
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Integration of a composite polymer electrolyte and Se/C cathodes toward high-performance all-solid-state Li–Se batteries†
Tae Hwa
Hong‡
 a,
Jea Duk
Kim‡
a,
Jung Seok
Lee
a,
Yujin
Choi
a,
Han Young
Jung
a,
Yoon Hak
Lee
a,
Sung Yeon
Hwang
*a,
KwangSup
Eom
*b and
Jung Tae
Lee
*ac
a,
Jea Duk
Kim‡
a,
Jung Seok
Lee
a,
Yujin
Choi
a,
Han Young
Jung
a,
Yoon Hak
Lee
a,
Sung Yeon
Hwang
*a,
KwangSup
Eom
*b and
Jung Tae
Lee
*ac
aDepartment of Plant and Environmental New Resources, Kyung Hee University, 1732, Deogyeong-daero, Giheung-gu, Yongin-si, Gyeonggi-do 17104, Republic of Korea. E-mail: crew75@khu.ac.kr; jungtae@khu.ac.kr
bSchool of Materials Science & Engineering, Gwangju Institute of Science and Technology (GIST), 123 Cheomdangwagi-ro, Buk-gu, Gwangju, 61005, Republic of Korea. E-mail: keom@gist.ac.kr
cDepartment of Biotechnology, Kyung Hee University, 1732, Deogyeong-daero, Giheung-gu, Yongin-si, Gyeonggi-do 17104, Republic of Korea
First published on 15th January 2024
Abstract
The coupling of composite polymer electrolytes (CPEs) with Se/C cathodes was achieved in this study to address the needs of modern solid-state lithium batteries, including high-energy density, low temperature (30 °C) operation, electrochemical stability, and ease of manufacture. Our solid-state Li–Se battery exhibited exceptional rate performance at 30 °C and even surpassed contemporary solid-state Li–S batteries, which operate at elevated temperatures, in terms of performance. Excellent battery performance was exemplified by a sustained capacity of 250 mA h g−1 over 200 cycles at 2C. We also fabricated pouch-type solid-state Li–Se batteries using CPEs and demonstrated their ease of manufacture relative to previous solid-state Li–Se batteries. This paper presents a promising approach for advancing all-solid-state batteries through the synergistic utilization of CPEs and Li–Se chemistry.
With increasing demand for energy storage devices exhibiting enhanced performance and safety, researchers have started focusing on the development of all-solid-state batteries (ASSBs).1–3 ASSBs are considered an attractive power source because they exhibit superior safety and energy densities compared with liquid-electrolyte batteries. Solid electrolytes have received attention due to their non-flammable properties and the ability to prevent the growth of lithium dendrites.4,5 Solid-state batteries can achieve greater energy densities by employing lithium metal anodes, which possess a theoretical capacity of 3860 mA h g−1.6,7 In addition to high-energy-density Li metal anodes, research has focused on high-energy-density cathode materials in solid-electrolyte systems.8–10 Lithium–sulfur (Li–S) chemistry is prominent in this regard due to its exceptional theoretical energy density (1675 mA h g−1) and the utilization of cost-effective and eco-friendly cathode resources. However, contemporary all-solid-state Li–S batteries face challenges such as sluggish reaction kinetics, voltage decay, and limited electrochemical stability.11,12 These problems also exist in liquid-electrolyte systems, which highlights the need for innovative solutions to unleash the full potential of all-solid-state Li–S batteries. Liquid-electrolyte systems based on lithium–selenium (Li–Se) chemistry may overcome the limitations of Li–S batteries.13–15 Li–Se batteries exhibit faster reaction kinetics than Li–S systems due to the greater electrical conductivity of elemental Se (1 × 10−3 S m−1) relative to that of S (5 × 10−18 S m−1). Furthermore, it exhibits more stable electrochemical features than Li–S batteries, contributing to a significantly extended cycle life. Despite its higher cost compared to S, Se is often considered a more economical option than commercially available cathode materials.16,17 These factors have made Li–Se chemistry a focal point in the development of all-solid-state Li–Se batteries (ASSLSeBs).18–26 However, existing ASSLSeBs largely employ inorganic solid electrolytes (ISEs), which suffer from poor interfacial contact that seriously limits their manufacture.27,28 Our objective is to enhance the promise of ASSLSeBs by overcoming the limitations of existing ISEs. Herein, we describe a solid-state Li–Se battery comprising a composite polymer electrolyte (CPE) and Se/C cathode, as