
Constructing Fe–N-doped porous carbon nanofibers for a pH-universal ORR and switchable, superior Zn–air batteries†
Yu
Ma
ab,
Yuhan
Xiao
a,
Yanfeng
Ge
 a,
Degong
Gao
c,
Yunge
Zhang
a,
Zepeng
Li
*a and
Yingdong
Han
*ab
a,
Degong
Gao
c,
Yunge
Zhang
a,
Zepeng
Li
*a and
Yingdong
Han
*ab
aCollege of Science, Civil Aviation University of China, Tianjin, P. R. China. E-mail: hansuo@126.com
bInstitute of Environment and Sustainable Development, Civil Aviation University of China, Tianjin, P. R. China
cShandong Daisheng Construction Co., Ltd, Taian, P. R. China
First published on 29th November 2023
Abstract
Mechanically stable air cathodes with exceptional oxygen reduction reaction (ORR) activities serve as a highly significant component of metal–air batteries. Herein, we have proposed and prepared a porous air electrode consisting of well-dispersed Fe nanoparticles (Zn assisted formation) embedded in nitrogen-rich 3D carbon nanofibers (called Fe(Zn)–N–C) derived from Fe, Zn–metal–organic frameworks (MOFs) by a facile and scalable approach. The obtained Fe(Zn)–N–C with a distinctly continuous porous structure presented a comparable or even better performance than the commercial 20 wt% Pt/C and previously reported non-noble electrocatalysts for Zn–air batteries, which includes (1) effective in the universal-pH range (E1/2 = 0.86, 0.74, and 0.67 V in alkaline, acidic, and neutral media, respectively), (2) extremely high stability (∼85%, 80%, and 82% activity retention for 86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 s in alkaline, acidic, and neutral media, respectively), (3) a high power density (193 and 48 mW cm−2 in alkaline and neutral media, respectively), and (4) an excellent specific capacity (800 and 688 mA h g−1 in alkaline and neutral media, respectively). This design strategy of porous non-precious metal-doped pH-universal ORR electrocatalysts could also be extended to fabricate other novel, stable, and easy-to-use multi-functional electrocatalysts for clean-energy technology.
400 s in alkaline, acidic, and neutral media, respectively), (3) a high power density (193 and 48 mW cm−2 in alkaline and neutral media, respectively), and (4) an excellent specific capacity (800 and 688 mA h g−1 in alkaline and neutral media, respectively). This design strategy of porous non-precious metal-doped pH-universal ORR electrocatalysts could also be extended to fabricate other novel, stable, and easy-to-use multi-functional electrocatalysts for clean-energy technology.
Introduction
Green power electronics, such as fuel cells and metal–air batteries, have sparked a rise in the demand for next-generation, high-performance energy-storage systems owing to their high energy densities and environmental friendliness.1–3 Among various energy-storage systems, Zn–air batteries (ZABs) are a prominent class of energy-storage devices that have drawn a lot of attention because of their high theoretical energy density of 1084 W h kg−1, inherent safety, environmental friendliness, and affordability.4,5 The oxygen reduction reaction (ORR) is recognized as the fundamental prerequisite for charge generation in these clean energy devices, yet, up till now, it faces the difficulty of limited efficiency caused by its sluggish kinetics,6 particularly in acidic and neutral electrolytes.7,8 To date, precious noble metals (e.g., Pt) are state-of-the-art electrocatalysts for the reaction in pH-universal media. Unfortunately, the large-scale development of these precious metal-based catalysts is severely restricted by their scarcity, high cost,9 unsatisfactory durability,10 and low activity for the corresponding reaction.11,12 Thus, significant efforts have been devoted to creating effective and reliable low-cost alternative catalysts in pH-universal media for high-performance ZABs.Recently, M–N–C (M = Fe, Ni, Co, etc.) materials have become a research hotspot owing to their promising ORR activities, robust interfacial conjugation, and quick adsorption of O2.13,14 In particular, Fe–N–C materials stand out as remarkably efficient and durable catalysts for ORR. Although great efforts have been devoted to preparing efficient Fe–N–C catalysts that are comparable to Pt/C, working conditions were mostly limited to alkaline conditions,15 which is still far from the pH-universal requirement for practical use. Thus, it is urgent to develop highly efficient electrocatalysts for application in acidic and neutral environments.
It has been documented that the durability of Fe–N–C moieties lowers when they are exposed to H2O2 in acidic media owing to the poor selectivity in the complex four proton-coupled electron transfer process of ORR catalysis.16 Additionally, Fe–N–C catalysts under long-term operation using acidic electrolytes accompany the effects of micropore flooding, demetallation, protonation of active sites, and/or carbon corrosion.17 Moreover, ORR activity in neutral environments is typically sluggish because of the a normally low ionic conductivity and an extremely low OH− concentration.18 In this regard, developing ORR catalysts with a high activity and good durability for all practical conditions (i.e., in alkaline, acid, and neutral media) is necessary and desirable for ZABs.
Carbon aerogels with the merits of low densities, large pore volumes, and high surface areas play an important role in the design of advanced catalysts. Particularly, their special superiority of excellent mechanical stability and metal nanoparticle compatibility brings in positively synergic and cooperative effects for elevating catalytic activity and durability, which potentially enables a lot of practical applications.19 Herein, we report a Zn-assisted Fe–N–C site catalyst loaded self-supported hierarchical porous carbon aerogel (Fe(Zn)–N–C) derived from Fe, Zn MOFs, which is provided with mechanically stable 3D catalyst-supporting architecture and electrochemical excellent conductivity. The composite aerogel imparts outstanding ORR activity and stability over the whole pH range (half-wave potential of 0.86, 0.74, and 0.67 V vs. reversible hydrogen electrode (RHE) in alkaline, acidic, and neutral environments, respectively). The Fe(Zn)–N–C catalyst shows superior durability in pH-universal electrolytes with little loss after 86400 s. This is attributed to the 3D network structure possessing improved oxygen release and mass transport properties, reduced diffusion barriers, and sustained exceptional structural stability upon long-term cycling. Moreover, we utilized the composite aerogel as an air electrode for ZABs, which reasonably show high power density (193 and 48 mW cm−2 in alkaline and neutral electrolytes, respectively), high specific capacity (800 and 688 mA h g−1 in alkaline and neutral electrolytes, respectively) and high discharging stability, making them the promising candidates for future energy-storage device applications.
