89d7-db01be018f5e/GlobalEnergyReviewCO2Emissionsin2021.pdf, https://www.iea.org/reports/global-energy-review-c, Accessed April 22th, 2023.
- H. S. Whang, J. Lim, M. S. Choi, J. Lee and H. Lee, BMC Chem. Eng., 2019, 1, 9 CrossRef.
- J. Ashok, S. Pati, P. Hongmanorom, Z. Tianxi, C. Junmei and S. Kawi, Catal. Today, 2020, 356, 471–489 CrossRef CAS.
- X. Su, J. Xu, B. Liang, H. Duan, B. Hou and Y. Huang, J. Energy Chem., 2016, 25, 553–565 CrossRef.
- C. Vogt, M. Monai, G. J. Kramer and B. M. Weckhuysen, Nat. Catal., 2019, 23(2), 188–197 CrossRef.
- P. A. U. Aldana, F. Ocampo, K. Kobl, B. Louis, F. Thibault-Starzyk, M. Daturi, P. Bazin, S. Thomas and A. C. Roger, Catal. Today, 2013, 215, 201–207 CrossRef CAS.
- A. Cárdenas-Arenas, A. Quindimil, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos, J. R. González-Velasco and A. Bueno-López, Appl. Catal., B, 2020, 265, 118538 CrossRef.
- R.-P. Ye, Q. Li, W. Gong, T. Wang, J. J. Razink, L. Lin, Y.-Y. Qin, Z. Zhou, H. Adidharma, J. Tang, A. G. Russell, M. Fan and Y.-G. Yao, Appl. Catal., B, 2020, 268, 118474 CrossRef CAS.
- W. Li, Y. Liu, M. Mu, F. Ding, Z. Liu, X. Guo and C. Song, Appl. Catal., B, 2019, 254, 531–540 CrossRef CAS.
- H. Liu, S. Xu, G. Zhou, K. Xiong, Z. Jiao and S. Wang, Fuel, 2018, 217, 570–576 CrossRef CAS.
- G. Zhou, T. Wu, H. Zhang, H. Xie and Y. Feng, Chem. Eng. Commun., 2014, 201, 233–240 CrossRef CAS.
- F. Wang, S. He, H. Chen, B. Wang, L. Zheng, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2016, 138, 6298–6305 CrossRef CAS PubMed.
- A. Aitbekova, L. Wu, C. J. Wrasman, A. Boubnov, A. S. Hoffman, E. D. Goodman, S. R. Bare and M. Cargnello, J. Am. Chem. Soc., 2018, 140, 13736–13745 CrossRef CAS PubMed.
- F. Feng, G. Song, J. Xiao, L. Shen and S. V. Pisupati, Fuel, 2019, 235, 85–91 CrossRef CAS.
- S. Rönsch, J. Schneider, S. Matthischke, M. Schlüter, M. Götz, J. Lefebvre, P. Prabhakaran and S. Bajohr, Fuel, 2016, 166, 276–296 CrossRef.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- G. Varvoutis, M. Lykaki, S. Stefa, V. Binas, G. E. Marnellos and M. Konsolakis, Appl. Catal., B, 2021, 297, 120401 CrossRef CAS.
- L. Shen, J. Xu, M. Zhu and Y. F. Han, ACS Catal., 2020, 10, 14581–14591 CrossRef CAS.
- C. Vogt, E. Groeneveld, G. Kamsma, M. Nachtegaal, L. Lu, C. J. Kiely, P. H. Berben, F. Meirer and B. M. Weckhuysen, Nat. Catal., 2018, 1, 127–134 CrossRef CAS.
- K. Yoshikawa, M. Kaneeda and H. Nakamura, Energy Procedia, 2017, 114, 2481–2487 CrossRef CAS.
- G. I. Siakavelas, N. D. Charisiou, A. AlKhoori, S. AlKhoori, V. Sebastian, S. J. Hinder, M. A. Baker, I. V. Yentekakis, K. Polychronopoulou and M. A. Goula, J. CO2 Util., 2021, 51, 101618 CrossRef CAS.