illustrated in Fig. 1a. To formulate a polymer electrolyte compatible with Se/C cathodes, we prepared CPEs using PEO as the polymer matrix, Li6.25La3Zr2Al0.25Nb0.25O12 (LLZANO) as the ceramic filler, lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) as the lithium salt, and succinonitrile (SN) as the plasticizer. Garnet-type LLZANO was chosen among various ISEs due to its exceptional characteristics, including high ionic conductivity at room temperature (10−3 to 10−4 S cm−1), excellent chemical stability against lithium metal, and a wide electrochemical stability window (at least ∼5.0 V vs. Li+/Li).29 The crystal structure of LLZANO was investigated by X-ray diffraction (XRD) analysis (Fig. S1†), revealing a cubic structure which exhibits superior ionic conductivity.30,31 Although the impurity peaks were observed in the 2θ range of 20–30°, a trace of impurities which were largely caused by Li0.5La2Al0.5O4 did not significantly influence LLZANO performance.32 The morphology and microstructure of LLZANO were examined using scanning electron microscopy (SEM) (Fig. S2a and b†) and EDS (Fig. S2c†), respectively. LLZANO consists of 5–8 μm particles with even contributions from La, Zr, Al, and Nb. Panel (i) in Fig. 1b contains a photograph of a 75.8 μm thick, 19 mm diameter LLZANO-dispersed CPE disk. Panel (ii) shows EDS mapping of the disk, which verifies the uniform distributions of C and O (from PEO), S and N (from LiTFSI and succinonitrile), and La, Al, and Nb (from LLZANO). Fig. S3† establishes the smoothness of the CPE surface, which enhances contact with the electrode relative to that of ISE surfaces. Differential scanning calorimetry (DSC) was used to examine the impact of LLZANO on the thermal properties of the CPE. LLZANO significantly lowers the melting point and glass transition temperature of the PEO-based electrolyte relative to that of a solid polymer electrolyte (SPE) without LLZANO (Fig. S4†). The reduction in crystalline area increases the amorphous content, promotes greater segmented polymer motion, and enhances ionic conductivity. Thus, LLZANO addition improves the ionic conductivity of the solid electrolyte. Electrochemical impedance spectroscopy (EIS) measurements were performed at 10, 15, 20, and 25 wt% LLZANO in the CPE to determine the optimal LLZANO concentration (Fig. S5a†). A CPE containing 15 wt% LLZANO exhibited the greatest ionic conductivity (1.91 × 10−4 S cm−1) making it the best candidate for use in a solid-state Li–Se battery (Fig. S5b†). A transference number of tLi+ = 0.3 was obtained using a Li symmetric cell fabricated with 15 wt% LLZANO in the CPE (Fig. S6†). The oxidative stability of the CPE was evaluated by linear sweep voltammetry (LSV) (Fig. S7†). When comparing SPE and CPE, the introduction of LLZANO successfully increased oxidation stability. The range of voltage stability extends to 4.8 V vs. Li+/Li, which supports the use of the CPE in solid-state Li–Se batteries. We investigated two aspects of Li–Se chemistry to identify the most favorable conditions for CPE utilization. Commercial mesoporous carbon Ketjen Black EC600JD (KB) and synthetic microporous carbon (MPC) were examined as host materials for Se confinement. The properties of nanodimensional pores in the host materials were characterized by N2 adsorption/desorption analysis at 77 K (Fig. S8a†). The results indicate that KB and MPC possess abundant meso/micropores suitable for nano-sized Se confinement. The isotherms of Se/KB and Se/MPC composites approach near-zero adsorption, which suggests effective filling of nano-sized Se into the meso/micropores of the hosts via a melt-diffusion process (Fig. S8b†). The pore size distributions of the host materials and Se/C composites also indicate the successful infiltration of Se into the host-material pores (Fig. S8c and d†). The micromorphologies of the Se/C composites were identified by SEM. The corresponding EDS results establish uniform dispersion of Se throughout the host material (Fig. 1c). The XRD patterns of the Se/C composites are amorphous (like those of the host materials) and lack the distinctive crystalline peaks of elemental Se (Fig. S9†). Additional insight into the chemical state of Se confined in the host materials was obtained by X-ray photoelectron spectroscopy (XPS) (Fig. S10†). The selenium 3d spectra comprise a well-defined 3d3/2–3d5/2 doublet and a Se–O interaction component in both composites. Accurate quantification of the Se loading in the cathodes was established by electrochemical measurements and thermogravimetric analysis. The TGA results in Fig. S11† indicate that the KB/Se and Se/MPC composites contain 70.1 and 49.7 wt% Se, respectively.