Results and discussion
The formation process of the Fe(Zn)–N–C carbon aerogel catalyst is schematically shown in Fig. S1.† We additionally prepared more samples for comparison (i.e., CF, Fe–N–C, (Zn)–N–C, and Fe(Zn)–N–C-x samples). As a depiction, we concentrate on the unique advantages of the Fe(Zn)–N–C synthesis process. Recyclable, environmental-friendly, and easily processable bacterial cellulose (BC), after being treated by 2,2,6,6-tetramethylpiperidinooxy (TEMPO) oxidation, is a special candidate for building three-dimensional (3D) network-structure blocks due to the strong mechanical properties.20 Meanwhile, it is feasible to interface with transition-metal molecular clusters through ion-exchanged reactions because of their abundant surface functional groups (i.e. –COOH) on the nanofiber surface, which can act as the anchoring sites for metal and facilitate water-dispersion ability as an added benefit.21 Moreover, such abundant surface functional groups can prevent the metal particle (i.e., Fe3+) from aggregation and enhance the interaction between the nanofibers and N source.22 Except for these surface functional groups, the unique skeleton structure of Fe(Zn)–N–C is also crucial for improving the dispersion of Fe ions. During the pyrolysis, Fe ions can lead to chemical etching of the disordered carbon and enhance crystallinity. After the self-assembly and carbonization, the Fe particles migrated to the partial vacancies of Zn(NO3)2 and 2-methylimidazole (2-MIM) formed after the volatilization of zinc atoms.23 As a result, the robust, atomically Fe-site-dispersed Fe(Zn)–N–C carbon aerogel with rich defects was obtained.The transmission electron microscopy (TEM) images in Fig. 1a–c reveal that the patterns of Fe(Zn)–N–C are porous carbon nanofibers with crossed interconnections, indicating a low mass density and a high specific surface area.24 In addition, the Fe particles are successfully embedded into the carbon nanofibers. The nano-size and uniform distribution of the catalyst particles within the 3D porous network can significantly expose more active sites, which plays a critical role in improving the electrocatalytic behavior. Such a structure could provide abundant active sites and a large contact area between the catalyst and electrolyte/air for enhanced electrocatalytic performance. Interestingly, some bamboo-like carbon nanotubes with small nanoparticles existing either at the tip or inside are visible, indicating that the presence of iron salt during the pyrolysis could promote graphitization of the surrounding C atoms.25 From the HRTEM image, the Fe-based nanoparticles with darker contrast can be clearly identified in carbon spheres (Fig. 1d), which display a fringe spacing of 0.203 nm corresponding to the (110) plane of crystalline Fe. The size of the Fe-associated nanoparticles is about 10–30 nm. These nanoparticles were encapsulated in well-crystallized graphene layers with a lattice distance of 0.35 nm. The graphitized network structure is helpful for improving the electron transfer rate in the process of catalysis. The uniform distribution of Fe(Zn)–N–C was further confirmed from EDX element mappings (Fig. 1e–i), which show a homogeneous distribution of C, N, O, and Fe atoms on the carbonaceous framework throughout the carbon aerogels catalyst. Fig. 2a shows the N2 adsorption–desorption isotherms of Fe(Zn)–N–C sample with a type IV isotherm of H3-type hysteresis loop, suggesting mesoporous characteristics, which are favorable for the accessibility of the catalytic active centers. The BET-specific surface area (SSA) of Fe(Zn)–N–C is 704 m2 g−1. Pore size distributions of the Fe(Zn)–N–C catalyst are presented in Fig. 2b. Multiple pore structures including both mesoporous and macroporous pores were observed in the Fe(Zn)–N–C catalyst because the typical diverging adsorptions at both low and high relative pressure can be observed. Accordingly, the pore distribution information of Fe(Zn)–N–C showed that it comprised narrow mesopore-size structures centered at 10 nm. Fe(Zn)–N–C possesses a large pore volume of 0.60 m2 g−1 and a pore size of 4.81 nm. The large specific surface area and hierarchical micro-mesoporous structures of Fe(Zn)–N–C catalyst are in favor of iron dispersion and mass transport and thus are expected to display desirable ORR performance.
