DOI:
10.1039/D3TA06988E
(Paper)
J. Mater. Chem. A, 2024,
12, 5319-5330
Electrosynthesis of ruthenium nanocluster incorporated nickel diselenide for efficient overall water splitting†
Received
13th November 2023
, Accepted 20th January 2024
First published on 2nd February 2024
Abstract
Design of effective catalyst activation strategies that enable efficient electrocatalytic activity towards overall water splitting is necessary for the development of clean energy conversion technologies. Incorporation of metal nanoclusters effectively increases the active site exposure leading to enhanced electrocatalytic activity. Here, we present an energy-efficient and scalable single-step approach for the electrosynthesis of ruthenium nanocluster decorated nickel diselenide catalysts for high-performance and stable alkaline water splitting application. The catalyst exhibits exceptional bifunctional activity for both hydrogen and oxygen evolution, and we demonstrate remarkable full-cell performance with a cell potential of 1.45 V to deliver a current density of 10 mA cm−2, along with impressive long-term stability over 400 h. Density functional theory (DFT) calculations are carried out, which demonstrate that the ruthenium nanocluster decoration facilitates the exothermic dissociation of water into H and OH species, while also optimizing the adsorption energies of H+ and OH− when compared to bare NiSe2. The present approach could be extended to a variety of catalytically active materials that would potentially be of interest for alkaline water-splitting applications.
Introduction
Hydrogen is considered as a promising energy carrier to replace the existing energy infrastructure, owing to its high gravimetric energy density (120 kJ kg−1) and zero carbon emission features.1,2 There are several methods for hydrogen production, based on the applied energy sources, such as high-temperature thermolysis, coal refining, electrocatalysis, photocatalysis, etc.3–8 The electrochemical water cycle, which involves electrolysis of water for H2 generation and subsequent utilization in fuel cells, is a significant approach towards a sustainable energy future. The energy conversion process involves two half-cell reactions, namely, the hydrogen evolution reaction (HER) happening at the cathode side, and the oxygen evolution reaction (OER) happening at the anode side.9–12 Owing to the multistep reaction processes in alkaline media, the HER and OER suffer from high activation barriers, resulting in large overpotentials leading to the slow kinetics of water electrolysis.13,14 Noble metal (Pt, Ir)-based electrocatalysts are currently regarded as the most active materials for the HER and OER. However, their high cost, limited availability, and poor stability hinder their large-scale applications.15–17 Therefore it is highly essential to develop cost-effective, highly active, and highly stable commercial bifunctional electrocatalysts for overall water splitting.18,19
Currently, transition metal compounds including chalcogenides, oxides, hydroxides, carbides, phosphides, etc., are studied as potential candidates for HER and OER activity towards water splitting.20–26 Recently, transition metal selenides have gained much more attention because of their d-orbital configuration and better conductivity.27–30 However, their electrochemical activity is still far from that of the benchmark catalysts because of their high adsorption energy, low number of electrochemically active sites, etc. In this context, several approaches such as forming heterostructures, creating defects, doping with other elements, strain modulation, etc., are adopted to improve the overall activity of such catalysts.31–35 Most importantly it is essential to improve the water dissociation ability to achieve enhanced alkaline HER activity.36 Incorporation of another component, such as a sub-nanometer-sized metal or metal oxide cluster with strong water dissociation ability is an effective strategy for hydrogen production because of its strong quantum confinement effects.37,38 Sub-nanometer metal nanocluster decoration effectively increases the active site exposure leading to enhanced electrocatalytic activity for overall water splitting.39–42 There have been efforts devoted to developing non-platinum-based bifunctional electrocatalysts for water splitting. Ruthenium, belonging to the class of noble metal elements, exhibits similar metal–hydrogen-bond strength of ∼65 kcal mol−1 activity to that of platinum and has a much lower price (∼4% of platinum).43,44 Ruthenium nanoclusters were shown to exhibit good ability for water dissociation by shifting the d-band center closer to the Fermi level, which is an important parameter for efficient alkaline water splitting.45 Ruthenium nanocluster decoration over nickel diselenide following a two-step process of hydrothermal and selenization methods has been reported to lead to enhanced electrocatalytic water splitting in alkaline media.46 However, the reported synthesis method involves multistep processes and high-temperature requirements, posing severe concerns toward practical implementation. Pu et al., reported the electrodeposition of nickel diselenide nanoparticles over titanium foil for overall water-splitting application.47 Ru-nanocluster growth on NiSe2 over conducting substrates such as nickel foam (NF) or carbon material via a single-step process would enable high metal utilization and improved electronic conductivity, resulting in efficient catalytic activity towards overall water splitting.
Herein, we demonstrate an energy-efficient and scalable single-step approach for the electrosynthesis of ruthenium nanocluster decorated nickel diselenide catalysts for high-performance and stable alkaline water splitting application. We optimize catalyst synthesis, and the sample with 50 mmol Ru (50-Ru–NiSe2) is shown to exhibit excellent bifunctional electrocatalytic activity with the lowest overpotential of 13 mV at a current density of 10 mA cm−2 for the HER and 260 mV at a current density of 30 mA cm−2 for the OER. We further demonstrate remarkable full-cell performance with a cell potential of 1.45 V to deliver a current density of 10 mA cm−2, along with impressive long-term stability over 400 h. Density functional theory (DFT) calculations are carried out, which demonstrate that the ruthenium nanocluster decoration facilitates the exothermic dissociation of water into H and OH species, while also optimizing the adsorption energies of H+ and OH− when compared to that of bare NiSe2.
Experimental section
Materials and reagents
Nickel acetate tetrahydrate (Ni(CH3COOH)2·4H2O, 98%), selenium dioxide (SeO2, 99.8%), ruthenium(III) chloride hydrate (RuCl3·xH2O, 99.98%), nickel foam (NF, 1.6 mm thickness), hydrochloric acid (HCl, 37%), potassium hydroxide pellets (KOH, 99.95%), platinum over carbon (20 wt% Pt/C), and iridium oxide (IrO2, 99.9%) were purchased from Sigma-Aldrich. High-purity Milli-Q water was used for preparing the samples.
Synthesis of Ru–NiSe2
A piece of NF (0.5 cm × 1 cm) was initially treated with HCl for 10 min under sonication and then washed with DI water followed by drying at 60 °C overnight. For electrodeposition, we used a typical steady-state three-electrode setup in which HCl pretreated NF is used as the working electrode, a platinum coil is used as the counter electrode, and silver/silver chloride (Ag/AgCl) is used as the reference electrode. A constant potential of −1 V vs. Ag/AgCl is applied for 10 min at the working electrode for electrodeposition. First, we synthesized nickel diselenide (NiSe2) using an electrolyte of 65 mmol of Ni(CH3COOH)2·4H2O, and 35 mmol of SeO2 was dissolved in 40 mL DI water and stirred for 5 min. After this, we added a few drops of 37% HCl to maintain the pH in the range of 2–3. The electrodeposited sample was washed several times with DI water and then dried at 60 °C overnight. Furthermore, the dried sample was annealed at 200 °C in an argon environment for 2 h. Similarly, we fabricated a ruthenium cluster decorated nickel diselenide sample under similar experimental conditions. Additionally, the concentration of RuCl3·xH2O in the solution was changed to 20 mmol, 30 mmol, 40 mmol, 50 mmol, and 60 mmol. Ru nanocluster decorated nickel diselenide samples are denoted as 20-Ru–NiSe2, 30-Ru–NiSe2, 40-Ru–NiSe2, 50-Ru–NiSe2, and 60-Ru–NiSe2, respectively.
Materials characterization
An X-Ray diffractometer (Empyrean, PANalytical) instrument with Cu Kα 1.54 Å is used to characterize the phase composition of all the prepared samples with a scan rate of 0.5 degree per min in the range of 20 to 80 degrees. A Nova scanning electron microscope (Nova Nano SEM 450) and transmission electron microscope (FEI Tecnai G2 F30 S-Twin TEM 300 kV) are used to analyze the surface morphology and elemental mapping of the samples. X-ray photoelectron spectroscopy (Omicron Nano Tech. XPS) with a Mg Kα source was performed to investigate the elemental composition and electronic state of all present elements. Finally, we performed inductively coupled plasma optical emission spectroscopy (PerkinElmer Optima 5300 DV ICP-OES) to analyze the ratio of ruthenium to nickel diselenide.
Electrochemical characterization
The electrochemical performance of all the catalysts for the HER and OER was evaluated using a Biologic SAS VMP3 electrochemical workstation in nitrogen-saturated 1.0 M KOH solution, at room temperature, using a typical three-electrode setup. All prepared electrodes are used as working electrodes, a graphite rod, as the counter electrode, and a Hg/HgO electrode, as the reference electrode. The working electrode potential was converted to RHE using the Nernst equation (ERHE = E0Hg/HgO + 0.059 × pH).9 All the polarization curves were recorded with 85% IR corrected by LSV at a scan rate of 2 mV s−1. We carried out electrochemical impedance spectroscopy to calculate the charge transfer resistance of the materials in the frequency range of 50 mHz to 100 kHz. We obtained the electrochemically active surface area (ECSA) of all the prepared electrodes from the double-layer capacitance (Cdl), which was evaluated by scanning the CV in the non-faradaic potential region vs. Hg/HgO with different scan rates from 10 mV s−1 to 100 mV s−1. Pt/C and IrO2 electrodes were studied as benchmark catalysts for the HER and OER, respectively, with 4 mg of Pt/C or IrO2 in 1 mL solution (750 μL of DI water, 200 μL of IPA, and 50 μL of Nafion). The solution is sonicated for 2 h to make a proper dispersion and then drop-coated over HCl-pretreated NF.