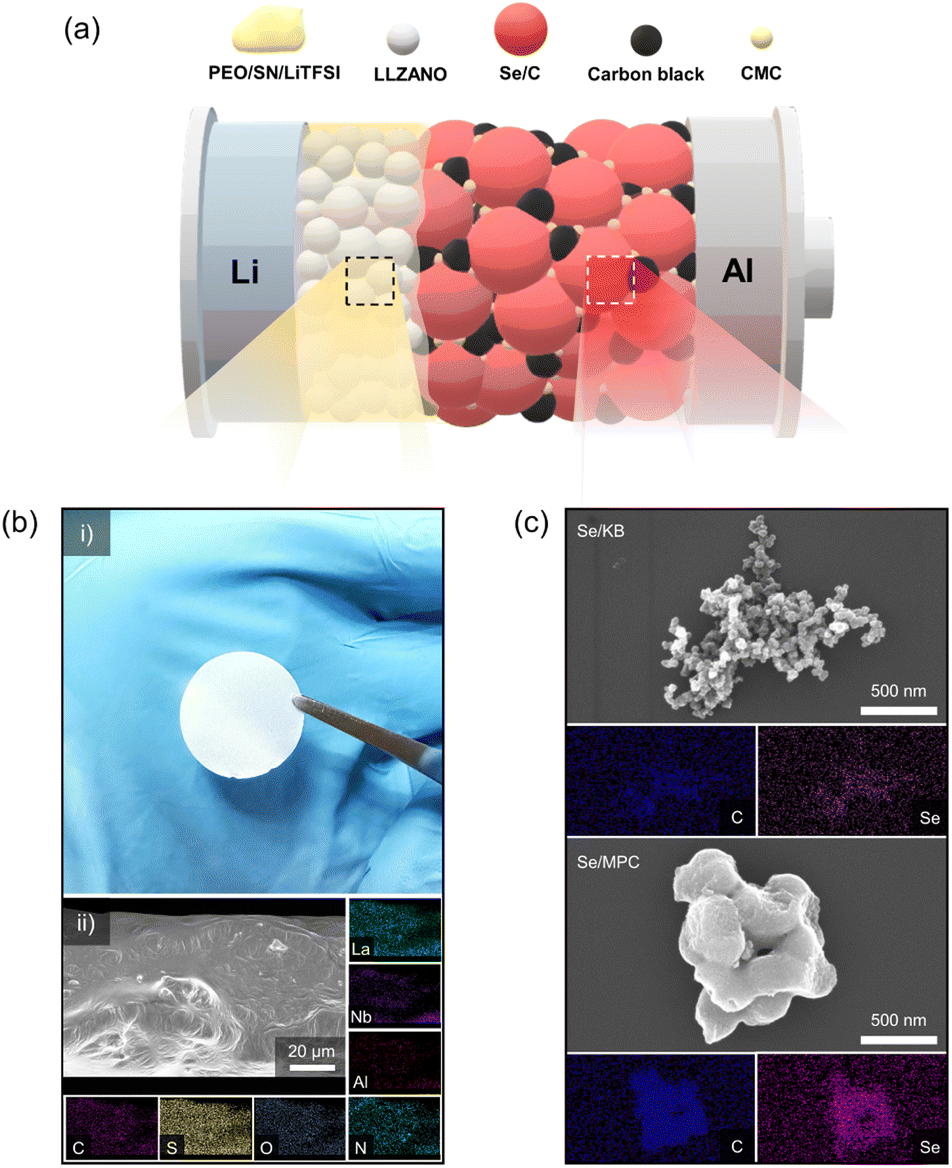 | ||
| Fig. 1 (a) Illustration of an ASSLSeB comprising a CPE and a Se/C cathode. (b) (i) Visual image of a polyethylene oxide (PEO)/LLZANO/LiTFSI/SN-CPE disk; (ii) CPE surface morphology and elemental mapping by energy dispersive X-ray spectroscopy (EDS). (c) Micromorphologies and EDS elemental mapping results of Se/KB and Se/MPC cathode materials. | ||
Electrochemical evaluation conducted on ASSLSeBs assembled from the CPE and the two Se/C cathodes is described in the following material. The electrochemical behavior of ASSLSeB cells was investigated using a galvanostatic charge–discharge protocol at 0.2C, following a 3-cycle formation protocol at 0.05C (Fig. 2a). The cells exhibited similar specific capacities that are close to the theoretical value for Se (675 mA h g−1) after the first discharge at a rate of 0.05C. The capacities decrease significantly to 310 mA h g−1 for Se/KB and 550 mA h g−1 for Se/MPC after three formation cycles at 0.05C. The differences in capacity retention indicate rapid fall-off for Se/KB and more sustained retention for Se/MPC. The disparity is attributed to the solid–liquid–solid conversion mechanism of Se/KB, which generates soluble high-order polyselenides during the (dis)charging process33,34 (voltage profiles of Se/KB and Se/MPC are shown in Fig. S12†). High-order polyselenides are known to be transformed into polymer chains.35 This conversion is confirmed by optical micrographs of the CPE after lithiation to 1.0 V (Fig. 2b). The CPE paired with Se/KB (upper image) becomes discolored with a dark-brown hue occurring around the electrode due to polyselenide permeation. Conversely, no color change is observed in the CPE assembled with Se/MPC (bottom image). To verify the permeation of polyselenides within the CPE in each system (Se/KB or Se/MPC), we conducted EDS elemental mapping analysis on each CPE obtained after electrochemical reactions. As depicted in Fig. S13a,† the presence of Se species was confirmed in the CPE surrounding the KB/Se electrode. Conversely, no Se species were detected in the CPE around the MPC/Se electrode (Fig. S13b†). The visually evident observations, based on both optical images and EDS analysis, unequivocally demonstrate the existence of permeated polyselenides in the CPE for each system leading to the clear conclusion that soluble polyselenides do not form during the discharging/charging process in the MPC/Se cell.36–38 EIS studies demonstrate how the presence of polyselenides is crucial to the performance of solid-state Li–Se batteries with a CPE. Fig. 2c shows the Nyquist plots and fitting results (fitted using an equivalent circuit as shown in the inset) for both cells in a fully lithiated state (1.0 V) after three formation cycles. The equivalent circuit for the fitting is shown in the inset. Compared to Se/KB, Se/MPC demonstrated lower bulk/SEI/charge transfer resistances (Rb, RSEI, and Rct) and a higher Li ion diffusion coefficient (DLi+) (Fig. 2d). The Rb values of Se/MPC and Se/KB before the electrochemical reaction (i.e., polyselenides are absent) are similar indicating similar ohmic resistances for Se/MPC and Se/KB (see Fig. S14a and b†). However, when polyselenides are introduced into the CPE, the Rb of Se/KB becomes larger than that of Se/MPC owing to polyselenide permeation into the polymer matrix. Moreover, RSEI and Rct are significantly larger than the corresponding values for Se/KB.39 The diffusional properties of Se/KB cathodes are also negatively impacted by the presence of polyselenides. Thus, polychalcogenide (polyselenide) regulation plays a crucial role in solid-electrolyte systems, where a fundamental understanding of reaction mechanisms is crucial for achieving rapid reaction kinetics. Consequently, elaboration of a direct solid–solid conversion mechanism that excludes the formation of soluble polychalcogenides is a promising approach for developing solid-electrolyte systems in improved solid-state Li–chalcogen batteries.