 | ||
| Fig. 1 (a)–(c) TEM images of sample Fe(Zn)–N–C, (d) HRTEM images showing the metallic Fe core and graphite shell, and (e)–(i) STEM image and the corresponding mapping images. | ||
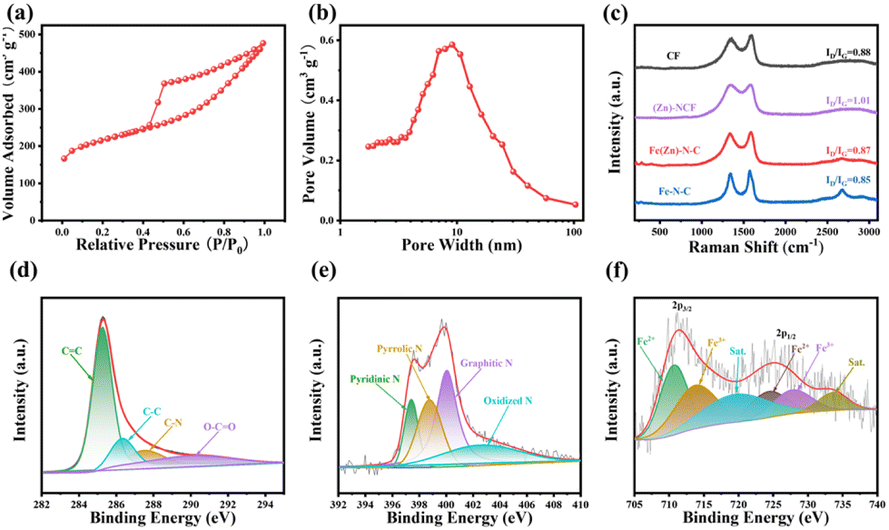 | ||
| Fig. 2 (a) N2 adsorption–desorption isotherms and (b) pore diameter distribution of Fe(Zn)–N–C. (c) Raman spectra of CF, Fe–N–C, (Zn)–N–C, and Fe(Zn)–N–C. (d) High-resolution XPS spectra of C 1s, (e) N 1s, and (f) Fe 2p for the Fe(Zn)–N–C catalyst. | ||
Additionally, the X-ray diffraction (XRD) patterns of CF, Fe–N–C, (Zn)–N–C and Fe(Zn)–N–C samples are shown in Fig. S2.† The broad diffraction peaks at ∼30° and 44.5° can be attributed to the (002) and (101) planes of graphitic carbon and amorphous carbon, respectively.26 All carbonized samples contain highly graphitic species in XRD, which are beneficial for enhanced catalyst durability. The broad hump can be attributed to the amorphous characteristic in partially graphitized materials. As for the Fe–N–C catalyst, the XRD pattern shows the characteristic peaks at 44.68° and 65.03°, which are assigned to the crystal planes (110) and (200) of Fe (PDF#97-004-4863), respectively. There are no detectable peaks in the XRD pattern of Fe(Zn)–N–C sample, implying that metal nanoparticles might be smoothly incorporated into the carbon framework vacancy formed in the volatilization sites of zinc atoms, which also agrees with the TEM results. Furthermore, Raman spectra were recorded to assess the graphitization degree of the samples (Fig. 2c). All the Raman spectra display two prominent peaks, D-band (1358 cm−1) and G-band (1594 cm−1). Generally, the intensity ratio of the D-band to the G-band (ID/IG) indicates different amounts of active defects on the catalysts. Specifically, the ID/IG ratio value of 1.01 of (Zn)–N–C is much higher than that of CF (i.e., 0.88), Fe–N–C (i.e., 0.85), and Fe(Zn)–N–C (i.e., 0.87), which is ascribed to its poor crystallization due to the dopants of N and Zn.27 In addition, the typical graphitic carbon characteristic peak of the 2D band at 2720 cm−1 can be observed within the Fe(Zn)–N–C and Fe–N–C samples, representing their high graphitization degree, which should be correlated with the formed Fe particles in comparison to the other samples.28 A balance is reached between doping and graphitization during carbonization, which means that the Fe(Zn)–N–C sample simultaneously has excellent graphitization and a large number of active distortions.
The elemental compositions and chemical state of the surface also have a great influence on the ORR activity. X-ray photoelectron spectroscopy (XPS) was performed to further investigate the surface chemistry and the valence state of the elements in Fe(Zn)–N–C. As revealed by XPS survey spectra shown in Fig. S3a,† C, O, N, Fe, and Zn are presented on the surface of Fe(Zn)–N–C. Quantitative analysis reveals the chemical composition to be 88.53 with atomic% values of C, O, N, Fe, and Zn as 8.61, 2.13, 0.71, and 0.02, respectively. The C 1s XPS shown in Fig. 2d indicates that four types of C species exist in Fe(Zn)–N–C, which was verified from the XPS peaks centered at 284.8 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 285.6 eV (C–C), 286.6 eV (C–N), and 289.0 eV (CO).29 As shown in Fig. 2e, the existing N 1s peaks of Fe(Zn)–N–C can be deconvoluted into four peaks, i.e., representative peaks centered at 398.3, 399.8, 400.2, 401.1, and 404.1 eV, which are attributed to pyridinic-N, pyrrolic-N, graphite-N, and oxidized-N, respectively.30 Notably, the peak located at 398.3 eV should also include a contribution of the Fe–Nx bond due to the close binding energy between Fe–Nx and pyridinic-N.31 Specifically, the higher percentage of pyridinic-N and graphitic-N can improve the onset potential (Eonset) and this is decisive on the diffusion limiting current of the ORR.32 The Fe–N group can also greatly facilitate the ORR process.33 The Fe 2p XPS spectra (Fig. 2f) exhibit two groups of peaks, at 710.8 and 724.3 eV for Fe2+, and 713.1 and 726.5 eV for Fe3+ along with two satellite peaks.34,35 The 2p peak in Zn XPS spectra cannot be divided into more peaks because of the low content (Fig. S3b†); however, the residual Zn species possibly catalyze the ORR performance, which was also verified in previous literatures.36,37
C), 285.6 eV (C–C), 286.6 eV (C–N), and 289.0 eV (CO).29 As shown in Fig. 2e, the existing N 1s peaks of Fe(Zn)–N–C can be deconvoluted into four peaks, i.e., representative peaks centered at 398.3, 399.8, 400.2, 401.1, and 404.1 eV, which are attributed to pyridinic-N, pyrrolic-N, graphite-N, and oxidized-N, respectively.30 Notably, the peak located at 398.3 eV should also include a contribution of the Fe–Nx bond due to the close binding energy between Fe–Nx and pyridinic-N.31 Specifically, the higher percentage of pyridinic-N and graphitic-N can improve the onset potential (Eonset) and this is decisive on the diffusion limiting current of the ORR.32 The Fe–N group can also greatly facilitate the ORR process.33 The Fe 2p XPS spectra (Fig. 2f) exhibit two groups of peaks, at 710.8 and 724.3 eV for Fe2+, and 713.1 and 726.5 eV for Fe3+ along with two satellite peaks.34,35 The 2p peak in Zn XPS spectra cannot be divided into more peaks because of the low content (Fig. S3b†); however, the residual Zn species possibly catalyze the ORR performance, which was also verified in previous literatures.36,37