Density functional theory calculations
All density functional theory calculations were performed using the Vienna ab initio simulation package (VASP)48–51 within the framework of the generalized gradient approximation (Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional).52 To account for the core–valence electron interactions, we employed the projector augmented wave (PAW) method.53 Furthermore, we incorporate the effects of van der Waals interactions via Grimme's D3 dispersion correction.54 The kinetic energy cutoff for plane wave expansions, convergence criteria for energy, and forces on each atom were set to 550 eV, 10–5 eV and 0.01 eV Å−1, respectively. To minimize possible interactions between the periodic images, a vacuum layer of 16 Å was added in the non-periodic direction. For a pure NiSe2 system, a (210) surface slab with seven layers, consisting of 28 Ni and 56 Se atoms, was used. All layers were relaxed during geometry optimization. However, for the Ru nanocluster decorated NiSe2 surface, a Ru8 cluster was adsorbed on the NiSe2 (210) surface slab with five layers, comprising 60 Ni, 120 Se, and 8 Ru atoms. For the pure NiSe2 (210) and Ru8–NiSe2 (210) systems, the Brillouin zone integrations were carried out using Γ-centred k-point meshes of 4 × 9 × 1 and 5 × 5 × 1, respectively.
Results and discussion
Synthesis of Ru nanocluster decorated nickel diselenide (Ru–NiSe2) supported on nickel foam was performed through a single-step electrodeposition process, followed by low-temperature thermal annealing in an argon atmosphere, as illustrated in ESI Fig. S1.† The process involved a co-electrodeposition strategy using precursors of nickel and selenium along with varying concentrations of ruthenium(III) chloride of 20 mmol, 30 mmol, 40 mmol, 50 mmol, and 60 mmol, as detailed in the ESI Methods section.† We applied −1 V vs. Ag/AgCl for 10 min to obtain a mass loading of 2.42 mg cm−2. Pristine NiSe2 samples are grown under controlled conditions on Ni foam. All the electrodeposited samples are washed with DI water and annealed in an argon atmosphere at 200 °C for 2 h.
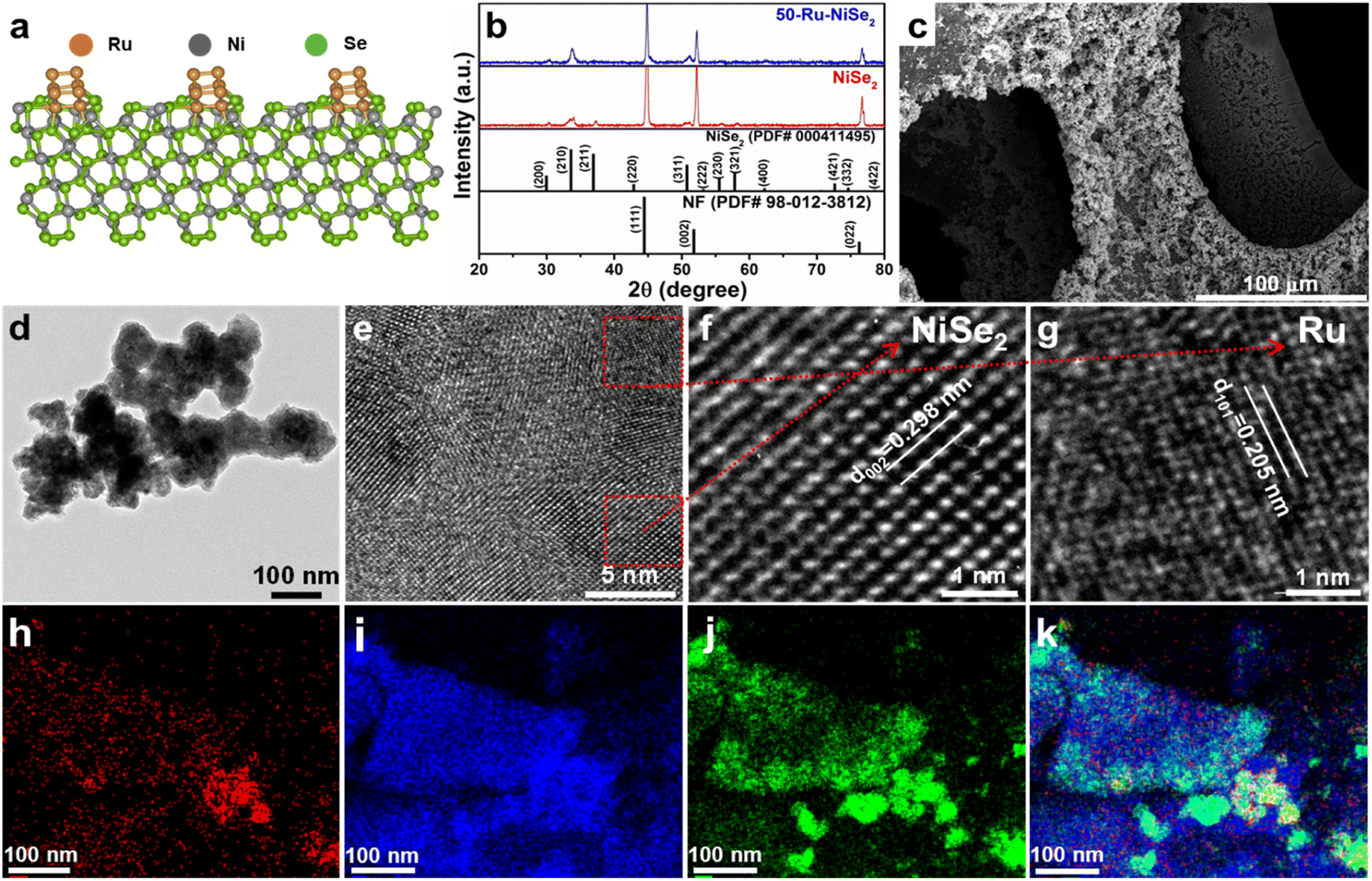
Fig. 1a shows the crystal structure of Ru nanocluster decorated nickel diselenide, which was confirmed from the X-ray diffraction (XRD) analysis (Fig. 1b). Three high-intensity peaks at 44.44, 51.78, and 76.27 correspond to the reflections from nickel foam (PDF no.-98-012-3812) (Fig. S2†).55 The other marked peaks correspond to the cubic phase of NiSe2 (PDF no.-00-041-1495),56 which seems to have remained unchanged with Ru decoration, indicative of no structural changes in NiSe2. We performed inductively coupled plasma optical emission spectroscopy (ICP-OES) to analyze the ruthenium content in nickel diselenide. For the 50-Ru–NiSe2 sample (with an initial concentration of 50 mmol ruthenium(III) chloride), a Ru content of 3.9 wt% was confirmed.