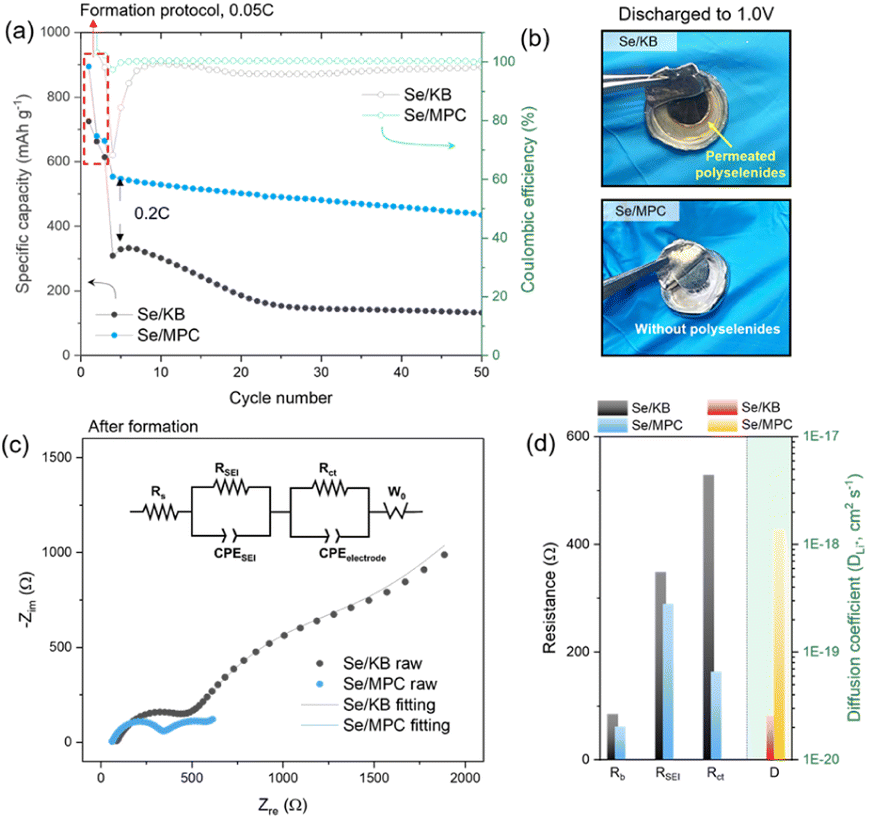 | ||
| Fig. 2 Comparison of Se/KB and Se/MPC cathode behavior. (a) Se/KB and Se/MPC capacities at 0.2C as a function of cycle number after a 3-cycle, 0.05C formation protocol. (b) Optical image of CPE in cells after discharge to 1.0 V. (c) Nyquist plots of Se/KB and Se/MPC cells after formation; the inset contains an equivalent circuit for the fitting. (d) Resistance parameters (R) and Li+ diffusion coefficient (DLi+) obtained by EIS. | ||
Our results establish that Se/MPC is the most favorable cathode material for solid-state Li–Se batteries based on its direct solid–solid conversion mechanism. Accordingly, ASSLSeBs, incorporating a Se/MPC and a CPE exhibit excellent rate performances, even at room temperature (RT) (Fig. 3a and b). The initial discharge of the cell at 0.05C produces a capacity of approximately 900 mA h g−1, which surpasses the theoretical capacity of Se (675 mA h g−1). This behavior is commonly observed for S or Se confined within a sub-nano spaces,40,41 which is recognized as an additional capacity arising from the reaction that forms the cathode-electrolyte interphase (CEI) layer.42 The cell subsequently displayed capacities of 640, 560, 500, 340, 210, and 150 mA h g−1 at rates of 0.05, 0.1, 0.2, 0.5, 1, and 2C, respectively. Fig. 3c shows the rate performance of our ASSLSeBs in comparison with chemically similar solid-state Li–S batteries comprising polymer-based solid electrolytes, which generally operate at 45–75 °C.43–59 The limited Li-ion kinetics imposed by the solid-electrolytes, combined with inherently sluggish reaction kinetics of Li–S chemistries, restricts the operation of solid-state Li–S batteries to the elevated temperature. Conversely, our ASSLSeBs demonstrate high reversible capacity and superior rate capability even at 30 °C. The exceptional reaction properties of our ASSLSeBs originated from the intrinsic difference in the reaction kinetics between Li–S and Li–Se chemistries. First, it's important to note that elemental selenium generally exhibits several orders of magnitude higher electrical conductivity than elemental sulfur, as mentioned earlier. This higher electrical conductivity facilitates faster charge transfer during electrochemical reactions, enhancing overall rate performance of electrochemical cells.60 Another primary factor that significantly enhances the solid-state reaction kinetics in Li–Se chemistries compared to Li–S chemistries is the superior ionic conductivity of the final reaction product, Li2Se, in contrast to Li2S. Previous studies have revealed that the migration barrier for Li-ions in Li2Se is much lower than that in Li2S.12,33 In solid-state battery systems, mass transport kinetics are pivotal factors influencing the rate of reaction.2,61 Consequently, the higher ionic conductivity of Li2Se formed within cathodes during the electrochemical reaction plays a substantial role in accelerating the reaction rates. Furthermore, the effective mitigation of polyselenide formation thorough confinement in sub-nano spaces further facilitated our ASSLSeBs to exhibit superior rate performance even under RT operation. These features highlight the potential of Li–Se chemistries, which achieve superior rate performance and the capability of RT operation in solid-electrolyte systems that are not attainable through Li–S chemistry. Furthermore, the excellent long-term cycling stability of our ASSLSeBs is confirmed by the cycle test in Fig. 3d. This ASSLSeB maintains a 250 mA h g−1 capacity at 2C, even after 200 cycles. To demonstrate the superior performance and potential of our ASSLSeB, we conducted a comparison with all-solid-state lithium batteries (ASSLBs) composed of CPE and conventional cathodes (NMC111 and LFP). In contrast to our ASSLSeB, the ASSLB employing the NMC111 cathode exhibits rapid capacity degradation within 50 cycles. The oxidation stability of PEO-based solid electrolyte was unsuitable with the operation voltage window of NMC-based cathodes without any stabilization treatment, resulting in degradation of PEO causing fatal damage to the electrolytes–electrodes interfaces.62,63 In contrast, the ASSLB composed of conventional LFP cathode and CPE shows stable capacity retention during the cycle test. However, its capacity was far lower than our ASSLSeB after 200 cycles (250 vs. 75 mA h g−1) based on the intrinsically lower theoretical capacities. These compelling results suggest the electrochemical superiority of Li–Se chemistry within polymer-based solid electrolyte systems when compared to conventional cathode materials under the same conditions.