As a result of enhanced mass/charge transfer and high active surface areas owing to the 3D network structure and incorporated metal nanoparticles, Fe(Zn)–N–C catalysts could be expected to display elevated ORR performance. The electrocatalytic activity of the Fe(Zn)–N–C was first examined by cyclic voltammetry (CV) measurements in an O2/N2-saturated 0.1 M KOH solution. For comparison, the performances of Fe–N–C, (Zn)–N–C, CF, and commercial Pt/C were also collected as shown in Fig. S4.† The curves of these catalysts exhibit oxygen reduction peaks at a scan rate of 20 mV s−1. The obviously well-defined cathodic peaks at 0.85 V vs. RHE are comparable to those of commercial Pt/C (0.86 V), and obviously higher than those of Fe–N–C (0.69 V), (Zn)–N–C (0.77 V), and CF (0.67 V), signifying the significant catalytic activity of oxygen reduction. To gain further insight into the ORR catalytic activity of these as-prepared samples, linear sweep voltammetry (LSV) measurements were performed using the rotating disk electrode (RDE) with a rotation rate of 1600 rpm (Fig. 3a). The pristine CF catalyst could also act as oxygen reduction catalyst at the electrode half-wave potential (E1/2) of 0.62 V vs. RHE, probably due to the similar defective structure, i.e., properties inherited from the 3D network nanofibers. After nitrogen treatment or Fe doping, the obtained (Zn)–N–C and Fe–N–C catalyst exhibited enhanced catalytic activity with an E1/2 of 0.76 and 0.65 V, respectively (Table S1†), indicating that the addition of Fe and N dopants results in the formation of more active sites during the pyrolysis process, in accordance with the XPS results. Fe(Zn)–N–C exhibited the most positive E1/2 of 0.86 V and the highest current density (Jk) of 6.16 mA cm−2, which is slightly better than those of the commercial Pt/C (E1/2 = 0.82 V, Jk = 5.68 mA cm−2), comparable to the control samples and recently reported catalysts in alkaline media (Table S2†). These results further confirm the synergistic effect of N doping, Fe nanoparticles incorporation, and Fe–Nx coordination on boosting the ORR activity. Furthermore, the superior ORR electrocatalytic activity of Fe(Zn)–N–C is ensured by the smallest Tafel slope of 66 mV dec−1, which is superior to 75 mV dec−1 of the commercial Pt/C and other samples (Fig. 3b), suggesting that Fe(Zn)–N–C and Pt/C have similar electron transfer rates and reactant diffusion in 0.1 M KOH solution. Additionally, the smallest charge transfer resistance of the Fe(Zn)–N–C catalyst also contributes to the high ORR activity (Fig. S5†). Then, LSVs of Fe(Zn)–N–C at a rotating speed of 400–2025 rpm on RDE measurements were conducted to gain insight into the pathway and kinetics of the ORR. It was clearly observed that the diffusion-limited current density was improved uniformly with the increase in rotating speed (Fig. S6a†), which proved that the ORR process is well controlled by oxygen diffusion. The analogous Koutecky–Levich (K–L) plots in Fig. S6b† at various potentials from 0.3 to 0.6 V showed good linear relationships, implying typical first-order reaction kinetics.38 The electron transfer number of Fe(Zn)–N–C is nearly 4, indicating that the ORR process on Fe(Zn)–N–C ideally follows the four-electron pathway. Therefore, the above results reveal that it is essential to simultaneously introduce both Fe and nitrogen into the catalyst for achieving such significant improvement in the ORR activity, which could produce intrinsically different active sites, namely, iron-nitrogen coordinating species (Fe–Nx).
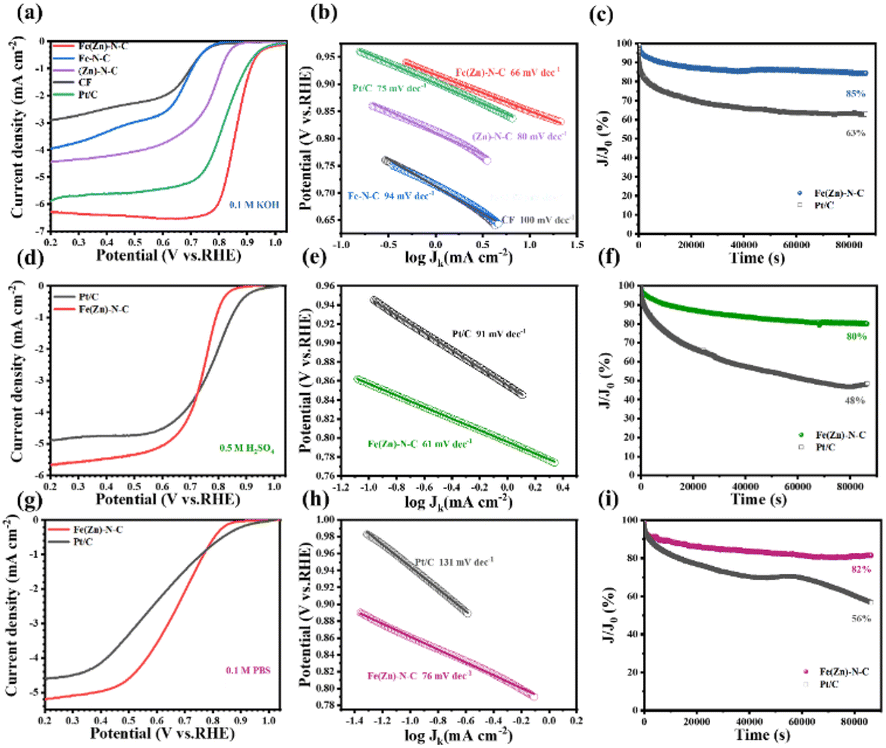 | ||
| Fig. 3 (a), (d) and (g) LSV polarization, (b), (e) and (h) Tafel slope, and (c), (f) and (i) chronoamperometric plots of the as-prepared catalysts and Pt/C in 0.1 M KOH, 0.5 M H2SO4, and 0.1 M PBS solutions, respectively. | ||
Fig. S7a–d† presents the CVs at various scan rates for the representative CF, (Zn)–N–C, Fe–N–C, and Fe(Zn)–N–C samples in N2 saturated 0.1 M KOH. These CVs were collected over a potential range from 1.092 to 1.192 V vs. RHE, where no faradaic processes were observed. The current here is entirely capacitive and increases due to the double-layer charging. This is also supported by the linear nature of the current vs. scan rate plots for various samples (Fig. S7e†). The catalyzing electrochemical surface area was reported to be proportional to the electrochemical double-layer capacitance (denoted as Cdl). Here, the Cdl value for Fe(Zn)–N–C (11.84 mF cm−2) is much higher than that for CF (4.48 mF cm−2), Fe–N–C (6.83 mF cm−2), and (Zn)–N–C (7.57 mF cm−2) samples, further revealing the advantage of Fe–Nx and the highest SSA in providing more catalytically active sites, and the highest SSA acting as a critical factor for the substantially enhanced ORR activity.
Higher electron transfer numbers and lower yield of H2O2 are highly preferred in the reduction of oxygen for energy conversion applications, which can be accessed simultaneously using the rotating ring-disk electrodes (RRDE). So, the ORR pathway was further confirmed by monitoring the transfer electron number (n) and peroxide yields (%) with a ring potential of 1.4 V in 0.1 M KOH solution at a rotation rate of 1600 rpm (Fig. S8a†). The H2O2 yield of Fe(Zn)–N–C was calculated to be 1.82–9.28% in the potential range from 0.1 V to 0.8 V, whereas it was calculated to be 2.04–10.08% for Pt/C. In addition, the n value for Fe(Zn)–N–C is 3.81–3.96 compared to 3.92–3.99 for Pt/C. Obviously, the above results suggest that the ORR process in Fe(Zn)–N–C mainly proceeds through a nearly 4e− pathway, which is consistent with the results obtained using the Koutecký–Levich (K–L) equation. Moreover, the Fe(Zn)–N–C catalyst exhibits robust tolerance to methanol compared to the commercial Pt/C (Fig. S9a†).
Durability is another important criterion for assessing electrocatalyst performance. To investigate the durability of the catalysts, the I–t chronoamperometric responses of the Fe(Zn)–N–C catalyst and Pt/C were detected under 0.1 M KOH solution over 86400 s. The results indicated that the current retention of Fe(Zn)–N–C was 85%, while that of commercial Pt/C was only 63% under the same cycling conditions (Fig. 3c). After the stability test, the structure of Fe(Zn)–N–C catalyst was characterized using TEM and XPS (Fig. S10 and S11,† respectively), from which we can observe that the Fe(Zn)–N–C still maintained its 3D porous structure with no obvious changes in morphology and microstructure, suggesting the superior electrochemical stability of the Fe(Zn)–N–C catalyst in the alkaline medium.
The excellent ORR activity of the Fe(Zn)–N–C catalyst was further ensured in acidic and neutral media. The CV curves of Fe(Zn)–N–C catalyst and Pt/C were obtained under different atmospheres (N2 or O2). For the Fe(Zn)–N–C catalyst, the distinct oxygen redox peak at 0.74 or 0.71 V vs. RHE was observed in O2-saturated 0.5 M H2SO4 or 0.1 M PBS solutions, which was a little lower than that of 20% Pt/C (0.79 or 0.78 V, respectively) (Fig. S12a and S13a,† respectively). It can be observed from Fig. 3d and g that Fe(Zn)–N–C shows competitive ORR activities (Eonset = 0.83 V, E1/2 = 0.74 V, and JL = 5.81 mA cm−2 in 0.5 M H2SO4 and Eonset = 0.83 V, E1/2 = 0.67 V, and JL = 5.26 mA cm−2 in 0.1 M PBS media), which are comparable to those of the commercial Pt/C (Eonset = 0.91 V, E1/2 = 0.78 V, and JL = 4.74 mA cm−2 in 0.5 M H2SO4 and Eonset = 0.89 V, E1/2 = 0.61 V and JL = 4.65 mA cm−2 in 0.1 M PBS) and most of the reported non-precious metal catalysts in acidic and neutral media (Tables S3 and S4†). Moreover, the Fe(Zn)–N–C catalyst displays lower Tafel slopes of 61 mV dec−1 and 76 mV dec−1 in 0.5 M H2SO4 and 0.1 M PBS solutions, Fig. 3e and h, in contrast to those of the commercial Pt/C, 91 mV dec−1 and 131 mV, dec−1, respectively. Besides, the good linear relationships of K–L plots at variable potentials in 0.5 M H2SO4 and 0.1 M PBS solutions indicate that the Fe(Zn)–N–C catalyst follows typical first-order reaction kinetics (Fig. S6c–f†). The RRDE tests manifest that the Fe(Zn)–N–C catalyst maintains higher n values meanwhile a lower percentage of H2O2 comparable to Pt/C in 0.5 M H2SO4 and 0.1 M PBS solutions (Fig. S8b and c†), verifying the efficient ORR process with a 4e− pathway again. Furthermore, the Fe(Zn)–N–C catalyst exhibited superior tolerance to methanol in both 0.5 M H2SO4 and 0.1 M PBS media compared to the commercial Pt/C (Fig. S9b and c†). Particularly, the Fe(Zn)–N–C catalyst also exhibits outstanding stability in both acidic and neutral media. After the continuous reaction for 86400 s in 0.5 M H2SO4 and 0.1 M PBS solutions, the Fe(Zn)–N–C catalyst still maintained 80% and 82% of its initial current values, respectively, which was superior to those of Pt/C (48% and 56%, respectively) (Fig. 3f and i). The XPS spectra and TEM images display negligible changes after long-term stability tests (Fig. S14–S17†), indicating the extraordinary stability for oxygen reduction in both acidic and neutral environments. In conclusion, the Fe(Zn)–N–C catalyst shows an outstanding pH-universal ORR performance compared to the reported non-precious electrocatalysts (Table S5†), portending the Fe(Zn)–N–C catalyst to be a widely potential candidate in universal media.
In order to explore the real active sites of the Fe(Zn)–N–C catalyst, thiocyanate ions (SCN−) were used to poison the Fe–Nx coordination sites.39 After adding 10 mM KSCN into O2-saturated 0.1 M KOH, 0.5 M H2SO4, and 0.1 M PBS, respectively, the negative shift of E1/2 and the decrease of the JL demonstrated that the Fe–Nx is the active site for ORR (Fig. S18a–c†). Moreover, to probe the effect of Fe particles on ORR performance, a hot acid etching experiment was carried out on the Fe(Zn)–N–C sample, where Fe particles were completely etched away in 0.5 M H2SO4 under 80 °C for 12 h. As observed from TEM images shown in Fig. S19,† left cavities confirmed the successful removal of Fe nanoparticles from the carbon matrix of Fe(Zn)–N–C. As shown in Fig. S18d–f,† the acid-treated sample shows reduced ORR activity with a negative shift of E1/2 and a much smaller decline of JL, indicating the positive effect of Fe nanoparticles on ORR catalysis.
The effect of the dosage of the introduced Fe on the electrocatalytic activity of Fe(Zn)–N–C catalysts was also explored. As shown in Fig. S20,† the electrocatalytic activity is not linearly dependent on the Fe dosage. Specifically, the catalyst Fe(Zn)–N–C (with moderate Fe dosage) shows the best performance in terms of the most positive E1/2 and the highest JL in 0.1 M KOH, 0.5 M H2SO4 and 0.1 M PBS. In contrast, when the dosage of Fe is too low (Fe(Zn)–N–C-1 sample) or too high (Fe(Zn)–N–C-3 sample), electrocatalytic activity will be lowered. The catalyst Fe(Zn)–N–C-1 exhibits reduced ORR activity with E1/2 values of 0.82 V, 0.63 V, and 0.54 V in 0.1 M KOH, 0.5 M H2SO4, and 0.1 M PBS, respectively, while the catalyst Fe(Zn)–N–C-3 also showed slightly reduced ORR activity with E1/2 values of 0.84 V, 0.69 V, and 0.59 V, respectively.
As a demonstration of the practical application, aqueous ZABs were assembled by applying Fe(Zn)–N–C as the air cathode in a static ambient atmosphere without purging air in both alkaline and neutral media (Fig. 4a and 5a). As shown in Fig. 4b, the assembled alkaline Zn–air battery showed an open circuit voltage (OCV) of 1.44 V for 10 h, comparable to the battery assembled using the Pt/C catalyst (1.46 V). As shown in Fig. 4c, the Fe(Zn)–N–C catalyst-based alkaline ZAB showed a peak power density of 193 mW cm−2, which is higher than Pt/C catalyst-based ZAB under the same conditions (111 mW cm−2). Galvanostatic discharge measurements (Fig. 4d) revealed that the Fe(Zn)–N–C catalyst-based alkaline ZAB exhibited very high voltages of 1.32 V and a specific capacity of 800 mA h g−1 at the discharge current density of 10 mA cm−2, surpassing that of the Pt/C catalyst (voltages of 1.29 V, specific capacity of 704 mA h g−1). Moreover, the Fe(Zn)–N–C catalyst-based alkaline ZAB displayed a high discharge voltage plateaus at current densities of 2–40 mA cm−2, and finally back to 2 mA cm−2 with no obvious voltage loss (Fig. 4e). Further, a light emitting diode panel showing “Zn–air” was successfully powered by the two alkaline ZABs in series (Fig. 4f), revealing the practical application capability of Fe(Zn)–N–C for environment-friendly energy devices.
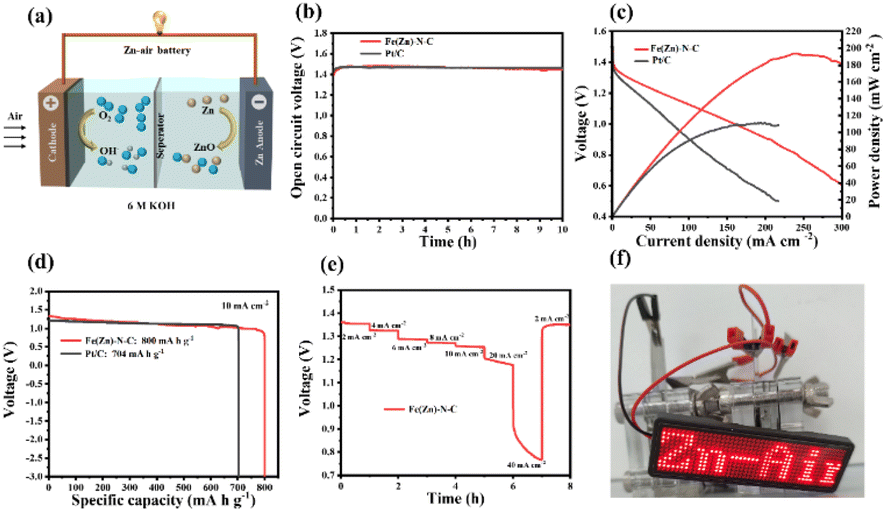 | ||
| Fig. 4 (a) Schematic illustration of the as-prepared ZABs in the alkaline electrolyte. (b) Open circuit plots of ZABs with Fe(Zn)–N–C and Pt/C cathodes. (c) Discharge polarization curves and the corresponding power density. (d) Specific discharging capacity plots at 10 mA cm−2. (e) Discharge curves of Fe(Zn)–N–C-based ZABs at various current densities. (f) Digital images of the LED screen powered by two Zn–air batteries in series based on the Fe(Zn)–N–C catalyst. | ||
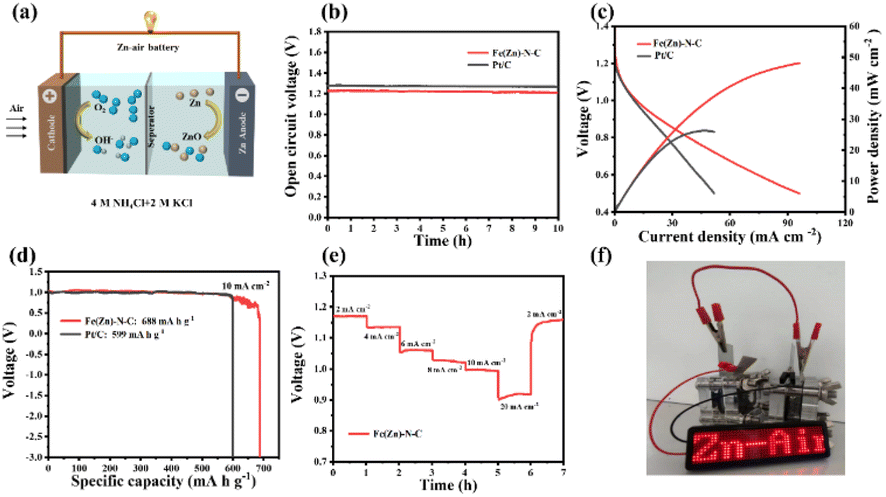 | ||
| Fig. 5 (a) A schematic illustration of the as-prepared ZABs in the neutral electrolyte. (b) Open circuit plots of ZABs with cathodes of Fe(Zn)–N–C and Pt/C. (c) Discharge polarization curves and the corresponding power densities. (d) Specific discharging capacity plots at 10 mA cm−2. (e) Discharge curves of the Fe(Zn)–N–C-based ZABs at various current densities. (f) Digital images of LED screen powered by two Zn–air batteries in series based on the Fe(Zn)–N–C catalyst. | ||
The Fe(Zn)–N–C-based neutral ZABs also exhibit better performance due to enhanced ORR activity in a neutral medium. Remarkably, Fe(Zn)–N–C-based ZAB exhibits a high and stable OCV of 1.23 V for more than 10 h (Fig. 5b). The Fe(Zn)–N–C-based ZAB demonstrated a peak power density of 48 mW cm−2 (Fig. 5c), much higher than that of Pt/C (26 mW cm−2), proving that Fe(Zn)–N–C is a potential Pt/C replacement material. Furthermore, the Fe(Zn)–N–C-based ZAB displayed a specific capacity of 688 mA h g−1 at 10 mA cm−2, surpassing that of the Pt/C catalyst (599 mA h g−1) (Fig. 5d). The Fe(Zn)–N–C catalyst-based neutral ZAB displayed a high discharge voltage plateaus at current densities of 2–20 mA cm−2, and finally back to 2 mA cm−2 without visible voltage loss (Fig. 5e). From Fig. 5f, two ZABs connected in series can light up the LED panel. Therefore, the homemade liquid ZABs with alkaline and neutral electrolytes exhibit excellent device-level performance, which is comparable to that of the most reported neutral/alkaline electrolytes and catalysts (Table S6†).
Conclusions
In summary, a highly efficient and long-term stable Fe site anchored-on (Zn-assisted formation) hierarchically porous N-doped carbon electrocatalyst (Fe(Zn)–N–C) derived from Fe, Zn MOFs was developed for excellent ORR over the whole pH range. The as-prepared Fe(Zn)–N–C electrocatalyst possesses a high density of Fe–N active sites and high BET surface area (704 m2 g−1). Importantly, the 3D nanofibrous network is highly favourable for the rapid transportation of ORR species. Such a combination of large reactive surface area and fast transportation path brought about superior ORR activity in a wide pH range and surpassed or showed comparable performance to that of the commercial Pt/C or the reported non-precious metal catalysts. Additionally, the Fe(Zn)–N–C showed great potential as an air cathode for zinc–air batteries in alkaline and neutral electrolytes. This work provides novel insight into structural engineering to fabricate highly efficient and durable pH-universal ORR catalysts, which are significant for advanced renewable energy conversion and storage devices. We expect this synthetic strategy for air electrode materials to be further extended to other 3D monolithic composites for diverse energy conversions and storage systems, such as other metal–air batteries, sodium-ion batteries, and supercapacitors.Author contributions
Yu Ma: investigation, methodology, formal analysis, and writing-original draft. Yuhan Xiao: methodology. Yanfeng Ge: methodology. Degong Gao: methodology. Yunge Zhang: methodology. Zepeng Li: writing-review and editing and resources. Yingdong Han: writing-review, editing and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (Grant No. 3122023QD20).Notes and references
- T. Xia, K. Zhao, Y. Zhu, X. Bai, H. Gao, Z. Wang, Y. Gong, M. Feng, S. Li, Q. Zheng, S. Wang, R. Wang and H. Guo, Adv. Mater., 2023, 35, 2206508 CrossRef CAS PubMed.
- Y. Xu, H. Xue, X. Li, X. Fan, P. Li, T. Zhang, K. Chang, T. Wang and J. He, Nano Res. Energy, 2023, 2, e9120052 CrossRef.
- X. J. Zhang, L. Zhou, K. P. Hu, D. D. Gao, S. Tang, L. He, Y. F. Chen, P. Zhang and Z. G. Zhang, Chem. Eng. J., 2023, 476, 146612 CrossRef CAS.
- W. F. Wu, X. Yan and Y. Zhan, Chem. Eng. J., 2023, 451, 138608 CrossRef CAS.
- K. Srinivas, Z. Chen, F. Ma, A. Chen, Z. H. Zhang, Y. Wu, M. Q. Zhu and Y. F. Chen, Appl. Catal., B, 2023, 335, 122887 CrossRef CAS.
- R. Cheng, M. Jiang, K. Li, M. Guo, J. Zhang, J. Ren, P. Meng, R. Li and C. Fu, Chem. Eng. J., 2021, 425, 130603 CrossRef CAS.
- L. Peng, J. Yang, Y. Yang, F. Qian, Q. Wang, D. Sun-Waterhouse, L. Shang, T. Zhang and G. I. N. Waterhouse, Adv. Mater., 2022, 34, 2202544 CrossRef CAS PubMed.
- X. Jiang, J. X. Wang, T. Huang, G. T. Fu, Y. W. Tang, X. Y. Qiu, J. C. Zhou and J. M. Lee, J. Mater. Chem. A, 2019, 7, 26243–26249 RSC.
- K. Srinivas, D. W. Liu, F. Ma, A. Chen, Z. H. Zhang, Y. Wu, Q. Wu and Y. F. Chen, Small, 2023, 19, 2301589 CrossRef CAS PubMed.
- B. Wang, K. Srinivas, Y. F. Liu, D. W. Liu, X. J. Zhang, W. L. Zhang and Y. F. Chen, Nano Res., 2022, 15, 3971–3979 CrossRef CAS.
- X. Wang, J. W. Wang, P. Wang, L. C. Li, X. Y. Zhang, D. M. Sun, Y. F. Li, Y. W. Tang, Y. Wang and G. T. Fu, Adv. Mater., 2022, 34, 2206540 CrossRef CAS PubMed.
- R. P. Zhao, Z. H. Chen, Q. H. Li, X. Wang, Y. W. Tang, G. T. Fu, H. Li, J. M. Lee and S. M. Huang, Chem Catal., 2022, 2, 3590–3606 CrossRef CAS.
- M. Tobisu, K. Yamakawa, T. Shimasaki and N. Chatani, Chem. Commun., 2011, 47, 2946–2948 RSC.
- T. Cui, Y. P. Wang, T. Ye, J. Wu, Z. Chen, J. Li, Y. Lei, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61, e202115219 CrossRef CAS PubMed.
- G. Wu and P. Zelenay, Acc. Chem. Res., 2013, 46, 1878–1889 CrossRef CAS PubMed.
- Y. Dong, Z. Fang, D. Ou, Q. Shi, Y. Ma, W. Yang, B. Tang and Q. Liu, Chem. Eng. J., 2022, 444, 136433 CrossRef CAS.
- H. Singh, S. Zhuang, B. Ingis, B. B. Nunna and E. S. Lee, Carbon, 2019, 151, 160–174 CrossRef CAS.
- L. An, Z. Zhang, J. Feng, F. Lv, Y. Li, R. Wang, M. Lu, R. B. Gupta, P. Xi and S. Zhang, J. Am. Chem. Soc., 2018, 140, 17624–17631 CrossRef CAS PubMed.
- D. Kobina Sam, E. Kobina Sam and X. Lv, ChemElectroChem, 2020, 7, 3695–3712 CrossRef CAS.
- Y. Ma, Q. Liu, W. Li, Y. Zheng, Q. Shi, Z. Zhou, G. Shao, W. Yang, D. Chen and X. Fang, J. Mater. Chem. A, 2021, 9, 900–907 RSC.
- C. Lai, L. Sheng, S. Liao, T. Xi and Z. Zhang, Surf. Interface Anal., 2013, 45, 1673–1679 CrossRef CAS.
- A. Isogai and L. Bergström, Curr. Opin. Green Sustainable Chem., 2018, 12, 15–21 CrossRef.
- X. Xu, X. Zhang, Z. Xia, R. Sun, H. Li, J. Wang, S. Yu, S. Wang and G. Sun, J. Energy Chem., 2021, 54, 579–586 CrossRef CAS.
- J. G. Wang, Y. Yang, Z. H. Huang and F. Kang, Electrochim. Acta, 2011, 56, 9240–9247 CrossRef CAS.
- Q. Wang, Y. Lei, Z. Chen, N. Wu, Y. Wang, B. Wang and Y. Wang, J. Mater. Chem. A, 2018, 6, 516–526 RSC.
- Q. Shi, Y. Ma, L. Qin, B. Tang, W. Yang and Q. Liu, ChemElectroChem, 2019, 6, 2924–2930 CrossRef CAS.
- Z. Zhao, Y. Xiong, S. Yu, T. Fang, K. Yi, B. Yang, Y. Zhang, X. Yang, X. Liu and X. Jia, J. Colloid Interface Sci., 2023, 650, 934–942 CrossRef CAS PubMed.
- Q. Shi, Q. Liu, Y. Ma, Z. Fang, Z. Liang, G. Shao, B. Tang, W. Yang, L. Qin and X. Fang, Adv. Energy Mater., 2020, 10, 1903854 CrossRef CAS.
- C. Hu, S. Bai, L. Gao, S. Liang, J. Yang, S.-D. Cheng, S. B. Mi and J. Qiu, ACS Catal., 2019, 9, 11579–11588 CrossRef CAS.
- L. Gao, M. Xiao, Z. Jin, C. Liu, J. Ge and W. Xing, J. Energy Chem., 2019, 35, 17–23 CrossRef.
- Z. Y. Wu, X. X. Xu, B. C. Hu, H. W. Liang, Y. Lin, L. F. Chen and S. H. Yu, Angew. Chem., Int. Ed., 2015, 54, 8179–8183 CrossRef CAS PubMed.
- P. Yan, J. Liu, S. Yuan, Y. Liu, W. Cen and Y. Chen, Appl. Surf. Sci., 2018, 445, 398–403 CrossRef CAS.
- K. Artyushkova, I. Matanovic, B. Halevi and P. Atanassov, J. Phys. Chem. C, 2017, 121, 2836–2843 CrossRef CAS.
- T. F. Li, Y. J. Hu, K. H. Liu, J. W. Yin, Y. Li, G. T. Fu, Y. W. Zhang and Y. W. Tang, Chem. Eng. J., 2022, 427, 131992 CrossRef CAS.
- M. N. Chen, Z. J. Lin, Y. Ren, X. Wang, M. Li, D. M. Sun, Y. W. Tang and G. T. Fu, Mater. Chem. Front., 2023, 7, 4132–4141 RSC.
- D. W. Liu, K. Srinivas, A. R. Chen, F. Ma, H. S. Yu, Z. H. Zhang, M. Y. Wang, Y. Wu and Y. F. Chen, J. Colloid Interface Sci., 2023, 635, 578–587 CrossRef CAS PubMed.
- D. W. Liu, K. Srinivas, X. Chen, F. Ma, X. J. Zhang, X. Q. Wang, B. Wang and Y. F. Chen, J. Colloid Interface Sci., 2022, 624, 680–690 CrossRef CAS PubMed.
- X. Liu, H. Gong, T. Wang, H. Guo, L. Song, W. Xia, B. Gao, Z. Jiang, L. Feng and J. He, Chem.–Asian J., 2018, 13, 528–535 CrossRef CAS PubMed.
- L. Jiao, G. Wan, R. Zhang, H. Zhou, S. H. Yu and H. L. J. A. C. Jiang, Angew. Chem., Int. Ed., 2018, 130, 8661–8665 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06537e |
| This journal is © The Royal Society of Chemistry 2024 |