- P. H. Ho, G. S. de Luna, S. Angelucci, A. Canciani, W. Jones, D. Decarolis, F. Ospitali, E. R. Aguado, E. Rodríguez-Castellón, G. Fornasari, A. Vaccari, A. M. Beale and P. Benito, Appl. Catal., B, 2020, 278, 119256 CrossRef CAS.
- G. Tang, D. Gong, H. Liu and L. Wang, Catalysts, 2019, 9, 442 CrossRef CAS.
- V. Alcalde-Santiago, A. Davó-Quiñonero, D. Lozano-Castelló, A. Quindimil, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos, J. R. González-Velasco and A. Bueno-López, ChemCatChem, 2019, 11, 810–819 CrossRef CAS.
- K. Kousi, C. Tang, I. S. Metcalfe, D. Neagu, K. Kousi, S. Metcalfe, C. Tang and D. Neagu, Small, 2021, 17, 2006479 CrossRef CAS PubMed.
- Q. Yang, G. Liu and Y. Liu, Ind. Eng. Chem. Res., 2018, 57, 1–17 CrossRef CAS.
- K. Kousi, D. Neagu, L. Bekris, E. Calì, G. Kerherve, E. I. Papaioannou, D. J. Payne and I. S. Metcalfe, J. Mater. Chem. A, 2020, 8, 12406–12417 RSC.
- J. Zhang, M. R. Gao and J. L. Luo, Chem. Mater., 2020, 32, 5424–5441 CrossRef CAS.
- S. Bhattar, M. A. Abedin, S. Kanitkar and J. J. Spivey, Catal. Today, 2021, 365, 2–23 CrossRef CAS.
- J. Wu, R. Ye, D. J. Xu, L. Wan, T. R. Reina, H. Sun, Y. Ni, Z. F. Zhou and X. Deng, Front. Chem., 2022, 10, 961355 CrossRef CAS PubMed.
- O. Kwon, S. Joo, S. Choi, S. Sengodan and G. Kim, JPhys Energy, 2020, 2, 032001 CrossRef CAS.
- M. A. Salaev, L. F. Liotta and O. V. Vodyankina, Int. J. Hydrogen Energy, 2022, 47, 4489–4535 CrossRef.
- X. Bao, M. Behrens, G. Ertl, Q. Fu, A. Knop-Gericke, T. Lunkenbein, M. Muhler, C. M. Schmidt and A. Trunschke, ACS Catal., 2021, 11, 6243–6260 CrossRef CAS.
- H. S. Lim, G. Kim, Y. Kim, M. Lee, D. Kang, H. Lee and J. W. Lee, Chem. Eng. J., 2021, 412, 127557 CrossRef CAS.
- P. Steiger, O. Kröcher and D. Ferri, Appl. Catal., A, 2020, 590, 117328 CrossRef CAS.
- A. P. Ramon, X. Li, A. H. Clark, O. V. Safonova, F. C. Marcos, E. M. Assaf, J. A. van Bokhoven, L. Artiglia and J. M. Assaf, Appl. Catal., B, 2022, 315, 121528 CrossRef CAS.
- D. D. Athayde, D. F. Souza, A. M. A. Silva, D. Vasconcelos, E. H. M. Nunes, J. C. D. Da Costa and W. L. Vasconcelos, Ceram. Int., 2016, 42, 6555–6571 CrossRef CAS.
- G. Wu, S. Li, C. Zhang, T. Wang and J. Gong, Appl. Catal., B, 2014, 144, 277–285 CrossRef CAS.
- Y. Gao, D. Chen, M. Saccoccio, Z. Lu and F. Ciucci, Nano Energy, 2016, 27, 499–508 CrossRef CAS.
- W. Smekal, W. S. M. Werner and C. J. Powell, Surf. Interface Anal., 2005, 37, 1059–1067 CrossRef CAS.
- G. Greczynski and L. Hultman, Angew. Chem., Int. Ed., 2020, 59, 5002–5006 CrossRef CAS PubMed.
- S. Turczyniak, D. Teschner, A. Machocki and S. Zafeiratos, J. Catal., 2016, 340, 321–330 CrossRef CAS.
- F. Polack, M. Silly, C. Chauvet, B. Lagarde, N. Bergeard, M. Izquierdo, O. Chubar, D. Krizmancic, M. Ribbens, J. -P. Duval, C. Basset, S. Kubsky and F. Sirotti, AIP Conf. Proc., 2010, 1234, 185 CrossRef CAS.
- R. Follath, M. Hävecker, G. Reichardt, K. Lips, J. Bahrdt, F. Schäfers and P. Schmid, J. Phys.: Conf. Ser., 2013, 212003 CrossRef.
- S. Hendel, F. Schäfers, M. Hävecker, G. Reichardt, M. Scheer, J. Bahrdt and K. Lips, AIP Conf. Proc., 2016, 1741, 030038 CrossRef.
- O. Mathon, A. Beteva, J. Borrel, D. Bugnazet, S. Gatla, R. Hino, I. Kantor, T. Mairs, M. Munoz, S. Pasternak, F. Perrin and S. Pascarelli, J. Synchrotron Radiat., 2015, 22, 1548–1554 CrossRef CAS PubMed.
- D. Bellet, B. Gorges, A. Dallery, P. Bernard, E. Pereiro and J. Baruchel, J. Appl. Crystallogr., 2003, 36, 366–367 CrossRef CAS.
- M. Newville, J. Phys.: Conf. Ser., 2013, 430, 012007 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. Martini, I. A. Pankin, A. Marsicano, K. A. Lomachenko and E. Borfecchia, Radiat. Phys. Chem., 2020, 175, 108333 CrossRef CAS.
- A. Quindimil, J. A. Onrubia-Calvo, A. Davó-Quiñonero, A. Bermejo-López, E. Bailón-García, B. Pereda-Ayo, D. Lozano-Castelló, J. A. González-Marcos, A. Bueno-López and J. R. González-Velasco, J. CO2 Util., 2022, 57, 101888 CrossRef CAS.
- A. Olafsen, A. K. Larsson, H. Fjellvåg and B. C. Hauback, J. Solid State Chem., 2001, 158, 14–24 CrossRef CAS.
- X. Li, Z. J. Zhao, L. Zeng, J. Zhao, H. Tian, S. Chen, K. Li, S. Sang and J. Gong, Chem. Sci., 2018, 9, 3426–3437 RSC.
- H. Provendier, C. Petit, C. Estournès, S. Libs and A. Kiennemann, Appl. Catal., A, 1999, 180, 163–173 CrossRef CAS.
- H. Hojo, Y. Inohara, R. Ichitsubo and H. Einaga, Catal. Today, 2023, 410, 127–134 CrossRef CAS.
- S. Ramesh and N. J. Venkatesha, ACS Sustain. Chem. Eng., 2017, 5, 1339–1346 CrossRef CAS.
- S. Singh, D. Zubenko and B. A. Rosen, ACS Catal., 2016, 6, 4199–4205 CrossRef CAS.
- W. Nabgan, T. A. T. Abdullah, R. Mat, B. Nabgan, A. A. Jalil, L. Firmansyah and S. Triwahyono, Int. J. Hydrogen Energy, 2017, 42, 8975–8985 CrossRef CAS.
- P. Cao, P. Tang, M. F. Bekheet, H. Du, L. Yang, L. Haug, A. Gili, B. Bischoff, A. Gurlo, M. Kunz, R. E. Dunin-Borkowski, S. Penner and M. Heggen, J. Phys. Chem. C, 2022, 126, 786–796 CrossRef CAS PubMed.
- G. Agostini, A. Piovano, L. Bertinetti, R. Pellegrini, G. Leofanti, E. Groppo and C. Lamberti, J. Phys. Chem. C, 2014, 118, 4085–4094 CrossRef CAS.
- D. Salusso, E. Borfecchia and S. Bordiga, J. Phys. Chem. C, 2021, 125, 22249–22261 CrossRef CAS.
- M. F. Sunding, K. Hadidi, S. Diplas, O. M. Løvvik, T. E. Norby and A. E. Gunnæs, J. Electron Spectrosc. Relat. Phenom., 2011, 184, 399–400 CrossRef CAS.
- P. Petzoldt, M. Eder, S. Mackewicz, M. Blum, T. Kratky, S. Günther, M. Tschurl, U. Heiz and B. A. J. Lechner, J. Phys. Chem. C, 2022, 126, 16127–16139 CrossRef CAS.
- C. J. Pan, M. C. Tsai, W. N. Su, J. Rick, N. G. Akalework, A. K. Agegnehu, S. Y. Cheng and B. J. Hwang, J. Taiwan Inst. Chem. Eng., 2017, 74, 154–186 CrossRef CAS.
- Y. Pang, X. Zhou, E. I. Vovk, C. Guan, S. Li, A. P. van Bavel and Y. Yang, Appl. Surf. Sci., 2021, 548, 149214 CrossRef CAS.
- S. Mickevičius, S. Grebinskij, V. Bondarenka, B. Vengalis, K. Šliužiene, B. A. Orlowski, V. Osinniy and W. Drube, J. Alloys Compd., 2006, 423, 107–111 CrossRef.
- J. P. H. Li, X. Zhou, Y. Pang, L. Zhu, E. I. Vovk, L. Cong, A. P. Van Bavel, S. Li and Y. Yang, Phys. Chem. Chem. Phys., 2019, 21, 22351–22358 RSC.
- W. Gu, J. Liu, M. Hu, F. Wang and Y. Song, ACS Appl. Mater. Interfaces, 2015, 7, 26914–26922 CrossRef CAS PubMed.
- D. R. Inns, X. Pei, Z. Zhou, D. J. M. Irving and S. A. Kondrat, Mater. Today Chem., 2022, 26, 101230 CrossRef CAS.
- D. R. Inns, A. J. Mayer, V. Skukauskas, T. E. Davies, J. Callison and S. A. Kondrat, Top. Catal., 2021, 64, 992–1009 CrossRef CAS.
- X. Zhou, Y. Pang, Z. Liu, E. I. Vovk, A. P. van Bavel, S. Li and Y. Yang, J. Energy Chem., 2021, 60, 649–659 CrossRef CAS.
- L. Zhong, M. Barreau, V. Caps, V. Papaefthimiou, M. Haevecker, D. Teschner, W. Baaziz, E. Borfecchia, L. Braglia and S. Zafeiratos, ACS Catal., 2021, 11, 5369–5385 CrossRef CAS.
- M. Li and A. C. van Veen, Appl. Catal., B, 2018, 237, 641–648 CrossRef CAS.
- S. Bernal, J. J. Calvino, M. A. Cauqui, J. M. Gatica, C. López Cartes, J. A. Pérez Omil and J. M. Pintado, Catal. Today, 2003, 77, 385–406 CrossRef CAS.
- S. Bernal, J. J. Calvino, C. López-Cartes, J. M. Pintado, J. A. Pérez-Omil, J. M. Rodríguez-Izquierdo, K. Hayek and G. Rupprechter, Catal. Today, 1999, 52, 29–43 CrossRef CAS.
- G. Chen, B. Han, S. Deng, Y. Wang and Y. Wang, Electrochim. Acta, 2014, 127, 355–361 CrossRef CAS.
- D. Ding, W. Lu, Y. Xiong, X. Pan, J. Zhang, C. Ling, Y. Du and Q. Xue, Appl. Surf. Sci., 2017, 426, 725–733 CrossRef CAS.
- K. A. Stoerzinger, R. Comes, S. R. Spurgeon, S. Thevuthasan, K. Ihm, E. J. Crumlin and S. A. Chambers, J. Phys. Chem. Lett., 2017, 8, 1038–1043 CrossRef CAS PubMed.
- H. Chen, H. Yu, F. Peng, H. Wang, J. Yang and M. Pan, J. Catal., 2010, 269, 281–290 CrossRef CAS.
- L. M. Cornaglia, J. Múnera, S. Irusta and E. A. Lombardo, Appl. Catal., A, 2004, 263, 91–101 CrossRef CAS.
- X. E. Verykios, Int. J. Hydrogen Energy, 2003, 28, 1045–1063 CAS.
- Y. Dai, M. Xu, Q. Wang, R. Huang, Y. Jin, B. Bian, C. Tumurbaatar, B. Ishtsog, T. Bold and Y. Yang, Appl. Catal., B, 2020, 277, 119271 CrossRef CAS.
- R. P. Turcotte, J. O. Sawyer and L. Eyring, Inorg. Chem., 1969, 8, 238–246 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta07391b |
| This journal is © The Royal Society of Chemistry 2024 |