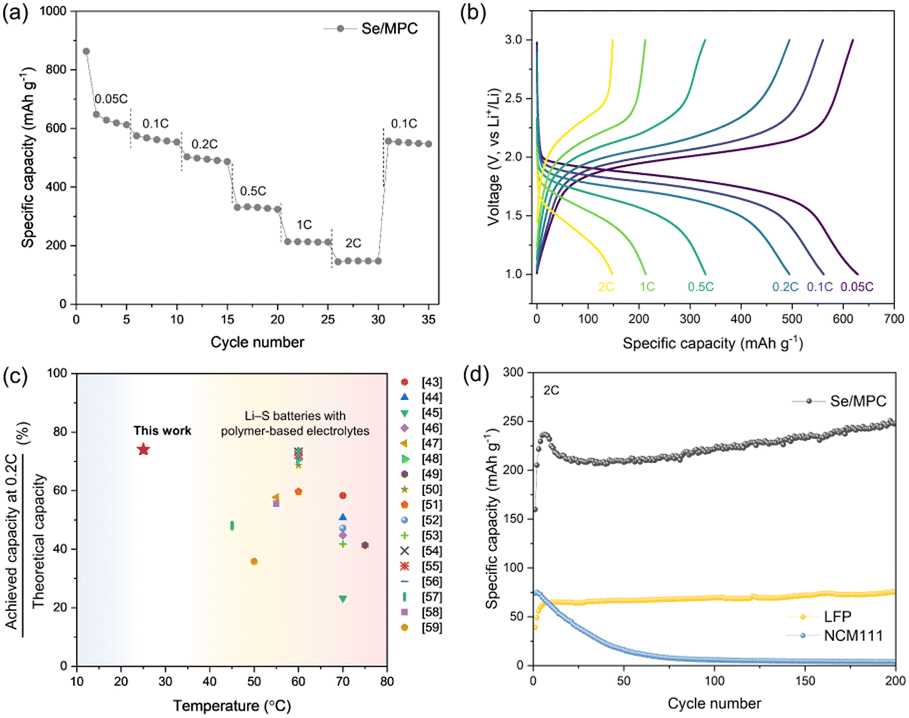 | ||
| Fig. 3 Electrochemical performance of ASSLSeBs with Se/MPC. (a) Capacity profiles and (b) voltage profiles at different current densities. (c) Comparison of operating temperature and practical-to-theoretical capacity ratio at 0.2C for the Li–Se battery with a CPE and the corresponding parameters of contemporary solid-state Li–S batteries with polymer-based electrolytes. (d) Cycle performance (at 2C) of Se/MPC and conventional LFP and NCM111 cathodes (at 0.05C for 3 cycles). | ||
As highlighted in our previous discussion, the primary advantages of our CPE-enabled ASSLSeBs lie in their ease of manufacture, which distinguishes them from previously reported ASSLSeBs.18–26 To showcase these advantages, we fabricated a pouch-type, solid-state Li–Se battery with 4 cm × 4 cm electrodes, as shown in Fig. 4a. A preliminary open-circuit-voltage (OCV) test using a multimeter confirmed successful fabrication of the cell (Fig. 4b(i)). The powering of a red LED upon connection further confirms its normal operational capability (Fig. 4b(ii)). The pouch cell maintains an exceptionally stable OCV of approximately 3.0 V throughout a 14-day rest period at 30 °C (Fig. S15†). There is no evidence of self-discharge, the absence of which is a necessary component of stable storage. However, it must be noted that Li–Se batteries operate at a lower voltage (∼2.0 V) than conventional Li-ion batteries (3.5–4.0 V). To overcome this limitation, we connected two solid-state Li–Se pouch cells in series and obtained an OCV output exceeding 5 V. This configuration enables the successful operation of a white LED with a threshold voltage of >3 V (panels (iii) and (iv) in Fig. 4b). Our solid-state Li–Se battery with a CPE is also resistant to physical stress, as evidenced by its ability to withstand folding without detrimental effects (Fig. 4c and Video S1†). Finally, Fig. 4d shows that our solid-state Li–Se pouch cell achieves a specific capacity of 450 mA h g−1 at a current density of 0.2C for up to 40 cycles. This work illustrates the immense potential of employing a CPE in solid-state Li–Se batteries. However, additional research focusing on cathode and CPE composition is needed to fully optimize the overall performance of these systems. A greater understanding of the synergism between a CPE and Li–Se chemistry should foster the design of more advanced ASSLSeBs.
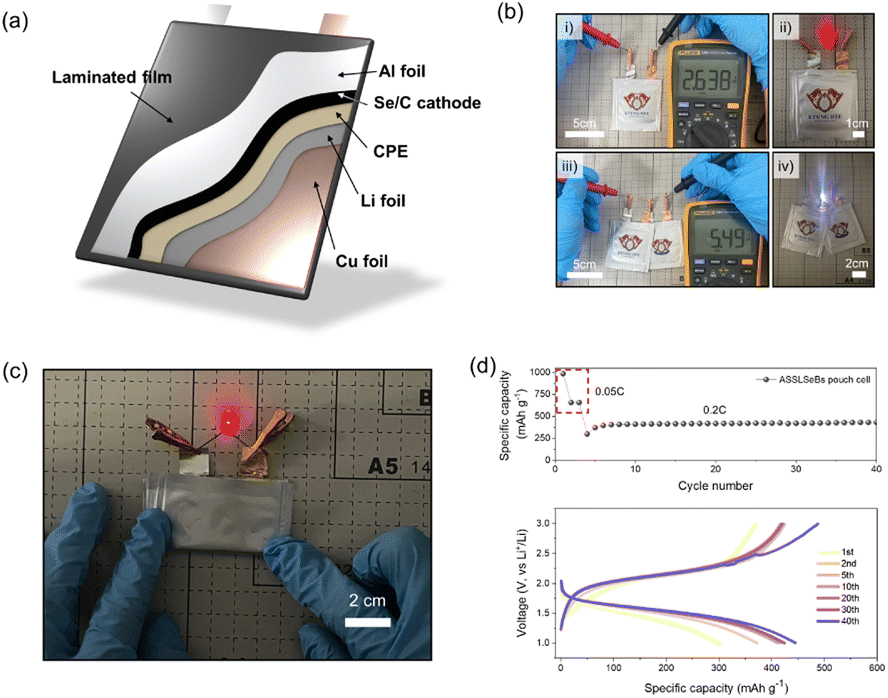 | ||
| Fig. 4 Design of the pouch-type ASSLSeBs. (a) Schematic of the structure of the solid-state Li–Se pouch cell. (b) Images showing successful operation of the solid-state Li–Se pouch cell; (i) open-circuit voltage confirmation and (ii) red LED operation of a single pouch cell. (iii) Open-circuit voltage confirmation and (iv) white LED operation of two pouch cells connected in series. (c) Image of a completely folded solid-state Li–Se pouch cell. (d) Electrochemical cycle properties of the solid-state Li–Se pouch cell. | ||
Conclusions
In summary, ASSLSeBs comprising a safe CPE and high-energy Se confined within nano-dimensional carbon pores provide solid-to-solid conversion that can fulfill the needs of modern solid-state lithium batteries including high-energy-density, RT operation, electrochemical stability, and ease of manufacture. Our study also reveals the detrimental effects of higher-order polyselenides on polymer-based electrolytes. Our ASSLSeB system, which is fabricated in the absence of higher-order polyselenides, exhibits satisfactory rate performance at 30 °C and outperforms previously reported solid-state Li–S batteries that operate at elevated temperatures (45–75 °C). The ASSLSeB also exhibits excellent cycle stability by maintaining a 230 mA h g−1 capacity over 200 cycles at a 2C-rate. Implementation of the CPE facilitated fabrication of the first pouch-type, solid-state Li–Se batteries. The integration of CPEs and Li–Se chemistries described in this work constitutes a promising approach for designing advanced ASSBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. T. Lee acknowledges the support of the National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (No. NRF-2020R1C1C1003656 and NRF-2021M3H4A3A02102349) and R&D Program for Forest Science Technology provided by Korea Forest Service (Korea Forestry Promotion Institute) (No. 2023489B10-2325-AA01).References
- Y. Xiao, Y. Wang, S.-H. Bo, J. C. Kim, L. J. Miara and G. Ceder, Nat. Rev. Mater., 2020, 5, 105–126 CrossRef CAS.
- J. Janek and W. G. Zeier, Nat. Energy, 2023, 8, 230–240 CrossRef.
- B. Qiu, F. Xu, J. Qiu, M. Yang, G. Zhang, C. He, P. Zhang, H. Mi and J. Ma, Energy Storage Mater., 2023, 102832 CrossRef.
- B. Wu, S. Wang, J. Lochala, D. Desrochers, B. Liu, W. Zhang, J. Yang and J. Xiao, Energy Environ. Sci., 2018, 11, 1803–1810 RSC.
- J. Kasemchainan, S. Zekoll, D. Spencer Jolly, Z. Ning, G. O. Hartley, J. Marrow and P. G. Bruce, Nat. Mater., 2019, 18, 1105–1111 CrossRef CAS PubMed.
- G. Cui, Matter, 2020, 2, 805–815 CrossRef.
- S. Randau, D. A. Weber, O. Kötz, R. Koerver, P. Braun, A. Weber, E. Ivers-Tiffée, T. Adermann, J. Kulisch and W. G. Zeier, Nat. Energy, 2020, 5, 259–270 CrossRef CAS.
- P. Minnmann, F. Strauss, A. Bielefeld, R. Ruess, P. Adelhelm, S. Burkhardt, S. L. Dreyer, E. Trevisanello, H. Ehrenberg and T. Brezesinski, Adv. Energy Mater., 2022, 12, 2201425 CrossRef CAS.
- R. Schlem, C. F. Burmeister, P. Michalowski, S. Ohno, G. F. Dewald, A. Kwade and W. G. Zeier, Adv. Energy Mater., 2021, 11, 2101022 CrossRef CAS.
- L. Li, H. Duan, J. Li, L. Zhang, Y. Deng and G. Chen, Adv. Energy Mater., 2021, 11, 2003154 CrossRef CAS.
- B. Ding, J. Wang, Z. Fan, S. Chen, Q. Lin, X. Lu, H. Dou, A. K. Nanjundan, G. Yushin and X. Zhang, Mater. Today, 2020, 40, 114–131 CrossRef CAS.
- F. Han, J. Yue, X. Fan, T. Gao, C. Luo, Z. Ma, L. Suo and C. Wang, Nano Lett., 2016, 16, 4521–4527 CrossRef CAS PubMed.
- J. Sun, Z. Du, Y. Liu, W. Ai, K. Wang, T. Wang, H. Du, L. Liu and W. Huang, Adv. Mater., 2021, 33, 2003845 CrossRef CAS PubMed.
- J. T. Lee, H. Kim, N. Nitta, K.-s. Eom, D.-C. Lee, F. Wu, H.-T. Lin, B. Zdyrko, W. I. Cho and G. Yushin, J. Mater. Chem. A, 2014, 2, 18898–18905 RSC.
- T. H. Hong, J. Kee, H. Jang, D. Kwon, D. Kim and J. T. Lee, J. Alloys Compd., 2023, 948, 169789 CrossRef CAS.
- C.-P. Yang, Y.-X. Yin and Y.-G. Guo, J. Phys. Chem. Lett., 2015, 6, 256–266 CrossRef CAS PubMed.
- J. Ding, Y. Wang, Z. Huang, W. Song, C. Zhong, J. Ding and W. Hu, ACS Appl. Mater. Interfaces, 2022, 14, 6828–6840 CrossRef CAS PubMed.
- F. Wu, J. T. Lee, Y. Xiao and G. Yushin, Nano Energy, 2016, 27, 238–246 CrossRef CAS.
- X. Li, J. Liang, X. Li, C. Wang, J. Luo, R. Li and X. Sun, Energy Environ. Sci., 2018, 11, 2828–2832 RSC.
- Q. Zhang, L. Cai, G. Liu, Q. Li, M. Jiang and X. Yao, ACS Appl. Mater. Interfaces, 2020, 12, 16541–16547 CrossRef CAS PubMed.
- X. Shi, Z. Zeng, H. Zhang, B. Huang, M. Sun, H. H. Wong, Q. Lu, W. Luo, Y. Huang and Y. Du, Small Methods, 2021, 5, 2101002 CrossRef CAS PubMed.
- H. Hu, F. Liu, Z. Shen, R. Yan and Z. Fu, ChemistryOpen, 2022, 11, e202100296 CrossRef CAS PubMed.
- B. Guo, Z. Wang, J. Chen, Y. Su, H. Li, H. Ye, X. Zhang, J. Yan, Z. Rong and J. Sun, ACS Nano, 2022, 16, 17414–17423 CrossRef CAS PubMed.
- C. Li, R. Liu, S. Zhang, Q. Li, C. Wang, Z. Zhang, C. Wang, L. Yin and R. Wang, Chin. Chem. Lett., 2023, 34, 108083 CrossRef CAS.
- X. Li, J. Liang, J. T. Kim, J. Fu, H. Duan, N. Chen, R. Li, S. Zhao, J. Wang and H. Huang, Adv. Mater., 2022, 34, 2200856 CrossRef CAS PubMed.
- J.-Y. Lin, S. Chen, J.-Y. Li, D. Yu, X.-L. Xu, C. Yu, S.-Q. Chen, X.-F. Miao, L.-F. Peng and C.-C. Wei, Rare Met., 2022, 41, 4065–4074 CrossRef CAS.
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 1–4 Search PubMed.
- T. Famprikis, P. Canepa, J. A. Dawson, M. S. Islam and C. Masquelier, Nat. Mater., 2019, 18, 1278–1291 CrossRef CAS PubMed.
- C. Wang, K. Fu, S. P. Kammampata, D. W. McOwen, A. J. Samson, L. Zhang, G. T. Hitz, A. M. Nolan, E. D. Wachsman and Y. Mo, Chem. Rev., 2020, 120, 4257–4300 CrossRef CAS PubMed.
- K. Meier, T. Laino and A. Curioni, J. Phys. Chem. C, 2014, 118, 6668–6679 CrossRef CAS.
- E. Rangasamy, J. Wolfenstine and J. Sakamoto, Solid State Ionics, 2012, 206, 28–32 CrossRef CAS.
- J. Košir, S. Mousavihashemi, B. P. Wilson, E.-L. Rautama and T. Kallio, Solid State Ionics, 2022, 380, 115943 CrossRef.
- J. T. Lee, H. Kim, M. Oschatz, D. C. Lee, F. Wu, H. T. Lin, B. Zdyrko, W. I. Cho, S. Kaskel and G. Yushin, Adv. Energy Mater., 2015, 5, 1400981 CrossRef.
- Z. Lei, Y. Lei, X. Liang, L. Yang and J. Feng, J. Power Sources, 2020, 473, 228611 CrossRef CAS.
- X. Gao, X. Zheng, Y. Tsao, P. Zhang, X. Xiao, Y. Ye, J. Li, Y. Yang, R. Xu and Z. Bao, J. Am. Chem. Soc., 2021, 143, 18188–18195 CrossRef CAS PubMed.
- S. Xin, L. Yu, Y. You, H.-P. Cong, Y.-X. Yin, X.-L. Du, Y.-G. Guo, S.-H. Yu, Y. Cui and J. B. Goodenough, Nano Lett., 2016, 16, 4560–4568 CrossRef CAS PubMed.
- D. B. Babu and K. Ramesha, Electrochim. Acta, 2016, 219, 295–304 CrossRef CAS.
- Y. Liu, L. Si, X. Zhou, X. Liu, Y. Xu, J. Bao and Z. Dai, J. Mater. Chem. A, 2014, 2, 17735–17739 RSC.
- T. Mutlu and R. Demir-Cakan, Electrochim. Acta, 2021, 390, 138825 CrossRef CAS.
- X. Li, J. Liang, K. Zhang, Z. Hou, W. Zhang, Y. Zhu and Y. Qian, Energy Environ. Sci., 2015, 8, 3181–3186 RSC.
- B. He, D. Liu, Z. Cheng, Z. Miao, Z. Rao, H. Zhang, L. Yuan, Z. Li and Y. Huang, Adv. Energy Mater., 2022, 12, 2102832 CrossRef CAS.
- M. Helen, T. Diemant, S. Schindler, R. J. r. Behm, M. Danzer, U. Kaiser, M. Fichtner and M. Anji Reddy, ACS Omega, 2018, 3, 11290–11299 CrossRef CAS PubMed.
- J. Gao, C. Sun, L. Xu, J. Chen, C. Wang, D. Guo and H. Chen, J. Power Sources, 2018, 382, 179–189 CrossRef CAS.
- H. Zhang, X. Judez, A. Santiago, M. Martinez-Ibañez, M. Á. Muñoz-Márquez, J. Carrasco, C. Li, G. G. Eshetu and M. Armand, Adv. Energy Mater., 2019, 9, 1900763 CrossRef.
- H. Zhang, U. Oteo, X. Judez, G. G. Eshetu, M. Martinez-Ibanez, J. Carrasco, C. Li and M. Armand, Joule, 2019, 3, 1689–1702 CrossRef CAS.
- X. Yin, L. Wang, Y. Kim, N. Ding, J. Kong, D. Safanama, Y. Zheng, J. Xu, D. V. M. Repaka and K. Hippalgaonkar, Adv. Sci., 2020, 7, 2001303 CrossRef CAS PubMed.
- L. Wang, X. Yin, C. Jin, C. Lai, G. Qu and G. W. Zheng, ACS Appl. Energy Mater., 2020, 3, 11540–11547 CrossRef CAS.
- M. Li, J. E. Frerichs, M. Kolek, W. Sun, D. Zhou, C. J. Huang, B. J. Hwang, M. R. Hansen, M. Winter and P. Bieker, Adv. Funct. Mater., 2020, 30, 1910123 CrossRef CAS.
- Y. Wang, G. Wang, P. He, J. Hu, J. Jiang and L.-Z. Fan, Chem. Eng. J., 2020, 393, 124705 CrossRef CAS.
- Y. Shi, Z. Fan, B. Ding, Z. Li, Q. Lin, S. Chen, H. Dou and X. Zhang, J. Electroanal. Chem., 2021, 881, 114916 CrossRef CAS.
- P. Zhai, N. Peng, Z. Sun, W. Wu, W. Kou, G. Cui, K. Zhao and J. Wang, J. Mater. Chem. A, 2020, 8, 23344–23353 RSC.
- I. i. Garbayo, A. Santiago, X. Judez, A. S. e. de Buruaga, J. Castillo and M. A. Muñoz-Márquez, ACS Appl. Energy Mater., 2021, 4, 2463–2470 CrossRef CAS.
- A. Santiago, J. Castillo, I. i. Garbayo, A. Saenz de Buruaga, J. A. Coca Clemente, L. Qiao, R. Cid Barreno, M. Martinez-Ibañez, M. Armand and H. Zhang, ACS Appl. Energy Mater., 2021, 4, 4459–4464 CrossRef CAS.
- Y. Wang, H. Ji, X. Zhang, J. Shi, X. Li, X. Jiang and X. Qu, ACS Appl. Mater. Interfaces, 2021, 13, 16469–16477 CrossRef CAS PubMed.
- Y. Ji, K. Yang, M. Liu, S. Chen, X. Liu, B. Yang, Z. Wang, W. Huang, Z. Song and S. Xue, Adv. Funct. Mater., 2021, 31, 2104830 CrossRef CAS.
- X. Zhang, H. Zhang, Y. Geng, Z. Shi, S. Zhu, Q. Xu and Y. Min, Chem. Eng. J., 2022, 444, 136328 CrossRef CAS.
- F. Chen, P. G. Puente, Y. Zhang, S. Cao, X. Lu, Z. Yi, Q. Shen and J. Li, Solid State Ionics, 2022, 380, 115926 CrossRef CAS.
- B. Wei, S. Huang, Y. Song, X. Wang, M. Liu, H. Jin and G. Cao, J. Mater. Chem. A, 2023, 11, 11426–11435 RSC.
- J. Li, H. Zhang, Y. Cui, H. Da, H. Wu, Y. Cai and S. Zhang, Chem. Eng. J., 2023, 454, 140385 CrossRef CAS.
- P. Bai and M. Z. Bazant, Nat. Commun., 2014, 5, 3585 CrossRef PubMed.
- Y. Kato, S. Shiotani, K. Morita, K. Suzuki, M. Hirayama and R. Kanno, J. Phys. Chem. Lett., 2018, 9, 607–613 CrossRef CAS PubMed.
- W. B. Hawley, Z. Du, A. J. Kukay, N. J. Dudney, A. S. Westover and J. Li, Electrochim. Acta, 2022, 404, 139579 CrossRef CAS.
- L. Seidl, R. Grissa, L. Zhang, S. Trabesinger and C. Battaglia, Adv. Mater. Interfaces, 2022, 9, 2100704 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta06151e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |