
Interfacial chemistry regulation using functional frameworks for stable metal batteries
Yaohui
Huang
 a,
Jiarun
Geng
a,
Tong
Zhang
a,
Zhuoliang
Jiang
a,
Hengyi
Fang
a,
Wei
Hu
a and
Fujun
Li
*ab
a,
Jiarun
Geng
a,
Tong
Zhang
a,
Zhuoliang
Jiang
a,
Hengyi
Fang
a,
Wei
Hu
a and
Fujun
Li
*ab
aState Key Laboratory of Advanced Chemical Power Sources, Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: fujunli@nankai.edu.cn
bHaihe Laboratory of Sustainable Chemical Transformations, Tianjin 300192, China
First published on 27th January 2024
Abstract
Rechargeable metal batteries (RMBs) stand out as an attractive energy storage technique owing to their high theoretical energy density. However, their unstable electrode–electrolyte interface, resulting from parasitic reactions between electrolytes and active metal anodes (Li/Na/Zn), leads to safety concerns and performance decay in RMBs. Constructing functional frameworks on metal anodes has been demonstrated for achieving stable interfacial chemistry. The frameworks can regulate cation desolvation and substrate metallic affinity to provide sufficient ion flux and abundant nucleation sites, thus realizing uniform metal deposition and dendrite suppression. This review focuses on material engineering in functional frameworks to improve reversible interfacial reactions. Furthermore, porous crystalline frameworks (PCFs), including metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and zeolites, are considered to tailor the solvation sheath and accelerate cation desolvation. Three-dimensional inorganic frameworks (IOFs), such as metal-based and carbon-based materials, are introduced to enhance ionic diffusion and metal nucleation for enhanced metal plating. Additionally, an outlook on the design strategies and challenges in the development of framework materials is provided for the future development of practical RMBs.
 Yaohui Huang | Yaohui Huang started his successive postgraduate and doctoral program at the College of Chemistry at Nankai University in 2020. He is currently a PhD candidate studying physical chemistry under the supervision of Prof. Fujun Li. His research interests are focused on electrolyte and interfacial chemistry in Li metal batteries and Na/Li-ion batteries. |
 Fujun Li | Fujun Li is a professor at State Key Laboratory of Advanced Chemical Power Sources, College of Chemistry, Nankai University. He obtained his PhD from the University of Hong Kong in 2011 and then worked as a postdoctoral fellow at the University of Tokyo and National Institute of Advanced Industrial Science and Technology (AIST, Tsukuba), Japan, till 2015. His research interests include energy materials chemistry, metal-air batteries, and Na/Li-ion batteries. |
1. Introduction
Rechargeable metal batteries (e.g., Li, Na, and Zn) are considered promising alternatives to commercial ion batteries owing to their high energy density.1–4 Metal anodes have low redox potential and high capacity (e.g., Li, 3860 mA h g−1) compared to traditional carbon-based anodes (e.g., graphite, 372 mA h g−1).5–7 However, reactive metal anodes suffer from chemical and electrochemical corrosion from electrolytes, which leads to irreversible metal plating/stripping on anodes.8,9 For example, hydrogen evolution reactions (HERs) usually occur in aqueous Zn metal batteries (ZMBs), increasing local electrolyte pH and internal pressure in sealed devices.10 Zn anodes in zinc sulfate (ZnSO4) and zinc triflate (Zn(OTf)2) electrolytes react with generated OH− to form basic zinc sulfates (Zn4(OH)6SO4·xH2O), which passivate the anodes.11 In lithium metal batteries (LMBs), organic solvents are prone to reduction by Li anodes, which further induces the formation of undesirable organic-rich solid electrolyte interphase (SEI).12–14 These parasitic reactions give rise to uneven ion flux and unstable interface, resulting in low coulombic efficiency, severe dendrite growth, and short cycle life.15–18 Therefore, suppressing side reactions and regulating reversible metal plating/stripping by constructing stable interfaces in practical RMBs is urgent.Electrolyte engineering and functional framework design have been demonstrated to effectively regulate interfacial chemistry.19–21 The main challenge in electrolyte regulation is modifying solvation structures for cation desolvation and SEI formation on electrode–electrolyte interface.22–24 However, this strategy is limited to the protection of the top surface of electrodes, especially at high current densities. The repeated expansion and contraction of metal anodes during charging/discharging break SEI integrity and lead to the continuous consumption of electrolytes.25–27 In addition, the original protective film in electrolytes typically has low mechanical strength, and it cannot accommodate lithium deposition or suppress metal dendrite growth.28–30 Compared to electrolyte engineering, specific artificial framework construction is more advantageous to inhibit side reactions in electrolytes. Sufficient ion flux and active nucleation sites are two critical factors for uniform metal deposition and stable interface.31–34 Artificial framework constructions specifically focus on designing protective surface layers and bulk internal structures.35–37 Notably, protective surface layers are closely related to cation desolvation and ionic diffusion, and bulk internal structures are associated with electron transport and metal nucleation. The designed functional frameworks can reduce the concentration polarization and nucleation barrier on anodes to suppress dendrite growth and improve metal plating/stripping.38–41
Porous crystalline frameworks (PCFs) material, including metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and zeolites, have been employed as ion conductors and protective layers for metal batteries.42–45 These porous host materials are famous for regular pore channels, large surface area, and uniform loading of active substances. Taking advantage of the PCFs topological variety, their tunable pores and channels are customized for ion diffusion, which enhances ion selectivity and improves metal deposition.46–48 The physical confinement of PCFs enables high density of charges in the channels and improves the cation-hopping mechanism for fast ion transport. Similar to PCFs, three-dimensional inorganic frameworks (IOFs), such as metal-based and carbon-based materials, have attracted significant attention for electrode-host frameworks.49,50 IOFs are utilized to provide plenty of active nucleation sites and sufficient space for metal deposits. They are required to have fast electron transport and high mechanical strength, which reduces nucleation polarization and tolerates the anode volume changes.51–53 Besides, IOFs can be endowed with various geometries and functional groups via different fabrication methods to facilitate metal deposition and dendrite suppression.7,54–56
Interfacial modification engineering is regarded as a complicated strategy due to the heterogeneity of mass transport and reactions from the bulk electrolyte to the electrode interface. Decoupling surface reactions and designing target functional frameworks should be systematically correlated to regulate interfacial reactions for stable RMBs. In this review, functional framework materials, including PCFs and IOFs, are comprehensively categorized to introduce their design concept and respective functionality in anode protection. First, the mechanism of PCFs and IOFs in regulating ion transport and dendrite suppression are explained in detail. Second, the applications of PCFs (MOFs, COFs, zeolites) and IOFs (metal-based, carbon-based) are carefully discussed to reveal the property–structure relationships in metal batteries. Finally, the challenges and prospects of functional framework materials are proposed for practical high-performance metal batteries. This review will provide a valuable guide on material design and a deep understanding of the mechanisms of interfacial reactions on metal anodes.
2. Mechanism of regulating interfacial reactions
Uniform and dense deposition is crucial for reversible interfacial reactions in metal batteries. Smaller metal surfaces reduce contact with electrolytes, leading to fewer side reactions and less active metal loss.57,58 In order to achieve homogeneous metal growth, two factors are crucial: sufficient metal ion flux and active nucleation sites. Dendrite growth is affected by these two factors as mossy dendrites arise from uneven and insufficient ion flux, and needle-like dendrites occur due to limited active nucleation sites.51,59–61 In this regard, anode protection using functional frameworks, including PCFs and IOFs, aims to facilitate ion diffusion and increase the number of active sites, realizing optimal metal deposition and dendrite-free metal batteries.PCFs can function as artificial SEI due to their high ionic and low electrical conductivity. Benefited by the rich function groups and cavity structures, PCFs can host and separate different ions in electrolytes.36,62 This feature can be exploited to enhance cation desolvation on the anode interface, where sluggish desolvation kinetics are often caused by excessive cation–solvent interactions.63,64 The facilitated desolvation is beneficial to the fast ion diffusion and even ion flux, which finally contributes to uniform metal deposition, as illustrated in Fig. 1. The ion selectivity of PCFs is highly associated with the relative size of ions and chemical environment of pores. The modified functional groups grafted on PCFs can interact with different electrolyte components to affect the framework sieving effect.
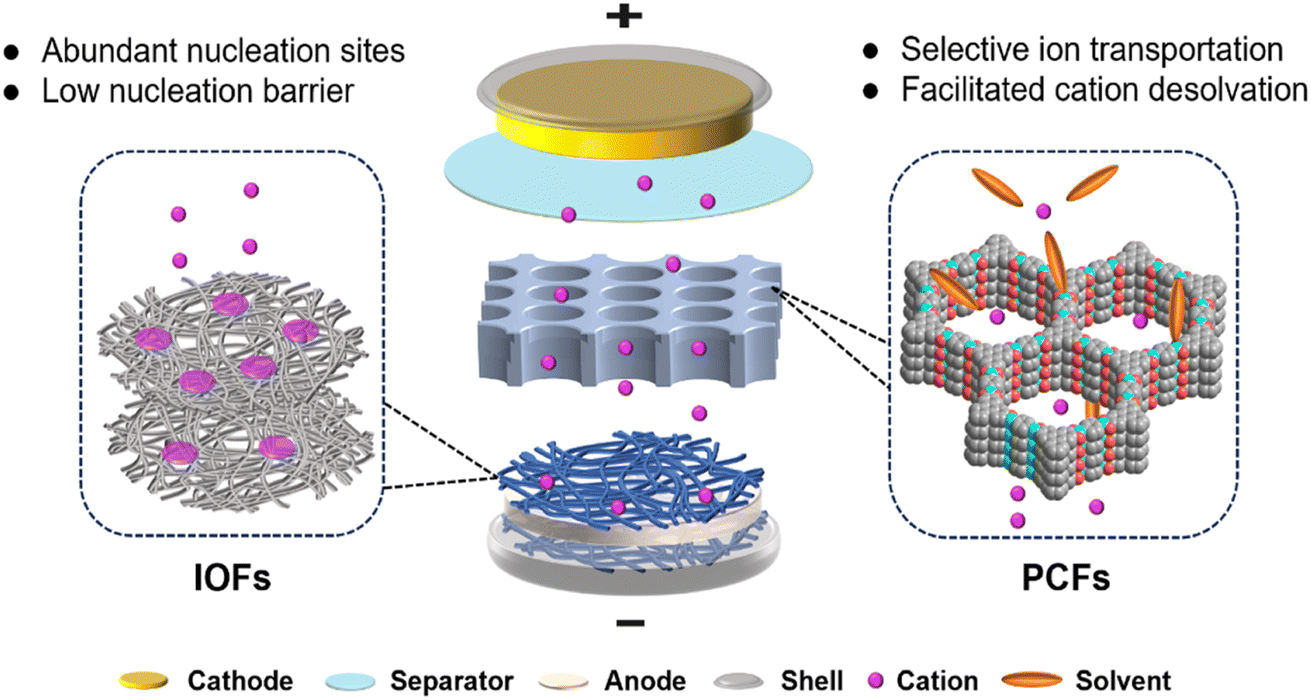 | ||
| Fig. 1 Schematic illustration of functional framework regulation for stable metal batteries. | ||
IOFs can serve as the electrode host to provide active nucleation sites and guide homogeneous metal deposition due to their high electrical conductivity.51,65,66 They are divided into metal-based (M-IOF) and carbon-based (C-IOF) materials according to the main components of the frameworks. M-IOF commonly includes nanostructure substrate conductors and metallophilic compounds, which have rapid electron transport and effective nucleation sites.41,67 C-IOF is based on different structured carbon matrices combined with various functional groups, including graphene nanofibers (GNFs) and carbon nanotubes (CNTs).50,68 Porosity and metallophilicity are two essential characteristics of IOFs for regulating metal growth. The porous structure of IOFs can accommodate repeated anode expansion and contraction, and their metallophilicity favors low energy barriers of metal nucleation.
3. Porous crystalline frameworks (PCFs)
PCFs, including MOFs, COFs, and zeolites, have been widely constructed on the surface of anodes in recent years.36,45 The periodic networks of PCFs are self-assembled employing various metal ions, ligands, or clusters, which exhibit their unique tunable features.47,69 The confined pores and channels of PCFs hinder the transport of solvated ions, leading to the sheathed solvent dissociation. This enables cation desolvation in advance on the surface of PCFs, mitigating the side reactions between metal anodes and solvents.62 To realize this vision, the exploitation of PCFs in metal batteries mainly focuses on the membrane modification and coatings on the anode surfaces.3.1 Metal–organic frameworks (MOFs)
Although LMBs have great potential for high energy density batteries, their practical application is restricted by Li dendrite growth and safety concerns. The conventional organic-rich SEI is usually inhomogeneous and low ionic conductive, which leads to nonuniform Li deposition.24,70,71 It is effective to regulate the ionic conductivity of SEI through additives, polymeric conductors, or ceramic particle modification.72–74 However, the obtained SEI still has the problems of low modulus and insufficient ionic conductivity. This endows an ideal artificial SEI with high cation transference number (tx+), high diffusion coefficient (Dx+), and high mechanical strength.75–77Fig. 2a illustrates the relationship among tx+, Dx+, and safe capacity, in which higher tx+ and Dx+ can lead to enhanced safe capacity of LMBs at a fixed current density. In a recent work, UiO-66 MOF was prepared by the solvothermal synthesis of zirconium oxide (ZrOx) clusters and 1,4-benzenedicarboxylic acid (H2BDC) to function as an artificial SEI on Li anodes in Fig. 2b.78 Hexafluorophosphate anions (PF6−) tend to be adsorbed by unsaturated “Zr-void” sites instead of “Zr–O′′ sites in the UiO-66 framework, and the binding of PF6− leads to increased tLi+, as evidenced by the density functional theory (DFT) calculations in Fig. 2c. The improved tLi+ of 0.59 and DLi+ of 3.23 × 10−7 cm2 s−1 reduce Li+ concentration gradient and enhance its ion flux at anode interface, effectively promoting even Li deposition and suppressing dendrite growth. In addition to the aprotic LMBs, UiO-66 has also been employed as an ion conductive interphase in aqueous ZMBs, which suffer from Zn dendrite growth and HER side reactions.79 The carboxyl groups are introduced in functionalized UiO-66-(COOH)2 MOF to enhance Zn2+ diffusion. Significantly, they enhance the Zn2+ affinity of UiO-66-(COOH)2 and induce its high concentration in the channels. This enables UiO-66-(COOH)2 with high ionic conductivity and inhibits water-induced side reactions on the anode. The resultant symmetric Zn–Zn cells based on UiO-66-(COOH)2@Zn anode exhibit a long cycle life of 2800 h at a current density of 2 mA cm−2, much longer than that of bare Zn anode (114 h).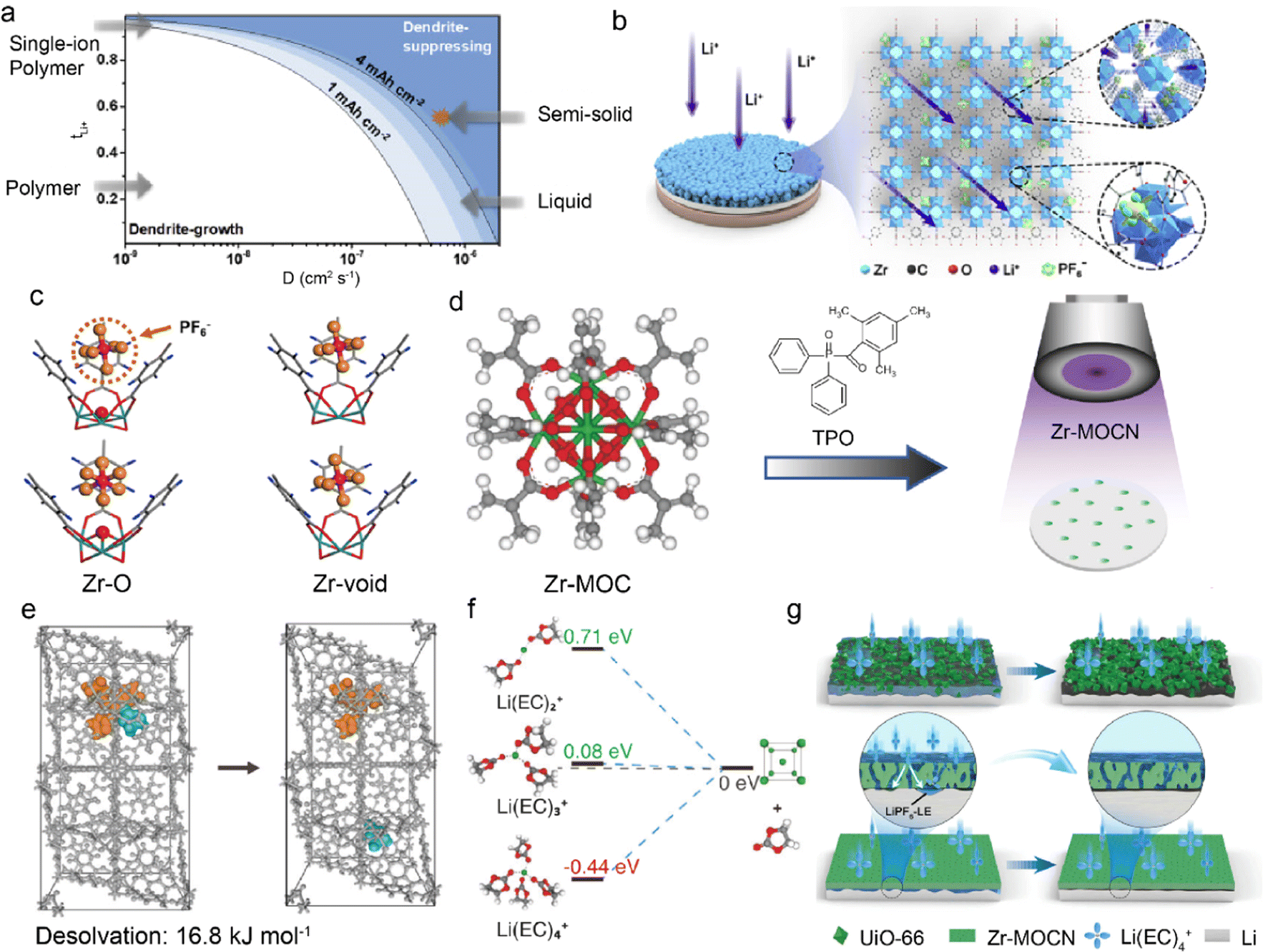 | ||
| Fig. 2 (a) Relationship between tLi+ and diffusion coefficient in suppressing Li dendrite growth. (b) Mechanism of regulating Li+ transport by UiO-66. (c) DFT calculation of binding energy between UiO-66 and PF6−. Reproduced with permission. Copyright 2020, Elsevier.78 (d) Construction of Zr-MOCN on a membrane via photopolymerization. (e) Dissociation energy of solvates in the UiO-66 structure. (f) Reduction energy of different solvates. (g) Li deposition in UiO-66 and Zr-MOCN. Reproduced with permission. Copyright 2022, Springer Nature.80 | ||
Separator modification with MOF is another promising way to suppress dendrite growth, in addition to surface coating. He et al. designed a nano-porous polymer separator (Zr-MOCN@PP) by the photopolymerization of multi-vinyl functionalized cluster (Zr-MOC), as shown in Fig. 2d.80 The Zr-MOCN framework exhibits lower dissociation energy of 16.8 kJ mol−1 between Li+ and ethylene carbonate (EC) to facilitate Li+ desolvation, as displayed in Fig. 2e. The less solvent-coordinated structures of Li(EC)2+ have a higher energy barrier of accepting electrons, indicating enhanced reduction stability of the electrolyte against Li anode, as depicted in Fig. 2f. The suppressed solvent decomposition further benefits inorganic-rich SEI formation and facilitates ion transport, which leads to uniform Li deposition and improved plating/striping reversibility. Remarkably, it is essential to synthesize smooth and dense protective layers (e.g., Zr-MOCN) in Fig. 2g as gaps and cracks inside the materials (e.g., UiO-66) can weaken the ion transport function. This necessitates rational synthesis methods for intact MOF structures on anodes. For example, crack-free zeolitic imidazolate framework-8 (ZIF-7x-8) is obtained by fast current-driven synthesis (FCDS) to improve the stability of ZMBs.81 The rigid 0.3 nm pores in the crack-free ZIF-7x-8 can not only accelerate Zn2+ desolvation but also block the contact between the Zn anode and active water, suppressing by-product accumulation. The regulated homogeneous Zn2+ flux leads to ordered dendrite-free Zn deposition and ultrahigh coulombic efficiency of 99.96%. Besides, a seamless MOF-based interphase is constructed by Ren et al. through a solid–vapor reaction between solid zinc oxide and 2-methylimidazol vapor.82 The obtained ZIF-8 layers are densely packed and defect-free to inhibit aqueous electrolytes from contacting with the Zn anode in ZMBs.