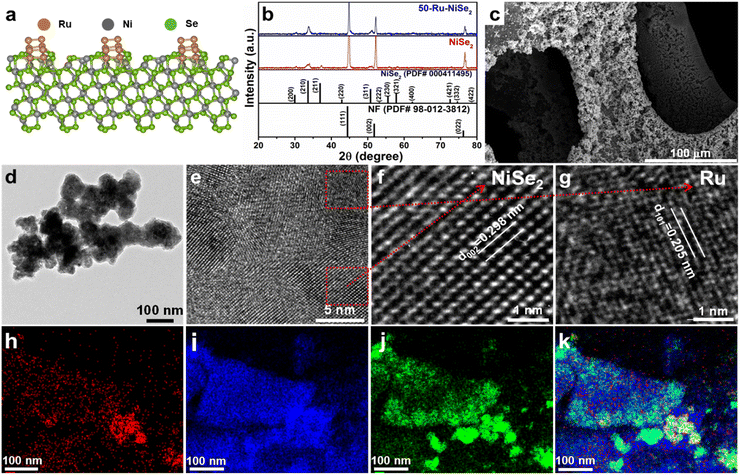 |
| | Fig. 1 Phase and morphology analyses of Ru nanocluster decorated nickel diselenide. (a) Crystal structure of ruthenium nanocluster decorated nickel diselenide. (b) XRD pattern of NiSe2 and 50-Ru–NiSe2. (c) SEM images of 50-Ru–NiSe2. (d) TEM image of 50-Ru–NiSe2. (e) HRTEM image of 50-Ru–NiSe2. (f and g) HRTEM images showing the interplanar spacing for NiSe2 and Ru nanoclusters, respectively. (h–j) Elemental mapping of Ru, Ni, and Se, respectively. (k) Elemental mapping of Ru, Ni, and Se, together. | |
The morphology of all the prepared catalysts was further analyzed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). For instance, Fig. 1c shows the SEM image of the surface of nickel foam which is fully covered with electrodeposited 50-Ru–NiSe2. Fig. 1d shows the TEM image of 50-Ru–NiSe2. The corresponding high-resolution TEM (HR-TEM) images given in Fig. 1e–g clearly reveal interplanar spacing of NiSe2 and Ru nanoclusters with the corresponding lattice planes.46,57 SEM images of electrodeposited NiSe2 over NF (Fig. S3†) with uniform growth of NiSe2 over the NF surface and the corresponding energy-dispersive spectroscopy (EDX) mapping showed uniform distribution of nickel and selenium over the surface (Fig. S4†). The TEM image shown in Fig. S5† displays the morphology of NiSe2 along with the corresponding d spacing. Besides the morphological studies, energy-dispersive spectroscopy (EDX) analysis is also performed in order to confirm the elemental composition in the sample. The uniform distribution of Ru, Ni, and Se over the surface was confirmed from the elemental mapping images shown in Fig. 1h–k, respectively. X-ray photoelectron spectroscopy (XPS) is a powerful technique to analyze the chemical composition and chemical state of the elements present in the sample. XPS measurements were carried out for NiSe2, and 50-Ru–NiSe2. Due to the spin–orbit coupling, Ni, Ru, and Se peaks split into Ni 2p1/2, Ni 2p3/2, Ru 3p1/2, Ru 3p3/2; Se 3d5/2, and Se 3d3/2, respectively.46,47,56 In Fig. 2a, the XPS survey spectrum of NiSe2 shows the presence of Ni and Se, while that of 50-Ru–NiSe2 shows the presence of Ru, Ni, and Se. Fig. 2b and c show the high-resolution XPS spectra of NiSe2, and Fig. 2d–f show the high-resolution XPS spectra of 50-Ru–NiSe2. The peaks at 854.5 eV and 872.3 eV are assigned to Ni2+ 2p3/2 and Ni2+ 2p1/2, respectively, while the two broad peaks at 859.4 eV and 877.8 eV correspond to the satellite peak of Ni 2p (Fig. 2b).58Fig. 2c shows the XPS peaks at 55.3 eV and 56.4 eV which correspond to Se 3d5/2 and Se 3d3/2, and a broad peak observed at 59.4 eV reveals partial oxidation of selenium.59,60Fig. 2d shows the high-resolution XPS peaks at 461.8 eV and 484.2 eV, corresponding to Ru0 3p3/2 and Ru0 3p1/2 from metallic Ru,61 which further indicates the formation of ruthenium nanoclusters. High-resolution XPS peaks observed at 854.8 eV and 872.6 eV in Fig. 2e are assigned to Ni2+ 2p3/2 and Ni2+ 2p1/2, respectively, and the two broad peaks at 860.2 eV and 878.6 eV correspond to the satellite peak of Ni 2p.58,62Fig. 2f shows XPS peaks at 55.3 eV and 56.4 eV which correspond to Se 3d5/2, and Se 3d3/2, respectively, and a broad peak seen at 59.3 eV reveals partial oxidation of selenium.59,60,63 A positive shift of +0.3 eV is observed in the high-resolution nickel spectra of 50-Ru–NiSe2 upon Ru incorporation. This indicates that there is charge transfer happening from nickel to ruthenium in 50-Ru–NiSe2, which is originated from the high electronegativity of Ru (2.5) compared to that of Ni (1.2).46,64 This further indicates the presence of an electrostatic interaction between Ni and Ru in the Ru nanocluster incorporated sample.
 |
| | Fig. 2 XPS analysis. (a) Survey spectra of NiSe2 and 50-Ru–NiSe2. (b and c) High-resolution XPS spectra of Ni and Se of NiSe2. (d–f) High-resolution XPS spectra of Ru, Ni, and Se of 50-Ru–NiSe2. | |
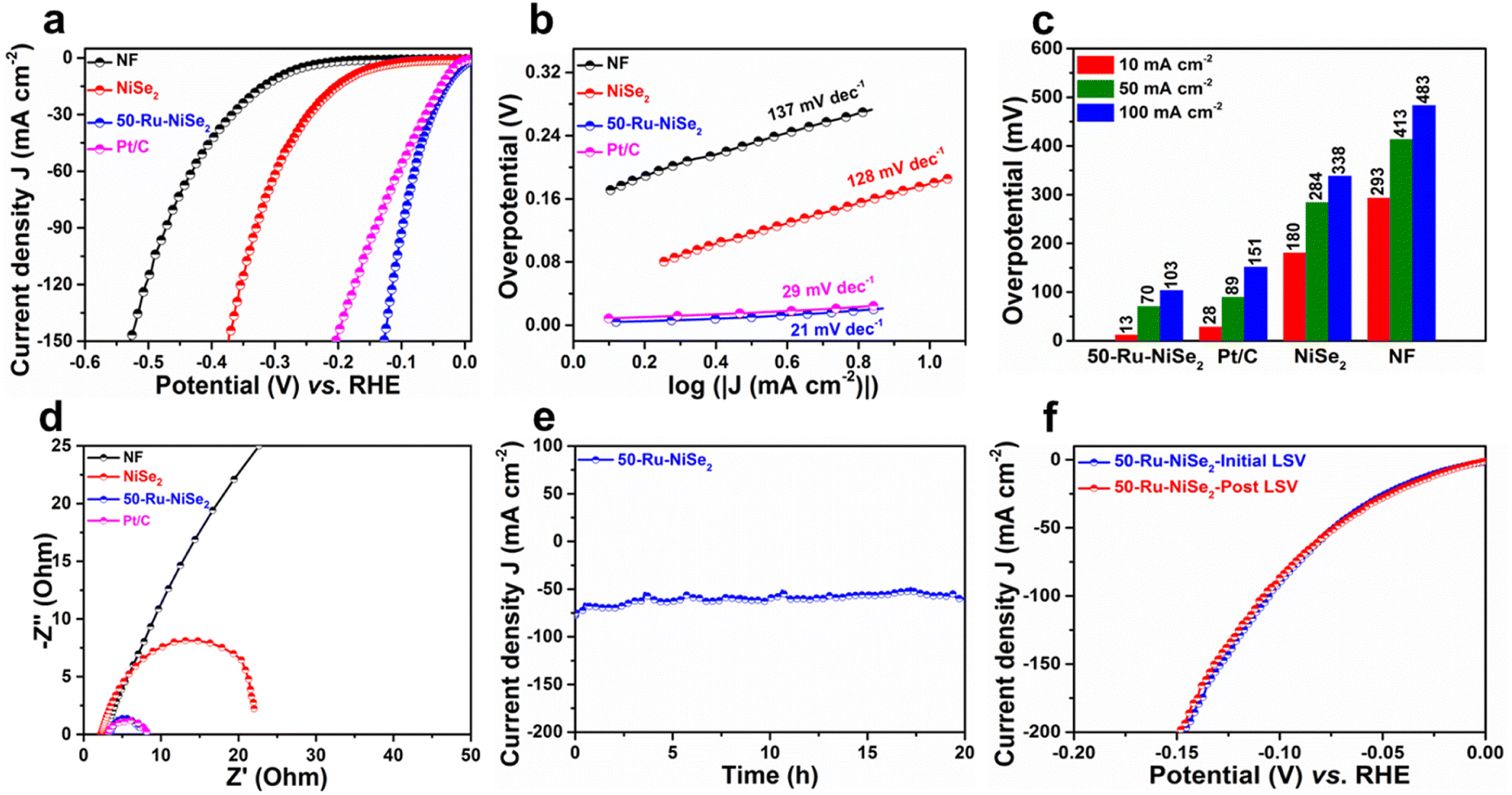
The electrochemical performance of the catalysts was studied in nitrogen-saturated 1.0 M KOH electrolyte using linear sweep voltammetry (LSV) at 2 mV s−1. Improvement in the electrocatalytic performance of the Ru-incorporated NiSe2 catalysts in comparison with their pristine counterpart towards the HER was clearly confirmed through detailed LSV studies performed on the catalysts with varying molar ruthenium contents (Ru = 10 mmol, 20 mmol, 30 mmol, 40 mmol, 50 mmol, and 60 mmol), as shown in Fig. S6.† The catalyst with an initial molar Ru content of 50 mmol (50-Ru–NiSe2) showed the best performance among all the studied catalysts. Based on this, detailed electrocatalytic performance evaluation on bare NF, NiSe2 sheets and 50-Ru–NiSe2 was carried out along with 20 wt% Pt/C (Pt/C) as the reference. As shown in Fig. 3a, IR-compensated linear sweep voltammetry was performed to analyze the alkaline HER activity of all the prepared catalysts. 50-Ru–NiSe2 showed excellent activity with a very low overpotential of 13 mV and 103 mV at current densities of 10 mA cm−2 and 100 mA cm−2, respectively. The obtained values are much better in comparison to those of the benchmark Pt/C catalyst, which showed overpotentials of 28 mV and 151 mV, at the respective current densities of 10 mA cm−2 and 100 mA cm−2. While NiSe2 showed an overpotential of 180 mV and 338 mV, NF exhibited poor activity with overpotentials of 293 mV and 483 mV at the respective current densities of 10 mA cm−2 and 100 mA cm−2. To understand the electrochemical kinetics for the HER, we investigated the Tafel slopes for all the catalysts. In general, there are three steps involved in the HER process;65
| | | M* + H2O + e− → M–H + OH− (Volmer step) | (1) |
| | | H2O + M–H + e− → M* + H2 + OH− (Heyrovsky step) | (2) |
| | | M–H + M–H → H2 + 2M* (Tafel step) | (3) |
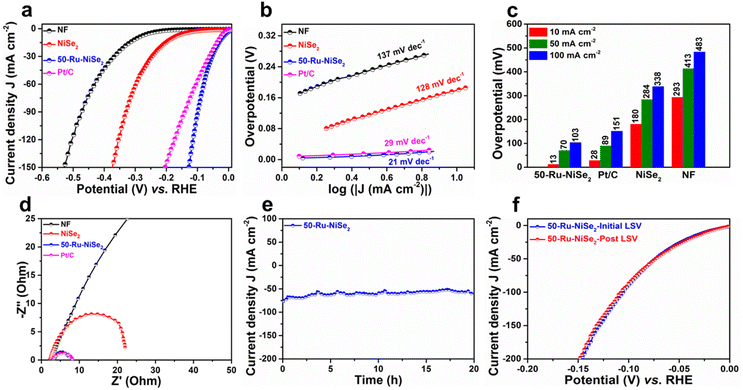 |
| | Fig. 3 HER studies. (a) LSV polarization curves recorded for NF, NiSe2, 50-Ru–NiSe2, and Pt/C. (b) The corresponding Tafel plots. (c) Overpotential measured at current densities of 10, 50, and 100 mA cm−2. (d) Comparison of Nyquist plots of NF, NiSe2, 50-Ru–NiSe2 and Pt/C. (e) Stability plot of 50-Ru–NiSe2 obtained using chronoamperometry measurements. (f) Initial and post LSV plots of 50-Ru–NiSe2. | |
The Tafel slopes obtained from the corresponding LSV plots of all the studied samples are depicted in Fig. 3b, which further confirmed the superior HER activity of 50-Ru–NiSe2 (21 mV dec−1), compared to Pt/C (29 mV dec−1), NiSe2 (128 mV dec−1), and, NF (137 mV dec−1). The low Tafel slopes of 50-Ru–NiSe2 and Pt/C reveal that the HER process for these two systems follows the Volmer–Tafel mechanism. As further indicated in Fig. 3c, from the comparison of overpotentials for the HER at different current densities of 10 mA cm−2, 50 mA cm−2, and 100 mA cm−2 for all the studied catalysts, 50-Ru–NiSe2 exhibits the lowest overpotential, which is better than all the best-reported values for alkaline HER (Table S1, ESI†). Electrochemical impedance spectroscopy was performed to understand the reaction kinetics for charge transfer between the interface of the catalyst surface and the electrolyte. 50-Ru–NiSe2 showed a lower charge transfer resistance (Rct) of 4.03 ohms, as compared to NiSe2, at an overpotential of −100 mV (Fig. 3d). Chronoamperometry was performed to understand the electrochemical stability of the catalyst. 50-Ru–NiSe2 showed excellent stability with negligible change in current density after a 20 h stability test at a current density of 65 mA cm−2 (Fig. 3e). This clearly indicates the catalyst's exceptional durability in alkaline media for high current density performance. In Fig. 3f, we showed a comparison of LSV plots recorded for 50-Ru–NiSe2 at the initial cycle and after the 20 h chronoamperometry test. The overlapping curves further indicate the high durability of the catalyst under long-term operating conditions, which is an essential parameter for the overall water-splitting process. High electrocatalytic activity and stability of 50-Ru–NiSe2 in alkaline media make this catalyst an efficient candidate for the HER.
We further explored the OER activity of all the catalysts in 1.0 M KOH electrolyte. The IR-compensated LSVs are plotted in Fig. 4a to analyze the OER activity of all the studied catalysts. 50-Ru–NiSe2 showed the best OER performance with a low overpotential of 313 mV and 352 mV at current densities of 50 mA cm−2 and 100 mA cm−2, respectively, as compared to NiSe2 (378 mV and 431 mV) and NF (403 mV and 488 mV). The OER performance of the studied catalyst was better than that of the benchmark catalyst IrO2. The electrochemical kinetics of these materials for the OER mechanism was investigated by using Tafel slopes.
 |
| | Fig. 4 OER studies. (a) LSV polarization curves of NF, NiSe2, 50-Ru–NiSe2, and IrO2 and (b) the corresponding Tafel plots. (c) Corresponding overpotential recorded at current densities of 10, 50, and 100 mA cm−2. (d) Nyquist plots of NF, NiSe2, IrO2 and 50-Ru–NiSe2. (e) Stability plots of 50-Ru–NiSe2 obtained using chronoamperometry measurements. (f) Initial and post LSV plots of 50-Ru–NiSe2. | |
Fig. 4b shows the Tafel slopes of all the studied catalysts. 50-Ru–NiSe2 exhibited a low Tafel slope (135 mV dec−1), compared to IrO2 (232 mV dec−1), NiSe2 (164 mV dec−1), and, NF (327 mV dec−1), indicative of its high OER kinetics. The overpotentials for the OER of all the catalysts recorded at different current densities of 50 mA cm−2, 100 mA cm−2, and 200 mA cm−2 are plotted in Fig. 4c, which showed that 50-Ru–NiSe2 exhibits the lowest overpotential compared to the other three electrodes (IrO2, NiSe2, and NF), which is also better than the recently reported values for alkaline OER from the literature (Table S2, ESI†). Electrochemical impedance spectroscopy was performed (Fig. 4d) to understand the reaction kinetics for charge transfer between the interface of the catalyst surface and the electrolyte. 50-Ru–NiSe2 showed a lower charge transfer resistance (Rct) of 0.34 ohm as compared to NiSe2 and IrO2 at an overpotential of 300 mV.
The chronoamperometry test was performed (Fig. 4e) to evaluate the stability of the catalyst. 50-Ru–NiSe2 showed a very small change in current density after a 20 h stability test at a current density of 50 mA cm−2, confirming its very good durability in alkaline media for high current density performance. In Fig. 4f, we compared the LSVs recorded for 50-Ru–NiSe2 after a 20 h chronoamperometry test along with that of the initial cycle, which did not show much significant change, clearly revealing the high durability of the catalyst under long-term operating conditions. High electrocatalytic activity and stability of 50-Ru–NiSe2 in alkaline media make this catalyst an efficient and stable candidate for the OER. After the stability test, we further performed XPS and SEM characterization studies to understand the surface reconstruction process. The post-cycling high-resolution XPS spectra show several changes in peak intensity and peak positions. The Ru 3p peaks show a slight positive shift as compared to those of metallic ruthenium (Fig. S7†), which indicates the oxidation of ruthenium.61 Similarly, the Ni 2p peak also exhibits a positive shift, indicating the formation of Ni3+ species from Ni2+ as shown in Fig. S8.†66 This positive shift confirms the conversion of the surface of nickel diselenide to nickel oxyhydroxide. Furthermore, there is a stronger oxidation peak observed in the Se 3d spectrum (Fig. S9†) compared to the initial state. This suggests that the selenium surface has undergone reconstruction to form SeOx species.67 To gain a better understanding of the surface reconstruction, the upper surface of the catalysts was etched with argon, and XPS analysis data were recorded from the inner layer of the catalysts. After 10 minutes of etching, the metallic ruthenium peak becomes clearly visible, indicating the presence of metallic ruthenium. The Ni2+ oxidation peaks corresponding to Ni 2p3/2 and Ni 2p1/2 are also observed. However, the oxidation peak for selenium disappears, indicating the absence of selenium oxide formation. Based on this XPS analysis, it can be concluded that the surface of NiSe2 has been converted from NiSe2 to Ni(OOH)x, while the inner core of the catalyst still remains in the NiSe2 phase. The Ni(OOH)x species serve as the active sites during the OER process, while the highly conductive inner core of NiSe2 facilitates charge migration.
Additionally, we performed the SEM analysis after the OER stability tests (Fig. S10†). Fig. S10a–d† show the SEM images of 50-Ru–NiSe2 before and after the OER stability test. Few changes occurred after the OER stability test during the surface reconstruction process. Inspired by the enhanced bifunctional electrochemical HER and OER activities of 50-Ru–NiSe2 in alkaline media, we designed an electrolyzer for overall alkaline water splitting with 50-Ru–NiSe2 as both the anode and cathode (50-Ru–NiSe2//50-Ru–NiSe2). The polarization curves recorded for the 50-Ru–NiSe2//50-Ru–NiSe2 and NiSe2//NiSe2 system are compared with that of Pt/C//IrO2, the benchmark catalyst (Fig. 5a). 50-Ru–NiSe2//50-Ru–NiSe2 showed a remarkably low cell potential of 1.45 V to achieve a current density of 10 mA cm−2, while Pt/C//IrO2 delivers the same current density at a higher potential of 1.58 V. NiSe2//NiSe2 required a much higher potential of 1.66 V to achieve 10 mA cm−2 current density. The full cell potentials required to obtain current densities of 10 mA cm−2, 50 mA cm−2, and 100 mA cm−2 for 50-Ru–NiSe2//50-Ru–NiSe2, NiSe2//NiSe2, and benchmark Pt/C//IrO2 are depicted in Fig. 5b. We performed the durability test for the full cell in Fig. 5c, and 50-Ru–NiSe2//50-Ru–NiSe2 showed impressive long-term durability with high current retention even after 400 h at an applied potential of 1.45 V, clearly revealing excellent performance for overall water splitting. We compared the performance of our system with that of most recently reported alkaline bifunctional electrocatalysts (at 10 mA cm−2), and the present data were found to be superior (Fig. 5d). The details are provided in Table S3 (ESI).† The long-term stability test was extended for a month with continuous operation, and 50-Ru–NiSe2//50-Ru–NiSe2 exhibited exceptional stability for alkaline water splitting (Fig. S7†). To understand the intrinsic activity of the catalyst, we calculated the electrochemically active surface area (ECSA) of the synthesized catalysts from the charge double layer capacitance Cdl (ESI, Note S2†). CV is carried out in the non-faradaic region of the potential with different scan rates in the alkaline electrolyte (Fig. S8†). The CVs recorded at 10 mV s−1 for 50-Ru–NiSe2 showed a much higher current density than those of NiSe2, suggesting higher Cdl of 50-Ru–NiSe2 compared to NiSe2. The Cdl of NF, NiSe2, and 50-Ru–NiSe2 is measured to be 1.2 mF cm−2, 11.6 mF cm−2, and, 71.3 mF cm−2, respectively (Table S4, ESI†).
 |
| | Fig. 5 Overall water splitting studies. (a) Comparison of LSV plots of 50-Ru–NiSe2//50-Ru–NiSe2, and NiSe2//NiSe2 with those of benchmark Pt/C//IrO2. (b) Cell potentials required to obtain current densities of 10 mA cm−2, 50 mA cm−2, and 100 mA cm−2 for 50-Ru–NiSe2//50-Ru–NiSe2, NiSe2//NiSe2 and Pt/C//IrO2. (c) Stability plots of 50-Ru–NiSe2//50-Ru–NiSe2 were obtained using a chronoamperometry test. (d) Comparison of the full-cell performance of our work with those of the best reported alkaline catalysts (Table S4, ESI†). | |
Theoretical studies
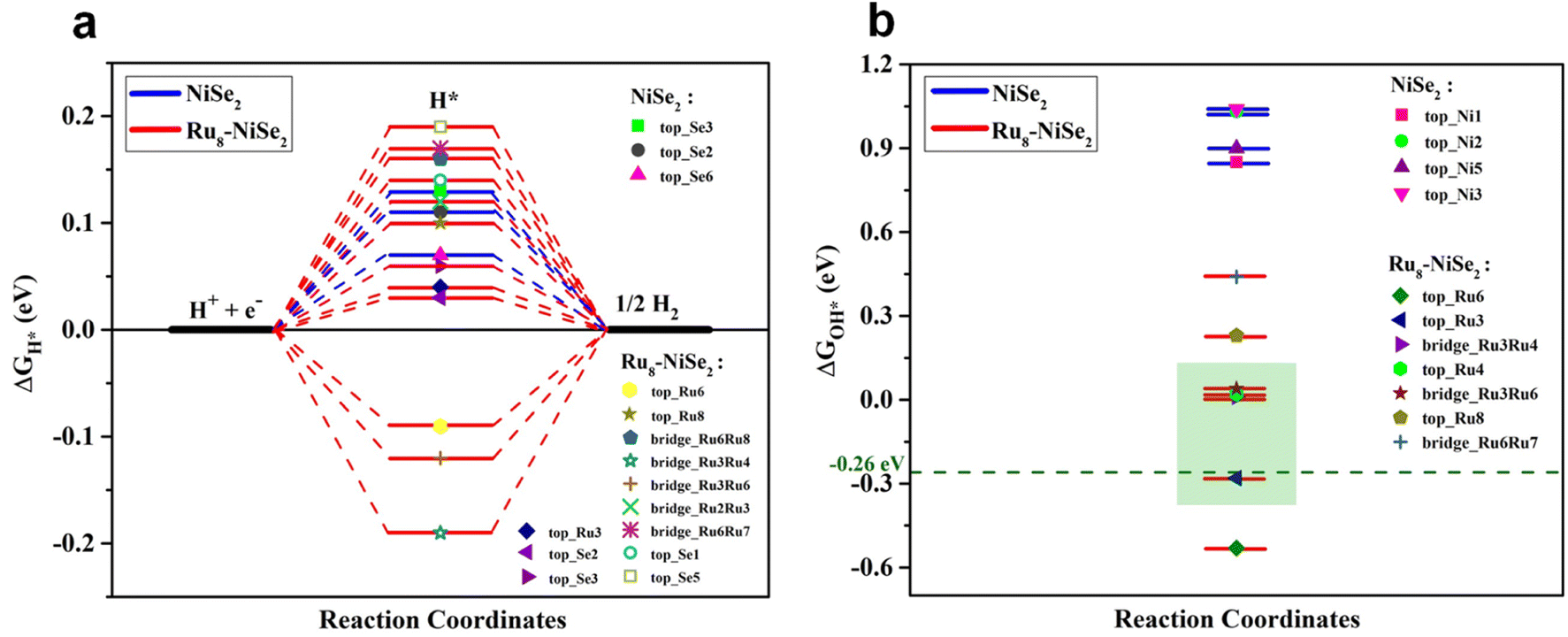
To obtain further insights on the remarkably enhanced bifunctional catalytic activity of Ru nanocluster-decorated NiSe2 over pristine NiSe2 towards the HER and OER in an alkaline medium, we employed first-principles DFT calculations. Since the as-prepared samples are large and computationally challenging to handle with DFT, we chose to study systems of reasonable sizes that can be feasibly analyzed. We believe that our model systems are sufficient to capture the fundamental chemical processes that underlie the observed catalytic behavior. Specifically, we investigated the HER and OER catalytic activities of the clean NiSe2 (210) surface and NiSe2 (210) surface decorated with a Ru nanocluster. The reason for choosing the (210) plane of NiSe2 is that the experimentally observed XRD pattern for NiSe2 indicates that the peak with the maximum intensity corresponds to the (210) plane (see Fig. 1b). Furthermore, it is clear from this figure that the XRD pattern remains unchanged with Ru nanocluster decoration. Therefore, for both pristine NiSe2 and Ru nanocluster-decorated NiSe2, we opted to study the (210) plane. Moreover, to examine the effect of Ru nanocluster decoration on the catalytic activity of NiSe2 (210) towards the HER and OER, we chose an 8-atom Ru nanocluster. In the absence of a clear indication from the experiments about how the Ru dopants are arranged on the surface, we consider a Ru8 nanocluster adsorbed NiSe2 (210) surface as a model catalyst. The choice of this particular size for the nanocluster is guided by the findings in published literature,68–70 which suggest that Ru8 is relatively more stable when compared to neighboring nanocluster sizes, and also takes into consideration the computational tractability. To compare the catalytic activities of clean and Ru8 adsorbed NiSe2 (210) surfaces, we consider the structures shown in Fig. S9.† We adsorbed all the relevant intermediate atoms/molecules involved in these reactions on all the available high-symmetry sites of these two systems. In the case of pure NiSe2, these sites include Se-top, Ni-top, and Se–Se bridge positions. However, in Ru8–NiSe2, there exists additional sites referred to as Ru-top and Ru–Ru bridge. All of these sites are marked in Fig. S9.† The complete HER in an alkaline medium is 4H2O + 4e− → 2H2 + 4OH−. As shown by Nørskov et al., the free energy of H adsorption, ΔGH*, is a good descriptor for characterizing the catalytic activity of transition metal-based materials towards the HER in acidic electrolytes.71,72 According to their results, adsorption-free energy of H, ΔGH* close to zero is a necessary criterion for a catalyst to be considered suitable for acidic HER. We adopt the criterion of |ΔGH*| ≤ 0.2 eV to identify the potential candidates for the HER in an acidic medium. However, in an alkaline medium, ΔGH* alone is insufficient to fully characterize the HER activity. This is because of the low availability of H+ ions in an alkaline medium, where the dissociation of H2O into H+ and OH− becomes an essential part of the HER and contributes additional energy to the overall reaction Gibbs free energy. Furthermore, there is currently no single descriptor in the literature that adequately describes the HER activity of a material in an alkaline medium. Therefore, in addition to the |ΔGH*| ≤ 0.2 eV criterion, we consider the following factors to assess the HER activity in alkaline environments:
(i) The binding energy of H2O with the material surface, (ii) the feasibility of H2O dissociation into H+ and OH−, and (iii) the free energy of OH adsorption, ΔGOH*, which should ideally be close to zero according to the Sabatier principle.73 First, we calculate the value of ΔGH* using the following equation:
| | | ΔGH* = Eads(H) + 0.24 eV, | (4) |
where
Eads(H) represents the adsorption energy of the H atom on either the pure or Ru
8 cluster decorated NiSe
2. Please check the ESI (Note S3)
† for more details. The visual representation of Δ
GH* at different sites on both pristine NiSe
2 and Ru
8–NiSe
2 is given in
Fig. 6a. We observe from this figure that in both the pristine and Ru
8–NiSe
2 materials, the value of Δ
GH* lies close to zero. The value of Δ
GH* closest to zero is 0.08 eV for the pristine material, while it is 0.04 eV for Ru
8–NiSe
2. We also note that, in the case of the pristine material, the |Δ
GH*| ≤ 0.2 eV criterion is satisfied only at the Se-top sites. On the other hand, for Ru
8–NiSe
2, in addition to a few Se-top positions, almost all sites, including Ru-top and Ru–Ru bridge positions of the Ru cluster, are HER active in terms of Δ
GH*. Therefore, based on the H adsorption free energy plot, we can conclude that both pristine and Ru
8–NiSe
2 (210) are HER active; however, the number of active sites is more in the case of the Ru cluster-decorated surface. Next, we examine the binding of H
2O molecules with the pristine and Ru
8-decorated NiSe
2 (210) surfaces. To characterize this interaction, we calculate the adsorption energy,
Eads(H
2O), of an H
2O molecule using the following equation.
| | | Eads(H2O) = E(H2O*) − E(*) − E(H2O) | (5) |
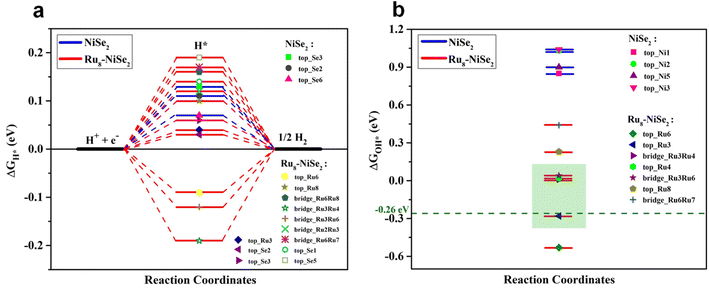 |
| | Fig. 6 Theoretical calculations for the HER. (a) Free energy plots for H adsorption and (b) OH adsorption on some of the high symmetry sites of pristine and Ru8–NiSe2 (210) surfaces. | |
In this equation, E(H2O*) represents the total energy of the adsorbed H2O molecule on either NiSe2 or Ru8–NiSe2. E(*) is the total energy of these systems prior to H2O adsorption, and E(H2O) represents the total energy of a free H2O molecule. A negative value of Eads(H2O) would indicate that the adsorption of H2O on the catalyst surface is exothermic. Values of Eads(H2O) at some of the possible sites of NiSe2 and Ru8–NiSe2 (210) are given in Table S5 (ESI).† From this table, it is clear that the maximum H2O adsorption energy is −0.83 eV on one of the Ni-top sites of pristine NiSe2. However, in the case of Ru8–NiSe2, the strongest binding (Eads(H2O) = −1.08 eV) occurs on one of the Ru-top positions. This shows that an H2O molecule is significantly more strongly adsorbed on Ru8–NiSe2 (210) compared to on the pristine material. While the optimal value is not known, it is conceivable that the strong binding energy of H2O could facilitate its dissociation at the surface, subsequently aiding the Heyrovsky or Tafel steps in the HER process. Next, we proceed to assess whether the dissociation of an H2O molecule is energetically favorable on NiSe2 (210) and Ru8–NiSe2 (210). The reaction energy, ΔE, determines whether a reaction is energetically favorable. We calculate the value of ΔE according to E(H* + OH*) − E(H2O*). The lowest possible pathways for H2O dissociation on the pristine and Ru8 cluster decorated NiSe2 surfaces are presented in Fig. 7. From this figure, it is evident that the value of ΔE on NiSe2 (210) is 0.76 eV. This significant positive value of ΔE indicates that the H2O dissociation into H and OH is an endothermic process. On the other hand, on the Ru8–NiSe2 system, the dissociation of a H2O molecule into H and OH fragments yields a reaction energy of −1.06 eV, which is significantly negative. This indicates that H2O dissociation is exothermic and thus energetically feasible on the cluster-decorated NiSe2 surface. As we have established that H2O is adsorbed more strongly on the Ru8–NiSe2 system and its dissociation into H and OH is energetically favorable on this system, we expect that the OH fragment will also be adsorbed more strongly on the Ru cluster-decorated surface compared to on pristine NiSe2, where H2O adsorption is weaker and H2O → H + OH is endothermic. To verify this assumption, we will now proceed to calculate the adsorption-free energy of OH, ΔGOH*, on both the pristine and Ru8–NiSe2 materials. To calculate ΔGOH*, we use the following equation:
| | | ΔGOH* = Eads(OH) + 0.35 eV. | (6) |
Here,
Eads(OH) denotes the adsorption energy of OH on either pure or Ru
8–NiSe
2 (210). More details are given in the ESI.
† The values of Δ
GOH* at various possible positions on both the systems are provided in Table S6 (ESI)
† and the corresponding plot is presented in
Fig. 6b. From this figure, it is clear that the value of Δ
GOH* is highly positive, nearly 1.0 eV, close to zero and even slightly negative at some of the positions on the Ru
8–NiSe
2 surface. For example, at the Ru
3-top site, Δ
GOH* = −0.28 eV. Koper
et al.73 showed that transition metal decorated stepped surfaces of Pt have the best HER performance in alkaline media when Δ
GOH* = −0.26 eV. While there is no knowledge of what would be an optimal Δ
GOH* on pristine or decorated NiSe
2 surfaces, one can take −0.26 eV as a guide. Since Δ
GOH* on the Ru cluster-decorated surface is very close to the optimum value (see
Fig. 6b), we expect Ru
8–NiSe
2 (210) to perform much better for the HER in alkaline media compared to pristine NiSe
2 (210). From the above discussion, we find that in terms of H adsorption, Δ
GH* is approximately zero for both pristine and Ru cluster-decorated NiSe
2; however Ru
8–NiSe
2 (210) has more active sites compared to the pristine material. Moreover, the adsorption of an H
2O molecule is comparatively stronger on Ru
8–NiSe
2 (210) than on pristine NiSe
2 (210). The most crucial step of H
2O dissociation into H and OH is an exothermic process on Ru
8–NiSe
2, whereas it is endothermic on the pristine catalyst surface. Additionally, Δ
GOH* is closer to the optimum value of −0.26 eV on Ru
8–NiSe
2 (210) while it is large and positive on the pristine material. These findings collectively suggest that Ru cluster-decorated NiSe
2 (210) is a significantly superior catalyst for alkaline HER compared to pristine NiSe
2 (210).
 |
| | Fig. 7 Representation of reaction pathways. (a) Pristine and (b) Ru8–NiSe2 (210) materials. Green, grey, golden yellow, red, and blue coloured balls represent Se, Ni, Ru, O, and H atoms, respectively. | |
The OER in an alkaline medium involves the transfer of four electrons, accompanied by the formation of the O–O bond and the breaking of the O–H bond. The complete oxygen evolution reaction in an alkaline medium can be represented as 4OH− ↔ 2H2O + O2 + 4e− and the intermediate reaction steps are as follows:
| | | OH* + OH− → O* + H2O + e− | (8) |
| | | OOH* + OH− → O2 + * + H2O + e− | (10) |
In summary, the OER follows the following path: OH− → OH* → O* → OOH* → O2. The free energy change at each of these reaction steps is calculated using the following equations. A more detailed discussion of these steps is provided in the ESI.†
| | | ΔG1 = G(OH*) − G(*) − G(OH− − e−) | (11) |
| | | ΔG2 = G(O*) + G(H2O) − G(OH*) − G(OH− − e−) | (12) |
| | | ΔG3 = G(OOH*) − G(O*) − G(OH− − e−) | (13) |
| | | ΔG4 = G(*) + G(O2) + G(H2O) − G(OOH*) − G(OH− − e−) | (14) |
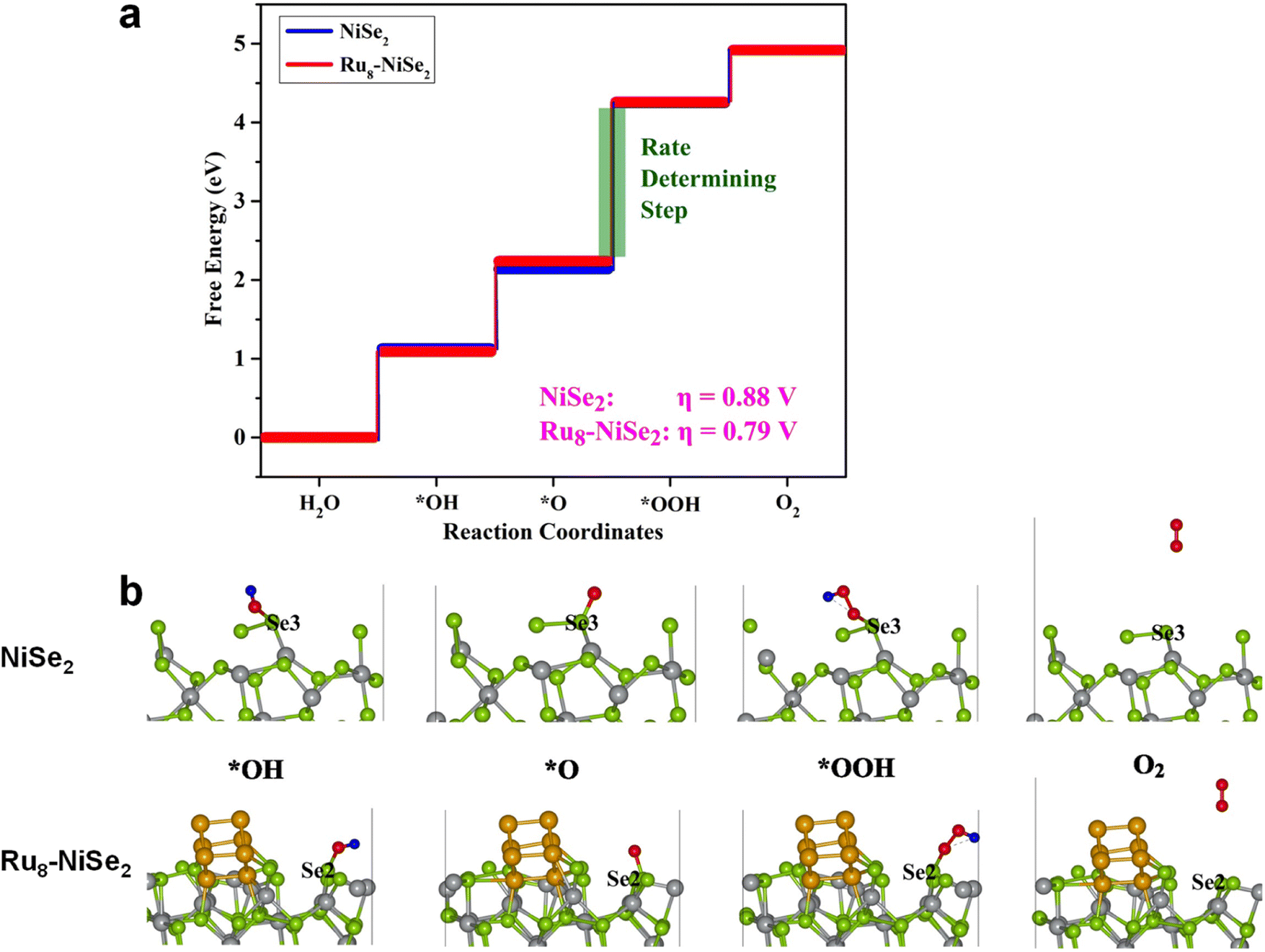
Once we have the values of ΔG1, ΔG2, ΔG3, and ΔG4, we can calculate the overpotential, η, using the following equation: η = max[ΔG1, ΔG2, ΔG3, ΔG4]/e − 1.23 V. We studied the adsorption of all the intermediates, that is, OH, O, and OOH, at all the possible non-equivalent positions on the pristine as well as the Ru8–NiSe2 systems. The calculated values of these free energy changes, along with η, at various positions on the pristine and Ru8–NiSe2 systems, are compiled in Table S7 (ESI).† From this table we observe that the value of ΔG3 is maximum at all the possible positions, indicating that the O* → OOH* step is the rate-determining as well as the potential-determining step.
The free energy plots for the OER on pristine and Ru8–NiSe2 surfaces, at the positions where the overpotential is minimum, are presented in Fig. 8. In this figure, we also demonstrate the structural evolution of the various intermediates on the pristine and Ru cluster-decorated surfaces. It is noteworthy that on both the clean and Ru8–NiSe2 (210) surfaces, the OER active sites are the Se-top positions. From Table S7 (ESI)† and Fig. 8, it is clear that the minimum OER overpotential is 0.88 V at one of the Se-top sites on the pristine NiSe2 surface, while this value decreases to 0.79 V after Ru cluster decoration. Due to this reduction in the overpotential value caused by Ru cluster decoration, we expect that Ru8–NiSe2 may serve as a marginally better catalyst for the OER than pure NiSe2.
 |
| | Fig. 8 Theoretical calculations for the OER. (a) Free energy plot and (b) intermediate structures involved in the OER at the minimum overpotential sites of pure and Ru8–NiSe2 (210) systems. | |
Conclusions
In summary, we have demonstrated an energy-efficient and scalable electrosynthesis approach for the synthesis of ruthenium nanocluster decorated nickel diselenide catalysts for high-performance and stable alkaline water splitting application. We optimized the catalyst synthesis, and 50-Ru–NiSe2 was found to exhibit a very low HER overpotential of 13 mV at a current density of 10 mA cm−2 and an OER overpotential of 260 mV at a current density of 30 mA cm−2. A full cell has been designed using 50-Ru–NiSe2 as both the anode and cathode, which showed a cell potential of 1.45 V to deliver a current density of 10 mA cm−2, and we performed a long-term 400 h stability test which showed negligible degradation in current density. The enhancement in the catalytic activity of NiSe2 towards the HER and OER, after Ru cluster decoration, was also investigated via density functional theory calculations, and the results of these calculations fully explain the experimental findings. The present work thus provides a new approach to design metal nanocluster-decoration over transition metal selenide-based electrocatalysts, resulting in high electrocatalytic activity, improved electrochemically active sites, and enhanced stability for the alkaline water splitting process.
Author contributions
V. Y. and M. M. S. conceived the idea and designed the experiments. V. Y. performed all the experiments including synthesis of the catalysts, characterization of the materials, and electrochemical studies. V. Y. and M. M. S. analysed the experimental data. P. S. designed the computational part of the work based on discussions with others. P. S. and M. S. performed the theoretical studies, analysed the computational data, and wrote the portions of the manuscript related to the computational studies. V. Y. wrote the manuscript with inputs from others. M. M. S. and P. S. reviewed and edited the manuscript. M. M. S. supervised all aspects of the experimental work.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
M. M. S. acknowledges the Department of Science & Technology, Govt. of India (DST/TMD/HFC/2k18/136), Science and Engineering Research Board, Department of Science and Technology, India, (CRG/2021/006246), and IISER Thiruvananthapuram, for the financial support. V. Y. is grateful to the Govt. of India, for a PhD scholarship through the Prime Minister's Research Fellowship.
References
- J. Zhao, J. J. Zhang, Z. Y. Li and X. H. Bu, Small, 2020, 16, 2003916 CrossRef CAS PubMed.
- M. Ďurovič, J. Hnát and K. Bouzek, J. Power Sources, 2021, 493, 229708 CrossRef.
- I. Lucentini, X. Garcia, X. Vendrell and J. Llorca, Ind. Eng. Chem. Res., 2021, 60, 18560–18611 CrossRef CAS.
- X. Chu, C. I. Sathish, J.-H. Yang, X. Guan, X. Zhang, L. Qiao, K. Domen, S. Wang, A. Vinu and J. Yi, Small, 2023, 2302875 CrossRef CAS PubMed.
- C. N. R. Rao and S. Dey, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 13385–13393 CrossRef CAS PubMed.
- P. V. Sarma, T. V. Vineesh, R. Kumar, V. Sreepal, R. Prasannachandran, A. K. Singh and M. M. Shaijumon, ACS Catal., 2020, 10, 6753–6762 CrossRef CAS.
- Z.-N. Zhang, X.-H. Wang, X.-L. Tian, Y. Chen and S.-N. Li, J. Mater. Chem. A, 2023, 11, 21628–21635 RSC.
- Q. Xue, X.-Y. Bai, Y. Zhao, Y.-N. Li, T.-J. Wang, H.-Y. Sun, F.-M. Li, P. Chen, P. Jin, S.-B. Yin and Y. Chen, J. Energy Chem., 2022, 65, 94–102 CrossRef CAS.
- N. P. Dileep, P. V. Sarma, R. Prasannachandran, V. Surendran and M. M. Shaijumon, ACS Appl. Nano Mater., 2021, 4, 7206–7212 CrossRef CAS.
- N. P. Dileep, T. V. Vineesh, P. V. Sarma, M. V. Chalil, C. S. Prasad and M. M. Shaijumon, ACS Appl. Energy Mater., 2020, 3, 1461–1467 CrossRef CAS.
- P. V. Sarma, T. V. Vineesh, R. Kumar, V. Sreepal, R. Prasannachandran, A. K. Singh and M. M. Shaijumon, ACS Catal., 2020, 10, 6753–6762 CrossRef CAS.
- R. Prasannachandran, T. V. Vineesh, M. B. Lithin, R. Nandakishore and M. M. Shaijumon, Chem. Commun., 2020, 56, 8623–8626 RSC.
- Y. Bai, Y. Wu, X. Zhou, Y. Ye, K. Nie, J. Wang, M. Xie, Z. Zhang, Z. Liu, T. Cheng and C. Gao, Nat. Commun., 2022, 13, 6094 CrossRef CAS PubMed.
- T. V. Vineesh, U. V. Anagha, N. P. Dileep, H. Cheraparambil, J. Nambeesan and M. M. Shaijumon, ChemElectroChem, 2020, 7, 3319–3323 CrossRef.
- L. Li, B. Wang, G. Zhang, G. Yang, T. Yang, S. Yang and S. Yang, Adv. Energy Mater., 2020, 10, 2001600 CrossRef CAS.
- A. Shan, X. Teng, Y. Zhang, P. Zhang, Y. Xu, C. Liu, H. Li, H. Ye and R. Wang, Nano Energy, 2022, 94, 106913 CrossRef CAS.
- J. Li, J. Hu, M. Zhang, W. Gou, S. Zhang, Z. Chen, Y. Qu and Y. Ma, Nat. Commun., 2021, 12, 3502 CrossRef CAS PubMed.
- N. K. Dang, J. N. Tiwari, S. Sultan, A. Meena and K. S. Kim, Chem. Eng. J., 2021, 404, 126513 CrossRef CAS.
- P. Thangavel, G. Kim and K. S. Kim, J. Mater. Chem. A, 2021, 9, 14043–14051 RSC.
- B. Kim, T. Kim, K. Lee and J. Li, ChemElectroChem, 2020, 7, 3578–3589 CrossRef CAS.
-
B. M. Leonard, in Encyclopedia of Inorganic and Bioinorganic Chemistry, 2021, pp. 1–27 Search PubMed.
- S. Kumaravel, K. Karthick, S. S. Sankar, A. Karmakar, R. Madhu, K. Bera and S. Kundu, Sustain. Energy Fuels, 2021, 5, 6215–6268 RSC.
- F. Song, L. Bai, A. Moysiadou, S. Lee, C. Hu, L. Liardet and X. Hu, J. Am. Chem. Soc., 2018, 140, 7748–7759 CrossRef CAS PubMed.
- Y. Liu, Y. Guo, Y. Liu, Z. Wei, K. Wang and Z. Shi, Energy Fuels, 2023, 37, 2608–2630 CrossRef CAS.
- P. V. Sarma, A. Kayal, C. H. Sharma, M. Thalakulam, J. Mitra and M. M. Shaijumon, ACS Nano, 2019, 13, 10448–10455 CrossRef CAS PubMed.
- R. Prasannachandran, T. V. Vineesh, A. Anil, B. M. Krishna and M. M. Shaijumon, ACS Nano, 2018, 12, 11511–11519 CrossRef CAS PubMed.
- Y. Zhao, B. Jin, Y. Zheng, H. Jin, Y. Jiao and S.-Z. Qiao, Adv. Energy Mater., 2018, 8, 1801926 CrossRef.
- Z. Zou, X. Wang, J. Huang, Z. Wu and F. Gao, J. Mater. Chem. A, 2019, 7, 2233–2241 RSC.
- D. Damien, A. Anil, D. Chatterjee and M. M. Shaijumon, J. Mater. Chem. A, 2017, 5, 13364–13372 RSC.
- B. Sun, G. Dong, J. Ye, D. Chai, X. Yang, S. Fu, M. Zhao, W. Zhang and J. Li, Chem. Eng. J., 2023, 459, 141610 CrossRef CAS.
- M. Singh, T. T. Nguyen, J. Balamurugan, N. H. Kim and J. H. Lee, Chem. Eng. J., 2022, 430, 132888 CrossRef CAS.
- M. Ahmad, B. Xi, Y. Gu, H. Zhang and S. Xiong, Inorg. Chem. Front., 2022, 9, 448–457 RSC.
- Q. Zhao, D. Zhong, L. Liu, D. Li, G. Hao and J. Li, J. Mater. Chem. A, 2017, 5, 14639–14645 RSC.
- K. Chang, D. T. Tran, J. Wang, N. H. Kim and J. H. Lee, J. Mater. Chem. A, 2022, 10, 3102–3111 RSC.
- B. Xu, H. Yang, L. Yuan, Y. Sun, Z. Chen and C. Li, J. Power Sources, 2017, 366, 193–199 CrossRef CAS.
- Q. Hu, K. Gao, X. Wang, H. Zheng, J. Cao, L. Mi, Q. Huo, H. Yang, J. Liu and C. He, Nat. Commun., 2022, 13, 3958 CrossRef CAS PubMed.
- M. P. de Lara-Castells, C. Puzzarini, V. Bonačić-Koutecký, M. A. López-Quintela and S. Vajda, Phys. Chem. Chem. Phys., 2023, 25, 15081–15084 RSC.
- J. Cai, R. Javed, D. Ye, H. Zhao and J. Zhang, J. Mater. Chem. A, 2020, 8, 22467–22487 RSC.
- L. Chen, B. Lu, J. Zhang, R. Wu and Y. Guo, J. Colloid Interface Sci., 2022, 623, 897–904 CrossRef CAS PubMed.
- H. Hu, F. M. D. Kazim, Q. Zhang, K. Qu, Z. Yang and W. Cai, ChemCatChem, 2019, 11, 4327–4333 CrossRef CAS.
- J. Zhu, R. Lu, W. Shi, L. Gong, D. Chen, P. Wang, L. Chen, J. Wu, S. Mu and Y. Zhao, Energy Environ. Mater., 2023, 6, e12318 CrossRef CAS.
- X. Gu, M. Yu, S. Chen, X. Mu, Z. Xu, W. Shao, J. Zhu, C. Chen, S. Liu and S. Mu, Nano Energy, 2022, 102, 107656 CrossRef CAS.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2016, 138, 16174–16181 CrossRef CAS PubMed.
- J. Yu, Q. He, G. Yang, W. Zhou, Z. Shao and M. Ni, ACS Catal., 2019, 9, 9973–10011 CrossRef CAS.
- Q. Hu, K. Gao, X. Wang, H. Zheng, J. Cao, L. Mi, Q. Huo, H. Yang, J. Liu and C. He, Nat. Commun., 2022, 13, 3958 CrossRef CAS PubMed.
- R. Qin, P. Wang, Z. Li, J. Zhu, F. Cao, H. Xu, Q. Ma, J. Zhang, J. Yu and S. Mu, Small, 2022, 18, 2105305 CrossRef CAS PubMed.
- Z. Pu, Y. Luo, A. M. Asiri and X. Sun, ACS Appl. Mater. Interfaces, 2016, 8, 4718–4723 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- X. Hu, X. Tian, Y.-W. Lin and Z. Wang, RSC Adv., 2019, 9, 31563–31571 RSC.
- C. Gu, S. Hu, X. Zheng, M.-R. Gao, Y.-R. Zheng, L. Shi, Q. Gao, X. Zheng, W. Chu, H.-B. Yao, J. Zhu and S.-H. Yu, Angew. Chem., Int. Ed., 2018, 57, 4020–4024 CrossRef CAS PubMed.
- S. K. Ramesh, V. Ganesan and J. Kim, ACS Appl. Energy Mater., 2021, 4, 12998–13005 CrossRef CAS.
- H. Huang, Y. Zhao, Y. Bai, F. Li, Y. Zhang and Y. Chen, Adv. Sci., 2020, 7, 2000012 CrossRef CAS PubMed.
- B. Xu, H. Yang, L. Yuan, Y. Sun, Z. Chen and C. Li, J. Power Sources, 2017, 366, 193–199 CrossRef CAS.
- Z. Zou, X. Wang, J. Huang, Z. Wu and F. Gao, J. Mater. Chem. A, 2019, 7, 2233–2241 RSC.
- H. Wang, X. Li, Q. Ruan and J. Tang, Nanoscale, 2020, 12, 12329–12335 RSC.
- K. Fan, H. Chen, Y. Ji, H. Huang, P. M. Claesson, Q. Daniel, B. Philippe, H. Rensmo, F. Li, Y. Luo and L. Sun, Nat. Commun., 2016, 7, 11981 CrossRef CAS PubMed.
- B. Xu, H. Yang, L. Yuan, Y. Sun, Z. Chen and C. Li, J. Power Sources, 2017, 366, 193–199 CrossRef CAS.
- I. S. Kwon, T. T. Debela, I. H. Kwak, Y. C. Park, J. Seo, J. Y. Shim, S. J. Yoo, J. G. Kim, J. Park and H. S. Kang, Small, 2020, 16, 2000081 CrossRef CAS PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- X. Luo, X. Tan, P. Ji, L. Chen, J. Yu and S. Mu, EnergyChem, 2023, 5, 100091 CrossRef CAS.
- S. Anantharaj and S. Noda, Int. J. Hydrogen Energy, 2020, 45, 15763–15784 CrossRef CAS.
- I. Demiroglu, K. Yao, H. A. Hussein and R. L. Johnston, J. Phys. Chem. C, 2017, 121, 10773–10780 CrossRef CAS.
- A. S. Chaves, M. J. Piotrowski and J. L. F. Da Silva, Phys. Chem. Chem. Phys., 2017, 19, 15484–15502 RSC.
- Y. Wu, L. Wang, T. Bo, Z. Chai, J. K. Gibson and W. Shi, Adv. Funct. Mater., 2023, 33, 2214375 CrossRef CAS.
- J. K. Noerskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, ChemInform, 2005, 152, J23–J26 Search PubMed.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- I. T. McCrum and M. T. M. Koper, Nat. Energy, 2020, 5, 891–899 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Prasenjit
Sen
b,
Prasenjit
Sen