MOF skeletons with angstrom-level pores have been explored to manipulate solvation structures without any additives in the pristine liquid electrolytes.36,62 The cation desolvation usually has sluggish kinetics at the electrode–electrolyte interface due to the strong ion–dipole interactions between cations and solvents. The incomplete desolvation can induce undesirable solvent decomposition and formation of organic-rich SEI, which further influences ion conduction and metal deposition.83–85 Anion migration has great effects on the cation transport from the bulk electrolyte to the interface between electrode and electrolyte. Recent researches focus on regulated interactions between microporous MOFs and species in the liquid electrolyte, aiming to tune the electrolyte solvation structures and hence the interfacial chemistry.48,62 As a proof of concept, the HKUST-1 MOF (Cu3(BTC)2, BTC = 1,3,5-benzene tricarboxylate) is utilized to modify the conventional ether-based electrolyte and improve homogeneous Li electrodeposition.86 The spatial confinement of bis(fluorosulfonyl)imide anion (TFSI−) in the channels of MOF drives its migration direction change for favorable ion diffusion, as illustrated in Fig. 3a. The corresponding energy barriers of the two diffusion paths of TFSI− are evaluated by DFT calculations in Fig. 3b, indicating its direction preference for Path-1. More restrained TFSI− enables a higher Li+ transference number of 0.7 in the MOF-modified electrolyte compared to that in the pristine electrolyte (0.2–0.4), which is beneficial to homogeneous Li+ ion flux on the anode. The Li+ cations are commonly coordinated with organic solvents rather than anions in the bulk electrolyte. However, the MOF host tends to participate in the Li+ solvation in the restricted cavities, as schematically displayed in Fig. 3c. The MOF skeleton is enriched with highly mobile Li+ ions and confined TFSI− anions, and this leads to sufficient Li+ flux and stable Li electrodeposition.
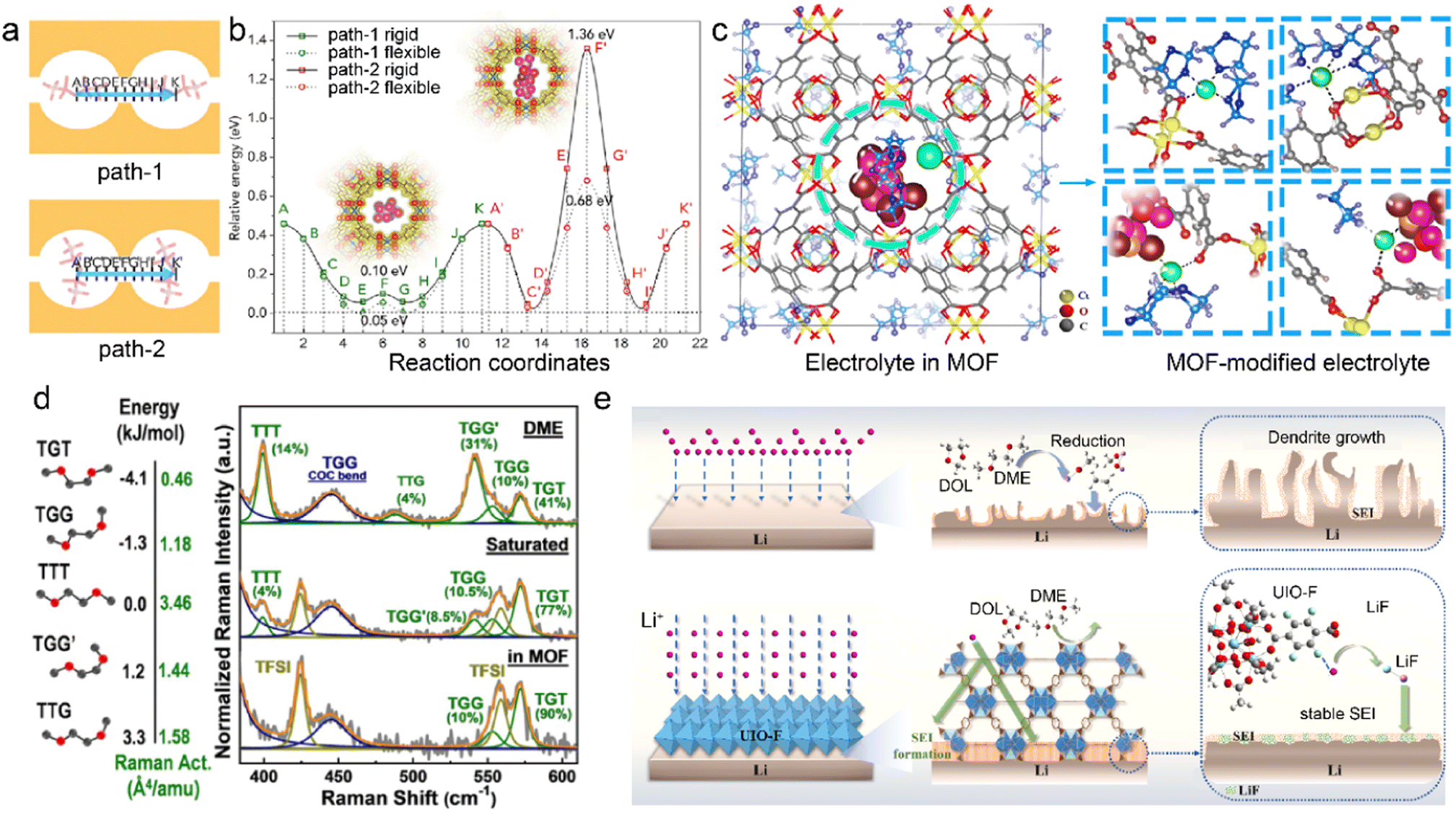 | ||
| Fig. 3 (a) Two paths of TFSI− migration and (b) corresponding energy barrier in an MOF. (c) Modified Li+ solvation structures in the MOF skeleton. Reproduced with permission. Copyright 2018, Elsevier.86 (d) Energy levels of DME in different conformers and Raman spectra of pristine and MOF-modified electrolytes. Reproduced with permission. Copyright 2021, Wiley-VCH.92 (e) Mechanism of UIO-F in regulating SEI reconstruction. Reproduced with permission. Copyright 2023, Elsevier.93 | ||
Inorganic species in the SEI promotes uniform Li deposition due to their high ionic conductivity. The inertness of inorganics gives rise to superior chemical and electrochemical stability of SEI, which mitigates SEI breakage and reconstruction. The robust inorganic-rich SEI leads to suppressed electrolyte decomposition and enhanced Li deposition. Anion-rich solvation structures have been recognized to benefit the formation of inorganic-rich SEI.87,88 However, the desired solvation structures in the high-concentration electrolyte (HCE) and localized HCE require high salt-to-solvent ratios, which limits their practical application.20,89–91 In order to effectively achieve the highly coordinated structures, the Cu-BTC/PSS (PSS = charged sulfonic polymer) MOF is adopted by Yang et al. to condense dilute electrolytes in the Li-sulfur (Li–S) batteries.92 The uniform pores and caves of Cu-BTC/PSS confine the pristine ether-based electrolyte, transforming the solvation structures from solvent-separated ion pairs (SSIP) to contact ion pairs (CIP) and aggregate ion pairs (AGG). More importantly, the solvent activity is suppressed in the MOF-modified electrolytes, as revealed by the distribution of five solvent conformers in Fig. 3d. Different conformers of dimethylether (DME) are distinguished according to the trans and gauge orientations of O–C–C–O. The high energy of TTG (trans-trans-gauge) and TGG′ (trans-gauge-gauge) conformers give rise to their low stability, and TGT (trans-gauge-trans) conformers with lowest energy level are more stable in the electrolytes. It can be found that a higher proportion of TGT conformers exists in the MOF-based electrolyte compared to the dominant TTG/TGG′ in the pristine dilute DME electrolyte. This enables the MOF-modified electrolyte with suppressed reactivity and high anti-oxidation capability, significantly reducing the side reactions at both the cathode and anode sides.
MOF can be employed to tailor both electrolyte solvation structures and SEI components on the metal anodes. An F-functionalized MOF (UIO-F) is reported to modify SEI and improve the performances of LMBs.93 The intrinsic adsorption sites in the UIO-F skeleton enhance Li+ desolvation kinetics and ion transport, which prevents solvent contact with the Li metal anode, as illustrated in Fig. 3e. The high reactive F functional groups in the UIO-F frameworks benefit the generation of uniform LiF-rich SEI, enhancing the ionic conductivity and mechanical strength of the Li anode interface. As a result, Li–Li symmetric cells based on UIO-F@Li anodes achieve an ultralong cycle life of more than 2000 h at a current density of 3 mA cm−2. Besides, the Cu2(BDC)2 with stereoscopic lithophilic sites (OA-MOF) is designed by Wu et al. as a dynamical SEI modulator to improve the interfacial stability in LMBs.94 The open-architecture and high electrolyte wettability of OA-MOF promote homogenized ion flux and enhance Li plating kinetics. The MOF-induced heterogeneous SEI is enriched with salt decomposition products, including Li2NSO2CF3/Li2CO3 in the outer layer and LiF/Li2O in the inner layer. The obtained inorganic-rich SEI reduces the energy barrier of Li diffusion and increases the exchange current density, leading to small deposition overpotentials and high CE in OA-MOF-based LMBs.
3.2 Covalent organic frameworks (COFs)
COFs are characteristic with similar features of MOFs, including abundant structures, large surfaces, high porosity, and tunable channels. COFs exhibit high chemical stability owing to their robust skeleton linked by strong covalent bonds.46,69,95 However, the functionalities of COFs are restricted by the lack of active metal centers, which can promote ion adsorption and guide uniform metal deposition. Recently, Ni-bis(dithiolene) centers are introduced into COFs to construct a stable interface to improve LMB stability.96 The Ni-TAP and Ni-TAA COFs are constructed by the extension of Ni(bded)2 linker (bded = bis[1,2-di(4-formylphenyl)ethylene-1,2-ditholate]) with 4-connected tetra(aminophenyl)pyrene (TAP) and 3-connected tris(aminophenyl)amine (TAA), respectively. Notably, the planar 4-connected building unit of TAP leads to the 2D network of Ni-TAP. At the same time, the 3-connected triangular TAA linker gives rise to the 3D framework of Ni-TAA in Fig. 4a. The uniform pore sizes and Ni-bis(dithiolene) bonds in the Ni-TAA and Ni-TAP enhance the lithiophilicity and electrolyte wettability. The Li deposition is therefore refined on the COF-modified anode, and faster reaction kinetics is facilitated for stable LMBs.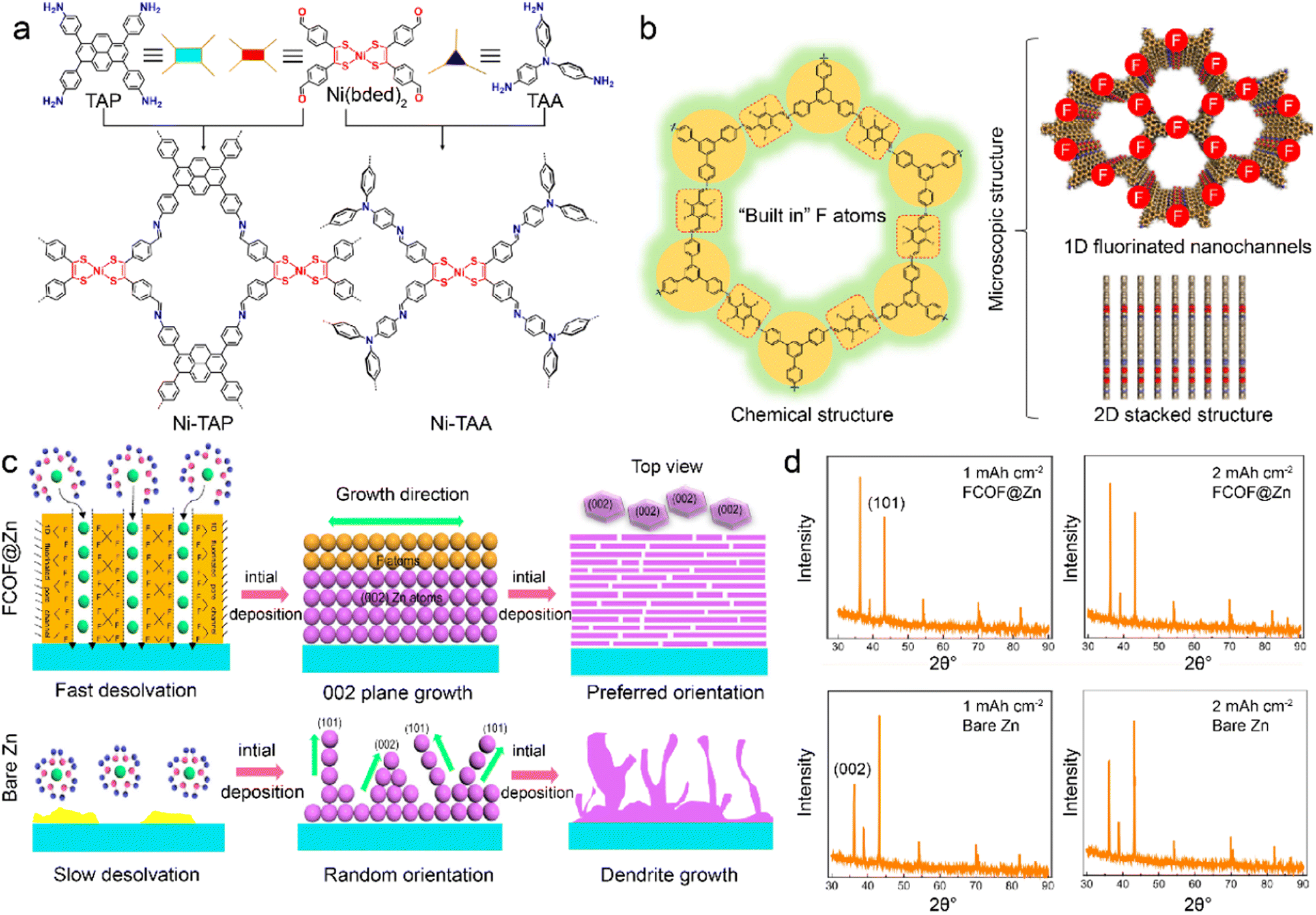 | ||
| Fig. 4 (a) Topological design of Ni-TAP and Ni-TAA COFs. Reproduced with permission. Copyright 2022, American Chemical Society.96 (b) Physicochemical structures of FCOFs. (c) Dendrite growth during Zn plating on the surface of FCOF@Zn and the bare Zn anode. (d) XRD patterns of deposited Zn on different surfaces. Reproduced with permission. Copyright 2021, Springer Nature.97 | ||
The interactions between COF hosts and electrolyte components are critical for interfacial chemistry. It is effective to modify the COF skeleton with functional groups to regulate the binding attractions with different species. The fluorinated COF is synthesized with 2,3,5,6-tetrafluoroterephthaldehyde (TFTA) and 1,3,5-tris(4-aminophenyl)benzene (TAPB) to act as a protective layer (FCOF) on Zn anode.97 The inherent 2D stacked frameworks and 1D nanochannels endow FCOF with excellent mechanical strength and fast ion diffusion in Fig. 4b. The conventional Zn2+ ions have low desolvation kinetics and deposit in random plane orientations, including (101) and (002) crystal planes, which induces negative deposition and dendrite formation. In contrast, FCOF promotes Zn2+ desolvation and enables its grains along a preferred orientation of the (002) crystal plane, as depicted in Fig. 4c. The strong interactions between F atoms in FCOF and Zn atoms change the relative surface energy of different crystal planes, and leads to more orientation exposure of (002) than (101) planes. The preferential anisotropic growth of Zn along the (002) crystal plane is parallel to the substrate and results in a platelet Zn deposition, as demonstrated by XRD patterns in Fig. 4d. The Zn dendrites are hence inhibited, and the FCOF-modified Zn anode contributes to high-safety and long-cycled ZMBs. Benefiting by fast ion transport in fluoride channels, the Zn–Zn cells afford a long cycle life up to 750 h at a high current density of 40 mA cm−2. In addition to the interactions with metal atoms, COFs can interact with solvents and anions to change the electrolyte aggregation states. A self-supported TPB-BD(OH)2-COF, which is synthesized with 2,4,6-trihydroxy-1,3,5-benzenetricarboxaldehyde, 3,3′-dihydroxy-4,4′-diaminobiphenyl, and p-toluenesulfonic acid (PTSA), are developed as a separator in LMBs.98 The hydroxyl groups in TPB-BD(OH)2-COF promote the unique construction of hydrogen bond networks between COF and EC/EMC (ethylmethyl carbonate) electrolyte. The formation of hydrogen bonds between O atoms of EC/DEC and –OH groups in COFs gives rise to strong solvent–COF interactions. This reduces free solvent coordination in the solvation sheath and induces more ion-pair aggregates than in dilute electrolytes. Consequently, the COF-modified electrolyte has fewer side reactions and favors even Li deposition.
Compared to traditional SEI modification, COF-induced SEI can enhance its stability under harsh conditions, such as high/low temperatures, large current density, and low electrolyte content. Functional groups have been widely introduced into the COF structures owing to their potential effects on the SEI components and ion diffusion. The COF incorporated with nitrate groups, which is synthesized with 1,3,5-triformylphloroglucinol and ethidium bromide (denoted as EB-COF:NO3), is designed by Wen et al. to facilitate the inorganic-rich SEI formation in LMB.99 LiNO3 has been employed to modify SEI components in ether-based electrolytes due to its inorganic decomposition products of Li3N and LiNxOy on the Li anode. However, its low solubility restricts the universal application in ester-based electrolytes.100–102 To overcome these challenges, NO3− is introduced into the MOF skeleton by the ion-exchange process, which realizes the desirable formation of NO3−-derived SEI in ether-based electrolytes. The synthesized EB-COF:NO3 is characteristic with positively charged channels, which immobilize anion clusters. The intensive interaction between Li+ and the anchored NO3− reduces solvent coordination in the solvation sheath. Due to the facilitated Li+ desolvation and released NO3− from EB-COF:NO3, the obtained Li3N and LiNxOy-rich SEI is highly ionic conductive and robust and suppresses side reactions and Li dendrite growth.
F-containing species have also been reported to enhance the SEI stability, in addition to nitride compounds.87,103–105 The EB-COF:NO3 is further modified by nano-selenium fluoride (SnF2) as a functional separator for LMBs in Fig. 5a.106 The SnF2 is encapsulated in EB-COF:NO3, and its favorable decomposition products contribute to the reinforced SEI. SnF2 is transformed into Li5Sn2 alloy and LiF during the Li deposition. The resultant Li5Sn2 alloy facilitates Li+ nucleation and diffusion at the interface, and LiF significantly enhances the mechanical robustness of SEI. The synergetic effects of NO3− and SnF2 nanoparticles ultimately lead to the ultra-stable inorganic-rich interface, which is enriched with Li3N, LiNxOy, LiF, and Li5Sn2 alloy. Due to the fast Li+ desolvation and highly ionic conductive SEI, the symmetric Li–Li cells are rewarded with reversible Li plating/striping at high current density and large areal capacity of 15 mA cm−2 and 30 mA h cm−2, respectively. Furthermore, the EB-COF:NO3@SnF2-coated separator can enable a practical Li anode under harsh conditions of high temperature, high voltage, low electrolyte content, and low N/P (negative/positive) ratios. For example, the assembled Li-NCM811 cells exhibit high capacity retention of 87.6% from 212.8 to 186.4 mA h g−1 at a high cutoff voltage of 4.5 V under 60 °C. A specific capacity of 190.4 mA h g−1 with high capacity retention of 95.9% can be achieved at 3.0–4.4 V using only 6 mL mA h−1 electrolyte and low N/P of 1.2 (3.9 mA h cm−2 for cathode). Moreover, Li symmetric cells deliver up to over 270 h stable cycles at a high current density of 15 mA cm−2 compared to the large voltage polarization of original separator within 45 h.
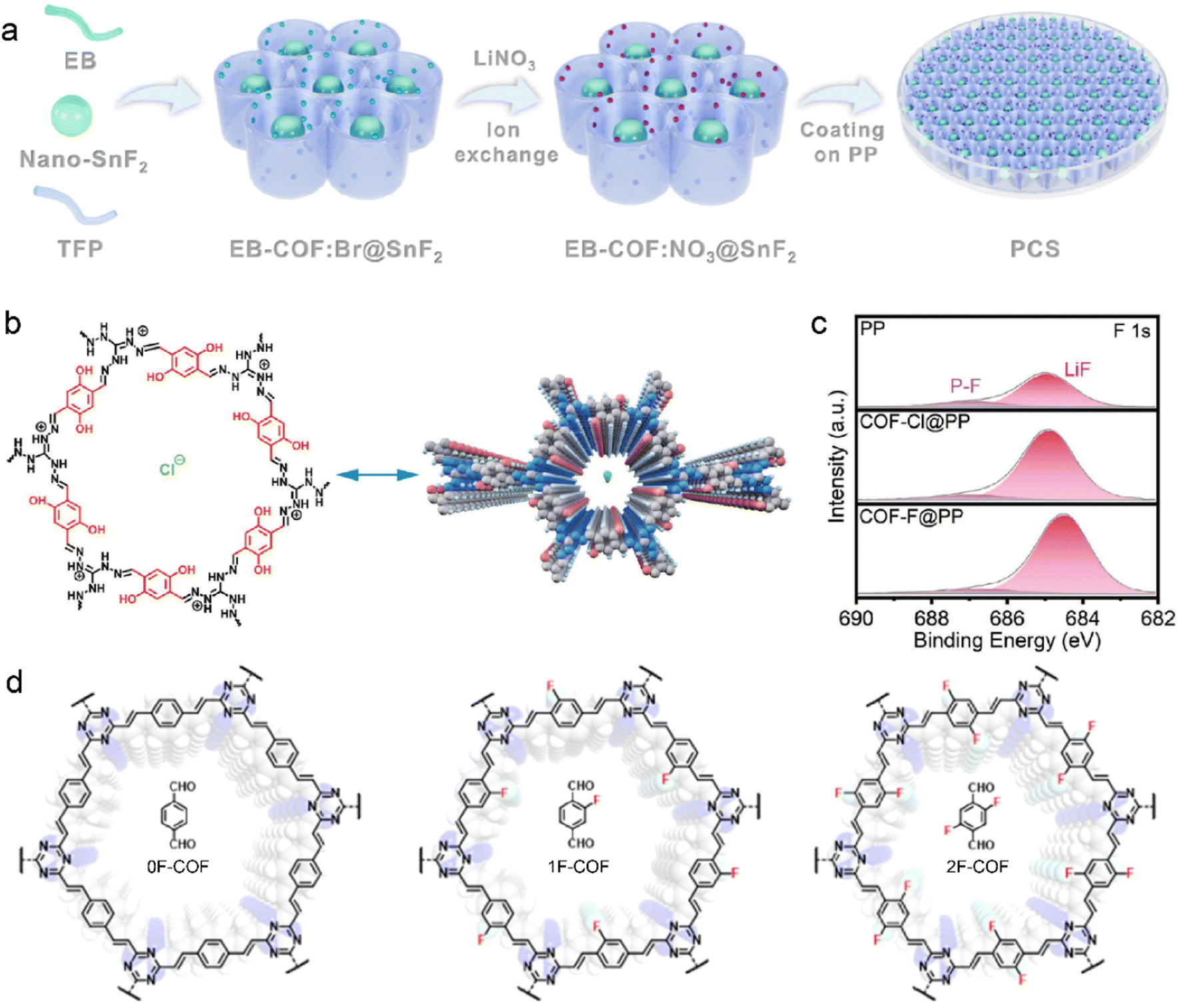 | ||
| Fig. 5 (a) Schematic illustration of EB-COF:NO3@SnF2 synthesis. Reproduced with permission. Copyright 2023, the Royal Society of Chemistry.106 (b) Molecular structure of COF–Cl. (c) F 1s XPS spectra of the Li anode with different separators after cycling. Reproduced with permission. Copyright 2023, Wiley-VCH.107 (d) Various fluorinated F-COF structures with different linkers. Reproduced with permission. Copyright 2023, Wiley-VCH.108 | ||
F-containing COF structures are constructed by introducing fluoride anions in the skeleton. The COF with F− anions (COF–F) is developed from cationic COF–Cl, which was synthesized with 2,5-dihydroxyterephthalaldehyde (Dha) and triaminoguanidinium chloride (TGCl), as shown in Fig. 5b.107 The Cl− anions can be easily exchanged with F− anions owing to the weak binding strength between Cl− and COF host. During Li deposition, the F− anions can be released from the as-prepared F-COF and participate in SEI formation, leading to LiF-riched anode interface in Fig. 5c. Meanwhile, the positively charged organic units in COF-F/Cl exhibit considerable attraction with solvent molecules in electrolytes, and it hinders the solvent decomposition on the Li anode. In this respect, robust SEI and fast Li+ desolvation kinetics can be expected in F-containing COF to improve ion transport and prevent Li dendrite growth for stable LMBs.
The F-containing COFs favor the fluorination of SEI and their syntheses need extra post-processing because the alien species cannot change their inherent properties. It is facile to construct an F-containing COF skeleton by directly employing the linkers with fluorinated groups. Olefin-linked COF has been reported by Li et al. to enhance Li+ diffusion and fabricate stable anode interface.108 The fluorination degree of COF is controlled by varied numbers of F atoms in the terepthalaldehyde linkers, as displayed in Fig. 5d. The obtained SEI in the 2F-COF system has the highest LiF content compared with 0F-COF and 1F-COF due to the abundant F atoms in the olefin linkage of 2-FCOF. In addition, electron-rich triazine rings and F atoms in the skeleton lead to the enhanced electronegativity of COF channels. This facilitates selective Li transport and improves salt dissociation within the channels, which gives rise to a high tLi+ of 0.85 and high ionic conductivity of 1.78 mS cm−1. A high capacity retention of 99% is consequently achieved after 100 cycles in the Li–LiFePO4 cells.
3.3 Zeolites
Zeolites are crystalline aluminosilicate materials, featured with periodic 3D networks, intersectional channels, and cavities. The open framework of zeolites is composed of corner-sharing TO4 tetrahedra (T = Si, Al, or P), the pore size of which is determined by the numbers of surrounding rings, as shown in Fig. 6a.109,110 The nanoscale dimensions of zeolites bring about high porosity and large surface area, creating confined space for ion diffusion and catalytic sites. Compared with MOFs and COFs, zeolites benefit from their low production cost, high thermal stability, and high corrosion resistance.111 They have been commercially utilized in industries, including gas separation, cracking catalysts, and sewage treatment.112,113 It can be anticipated that zeolites are playing an important role in renewable energy and sustainable chemistry.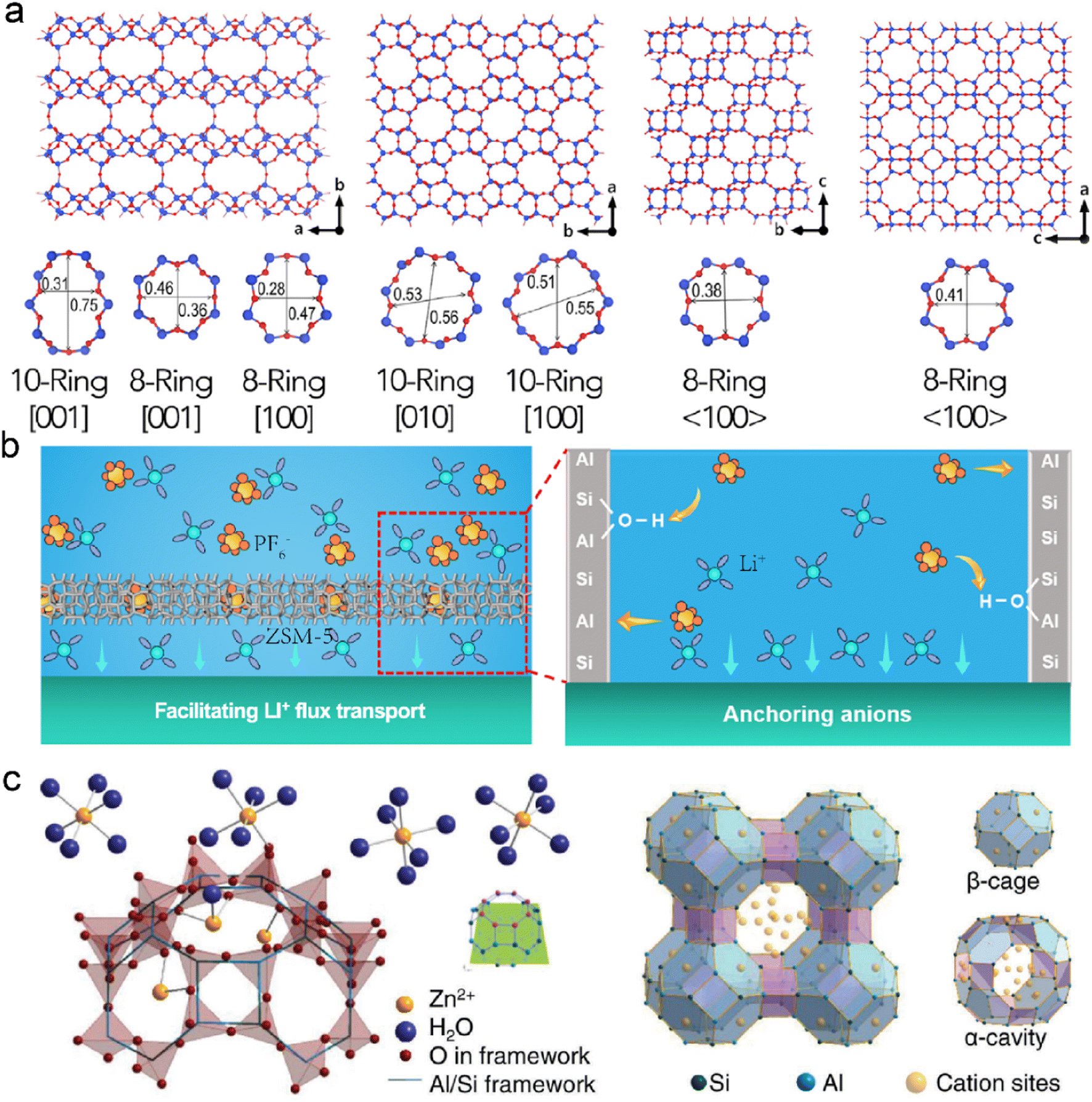 | ||
| Fig. 6 (a) Molecular structures of several zeolite frameworks. Reproduced with permission. Copyright 2017, Elsevier.109 (b) Mechanisms of ZSM-5 on protecting the Li anode. Reproduced with permission. Copyright 2023, American Chemical Society.115 (c) Schematic illustration of ion exchange and framework structure of LTA zeolite. Reproduced with permission. Copyright 2023, Wiley-VCH.117 | ||
Zeolites are implanted with Lewis acid sites due to the substitution of Si atoms with metal cations (e.g., Zr, Ti, and Sn), which can interact with electron-rich molecules.114 They can modify electrolytes through acid sites since solvents and anions are nucleophilic in nature. Zeolite Socony Mobil-5 (ZSM-5) has been employed as a functional interface layer on the Li anode. Abundant acid sites in ZSM-5 show strong acid–base interactions with PF6− anions, and it leads to fast cation–anion dissociation in the ordered nanochannels, as displayed in Fig. 6b.115 The enhanced mass transfer of Li+ reduces concentration polarization and induces dense Li deposition. Besides, chemically stable ZSM-5 with high mechanical strength maintains the excellent structural integrity of the Li anode, resulting in the prolonged cycling of LMBs.
The limited pore size of channels in zeolites often exhibits great spatial confinement to the solvents and ions in electrolytes. The molecular sieving effect provides selective entrance and diffusion of guest species in the frameworks, which significantly depends on the size, geometry, and polarity of the substances.116 LTA zeolite consisting of α-cavities and β-cages is reported to protect the Zn anode, as shown in Fig. 6c.117 Their frameworks are usually negatively charged due to the replacement of Al in the SiO4 tetrahedra. Extra cations with different valences are introduced to compensate for the unbalanced charges in the zeolites. Notably, it is essential to select suitable cations in the zeolite frameworks as different cations can influence the original charge carrier transport during deposition. For instance, Na+ cation is substituted with Zn2+ cation in an ion-exchange process to avoid the electrostatic shielding effect at the interface. The average pore size of the Zn zeolite (ZnA) is smaller than the diameter of sulfate ions, and it allows for the sieving effect in the electrolyte. The decreased contact between SO42− and Zn anode prohibits the generation of Zn4(OH)6SO4·xH2O and increases its coulombic efficiency. In addition, the negatively charged framework has strong Zn2+ affinity, and the concentration gradient can be mitigated near the anode surface. Based on the ZnA-modified anode, the symmetric Zn–Zn batteries exhibit an enhanced lifespan of 2400 h at 5 mA cm−2. The symmetric cells can even endure a high current density of 100 mA cm−2 for reversible Zn plating/stripping due to the reduced concentration gradient and fast interfacial kinetics. The resultant Zn–MnO2 full cells achieve high capacity retention of 76.4% after 7500 cycles.
Cations are commonly coordinated with excessive solvents in the conventional dilute electrolytes, which significantly increases solvent polarity and decreases solvent stability against metal anodes.9,15,48 In the case of aqueous ZMBs, the polarized water in the Zn2+ solvation sheath induced serious corrosion, as illustrated in Fig. 7a.118 Zeolites have been utilized to reduce the solvent reactivity: uniform zeolite coatings can physically inhibit the free solvent contact with meal anodes, and considerable interactions among cations, solvent, and zeolite frameworks can reduce solvent coordination in solvates. This is conducive to fast cation desolvation kinetics and suppression of side reactions between polarized solvents and metal anodes. 3 Å zeolite molecular sieves, which are widely used in gas separation and liquid drying, are exploited to manipulate the electrolyte solvation structures in ZMBs.111 The zeolite-modified electrolyte is formulated by the repeated soaking of zeolite membrane in the electrolyte of 2 M ZnSO4, followed by drying to remove the extra electrolyte.118 The obtained solvation configurations in the zeolite and pristine electrolytes are investigated by Raman spectra, as displayed in Fig. 7b. The new emerging peak at 306.7 cm−1 is attributed to the binding interactions between Zn2+ ions and Si/Al–O frameworks. The positive shift of the SO42− vibration peak indicates the transformation of electrolyte aggregate states from SSIP-dominant structures to CIP-enhanced structures. The absorbed water in the bulk zeolite is evidenced by the sharp peak of O–H stretching vibration at 3588 cm−1. The less coordinated solvents have lower reactivity compared to the complex solvent clusters, leading to suppressed parasitic reactions on the anode. Besides, the adsorbed water in the zeolite increases the electrolyte wettability and promotes fast Zn2+ transportation at the interface.
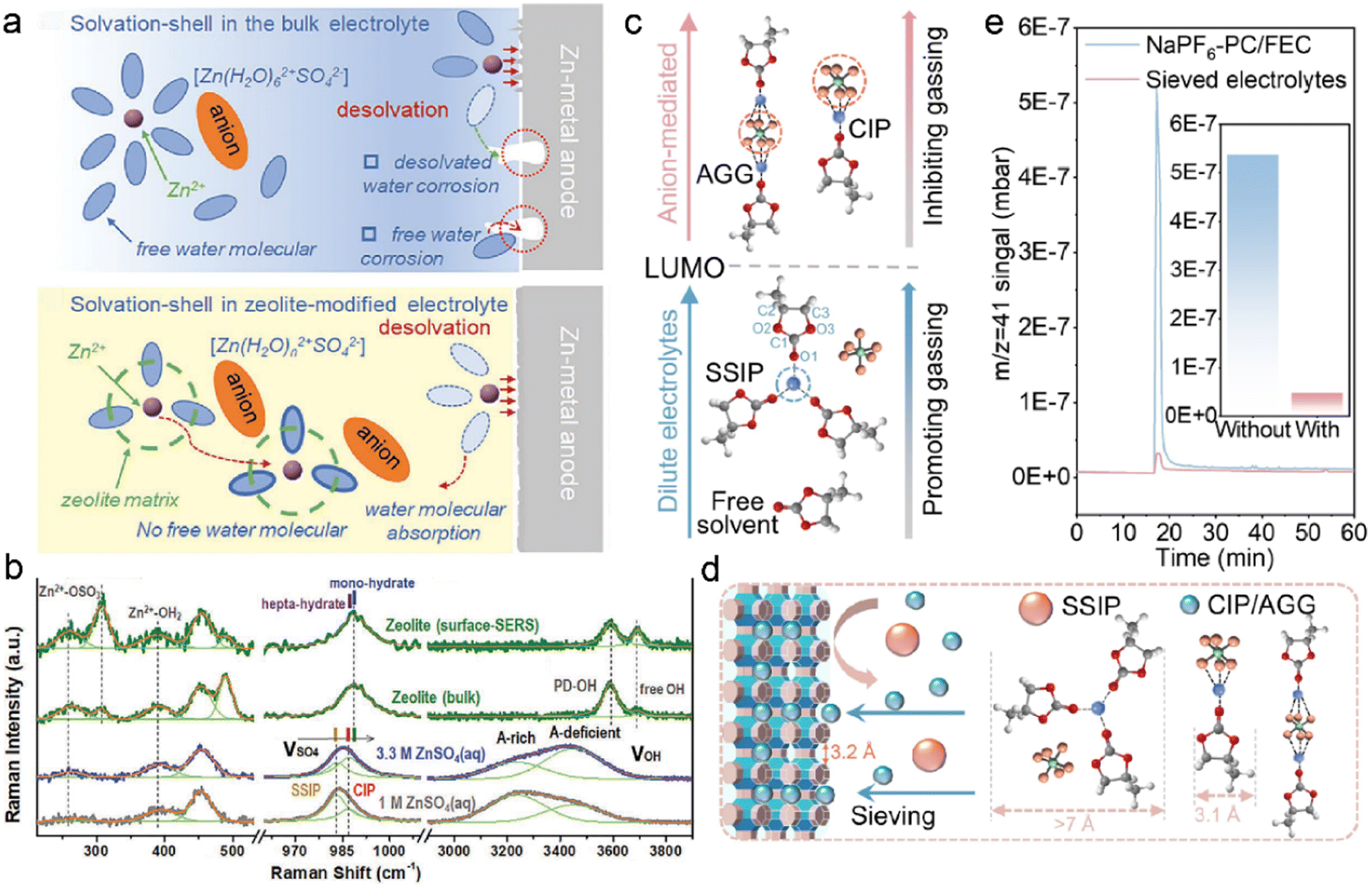 | ||
| Fig. 7 (a) Comparisons of water decomposition on the anode surface with/without a zeolite layer. (b) Raman spectra of pristine electrolytes and zeolite-modified electrolytes. Reproduced with permission. Copyright 2021, Wiley-VCH.118 (c) Relationship between solvation configurations and gas production. (d) Schematic of solvation regulation by zeolite framework. Reproduced with permission. Copyright 2022, Wiley-VCH.124 | ||
Benefited by zeolite-modified solvation structures, undesired solvent decompositions have been effectively suppressed in aqueous ZMB, and this further contributes to suppressing gas evolution. Ester-based electrolytes are widely applied in organic metal batteries due to their high salt solubility and high oxidation voltages.119–121 However, they are less compatible with the metal anode, and unstable SEI formation results in inferior side reactions.21,71 The continuous electrolyte consumption gives rise to low coulombic efficiency and safety concerns, including dendrite growth and flammable gas evolution.122,123 3 Å zeolite molecular sieves are studied by Lu et al. to investigate gassing suppression in sodium metal batteries (SMBs).124 The gas evolution depends on the reduction stability of electrolytes, which is closely related to the solvation structures. As depicted in Fig. 7c, CIP and AGG structures have higher LUMO energy for accepting electrons and inhibiting gas production. In contrast, lower LUMO energy enables the reduction of SSIP and promotes gas generation in electrolytes. The zeolite can change the proportion of CIP, AGG, and SSIP configurations in electrolytes through the sieving effect, as illustrated in Fig. 7d. The reactive SSIP structures are excluded by the zeolite owing to their larger cluster size (>7 Å) compared to the pore sizes. For comparison, smaller CIP and AGG complexes can diffuse in the zeolite channels, improving the electrolyte reductive stability at the anode interface. The functionality of zeolites in suppressing gas evolution is further demonstrated by gas chromatography (GC), as shown in Fig. 7e. The amount of C3H6 (m/z = 41) in the zeolite-modified electrolyte is one tenth of that in the blank electrolyte, which confirms the sieving effect for reduced parasitic reactions on anodes. The capacity of the Na-NVPF (sodium vanadium fluorophosphate) cells with the pristine electrolyte decays rapidly after 11 cycles at 60 °C, resulting from the enhanced side reactions and gas production at high temperatures. However, the zeolite-protected cells can afford high specific capacity of 91.5 mA h g−1 with capacity retention of 90.1% after 100 cycles under harsh conditions.
4. Three-dimensional inorganic frameworks (IOFs)
According to Sand's time theory, uniform metal deposition is highly associated with local current density and ion diffusion ability. The high current density induces ion concentration polarization and uneven surface potential, leading to uncontrollable dendrite growth.125–127 IOFs are featured with high surface area and excellent electron transport, which effectively decreases the local current density and prolongs the Sand's time. Importantly, metal anode suffers huge volume changes, and this results in the breakage and reconstruction of interfacial protective layers.128–130 The abundant porosity of IOFs provides internal space to accommodate the large volume change of anodes, and they should possess high mechanical strength to maintain the integrity of electrodes for stable RMBs.4.1 Metal-based three-dimensional inorganic frameworks (M-IOF)
The M-IOF mainly consists of 3D metallic frameworks with high electrical conductivity. However, insufficient active nucleation sites in the substrate lead to aggregated metal growth and needle-like dendrite formation.131,132 In order to solve the dilemma, metal compounds have been introduced into M-IOF to provide effective sites with lower nucleation barriers for metal deposition. The CF@ZnO skeleton is synthesized through the chemical precipitation of ZnO on the 3D Cu foam for dendrite-free SMBs.133 The hierarchical ZnO nanorod arrays grow vertically on the Cu foam to offer abundant Na nucleation sites in Fig. 8a. The cylindrical core–shell structure of CF@ZnO reduces the local current density and mitigates the severe volume change of Na anode. Due to the reduced nucleation barrier, Na metal homogeneously deposits on the ZnO arrays with low nucleation overpotentials. In contrast, Na particles tend to agglomerate on a few active nucleation sites and lead to the mossy deposition morphology and Na dendrites. COMSOL Multiphysics simulation is conducted to visualize the distribution of current density and Na+ concentration in Fig. 8b. Higher Na+ flux and current density are observed in the neck of nanorods, consistent with the preferential Na deposition on the ZnO arrays. The uneven distribution of Na+ ions and current density promotes self-amplified dendrite growth on the bare Cu surface. With the CF@ZnO anode, the full cells paired with Na3V2(PO4)3 (NVP) cathode achieve capacity retention of 93% after 500 cycles at 10C (1C = 110 mA g−1). In comparison, the original Cu-NVP cell only has 79.4% capacity retention with lower coulombic efficiency of 98.2%.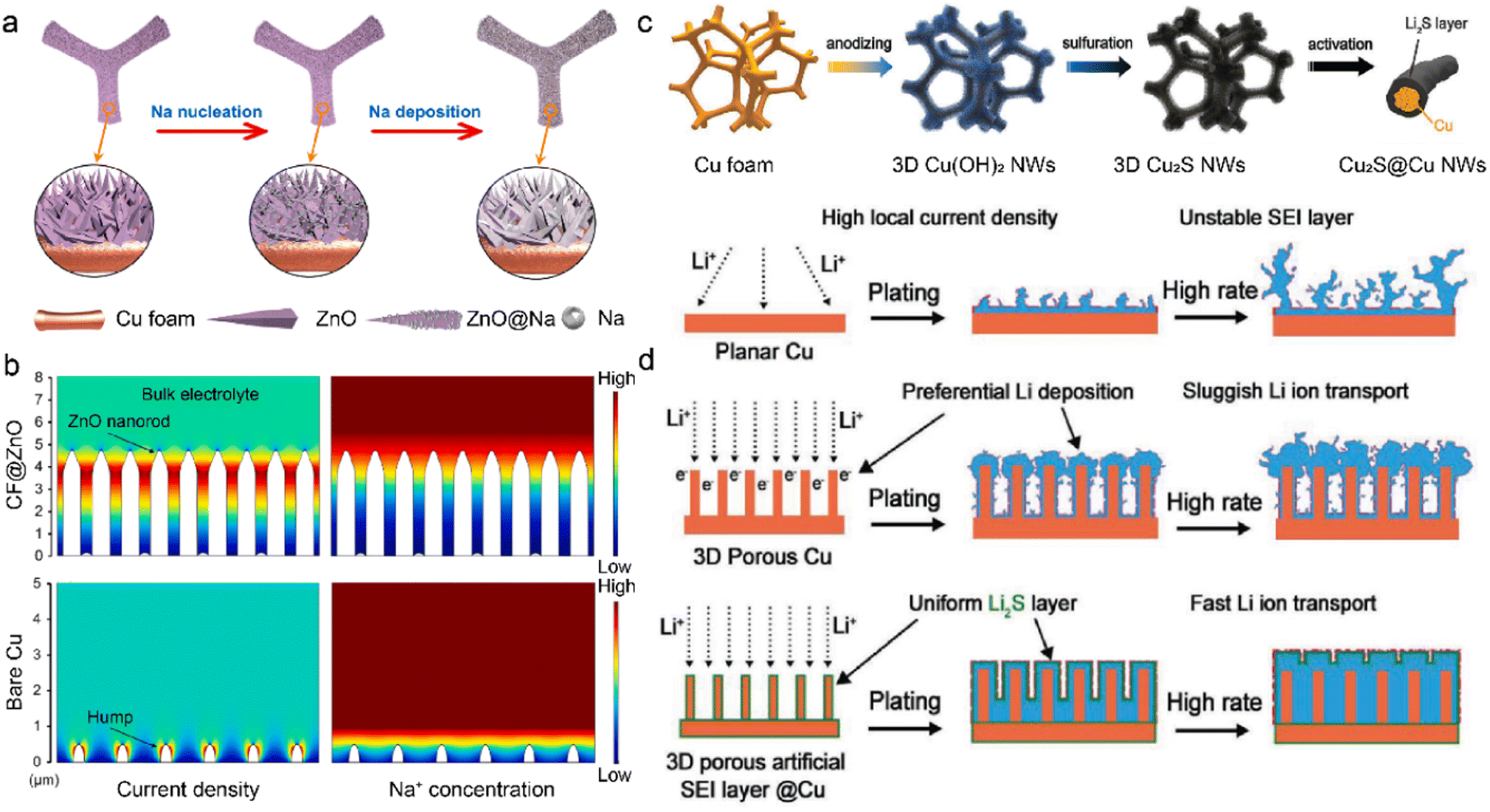 | ||
| Fig. 8 (a) Na nucleation and deposition on the CF@ZnO anode. (b) Current and Na+ concentration distribution at the interface using COMSOL simulation. Reproduced with permission. Copyright 2021, Elsevier.133 (c) Schematic of constructing Li2S @3DCu. (d) Li deposition behaviors on various anodes. Reproduced with permission. Copyright 2020, Wiley-VCH.134 | ||
Metal compounds can be employed as the artificial SEI on M-IOF, in addition to reducing the nucleation barrier. Lithium sulfide is in situ grown on the Cu foam to serve as a passivation layer in LMBs in Fig. 8c.134 The copper hydroxide (Cu(OH)2) nanowires are first introduced on the original Cu frameworks by anodization, followed by partial sulfurization with hydrogen sulfide. The obtained Cu2S nanowires are subsequently converted into Li2S in the initial galvanostatic electroplating. The homogeneous Li2S layer on the Cu framework (Li2S@3DCu) prohibits continuous contact between the substrate and deposited Li. This leads to fewer side reactions and improved Li deposition, as shown in Fig. 8d. On the 2D planar Cu substrate, the high local current density gives rise to huge volume change of Li anode, and this easily breaks the fragile SEI and constantly consumes electrolytes. Although surface polarization is decreased by enhanced electronic conductivity in 3D porous Cu, sluggish Li+ transport exhibits lower kinetics compared with electron transference, leading to selective deposition on the top of electrodes. In this regard, Li2S is constructed as the passivation layer on the surface of the 3D Cu framework. The as-prepared 3D Li2S @3DCu promotes Li+ conduction through the SEI and mitigates aggregated Li+ concentration. More importantly, the high ionic conductivity of Li2S on the Cu framework creates a balance in the transport kinetics of ions and electrons. This promotes uniform distribution of ions and electrons, avoids rapid local Li growth to suppress dendrite formation, and improves Li deposition.
It is essential to control the preferential region for metal deposition as the common top-growth mode easily leads to dendrite formation and separator penetration.135–137 The electroplating position fundamentally depends on multiphysical parameters, such as current density, cation concentration, and surface potential. Three interfacial resistances can be applied to decouple the Li deposition process on anodes, as shown in Fig. 9a.138 The electrode commonly shares similar electric resistance (Re) owing to the uniform electron conductivity. The Li+ transport resistance (RLi) is related to ion concentration, in which the anode-separator interface has the highest Li+ content and exhibits the lowest RLi. The charge transfer resistance (Rct) is determined by different reactions and their corresponding interfacial kinetics. The preferential Li deposition sites can be transformed from the top area to the bottom of anodes by manipulating the three interfacial resistances. Deposition-regulating framework (DRS), which manifests gradient Li+ conductivity and lithiophilicity, is designed by Pu et al. to realize bottom-growth deposition in LMBs, as shown in Fig. 9b.138 The nickel metal is initially plated on the Cu template, and the obtained Ni scaffold (BNS) is employed as the host framework after etching the Cu substrate. To prevent the risky Li deposition in the top region, alumina (Al2O3) with low electronic conductivity is further coated on the surface of BNS. The obtained Al2O3-modified BNS is then sputtered with a thin Au layer at the bottom of the framework. The insulating Al2O3 coating increases Re in the top region of DRS, and the Au layer decreases the local resistance of RLi at the bottom due to its low nucleation barrier. The inverse gradient of electron/ion conduction effectively guides Li+ deposition in the preferential regions of DRS, converting the deposition mode from top-growth to bottom-growth, as depicted in Fig. 9c. This is demonstrated by the color change in the top and bottom surfaces of the anode after Li plating of 5 mA h cm−2 in Fig. 9d. Benefited by the gradient structure of DRS, the symmetric Li–Li cells exhibit high cycling stability at both room or low temperatures (−15 °C).
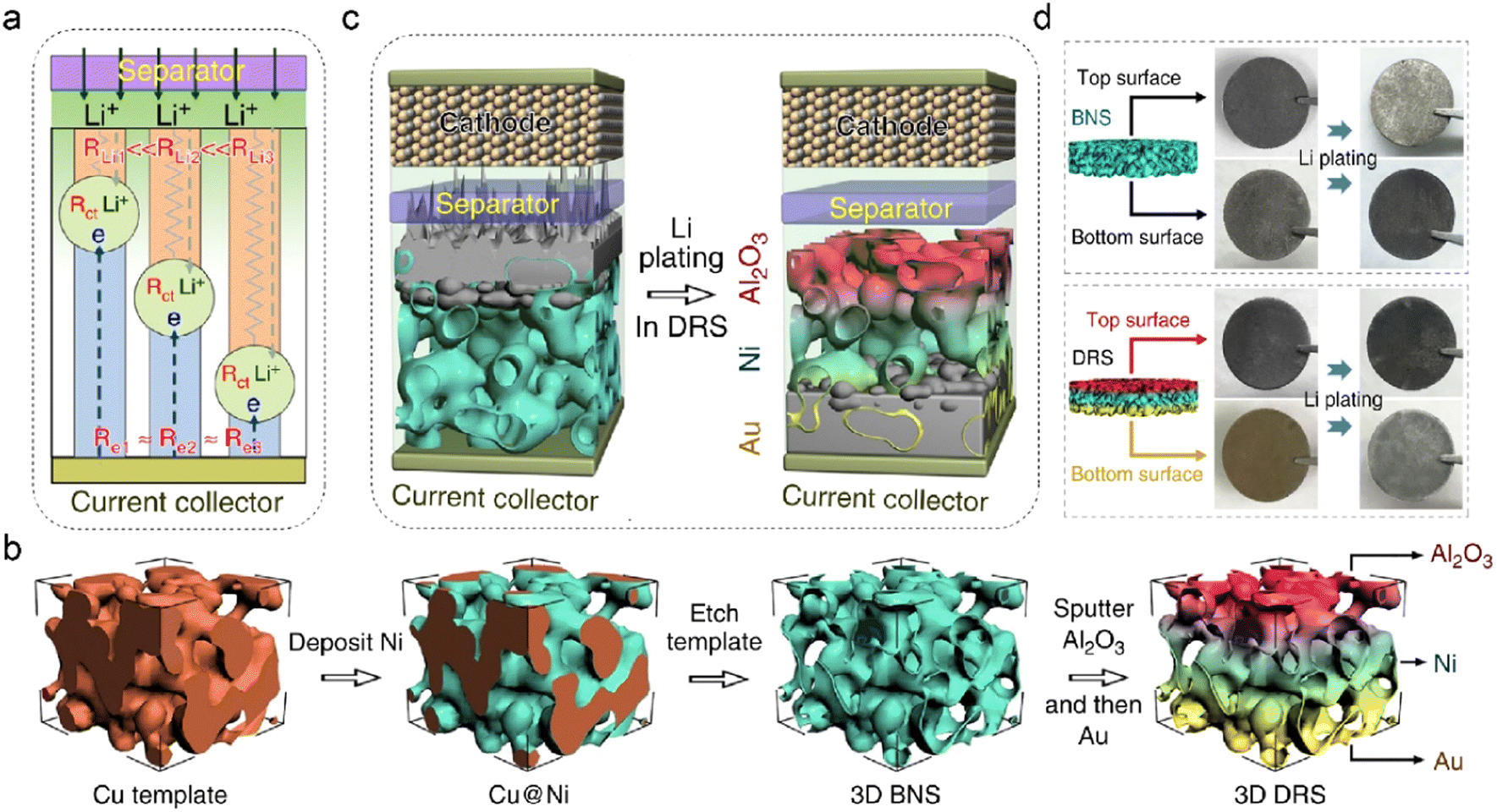 | ||
| Fig. 9 (a) Various transport resistances at the electrode/electrolyte interface. (b) Schematic illustration of fabricating DRS structures. (c) Different Li deposition modes in 3D Ni foam and DRS. (d) Photographs of Li plating on different anodes. Reproduced with permission. Copyright 2019, Springer Nature.138 | ||
4.2 Carbon-based three-dimensional inorganic frameworks (C-IOF)
3D carbon-based frameworks have achieved much attention due to their tunable properties, superior electron conductivity, and high surface area. They are considered as a promising alternative to heavy metal-based materials because of light mass density and low production cost.139,140 Due to the abundant carbon precursors, C-IOF can be prepared through diverse large-scale production methods, which exhibits more economical and environment-friendly advantages over other conductive materials. Various carbon matrices, including carbon nanofiber, carbon nanotube (CNT), and graphene, bring about numerous 3D nanostructures in the fabrication of C-IOF. Their unique structures play critical roles in reversible Li metal storage and electrochemical performance.141–144Graphene-supported materials have been widely reported to regulate metal deposition and construct 3D host structures for stable RMBs. The introduction of functional groups further provides active nucleation sites and modifies the metal affinities of the carbon matrix.145–147 Mu et al. develop a multichannel carbon matrix for high-rate and large-capacity ZMBs, which consists of nanofibers clusters of 3D nitrogen-doped graphene (GFs) and vertical graphene arrays (VGs).148 Longitudinal and radial 3D graphene-modified matrices (3D-LFGC and 3D-RFGC) are designed to investigate Zn plating/stripping behaviors in Fig. 10a. 3D-RFGC@Zn exhibits better cycling performance at high current density and areal capacities, compared to bare Zn foil and 3D-RFLFGC@Zn. The connected porous structures and radial channels in 3D-RFGC promote uniform Zn2+ flux and decrease local current density to inhibit Zn dendrites. The higher specific surface area and more zincophilic active sites benefit Zn nucleation and reversible plating/striping on anodes. Accordingly, 3D-RFGC@Zn anode survives for 3000 cycles with a coulombic efficiency of 99.67% under an ultrahigh current density of 120 mA cm−2. Besides, biomass-derived 3D-RFGC and woodblock-featured radial channels significantly reduce the cost and are sustainable for practical ZMBs, as illustrated in Fig. 10b. Dual heteroatom-doping is another effective way to modify graphene materials. Copper bromide and Br atoms are doped in the graphene on a conductive Cu skeleton (BGCF) to regulate Li nucleation and deposition.149 Enhanced Li+ adsorption is attributed to the strong interaction between Li+ ions and defect sites, and it guides the uniform Li+ flux in the Br-doped graphene. LiBr is derived from the reactions between CuBr and Li and provides a fast Li+ diffusion route for homogeneous nucleation sites. The uniformly dispersed Li seeds lead to granular Li morphology and suppress Li dendrite formation. Benefiting from the bromide intermediate in BGCF, the cells exhibit enhanced coulombic efficiency of 98.8% at 2.0 mA cm−2 after 300 cycles.
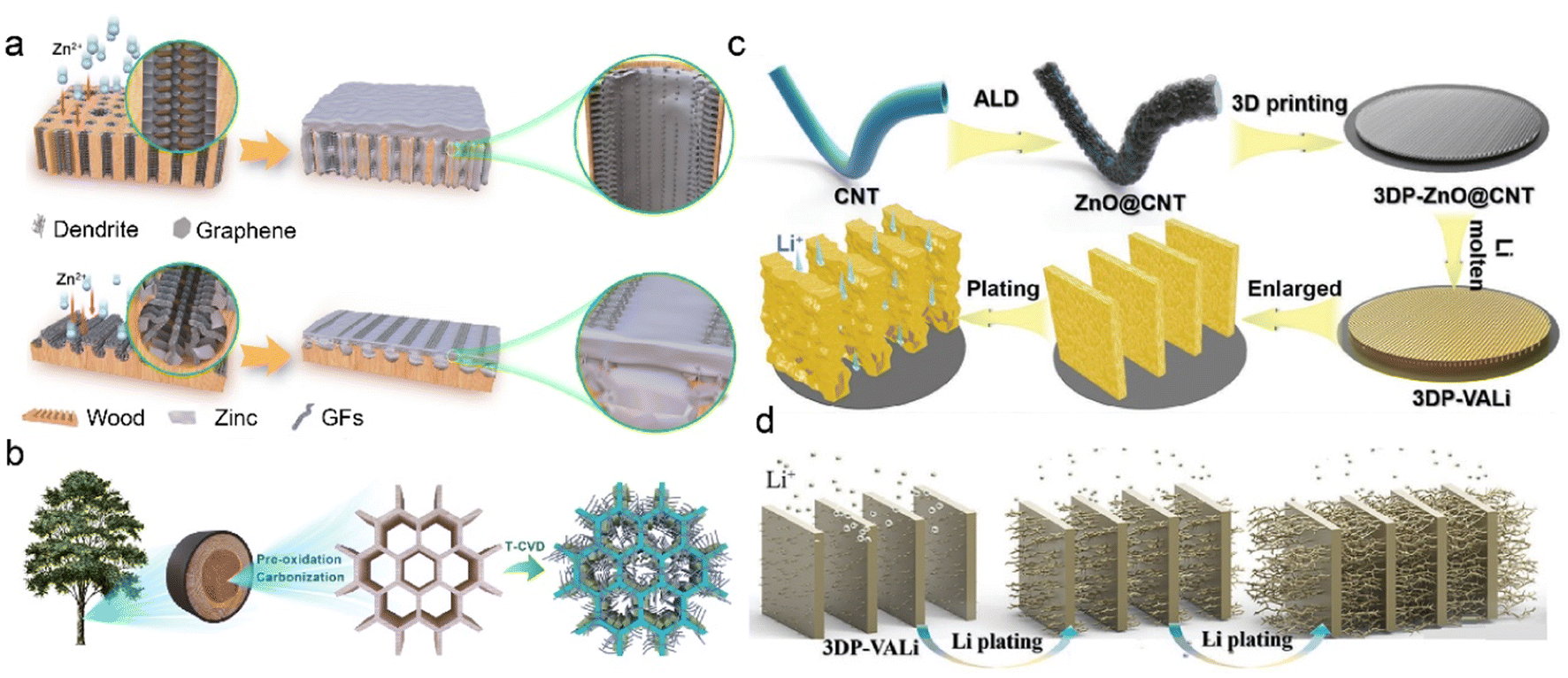 | ||
| Fig. 10 (a) Design of 3D longitudinal and radial graphene matrices. (b) Fabrication and structural features of 3D-LFGC and 3D-RFGC. Reproduced with permission. Copyright 2023, Springer Nature.148 (c) Schematic diagram of constructing 3DP-CNTLi via a 3D-printed method. (d) Illustration of Li deposition on the 3DP-CNTLi anode. Reproduced with permission. Copyright 2020, Wiley-VCH.152 | ||
3D CNT-based C-IOF is featured with high surface area and good electrical conductivity, which can be fabricated via a range of modifications and synthesis strategies.150,151 The CNT-modified Li anode (3DP-CNTLi) by a 3D-printed method is designed to achieve directional Li deposition in LMBs in Fig. 10c.152 ZnO layer with high lithiophilic capability is coated on CNT (ZnO@CNT) through atomic layer deposition (ALD). The mixture of aqueous binder and the obtained ZnO@CNT is printed into the carbon paper (CP), followed by freeze-drying treatment. Owing to the template effect induced by the anisotropic crystal growth of ice, the as-prepared 3DP-CNTLi has vertically aligned microwalls to accommodate Li deposits. This controlled microscale structure promotes fast Li+ diffusion and preferential deposition within microchannel walls instead of on the CP current collector. The unique microstructure of 3DP-CNTLi inhibits vertical dendrite growth at high Li plating capacity and high current density in Fig. 10d. The symmetric Li–Li cells based on 3DP-CNTLi exhibit high cycling stability over 3000 h at 1 mA cm−2, 1 mA h cm−2. Excellent performance is also verified at a high current density of 10 mA cm−2, 10 mA h cm−2 for 1500 h, and at a high capacity of 20 mA h cm−2, 5 mA cm−2 for 400 h.
Oxygenated functional groups often exhibit strong affinity with metal ions due to their higher electronegativity of oxygen atoms.153 They are introduced into the CNT network to afford active deposition sites and regulate metal nucleation barriers. The oxygen-functionalized CNT is employed as the sodiophilic interphase (Na@OCNT) for dendrite-free SMBs through chemical vapor deposition (CVD).154 The sodiophilic sites throughout the Na@OCNT network increase the interaction between Na+ and carbon substrate. This facilitates Na+ nucleation on Na@OCNT with reduced energy barriers and benefits subsequent Na growth. The continuous lateral Na deposition gradually fills the porous skeleton, compared to the vertical growth of Na dendrite on the conventional substrate in Fig. 11a. The obtained Na@OCNT anode affords revisable Na plating/striping over 6000 h with high coulombic efficiency of 99.7%. The resultant sodium-air batteries exhibit a quintuple enhancement in cycling performance compared to the original Na anode.
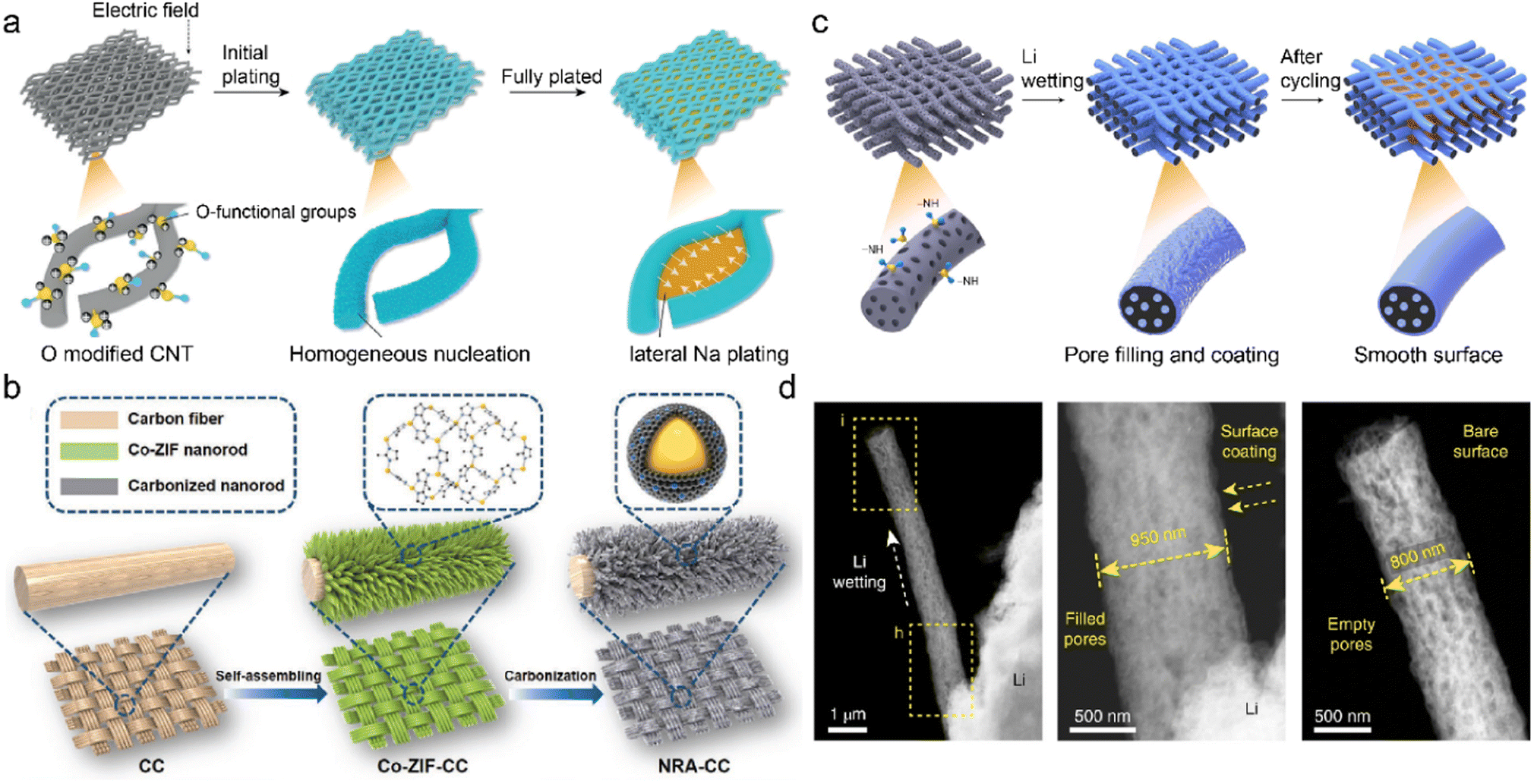 | ||
| Fig. 11 (a) Mechanism of Li deposition regulation on O-modified CNTs. Reproduced with permission. Copyright 2019, Wiley-VCH.154 (b) Fabrication of an MOF-derived carbon substrate. Reproduced with permission. Copyright 2020, Wiley-VCH.159 (c) Diagrams of constructing self-smoothing 3D mesoporous carbon fibers. (d) STEM images of single carbon fiber during Li plating. Reproduced with permission. Copyright 2019, Springer Nature.162 | ||
Carbon cloth is a promising substrate for C-IOF due to its good electrical conductivity, flexible frameworks, and high chemical stability. However, its application is restricted by poor lithiophilicity and insufficient porosity/surface area, which leads to high nucleation overpotentials and local current density during metal plating.155–157 Surface modification can create defects or nanostructures to increase the surface area as well as introduce lithiophilic components to enhance cation affinity. A 3D multifunctional anode (KNCC) is constructed by KOH-etching and nitrogen-doping of carbon cloth in a one-pot process.158 The redox etching of KOH promotes mesoporous defect formation and superior surface area in KNCC. The hydrothermal doping of nitrogen increases the substrate lithiophilicity and guides uniform Li deposition. It selectively catalyzes electrolyte decomposition on anodes to facilitate the formation of robust and insulating SEI. The obtained LiF/Li3N/N–O SEI suppresses electrolyte corrosion and prohibits Li dendrite growth. The Li–Cu cells thereby achieve extremely high coulombic efficiency of 99.8% after 1000 cycles at 2 mA h cm−2. Different precursors usually influence the effectiveness of functional groups and active sites in the carbon materials. A Co/N-modified carbon cloth is prepared from the carbonization of Co-ZIF nanorod arrays, as displayed in Fig. 11b.159 Abundant Co/N atoms in the MOF-derived lithiophilic nanorod provide adequate Li nucleation sites to reduce local current density and avoid Li+ accumulation. The 3D interconnected carbon cloth affords much space for a large amount of Li deposition, mitigating its volume change during repeated plating/stripping. These unique structures and active sites synergistically raise the cell endurance for high capacity/current density.
The energy density of metal batteries is limited by high N/P ratio, high E/C (electrolyte/cathode) ratio, and excessive metal anode.160,161 The 3D mesoporous carbon fibers with functional amine groups are designed for high-energy LMBs under realistic conditions. The incorporation of –NH groups regulates the substrate surface energy to improve the Li+ wettability of the framework, and simultaneously the strong interaction between electron-rich –NH groups and Li+ ions promotes Li deposition at defective sites of carbon nanofibers.162 The preferential Li nucleation in the pores/cavities induces a self-smoothing effect on the modified carbon nanofibers, as illustrated in Fig. 11c. This leads to the gradual smooth of the rough electrode surface, compared with the radial Li dendrite growth on the pristine carbon substrate in Fig. 11d. The full cells paired with nickel–manganese–cobalt oxide cathode (NMC 622/811) exhibit stable 200 cycles under realistic conditions, in which N/P and E/C ratio are smaller than 2 and 3 gA h−1, respectively. High energy densities of 353 and 381 W h kg−1 can be achieved in the NCM622/811 cells using the designed self-smoothing anode, respectively.
5. Challenges
Functional frameworks have been demonstrated to efficiently regulate interfacial chemistry in RMBs, resulting in a significant improvement in electrochemical performance, as shown in Table 1. However, their practical application is hindered by their structural instability and high production costs. There remain many challenges for framework materials that require to be addressed.| Materials | Fabrication methods | Anode cathode | Electrochemical performance | Improvements | Reference |
|---|---|---|---|---|---|
| UiO-66 | Solvothermal | Li–LiCoO2 | 3.0–4.5 V, charge 0.1C, discharge 0.5C, 80% retention after 250 cycles | Improving cation diffusion rate | 78 |
| Zr-MOCN | Photopolymerization | Li-NMC622 | 2.7–4.6 V, 0.2C, 94.6% retention after 100 cycles | Facilitating cation desolvation | 80 |
| HKUST-1 | GO-mediated self-assembly | Li–Li4Ti5O12 | 1.0–2.5 V, 5C, 95% retention after 250 cycles | Restricting anion migration | 86 |
| FCOF | Freeze–pump–thaw | Zn–MnO2 | 1.0–1.85 V, 5C, 92% retention after 1000 cycles | Regulating crystal plane growth | 97 |
| TPB-BD(OH)2-COF | Solid-state grinding and thermal curing | Li–LiNi0.5Mn1.5O4 | 1.0–1.85 V, 1C, 90.1% retention after 334 cycles | Hydrogen bond network formation | 98 |
| EB-COF: NO3@SnF2 | Solvothermal | Li-NCM811 | 3.0–4.5 V, 1C, 97.1% retention after 150 cycles | Promoting inorganic-rich SEI formation | 106 |
| ZnA zeolite | Hydrothermal with ion exchange | Zn–MnO2 | 0.8–1.8 V, 2 A g−1, 80.0% retention after 360 cycles | Repelling anions by sieving effect | 117 |
| Li2S@3DCu | Chronopotentiometry with sulfuration | Li–LiFePO4 | 2.2–4.0 V, 0.5C, 87.3% retention after 300 cycles | Balancing ion and electron transport | 134 |
| DRS | Electroplating and sputtering | Li–Cu | 1 mA cm−2, coulombic efficiency of 98.1% after 500 cycles | Constructing conductivity/lithiophilicity gradient | 138 |
| 3D-RFGC | Thermochemical vapor deposition | Zn–MnO2 | 1.0–1.8 V, 4 mA cm−2, 99.1% retention after 500 cycles | Decreasing local current density | 148 |
| 3DP-CNTLi | 3D-printing and ALD | Li–KB@S | 1.7–2.8 V, 1C, 81.0% retention after 250 cycles | Guiding nucleation in microchannels | 152 |
| Na@OCNT | Oxygen microwave plasma | Na–Cu | 1 mA cm−2, coulombic efficiency of 99.7% after 3000 cycles | Increasing Na+ affinity of substrate | 154 |
| KNCC | Hydrothermal | Li–LiFePO4 | 2.0–4.0 V, 1C, 86.0% retention after 500 cycles | Enhancing anionic decomposition and lithiophilicity | 158 |
(1) The reaction mechanisms of functional frameworks are still vague. The spatial confinement of porous PCFs has a great influence on ion diffusion, but the relationship between ionic conductivity and pore size of PCFs is not clear. The understanding of the ion transport mechanism in the channels of PCFs is constrained due to the challenging experimental detection of these processes. The cation desolvation and metal deposition at the electrode–electrolyte interfaces are complicated, which needs further investigation into the interactions among frameworks, solvents, anions, and cations.
(2) The framework structure tends to degrade during cycling. Numerous active species generated in electrolytes, such as superoxide and singlet oxygen in metal–oxygen batteries, have strong parasitic reactions with frameworks. This leads to the formation of insulating products in electrodes, which results in high interfacial resistance and fast performance decay. Meanwhile, the functional groups in frameworks could irreversibly oxidize at high charging voltages, and active metal centers in PCFs are prone to be reduced by metal anodes at low voltages. It urges to enhance the structural stability of framework materials for long-term cycling stability.
(3) The electrochemical performance is inferior under practical conditions. High-rate capabilities, high active mass loading, and less electrolyte usage are essential for high energy density of RMBs to realize practical application in vehicular power systems. Fast ion transport and reaction kinetics at interfaces are conducive to good rate performance. However, the insufficient ionic conductivity of framework materials results in sluggish ion diffusion and huge concentration polarization under large current densities. The high weight proportion of framework materials in the total mass of batteries decreases their energy density. In addition, the large surface area of frameworks increases the electrolyte–electrode contact to consume electrolytes for decreased initial coulombic efficiency.
(4) The production cost of framework materials is high. The functionalities of framework materials can be achieved only by the precise control of numerous variables in complicated synthetic steps. This hinders the high yields of functional frameworks with low impurity out of laboratories, giving rise to significant challenges for large-scale industrial production. In addition, the precursors of PCFs and IOFs are often specially designed, which further increases the production cost. Thus, scalable and sustainable synthetic methods should be developed to obtain cost-effective functional frameworks.
6. Summary and perspective
Rechargeable metal batteries have become an attractive energy storage option due to their high energy density. However, inferior side reactions and severe dendrite growth limit the practical application of high-capacity metal anode. PCFs and IOFs have been developed to regulate the interfacial chemistry by providing sufficient ion flux and adequate nucleation sites, respectively. Benefiting from periodic networks and high porosity, PCFs commonly serve as separators and protective coatings on anodes. The unique spatial confinement induces the selective transport of different components in electrolytes, which is dependent on the relative size of ions and framework channels. Moreover, the rich functional groups in the framework exhibit strong interactions with cations, anions, and solvents to facilitate fast desolvation kinetics on anodes. This gives rise to less solvent participation in the solvation structures, leading to more CIP and AGG structures in dilute electrolytes. The obtained PCF-modified electrolyte reduces solvent reactivity and inhibits physical contact with the electrode to prevent undesired solvent decomposition. Notably, uniform pore sizes and low electrical conductivity of PCFs are prerequisites to realizing the functions of cation desolvation and surface passivation. Besides, high mechanical and chemical stability are necessary for PCFs to prohibit dendrite growth and maintain self-stability.IOFs, including metal-based and carbon-based frameworks, are featured with high electron transport and large surface area. Their connected 3D networks can accommodate a large capacity of metal deposition and mitigate the repeated volume changes in anodes. However, conventional substrates have inadequate active nucleation sites, and it leads to aggregated ion concentration and vertical dendrite growth. Metallophilic nanoparticles, functional groups, and heteroatoms are incorporated into the IOFs to decrease the energy barrier of nucleation on the substrate. The uniformly distributed metal seeds reduce the local current density and promote dispersed ion diffusion on electrodes. This induces lateral metal growth along the substrate surface and suppresses unlimited dendrite growth, benefiting reversible Li plating/striping and cycling stability.
Numerous functional framework materials have been exploited to fabricate advanced electrolyte–electrode interfaces in metal batteries. However, practical applications of metal batteries are still restricted by moderate cycling performance, high production cost, and security concerns. More attention should be paid to the mechanisms of interfacial reactions, and several perspectives are proposed for the future development of functional framework materials.
(1) Increasing structure stability. Functional frameworks should be featured with high chemical stability against the active intermediates generated in electrolytes. The strong electrochemical stability is also essential for frameworks to achieve wide electrochemical windows. The robust skeletons enhance the mechanical strength of framework materials, which suppress structural degradation and metal dendrite growth. Therefore, selecting appropriate linkers or substrates before framework synthesis benefits electrode compatibility in the different battery systems.
(2) Optimizing fabrication parameters. The thickness and integrity of framework materials on electrodes are two crucial parameters, which significantly affect the cycling performance of batteries. For example, gaps between porous MOFs and COFs particles could weaken the selective transport of different species in electrolytes. Crack-free framework materials should be developed and uniformly coated on the anode/separator surface to fully realize sieving functions. Although protective layers are conducive to preventing side reactions, excessively thick coatings hinder mass transport and induce concentration polarization.
(3) Developing economical production methods. High-quality PCFs and IOFs can be efficiently synthesized in laboratories, but the current technologies such as chemical vapor deposition and magnetron sputtering are not suitable for large-scale production due to the strict conditions and complicated multi-step treatments. It is necessary to develop scalable and environment-friendly techniques to improve the yield of target materials.
(4) Testing batteries under harsh conditions. Practical anodes are expected to endure the high capacity and current density measurements without severe dendrite growth. The high-energy density of batteries should meet the requirement of high output voltage, high mass loading, and less electrolyte usage. Employing functional frameworks with low density is effective in reducing the proportion of inactive substances in the whole system. Moreover, it is essential to develop thermally stable frameworks for high- and low-temperature batteries.
(5) Exploiting high ion-conductive frameworks. A number of functional frameworks have been constructed to improve interfacial chemistry. However, their insufficient ionic conductivity limits interfacial kinetics. Pore size regulation in the frameworks can provide one- or two-dimensional channels, promoting the diffusion of solvated metal ions. High cation transference number can be realized by constructing defective sites in the secondary building units, which improve the interactions between anions and uncoordinated sites. Moreover, designing anionic skeleton is an effective strategy to enhance cation attraction to frameworks. This approach facilitates cation diffusion and reduces ion concentration polarization, finally contributing to improved interfacial chemistry.
(6) Deepening reaction mechanism understanding. Advanced in situ techniques are necessary to characterize molecular structure and redox reaction of functional frameworks. For example, in situ synchrotron powder X-ray diffraction (Synchrotron PXRD) and rotation electron diffraction (RED) of in situ Cryo-Transmission Electron Microscopy (Cryo-TEM) are utilized to determine the microcrystal structures of MOFs and COFs. Elemental valence state changes during the charge/discharge process can be monitored by in situ X-ray absorption near edge structure (XANES) spectra. Along with the advanced experimental characterizations, theoretical calculations are also considered as an important tool to study the reaction mechanisms. First-principles calculations are employed to explore a range of properties of framework materials at the atomic levels, including coordination structure, electronic structure, and molecular adsorption energy. However, the high time consumption of first-principles calculations limits its application in large-scale molecular systems. Therefore, classical molecular dynamics simulation, which is based on classical mechanics, is widely employed to investigate reactions between electrolytes and framework materials due to the large number of atoms in the systems.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22325902 and 52171215), and Haihe Laboratory of Sustainable Chemical Transformations.References
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- D. Du, Z. Zhu, K.-Y. Chan, F. Li and J. Chen, Chem. Soc. Rev., 2022, 51, 1846–1860 RSC.
- J. Geng, Y. Ni, Z. Zhu, Q. Wu, S. Gao, W. Hua, S. Indris, J. Chen and F. Li, J. Am. Chem. Soc., 2023, 145, 1564–1571 CrossRef CAS PubMed.
- Z. Jiang, Y. Huang, Z. Zhu, S. Gao, Q. Lv and F. Li, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2202835119 CrossRef CAS PubMed.
- W.-J. Kwak, Rosy, D. Sharon, C. Xia, H. Kim, L. R. Johnson, P. G. Bruce, L. F. Nazar, Y.-K. Sun, A. A. Frimer, M. Noked, S. A. Freunberger and D. Aurbach, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS PubMed.
- F. Li and J. Chen, Adv. Energy Mater., 2017, 7, 1602934 CrossRef.
- B. Acebedo, M. C. Morant-Miñana, E. Gonzalo, I. Ruiz De Larramendi, A. Villaverde, J. Rikarte and L. Fallarino, Adv. Energy Mater., 2023, 13, 2203744 CrossRef CAS.
- Y. Guo, H. Li and T. Zhai, Adv. Mater., 2017, 29, 1700007 CrossRef PubMed.
- H. Wang, D. Yu, C. Kuang, L. Cheng, W. Li, X. Feng, Z. Zhang, X. Zhang and Y. Zhang, Chem, 2019, 5, 313–338 CAS.
- W. Hu, J. Ju, N. Deng, M. Liu, W. Liu, Y. Zhang, L. Fan, W. Kang and B. Cheng, J. Mater. Chem. A, 2021, 9, 25750–25772 RSC.
- Y. Li, Z. Yu, J. Huang, Y. Wang and Y. Xia, Angew. Chem., Int. Ed., 2023,(62), e202309957 CAS.
- G. M. Hobold, J. Lopez, R. Guo, N. Minafra, A. Banerjee, Y. Shirley Meng, Y. Shao-Horn and B. M. Gallant, Nat. Energy, 2021, 6, 951–960 CrossRef CAS.
- S. Weng, X. Zhang, G. Yang, S. Zhang, B. Ma, Q. Liu, Y. Liu, C. Peng, H. Chen, H. Yu, X. Fan, T. Cheng, L. Chen, Y. Li, Z. Wang and X. Wang, Nat. Commun., 2023, 14, 4474 CrossRef.
- X. He, D. Bresser, S. Passerini, F. Baakes, U. Krewer, J. Lopez, C. T. Mallia, Y. Shao-Horn, I. Cekic-Laskovic, S. Wiemers-Meyer, F. A. Soto, V. Ponce, J. M. Seminario, P. B. Balbuena, H. Jia, W. Xu, Y. Xu, C. Wang, B. Horstmann, R. Amine, C.-C. Su, J. Shi, K. Amine, M. Winter, A. Latz and R. Kostecki, Nat. Rev. Mater., 2021, 6, 1036–1052 CrossRef CAS.
- Y. Huang, B. Wen, Z. Jiang and F. Li, Nano Res., 2022, 16, 8072–8081 CrossRef.
- D. Puthusseri, M. Wahid and S. Ogale, Energy Fuels, 2021, 35, 9187–9208 CrossRef CAS.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- F. Ding, W. Xu, G. L. Graff, J. Zhang, M. L. Sushko, X. Chen, Y. Shao, M. H. Engelhard, Z. Nie, J. Xiao, X. Liu, P. V. Sushko, J. Liu and J. G. Zhang, J. Am. Chem. Soc., 2013, 135, 4450–4456 CrossRef CAS PubMed.
- H. Wang, J. Liu, J. He, S. Qi, M. Wu, F. Li, J. Huang, Y. Huang and J. Ma, eScience, 2022, 2, 557–565 CrossRef.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Nat. Energy, 2019, 4, 269–280 CrossRef CAS.
- Z. Piao, R. Gao, Y. Liu, G. Zhou and H. M. Cheng, Adv. Mater., 2022, 35, 2206009 Search PubMed.
- Z. Tian, Y. Zou, G. Liu, Y. Wang, J. Yin, J. Ming and H. N. Alshareef, Adv. Sci., 2022, 9, 2201207 CrossRef CAS.
- N. Yao, X. Chen, Z. H. Fu and Q. Zhang, Chem. Rev., 2022, 122, 10970–11021 CrossRef CAS PubMed.
- Y. Huang, J. Geng, Z. Jiang, M. Ren, B. Wen, J. Chen and F. Li, Angew. Chem., Int. Ed., 2023, 62, e202306236 CrossRef CAS.
- G. Liu and W. Lu, J. Electrochem. Soc., 2017, 164, A1826–A1833 CrossRef CAS.
- A. Kushima, K. P. So, C. Su, P. Bai, N. Kuriyama, T. Maebashi, Y. Fujiwara, M. Z. Bazant and J. Li, Nano Energy, 2017, 32, 271–279 CrossRef CAS.
- H. Dai, X. Gu, J. Dong, C. Wang, C. Lai and S. Sun, Nat. Commun., 2020, 11, 643 CrossRef CAS.
- Q.-K. Zhang, X.-Q. Zhang, J. Wan, N. Yao, T.-L. Song, J. Xie, L.-P. Hou, M.-Y. Zhou, X. Chen, B.-Q. Li, R. Wen, H.-J. Peng, Q. Zhang and J.-Q. Huang, Nat. Energy, 2023, 8, 725–735 CrossRef CAS.
- P. Bai, J. Guo, M. Wang, A. Kushima, L. Su, J. Li, F. R. Brushett and M. Z. Bazant, Joule, 2018, 2, 2434–2449 CrossRef CAS.
- T. Naren, G. C. Kuang, R. Jiang, P. Qing, H. Yang, J. Lin, Y. Chen, W. Wei, X. Ji and L. Chen, Angew. Chem., Int. Ed., 2023, 62, e202305287 CrossRef CAS.
- L. Wang, N. Ren, Y. Yao, H. Yang, W. Jiang, Z. He, Y. Jiang, S. Jiao, L. Song, X. Wu, Z. S. Wu and Y. Yu, Angew. Chem., Int. Ed., 2023, 62, e202214372 CrossRef CAS PubMed.
- D. R. Ely and R. E. García, J. Electrochem. Soc., 2013, 160, A662–A668 CrossRef CAS.
- X. R. Chen, Y. X. Yao, C. Yan, R. Zhang, X. B. Cheng and Q. Zhang, Angew. Chem., Int. Ed., 2020, 132, 7817–7821 CrossRef.
- S. Zhang, R. Li, N. Hu, T. Deng, S. Weng, Z. Wu, D. Lu, H. Zhang, J. Zhang, X. Wang, L. Chen, L. Fan and X. Fan, Nat. Commun., 2022, 13, 5431 CrossRef CAS PubMed.
- Q. Wang, L. Ren, T. Lu, S. Wang, Z. Jiang, I. A. Chandio, L. Guan, L. Hou, H. Du, H. Wei, X. Liu, C. Yang, Y. Wei, W. Liu and H. Zhou, ACS Energy Lett., 2023, 8, 4441–4464 CrossRef CAS.
- Y. He, Y. Qiao, Z. Chang and H. Zhou, Energy Environ. Sci., 2019, 12, 2327–2344 RSC.
- S. Ni, S. Tan, Q. An and L. Mai, J. Energy Chem., 2020, 44, 73–89 CrossRef.
- Z. Miao, F. Zhang, H. Zhao, M. Du, H. Li, H. Jiang, W. Li, Y. Sang, H. Liu and S. Wang, Adv. Funct. Mater., 2022, 32, 2111635 CrossRef CAS.
- D. Lin, Y. Liu, A. Pei and Y. Cui, Nano Res., 2017, 10, 4003–4026 CrossRef CAS.
- S. Li, J. Huang, Y. Cui, S. Liu, Z. Chen, W. Huang, C. Li, R. Liu, R. Fu and D. Wu, Nat. Nanotechnol., 2022, 17, 613–621 CrossRef CAS PubMed.
- Z. Li, K. Zhu, P. Liu and L. Jiao, Adv. Energy Mater., 2022, 12, 2100359 CrossRef CAS.
- Z. Chang, Y. Qiao, H. Yang, X. Cao, X. Zhu, P. He and H. Zhou, Angew. Chem., Int. Ed., 2021, 60, 15572–15581 CrossRef CAS PubMed.
- T. Wei, J. Lu, M. Wang, C. Sun, Q. Zhang, S. Wang, Y. Zhou, D. Chen and Y. Q. Lan, Chin. J. Chem., 2023, 41, 1861–1874 CrossRef CAS.
- H. Zhao, L. Sheng, L. Wang, H. Xu and X. He, Energy Storage Mater., 2020, 33, 360–381 CrossRef.
- L. Zhang and Y. Hou, Adv. Energy Mater., 2023, 13, 2204378 CrossRef CAS.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS.
- B. Li, H. M. Wen, Y. Cui, W. Zhou, G. Qian and B. Chen, Adv. Mater., 2016, 28, 8819–8860 CrossRef CAS PubMed.
- X. Li, X. Wang, L. Ma and W. Huang, Adv. Energy Mater., 2022, 12, 2202068 CrossRef CAS.
- J. Cao, G. Qian, X. Lu and X. Lu, Small, 2023, 19, 2205653 CrossRef CAS.
- H. Sun, J. Zhu, D. Baumann, L. Peng, Y. Xu, I. Shakir, Y. Huang and X. Duan, Nat. Rev. Mater., 2018, 4, 45–60 CrossRef.
- S. Park, H. J. Jin and Y. S. Yun, Adv. Mater., 2020, 32, 2002193 CrossRef CAS.
- Y. Zhang, C. Wang, G. Pastel, Y. Kuang, H. Xie, Y. Li, B. Liu, W. Luo, C. Chen and L. Hu, Adv. Energy Mater., 2018, 8, 1800635 CrossRef.
- F. Ren, Z. Lu, H. Zhang, L. Huai, X. Chen, S. Wu, Z. Peng, D. Wang and J. Ye, Adv. Funct. Mater., 2018, 28, 1805638 CrossRef.
- D. Li, B. Chen, H. Hu and W. Y. Lai, Adv. Sustainable Syst., 2022, 6, 2200010 CrossRef CAS.
- X. Fu, H. Duan, L. Zhang, Y. Hu and Y. Deng, Adv. Funct. Mater., 2023,(33), 2308022 CrossRef CAS.
- P. Xue, S. Liu, X. Shi, C. Sun, C. Lai, Y. Zhou, D. Sui, Y. Chen and J. Liang, Adv. Mater., 2018, 30, e1804165 CrossRef.
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M.-H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed.
- B. Thirumalraj, T. T. Hagos, C. J. Huang, M. A. Teshager, J. H. Cheng, W. N. Su and B. J. Hwang, J. Am. Chem. Soc., 2019, 141, 18612–18623 CrossRef CAS PubMed.
- J. X. K. Zheng, J. Yin, T. Tang and L. A. Archer, ACS Energy Lett., 2023, 8, 2113–2121 CrossRef CAS.
- J. Xiao, Science, 2019, 366, 426–427 CrossRef CAS PubMed.
- D. Wang, W. Zhang, W. Zheng, X. Cui, T. Rojo and Q. Zhang, Adv. Sci., 2017, 4, 1600168 CrossRef PubMed.
- Z. Chang, H. Yang, Y. Qiao, X. Zhu, P. He and H. Zhou, Adv. Mater., 2022, 34, 2201339 CrossRef CAS PubMed.
- Z. Yu, P. E. Rudnicki, Z. Zhang, Z. Huang, H. Celik, S. T. Oyakhire, Y. Chen, X. Kong, S. C. Kim, X. Xiao, H. Wang, Y. Zheng, G. A. Kamat, M. S. Kim, S. F. Bent, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2022, 7, 94–106 CrossRef CAS.
- J. Wu, Z. Gao, Y. Tian, Y. Zhao, Y. Lin, K. Wang, H. Guo, Y. Pan, X. Wang, F. Kang, N. Tavajohi, X. Fan and B. Li, Adv. Mater., 2023, 35, e2303347 CrossRef PubMed.
- G. Li, Z. Liu, Q. Huang, Y. Gao, M. Regula, D. Wang, L.-Q. Chen and D. Wang, Nat. Energy, 2018, 3, 1076–1083 CrossRef CAS.
- L. Kong, H. Peng, J. Huang and Q. Zhang, Nano Res., 2017, 10, 4027–4054 CrossRef CAS.
- X. Zhang, Y. Yang and Z. Zhou, Chem. Soc. Rev., 2020, 49, 3040–3071 RSC.
- Y. Zhang, Z. Zeng, S. Yang, Y. Zhang, Y. Ma and Z. Wang, Energy Storage Mater., 2023, 57, 557–567 CrossRef.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H. C. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef PubMed.
- E. W. C. Spotte-Smith, R. L. Kam, D. Barter, X. Xie, T. Hou, S. Dwaraknath, S. M. Blau and K. A. Persson, ACS Energy Lett., 2022, 7, 1446–1453 CrossRef CAS.
- S.-J. Yang, N. Yao, X.-Q. Xu, F.-N. Jiang, X. Chen, H. Liu, H. Yuan, J.-Q. Huang and X.-B. Cheng, J. Mater. Chem. A, 2021, 9, 19664–19668 RSC.
- Y. Gao, Z. Yan, J. L. Gray, X. He, D. Wang, T. Chen, Q. Huang, Y. C. Li, H. Wang, S. H. Kim, T. E. Mallouk and D. Wang, Nat. Mater., 2019, 18, 384–389 CrossRef CAS PubMed.
- S.-J. Zhang, Z.-W. Yin, Z.-Y. Wu, D. Luo, Y.-Y. Hu, J.-H. You, B. Zhang, K.-X. Li, J.-W. Yan, X.-R. Yang, X.-D. Zhou, S. Zanna, P. Marcus, F. Pan, J. Światowska, S.-G. Sun, Z. Chen and J.-T. Li, Energy Storage Mater., 2021, 40, 337–346 CrossRef.
- R. Guo, D. Wang, L. Zuin and B. M. Gallant, ACS Energy Lett., 2021, 6, 877–885 CrossRef CAS.
- X. Zhou, Q. Zhang, Z. Zhu, Y. Cai, H. Li and F. Li, Angew. Chem., Int. Ed., 2022, 61, e202205045 CrossRef CAS PubMed.
- R. Xu, X.-B. Cheng, C. Yan, X.-Q. Zhang, Y. Xiao, C.-Z. Zhao, J.-Q. Huang and Q. Zhang, Matter, 2019, 1, 317–344 CrossRef.
- L. Chen, J. Lai, Z. Li, H. Zou, J. Yang, K. Ding, Y.-P. Cai and Q. Zheng, Commun. Mater., 2023, 4, 18 CrossRef CAS.
- Y. Xu, L. Gao, L. Shen, Q. Liu, Y. Zhu, Q. Liu, L. Li, X. Kong, Y. Lu and H. B. Wu, Matter, 2020, 3, 1685–1700 CrossRef.
- W. Xin, J. Xiao, J. Li, L. Zhang, H. Peng, Z. Yan and Z. Zhu, Energy Storage Mater., 2023, 56, 76–86 CrossRef.
- L. Sheng, Q. Wang, X. Liu, H. Cui, X. Wang, Y. Xu, Z. Li, L. Wang, Z. Chen, G.-L. Xu, J. Wang, Y. Tang, K. Amine, H. Xu and X. He, Nat. Commun., 2022, 13, 172 CrossRef CAS PubMed.
- D. Xu, X. Ren, Y. Xu, Y. Wang, S. Zhang, B. Chen, Z. Chang, A. Pan and H. Zhou, Adv. Sci., 2023, 10, 2303773 CrossRef CAS PubMed.
- X. Luo, Q. Nian, Z. Wang, B.-Q. Xiong, S. Chen, Y. Li and X. Ren, Chem. Eng. J., 2023, 455, 140510 CrossRef CAS.
- Q. Wang, C. Zhao, S. Wang, J. Wang, M. Liu, S. Ganapathy, X. Bai, B. Li and M. Wagemaker, J. Am. Chem. Soc., 2022, 144, 21961–21971 CrossRef CAS PubMed.
- M. Qin, M. Liu, Z. Zeng, Q. Wu, Y. Wu, H. Zhang, S. Lei, S. Cheng and J. Xie, Adv. Energy Mater., 2022, 12, 2201801 CrossRef CAS.
- R. Xu, C. Yan and J.-Q. Huang, Trends Chem., 2021, 3, 5–14 CrossRef CAS.
- S. Bai, Y. Sun, J. Yi, Y. He, Y. Qiao and H. Zhou, Joule, 2018, 2, 2117–2132 CrossRef CAS.
- S. Tan, J.-M. Kim, A. Corrao, S. Ghose, H. Zhong, N. Rui, X. Wang, S. Senanayake, B. J. Polzin, P. Khalifah, J. Xiao, J. Liu, K. Xu, X.-Q. Yang, X. Cao and E. Hu, Nat. Nanotechnol., 2023, 18, 243–249 CrossRef CAS PubMed.
- S. Ko, T. Obukata, T. Shimada, N. Takenaka, M. Nakayama, A. Yamada and Y. Yamada, Nat. Energy, 2022, 7, 1217–1224 CrossRef CAS.
- Y. Yu, G. Huang, J. Du, J. Wang, Y. Wang, Z. Wu and X. Zhang, Energy Environ. Sci., 2020, 13, 3075–3081 RSC.
- J. Zheng, S. Chen, W. Zhao, J. Song, M. H. Engelhard and J.-G. Zhang, ACS Energy Lett., 2018, 3, 315–321 CrossRef CAS.
- J. Zheng, X. Fan, G. Ji, H. Wang, S. Hou, K. C. DeMella, S. R. Raghavan, J. Wang, K. Xu and C. Wang, Nano Energy, 2018, 50, 431–440 CrossRef CAS.
- H. Yang, Y. Qiao, Z. Chang, P. He and H. Zhou, Angew. Chem., Int. Ed., 2021, 60, 17726–17734 CrossRef CAS PubMed.
- Y. Li, W. Chen, T. Lei, H. Xie, A. Hu, F. Wang, J. Huang, X. Wang, Y. Hu, C. Yang and J. Xiong, Energy Storage Mater., 2023, 59, 102765 CrossRef.
- Q. Wu, Y. Zheng, X. Guan, J. Xu, F. Cao and C. Li, Adv. Funct. Mater., 2021,(31), 2101034 CrossRef CAS.
- A. Ghafari, A. G. Yeklangi, F. A. Sima and S. Akbari, J. Appl. Electrochem., 2024,(54), 215–243 CrossRef CAS.
- S.-W. Ke, Y. Wang, J. Su, K. Liao, S. Lv, X. Song, T. Ma, S. Yuan, Z. Jin and J.-L. Zuo, J. Am. Chem. Soc., 2022, 144, 8267–8277 CrossRef CAS PubMed.
- Z. Zhao, R. Wang, C. Peng, W. Chen, T. Wu, B. Hu, W. Weng, Y. Yao, J. Zeng, Z. Chen, P. Liu, Y. Liu, G. Li, J. Guo, H. Lu and Z. Guo, Nat. Commun., 2021, 12, 6606 CrossRef CAS PubMed.
- Y. Yang, S. Yao, Z. Liang, Y. Wen, Z. Liu, Y. Wu, J. Liu and M. Zhu, ACS Energy Lett., 2022, 7, 885–896 CrossRef CAS.
- Y. Wen, J. Ding, Y. Yang, X. Lan, J. Liu, R. Hu and M. Zhu, Adv. Funct. Mater., 2022, 32, 2109377 CrossRef CAS.
- S. Liu, J. Xia, W. Zhang, H. Wan, J. Zhang, J. Xu, J. Rao, T. Deng, S. Hou, B. Nan and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202210522 CrossRef CAS PubMed.
- Z. Piao, P. Xiao, R. Luo, J. Ma, R. Gao, C. Li, J. Tan, K. Yu, G. Zhou and H. M. Cheng, Adv. Mater., 2022, 34, e2108400 CrossRef PubMed.
- X. Li, R. Zhao, Y. Fu and A. Manthiram, eScience, 2021, 1, 108–123 CrossRef.
- L. Xia, H. Miao, C. Zhang, G. Z. Chen and J. Yuan, Energy Storage Mater., 2021, 38, 542–570 CrossRef.
- Y. Wang, Z. Li, Y. Hou, Z. Hao, Q. Zhang, Y. Ni, Y. Lu, Z. Yan, K. Zhang, Q. Zhao, F. Li and J. Chen, Chem. Soc. Rev., 2023, 52, 2713–2763 RSC.
- D. J. Yoo, Q. Liu, O. Cohen, M. Kim, K. A. Persson and Z. Zhang, Adv. Energy Mater., 2023, 13, 2204182 CrossRef CAS.
- Y. Wen, J. Ding, J. Liu, M. Zhu and R. Hu, Energy Environ. Sci., 2023, 16, 2957–2967 RSC.
- S. Yao, Y. Yang, Z. Liang, J. Chen, J. Ding, F. Li, J. Liu, L. Xi, M. Zhu and J. Liu, Adv. Funct. Mater., 2023, 33, 2212466 CrossRef CAS.
- Z. Li, L. Sun, L. Zhai, K. S. Oh, J. M. Seo, C. Li, D. Han, J. B. Baek and S. Y. Lee, Angew. Chem., Int. Ed., 2023, 62, e202303086 CrossRef PubMed.
- Y. Li, L. Li and J. Yu, Chem, 2017, 3, 928–949 CAS.
- M. E. Davis and R. F. Lobo, Chem. Mater., 1992, 4, 756–768 CrossRef CAS.
- L. H. Chen, M. H. Sun, Z. Wang, W. Yang, Z. Xie and B. L. Su, Chem. Rev., 2020, 120, 11194–11294 CrossRef CAS PubMed.
- Y. Li and J. Yu, Nat. Rev. Mater., 2021, 6, 1156–1174 CrossRef CAS.
- D. Kerstens, B. Smeyers, J. Van Waeyenberg, Q. Zhang, J. Yu and B. F. Sels, Adv. Mater., 2020, 32, 2004690 CrossRef CAS PubMed.
- M. Ravi, V. L. Sushkevich and J. A. Van Bokhoven, Nat. Mater., 2020, 19, 1047–1056 CrossRef CAS PubMed.
- J. Du, X. Duan, W. Wang, G. Li, C. Li, Y. Tan, M. Wan, Z. W. Seh, L. Wang and Y. Sun, Nano Lett., 2023, 23, 3369–3376 CrossRef CAS PubMed.
- J. Zhu, Z. Bie, X. Cai, Z. Jiao, Z. Wang, J. Tao, W. Song and H. J. Fan, Adv. Mater., 2022, 34, e2207209 CrossRef PubMed.
- R. Zhao, J. Yang, X. Han, Y. Wang, Q. Ni, Z. Hu, C. Wu and Y. Bai, Adv. Energy Mater., 2023, 13, 2203542 CrossRef CAS.
- H. Yang, Y. Qiao, Z. Chang, H. Deng, X. Zhu, R. Zhu, Z. Xiong, P. He and H. Zhou, Adv. Mater., 2021, 33, 2102415 CrossRef CAS PubMed.
- Z. Piao, H. R. Ren, G. Lu, K. Jia, J. Tan, X. Wu, Z. Zhuang, Z. Han, C. Li, R. Gao, X. Tao, G. Zhou and H. M. Cheng, Angew. Chem., Int. Ed., 2023, 62, e202300966 CrossRef CAS PubMed.
- L. Suo, W. Xue, M. Gobet, S. G. Greenbaum, C. Wang, Y. Chen, W. Yang, Y. Li and J. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 1156–1161 CrossRef CAS PubMed.
- X. Cao, H. Jia, W. Xu and J.-G. Zhang, J. Electrochem. Soc., 2021, 168, 010522 CrossRef CAS.
- H. Jia, Z. Yang, Y. Xu, P. Gao, L. Zhong, D. J. Kautz, D. Wu, B. Fliegler, M. H. Engelhard, B. E. Matthews, B. Broekhuis, X. Cao, J. Fan, C. Wang, F. Lin and W. Xu, Adv. Energy Mater., 2022, 13, 2203144 CrossRef.
- H. Zhao, J. Wang, H. Shao, K. Xu and Y. Deng, Energy Environ. Mater., 2021, 5, 327–336 CrossRef.
- Z. Lu, H. Yang, Y. Guo, P. He, S. Wu, Q. H. Yang and H. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202206340 CrossRef CAS PubMed.
- Q. Pang, X. Liang, A. Shyamsunder and L. F. Nazar, Joule, 2017, 1, 871–886 CrossRef CAS.
- P. Bai, J. Li, F. R. Brushett and M. Z. Bazant, Energy Environ. Sci., 2016, 9, 3221–3229 RSC.
- Z. Wu, H. Liu, J. Holoubek, C. Anderson, L. Shi, H. Khemchandani, D. Lu, D. Liu, C. Niu, J. Xiao and P. Liu, ACS Energy Lett., 2022, 7, 2701–2710 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, npj Comput. Mater., 2018, 4, 15 CrossRef.
- H. Ye, Y. Zhang, Y.-X. Yin, F.-F. Cao and Y.-G. Guo, ACS Cent. Sci., 2020, 6, 661–671 CrossRef CAS PubMed.
- Y. Feng, C. Zhang, X. Jiao, Z. Zhou and J. Song, Energy Storage Mater., 2020, 25, 172–179 CrossRef.
- X. Guan, A. Wang, S. Liu, G. Li, F. Liang, Y. W. Yang, X. Liu and J. Luo, Small, 2018, 14, e1801423 CrossRef PubMed.
- J. Wang, L. Li, H. Hu, H. Hu, Q. Guan, M. Huang, L. Jia, H. Adenusi, K. V. Tian, J. Zhang, S. Passerini and H. Lin, ACS Nano, 2022, 16, 17729–17760 CrossRef CAS.
- W. Yang, W. Yang, L. Dong, G. Shao, G. Wang and X. Peng, Nano Energy, 2021, 80, 105563 CrossRef CAS.
- P. Zhai, Y. Wei, J. Xiao, W. Liu, J. Zuo, X. Gu, W. Yang, S. Cui, B. Li, S. Yang and Y. Gong, Adv. Energy Mater., 2020, 10, 1903339 CrossRef CAS.
- J. Wu, Z. Ju, X. Zhang, A. C. Marschilok, K. J. Takeuchi, H. Wang, E. S. Takeuchi and G. Yu, Adv. Mater., 2022, 34, 2202780 CrossRef CAS PubMed.
- Y. Liu, Y. Li, J. Sun, Z. Du, X. Hu, J. Bi, C. Liu, W. Ai and Q. Yan, Nano Research Energy, 2023, 2, e9120048 CrossRef.
- L. Zhang, H. Zheng, B. Liu, Q. Xie, Q. Chen, L. Lin, J. Lin, B. Qu, L. Wang and D.-L. Peng, J. Energy Chem., 2021, 57, 392–400 CrossRef CAS.
- J. Pu, J. Li, K. Zhang, T. Zhang, C. Li, H. Ma, J. Zhu, P. V. Braun, J. Lu and H. Zhang, Nat. Commun., 2019, 10, 1896 CrossRef PubMed.
- H. Jiang, P. S. Lee and C. Li, Energy Environ. Sci., 2013, 6, 41–53 RSC.
- H. Liu, X. Liu, W. Li, X. Guo, Y. Wang, G. Wang and D. Zhao, Adv. Energy Mater., 2017, 7, 1700283 CrossRef.
- H. Tian, T. Wang, F. Zhang, S. Zhao, S. Wan, F. He and G. Wang, J. Mater. Chem. A, 2018, 6, 12816–12841 RSC.
- J. Han, I. Johnson and M. Chen, Adv. Mater., 2022, 34, 2108750 CrossRef CAS PubMed.
- C. Jin, J. Nai, O. Sheng, H. Yuan, W. Zhang, X. Tao and X. W. Lou, Energy Environ. Sci., 2021, 14, 1326–1379 RSC.
- S. Zhang, G. Wang, B. Wang, J. Wang, J. Bai and H. Wang, Adv. Funct. Mater., 2020, 30, 2001592 CrossRef CAS.
- Z. Wang, H. Gao, Q. Zhang, Y. Liu, J. Chen and Z. Guo, Small, 2019, 15, 1803858 CrossRef.
- Q. Xu, X. Yang, M. Rao, D. Lin, K. Yan, R. Du, J. Xu, Y. Zhang, D. Ye, S. Yang, G. Zhou, Y. Lu and Y. Qiu, Energy Storage Mater., 2020, 26, 73–82 CrossRef.
- Q. Yun, Y. Ge, B. Chen, L. Li, Q. Wa, H. Long and H. Zhang, Adv. Funct. Mater., 2022, 32, 2202319 CrossRef CAS.
- Y. Mu, Z. Li, B.-K. Wu, H. Huang, F. Wu, Y. Chu, L. Zou, M. Yang, J. He, L. Ye, M. Han, T. Zhao and L. Zeng, Nat. Commun., 2023, 14, 4205 CrossRef CAS PubMed.
- H. Duan, J. Zhang, X. Chen, X.-D. Zhang, J.-Y. Li, L.-B. Huang, X. Zhang, J.-L. Shi, Y.-X. Yin, Q. Zhang, Y.-G. Guo, L. Jiang and L.-J. Wan, J. Am. Chem. Soc., 2018, 140, 18051–18057 CrossRef CAS PubMed.
- S. Zhu, J. Sheng, Y. Chen, J. Ni and Y. Li, Natl. Sci. Rev., 2021, 8, nwaa261 CrossRef CAS PubMed.
- F. Tao, Y. Liu, X. Ren, A. Jiang, H. Wei, X. Zhai, F. Wang, H.-R. Stock, S. Wen and F. Ren, J. Alloys Compd., 2021, 873, 159742 CrossRef CAS.
- X. Gao, X. Yang, K. Adair, X. Li, J. Liang, Q. Sun, Y. Zhao, R. Li, T. K. Sham and X. Sun, Adv. Energy Mater., 2020, 10, 1903753 CrossRef CAS.
- A. R. Deline, B. P. Frank, C. L. Smith, L. R. Sigmon, A. N. Wallace, M. J. Gallagher, D. G. Goodwin, D. P. Durkin and D. H. Fairbrother, Chem. Rev., 2020, 120, 11651–11697 CrossRef CAS PubMed.
- L. Ye, M. Liao, T. Zhao, H. Sun, Y. Zhao, X. Sun, B. Wang and H. Peng, Angew. Chem., Int. Ed., 2019, 58, 17054–17060 CrossRef CAS PubMed.
- H. Shi, G. Wen, Y. Nie, G. Zhang and H. Duan, Nanoscale, 2020, 12, 5261–5285 RSC.
- C. Guan, W. Zhao, Y. Hu, Q. Ke, X. Li, H. Zhang and J. Wang, Adv. Energy Mater., 2016, 6, 1601034 CrossRef.
- S. Yang, Y. Cheng, X. Xiao and H. Pang, Chem. Eng. J., 2020, 384, 123294 CrossRef CAS.
- F. Cheng, X. Yang, O. Ka, L. Wen, X. Wang and W. Lu, J. Mater. Chem. A, 2023, 11, 4205–4219 RSC.
- T. S. Wang, X. Liu, Y. Wang and L. Z. Fan, Adv. Funct. Mater., 2020, 31, 2001973 CrossRef.
- X. Ren, L. Zou, X. Cao, M. H. Engelhard, W. Liu, S. D. Burton, H. Lee, C. Niu, B. E. Matthews, Z. Zhu, C. Wang, B. W. Arey, J. Xiao, J. Liu, J.-G. Zhang and W. Xu, Joule, 2019, 3, 1662–1676 CrossRef CAS.
- A. Mohammadi, S. Djafer, S. Sayegh, A. J. Naylor, M. Bechelany, R. Younesi, L. Monconduit and L. Stievano, Chem. Mater., 2023, 35, 2381–2393 CrossRef CAS.
- C. Niu, H. Pan, W. Xu, J. Xiao, J. G. Zhang, L. Luo, C. Wang, D. Mei, J. Meng, X. Wang, Z. Liu, L. Mai and J. Liu, Nat. Nanotechnol., 2019, 14, 594–601 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |