
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCharge compensation in a layered van der Waals NiPS3 host through various cationic intercalations†
Sebastian
Pazek
a,
Anna
Efimenko
b,
Roberto
Félix
 b,
Maria
Roslova
a,
Christine Joy
Querebillo
a,
Mikhail V.
Gorbunov
a,
Alexander
Ovchinnikov
c,
Andreas
Koitzsch
a,
Carlos
Escudero
d,
Yuliia
Shemerliuk
a,
Saicharan
Aswartham
a,
Bernd
Büchner
a,
Ahmad
Omar
*a and
Daria
Mikhailova
*a
b,
Maria
Roslova
a,
Christine Joy
Querebillo
a,
Mikhail V.
Gorbunov
a,
Alexander
Ovchinnikov
c,
Andreas
Koitzsch
a,
Carlos
Escudero
d,
Yuliia
Shemerliuk
a,
Saicharan
Aswartham
a,
Bernd
Büchner
a,
Ahmad
Omar
*a and
Daria
Mikhailova
*a
aLeibniz Institute for Solid State and Materials Research (IFW), Dresden e.V. Helmholtzstr. 20, 01069 Dresden, Germany. E-mail: d.mikhailova@ifw-dresden.de; a.omar@ifw-dresden.de
bDepartment Interface Design, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH (HZB), Albert-Einstein-Str. 15, 12489 Berlin, Germany
cDepartment of Chemistry and Food Chemistry, Technische Universität Dresden, 01062 Dresden, Germany
dALBA Synchrotron, Carrer de la Llum 2-26, 08290 Cerdanyola del Vallès, Barcelona, Spain
First published on 27th December 2023
Abstract
The layered structure of van der Waals compounds enables facile insertion of guest species between layers, resulting in material multifunctionality through easily modifying its physical properties. Isostructural MPS3 compounds with 3d transition metal cations such as Mn, Fe, Co and Ni can serve as hosts for relatively small alkali metals as well as larger organic molecules. NiPS3 is the most exotic representative among them, because despite 30 years of intense research, its electronic structure still evokes numerous questions, not to mention the electronic structure of intercalated NiPS3. There are two possibilities for electron transfer in semiconducting NiPS3 upon insertion of electron-donating species, either to a discrete Ni atomic level, or to a molecular level of the (P2S6)4− unit. We performed a systematic structural and spectroscopic study of NiPS3 upon electrochemical intercalation of Li, Na and 1-ethyl-3-methylimidazolium (EMIM) cations. Up to 0.5 Li or 0.5 Na per NiPS3 formula unit can be inserted into free octahedral spaces in the interlayers without visible changes in the diffraction pattern of the host. In contrast, more than 1 EMIM per NiPS3 unit can be intercalated between host layers leading to a significant interlayer distance expansion from 6.33 Å to 11.3 Å. The charge compensation was found to be different for the three intercalants: upon Li insertion, the electron density increases on the (P2S6)4− unit and Ni remains redox-inactive, while intercalation of Na leads to reduction of Ni. In contrast, uptake of larger EMIM cations does not result in any changes in Ni, S and P K-edge near edge XANES spectra of NiPS3 and results in only very little change in their extended X-ray absorption fine structure spectra. It is likely that there is an electrochemical reduction of EMIM cations to heterocyclic carbenes with their possible dimerization. The impact on magnetization of Li and EMIM intercalation was also studied.
1. Introduction
Layered transition metal phosphorus trisulfides (M2P2S6 or MPS3 where M = Ni, Mn, Fe etc.) have been of interest towards opto-electronic applications due to their versatile anisotropic properties1 and the possibility of novel ground states.2,3 In the recent decades, they have also been found to be promising for electrocatalytic applications.4 The family of MPS3 compounds crystallizes in a CdCl2-type structure with the monoclinic space group C2/m, as shown in Fig. 1a. In the layers, transition metal ions M, usually in the oxidation state 2+, are octahedrally surrounded by S anions, which are interconnected via P cations. Since there are P–P bonds in the structure, the correct presentation of the chemical formula corresponds to M2P2S6 showing (P2S6)4− units. There are competing inter- and intra-layer magnetic interactions in the structure. The relatively weaker van der Waals interaction between magnetic cations in neighboring layers can be controlled through change of the interlayer distance. Since it is not always feasible to synthesize the desired structures, post-mortem intercalation of cations may allow tuning of the structure.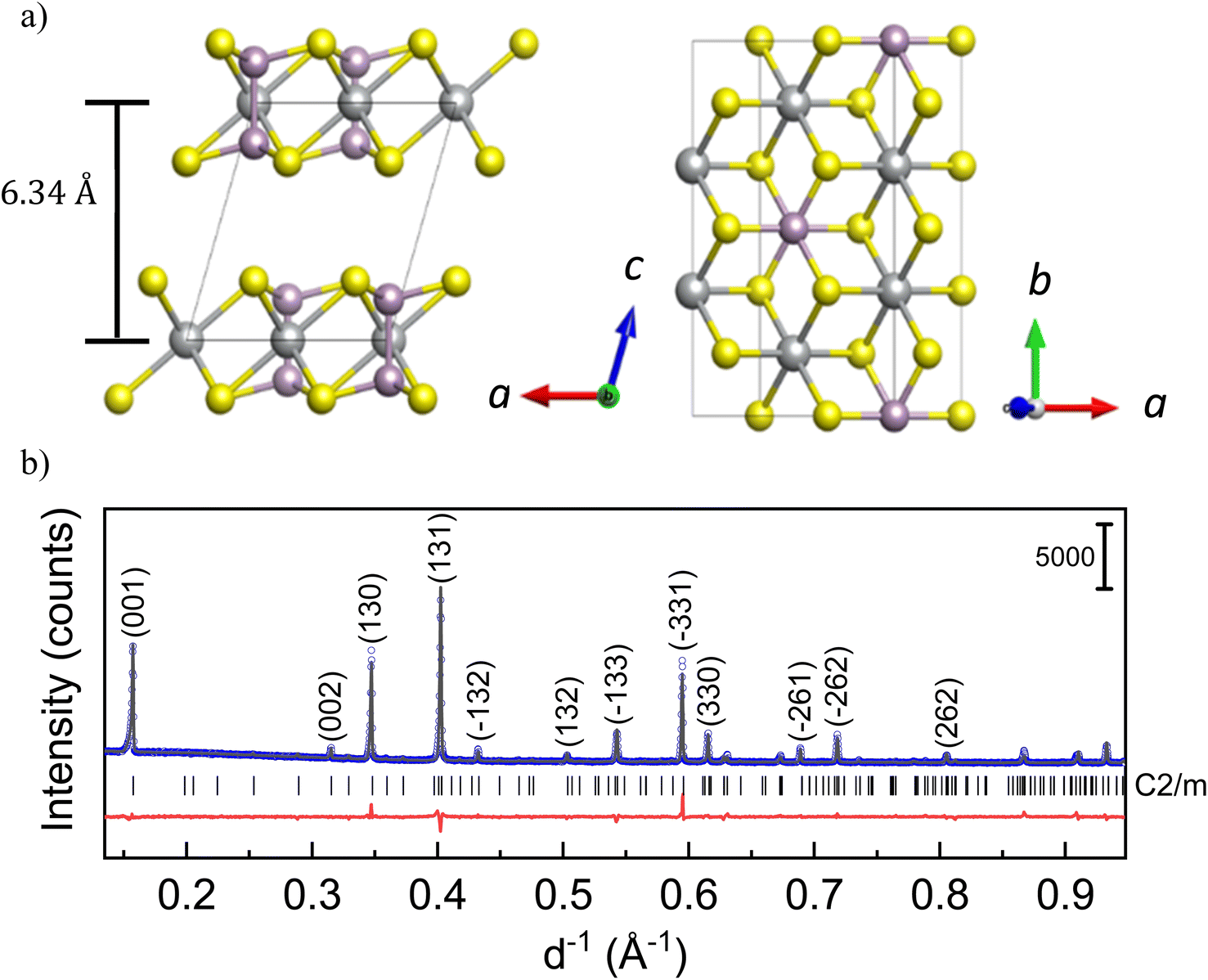 | ||
| Fig. 1 (a) Crystal structure of MPS3 compounds with M – Ni, Mn, and Fe; left – along the c-axis, and right – in the ab-plane. Metal cations (grey spheres) are octahedrally coordinated by S anions (yellow spheres). P cations (violet) are 4-fold-coordinated by 3S and 1P atoms, resulting in (P2S6)4− units. (b) X-ray powder diffraction pattern of NiPS3 with the observed (blue dots) and calculated (black solid line) curves together with their difference curve (red solid line) and Bragg positions given as black vertical lines. | ||
Intercalation of alkali ions in MPS3 and the corresponding structural and electronic changes have been extensively studied, especially with Li ions.5–10 Due to the limited absorption capacity of lithium by MPS3 and the modest influence on the physical properties, lithium is only suitable for controlled tuning of the magnetism to a certain extent.5,11,12 It is understood that the small alkali ions occupy two types of octahedral voids present within the layers, without affecting the interlayer spacing.11 An extended Li intercalation results in structure decomposition into mainly metal–sulfur and metal–phosphorus compounds likely due to the high reactivity of alkali ions with chalcogens.5,10 However, there is still a discrepancy in the literature regarding the electrochemical potential for intercalation as well as the composition of an intercalated compound with structural stability. Moreover, the charge compensation mechanism upon intercalation is still not completely understood. Therefore, as an alternative, intercalation of large cations or organic molecules, leading to detectable changes in the crystal structure, is also of interest for these compounds. For example, insertion of complex ammonium cations with different organic groups such as tetrabutyl ammonium (TBA+), tetrapropyl ammonium (TPA+) and cetyltrimethyl ammonium (CTA+) were demonstrated in NiPS3,13,14 while MnPS3 is known to be a host for poly(phenylene vinylene) and for pyridine.15,16
In the family of transition metal phosphorus trisulfides, different aspects of intercalation have been comprehensively studied in NiPS3.9,14,17,18 This compound attracts a special attention due to a big difference in the mechanistic behavior of organic molecule intercalation compared to that of other MPS3 systems regarding the charge compensation. For instance, it was reported that there is a strong tendency in MnPS3 and FePS3 for cation exchange, along with the creation of M2+ vacancies during intercalation.15,16,19–21 In contrast, no direct intercalation of pyridine was observed for NiPS3, although metal-to-ligand interaction would be of a similar bond strength.22 Pattayil et al. suggested that a possible reason for this is the difference in the crystal field energy, which stabilizes Ni in the crystal structure,22 thereby impeding the pyridine intercalation. Similarly, no reaction was reported with 1,10-phenanthroline on NiPS3.18 It should be mentioned that intercalation into NiPS3 was only possible either by Na-ion mediation (first Na intercalation, followed by cation replacement) or by NiPS3 doping with other transition metal cations such as Fe2+.18,23 During the electrochemical intercalation process with TBA+, a decreasing Raman band associated with octahedrally coordinated Ni2+ cations was observed, giving an indication of the reduction of nickel.14 Furthermore, it has been confirmed through electron energy loss spectroscopy (EELS) analysis that both Ni and P were reduced upon more than 1 Li intercalation per NiPS3 formula unit.10
Summing up, a solution for a controllable tuning of the interlayer spacing in MPS3 did not exist until now. The understanding of the charge compensation mechanism is in the nascent stage. Even for the intensively studied lithium intercalation into NiPS3, to the best of our knowledge, no structural monitoring via operando diffraction methods has been published to date.
Therefore, in the present work we focused on the intercalation principles of NiPS3 with regard to small alkali metal cations Li and Na as classical intercalation cations, along with a big organic cation. We aimed to understand structural changes in NiPS3 and the redox activity of each element upon intercalation, and to compare these with those of other MPS3 compounds. For this, we synthesized NiPS3 powder of high quality (see Fig. 1b) and performed a detailed operando X-ray diffraction and X-ray absorption spectroscopy study on Li and Na intercalation. We further presented successful organic cation intercalation in NiPS3 using 1-ethyl-3-methylimidazolium trifluoromethylsulfonylimide [EMIM]+[TFSI]− ionic liquid (IL). ILs are notable for their wide electrochemical stability window.24 EMIM-TFSI is one of the more commonly used ionic liquids, being in the liquid state at room temperature.25–27 More importantly, EMIM-TFSI has a high conductivity (0.96 S m−1) and low viscosity (24.1 mPa s), which is favorable for electrochemical intercalation.28 Detailed ex situ and operando characterization was performed to study the structure and morphology of the intercalated materials, towards understanding the mechanism and nature of charge storage upon intercalation in NiPS3.
2. Experimental section
2.1 Synthesis of pristine NiPS3
The NiPS3 sample was synthesized directly from the elements via a solid-state reaction. Ni (powder, Alfa Aesar, 99.8%), P (powder, Alfa Aesar, 99.999%), and S (powder, Alfa Aesar, 99.999%) were pressed into pellets (1 cm diameter at approximately 30 kN using a hydraulic press) in stoichiometric ratio and sealed in a quartz tube (12 mm inner diameter and 3 mm wall thickness) under 300 mbar Ar, after evacuation. After an initial heat treatment at 573 K for 24 h, the sample was heated to 973 K and held for 12 days. The heating rate was 50 K h−1. After this, the tube was cooled down to room temperature at the rate of 300 K h−1 and opened under an argon atmosphere. The NiPS3 structure was confirmed by powder X-ray diffraction measurements (Fig. 1b).2.2 X-ray diffraction (XRD) measurements
Lab-based powder XRD was performed in transmission geometry on a STOE Stadi P diffractometer with a curved Ge(111) crystal monochromator and Cu Kα1 radiation (λ = 1.54056 Å), equipped with a Dectris single-strip Mythen 1K detector. In order to avoid air exposure, the samples were sealed using Kapton tape inside an Ar-filled glovebox. The NiPS3 samples after cation intercalation for ex situ measurements were washed with acetonitrile and dried at 323 K under vacuum to remove all organic residues. All handling was undertaken inside an Ar-filled glovebox.Operando XRD measurements were performed in the lab on a STOE Stadi P diffractometer using Mo Kα1 radiation (λ = 0.70926 Å), at the synchrotron facilities DESY, Hamburg, Germany (P02.1 beamline, λ = 0.2074 Å) and ALBA, Barcelona, Spain (MSPD, λ = 0.41273 Å; and NOTOS, λ = 0.6894 Å, beamlines). All samples for operando measurements were prepared in special coin cells with fused silica or Kapton windows. This allows measurements in the transmission mode with simultaneous control of the potential of the cell. A multi-sample holder for parallel operando measurements was used.29 XRD analysis was performed by the Rietveld method using JANA2006 software.30
Elemental analysis was performed by inductively coupled plasma – optical emissions spectroscopy (ICP-OES) on an iCAP 6500 Duo View from ThermoFisher Scientific.
2.3 Electrochemical tests
A BioLogic VMP3 potentiostat was used for all electrochemical studies, including galvanostatic cycling with potential limitation (GCPL), cyclic voltammetry (CV), and the galvanostatic intermittent titration technique (GITT). For electrode preparation, the NiPS3 powder was mixed with Super P carbon (BASF) as a conductive agent, and polytetrafluoroethylene (PTFE) binder (Aldrich) for better contact with the current collector. The mixture, in a 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:5 weight ratio, was pressed on a copper mesh with an average loading of 11 mg cm−2. The electrodes were assembled in Swagelok-type cells, using lithium chips (12 mm diameter and 250 μm thickness, Chemetall), platinum mesh (12 mm diameter, 0.25 mm thickness, 99.9% purity, Alfa Aesar) or sodium metal (Alfa Aesar, 99.95%), which was rolled out into flat disks, as a counter electrode. Two glass fiber separators (Whatman GF/D, GE) were applied and soaked with electrolyte. Intercalation of alkali cations was performed with a LP30 electrolyte containing 1 M LiPF6 in ethylene carbonate (EC, BASF)/dimethyl carbonate (DMC, BASF) in a 1:1 ratio (LP30 Selectilyte, BASF), or with a home-made electrolyte composed of 1 M NaPF6 (abcr, 99%) dissolved in an EC/DMC mixture (1:1). For intercalation of organic species, the ionic liquid EMIM-TFSI from Sigma Aldrich was used as the electrolyte. Electrodes and cells were built in an Ar-filled glove box (H2O < 1 ppm, O2 < 1 ppm) to avoid contact with oxygen and water. All cells were operated in a climate chamber at room temperature. For diffusion coefficient determination, the galvanostatic intermittent titration technique (GITT) with a constant current pulse of 0.1C (1C corresponds to the current needed for intercalation of 1 EMIM ion per NiPS3 formula unit in 1 h) was applied, followed by an open circuit phase for several hours. The steps were repeated until the EMIM content exceeded 4 times the formula unit.
:20:5 weight ratio, was pressed on a copper mesh with an average loading of 11 mg cm−2. The electrodes were assembled in Swagelok-type cells, using lithium chips (12 mm diameter and 250 μm thickness, Chemetall), platinum mesh (12 mm diameter, 0.25 mm thickness, 99.9% purity, Alfa Aesar) or sodium metal (Alfa Aesar, 99.95%), which was rolled out into flat disks, as a counter electrode. Two glass fiber separators (Whatman GF/D, GE) were applied and soaked with electrolyte. Intercalation of alkali cations was performed with a LP30 electrolyte containing 1 M LiPF6 in ethylene carbonate (EC, BASF)/dimethyl carbonate (DMC, BASF) in a 1:1 ratio (LP30 Selectilyte, BASF), or with a home-made electrolyte composed of 1 M NaPF6 (abcr, 99%) dissolved in an EC/DMC mixture (1:1). For intercalation of organic species, the ionic liquid EMIM-TFSI from Sigma Aldrich was used as the electrolyte. Electrodes and cells were built in an Ar-filled glove box (H2O < 1 ppm, O2 < 1 ppm) to avoid contact with oxygen and water. All cells were operated in a climate chamber at room temperature. For diffusion coefficient determination, the galvanostatic intermittent titration technique (GITT) with a constant current pulse of 0.1C (1C corresponds to the current needed for intercalation of 1 EMIM ion per NiPS3 formula unit in 1 h) was applied, followed by an open circuit phase for several hours. The steps were repeated until the EMIM content exceeded 4 times the formula unit.
2.4 X-ray absorption spectroscopy (XAS) studies
Operando X-ray absorption spectroscopy measurements at the Ni K-edge were carried out at the DESY Synchrotron (P64 PETRA III beamline, Hamburg, Germany) and at the ALBA synchrotron (NOTOS beamline, Barcelona, Spain). The NOTOS beamline enables a combination of XAS-XRD measurements. The samples were assembled in electrochemical coin cells with Kapton windows. A multi-sample holder for parallel operando measurements was used.29 The measurements were performed in both transmission and fluorescence mode. Ex situ XAS measurements of the S, P and Ni K-edges were carried out at the BESSY II synchrotron (HiKE endstation located at the KMC-1 bending magnet beamline, Berlin, Germany).31,32In case of the P and S K-edge, the incident X-rays were monochromatized using a Si (111) double-crystal monochromator and focused by means of a piezo refocusing capillary to a size of 100 μm × 100 μm (H × V), if the beam is perpendicular to the sample. For the Ni K-edge, the Si (422) crystal pair was used. The X-ray absorption near edge structure (XANES) was collected in partial fluorescence yield (PFY) mode at a grazing angle of 45° with a Bruker XFlash 4010 fluorescence silicon drift detector (SDD). The incoming photon energy was calibrated using Au 4f spectrum of clean Au foil. The obtained data were processed with Athena software, including background subtraction and normalization.33
2.5 Magnetometry
Magnetization measurements were performed on a superconducting quantum interference device (SQUID) magnetometer (MPMS, Quantum Design) using powder samples. The magnetization was measured as a function of temperature at an applied magnetic field of 2 T in the field-cooled (FC) and zero-field-cooled (ZFC) mode, in the temperature range of 5 to 330 K.2.6 Single crystal X-ray diffraction (SCXRD)
SCXRD data acquisition was accomplished on a Bruker D8 Venture (Mo Kα, λ = 0.71073 Å) equipped with a PHOTON 100 CMOS detector. The measurement was performed at room temperature. Indexing was performed using APEX3 software.34 Data integration and absorption corrections were performed using the SAINT and SADABS34,35 software, respectively. The crystal structure was solved by dual-space methods implemented in the SHELXT36 program and refined by the full-matrix least-squares method on F2 with SHELXL.372.7 High-resolution transmission electron microscopy (HRTEM) and electron diffraction (ED)
HRTEM, ED and energy-dispersive X-ray (EDX) spectroscopy measurements were performed with an aberration-corrected FEI Titan 80–300 electron microscope (ThermoFisher, USA) equipped with an in-column EDX detector. Diffraction patterns were treated in the kinematical approximation.2.8 Raman spectroscopy
Raman measurements were performed using a S&I Monovista CRS+ confocal Raman spectrometer at 405 nm or 514 nm, with a 2400 grating. The laser was adjusted and focused through a Nikon 20× objective (N.A. 0.35; WD = 20 mm) to give an incident laser power of approximately 0.5–5 mW, depending on the sample being measured. Spectra were recorded for a total accumulation time of 75–500 s, depending on the sensitivity and spectral quality of the sample. The peak positions were corrected with respect to the Raman spectrum of a toluene–acetonitrile mixture. Baseline correction and spectral analysis were performed using Qpipsi (a Matlab-based software for spectral analysis) and OriginPro.3. Results
3.1 Structural evolution of NiPS3 upon alkali metal intercalation
According to the literature reports, NiPS3 can incorporate up to 1.5 Li per formula unit at a very low current density close to the open circuit voltage (OCV) regime.38 The pristine structure of NiPS3 contains two different octahedral gaps in the van der Waals interlayer, into which alkali-metal cations Li and Na can potentially be intercalated without affecting the interlayer distance.38 1/3 of the octahedral spaces belong to the 2d Wyckoff position and 2/3 to the 4h positions. However, the intercalation behavior of MPS3 (M – transition metal cation Fe, Mn or Ni) compounds in electrochemical cells can depend on the applied current density: a higher current abruptly decreases the cell potential, thus influencing the Li-ion mobility and structural changes.38,39We studied Li and Na intercalation behavior in NiPS3 at current densities significantly exceeding the OCV regime (current densities of 60 to 70 mA g−1). Fig. 2 shows operando XRD data collected during intercalation of (a) Li and (b) Na cations into NiPS3, in the form of a waterfall plot and a contour plot for better visualization, including the corresponding potential curve of an operando electrochemical cell.
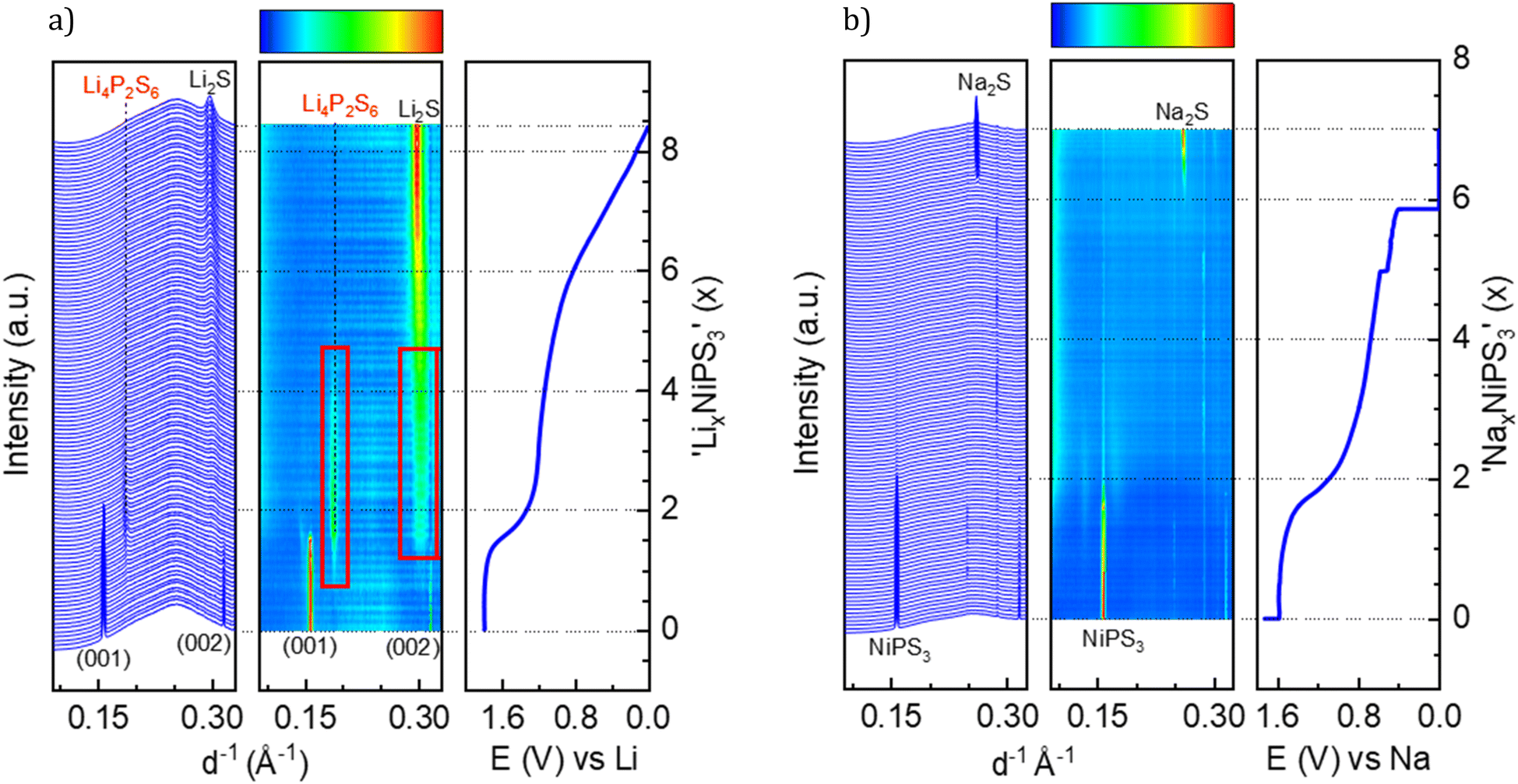 | ||
| Fig. 2 Waterfall plot, contour plot and potential profile from operando XRD. (a) LixNiPS3, with a current density of 71 mA g−1; red boxes highlight the intermediate phase, and (b) NaxNiPS3 with a current density of 61 mA g−1. | ||
At the beginning of Li-intercalation up to x(Li) = 0.5 (Li0.5NiPS3 composition), Bragg reflections in the diffraction patterns remain unaffected, likely due to insertion into free octahedral spaces.40 The second step up to x(Li) = ∼1.6 is accompanied by a continuous decrease in reflection intensities of the parent compound, until they completely disappeared, suggesting structural degradation. This behavior, clearly confirmed by evolution of the characteristic (001) and (002) reflections of NiPS3 shown in Fig. 2a, is in line with the DFT calculations of Choi et al., which show a significant structural distortion at x(Li) = 0.875 induced by Li intercalation.10 The authors also observed some changes in the near surrounding of nickel cations starting from Li0.8NiPS3, using pair distribution function (PDF) analysis. Evolution of reflections corresponding to Li2S is observed starting from x = 1.6. This marks the end of the decomposition process of NiPS3 according to the reaction 9Li + NiPS3 → 3Li2S + Li3P + Ni.41 The fact that no reflections of elemental nickel and lithium phosphide were observed could be due to an amorphous structure and/or a very small crystallite size. Additionally, we could also identify Li4P2S6 as an intermediate phase, based on a set of reflections developing at 0.187 and 0.301 Å−1.42 This phase is observed at a Li-content of 0.8 < x < 4.5 as a pathway for decomposition of the parent structure to Li2S. The Li4P2S6 phase is not stable during the subsequent lithiation process, as demonstrated by the disappearance of the reflection at 0.187 Å−1 and a slight shift of the reflection at 0.301 Å−1.
Choi et al.,10 based on the results of XPS and Raman measurements, also concluded a multistep reduction process of Ni2+, which includes partially reduced inorganic intermediates such as NixPSy and LixPSy that eventually leads to Li2S as the final decomposition product. Formation of Li2S was also observed by Foot et al.38 during intercalation at a low current density for x > 1.6.
In a comparable study of Brec et al.,40 performed at a lower current rate, two separate plateaus are visible in the range of 0 < x(Li) < 1.5 and above 2.25 V vs. Li+/Li (in contrast to the first plateau at 1.75 V vs. Li+/Li in our case). These plateaus were attributed to two stages of intercalation arising from filling two different Wyckoff positions. Under those conditions close to the equilibrium state, some metastable, partially ordered phases can be formed, which are not detectable when the current density is high. Note that the authors39 measured single crystals in the presence of a different electrolyte, which can slightly impact the total potential vs. Li+/Li.
This brought us to the conclusion that the high galvanostatic currents applied to NiPS3, result in aggravate filling of interlayers in the structure probably due to a kinetic reason, causing an abrupt potential drop that leads to structural decomposition of NiPS3, since other phases become thermodynamically more stable at such a low cell potential. Therefore, we observed the loss of crystallinity of the initial compound and the formation of lithium decomposition products at much lower states of lithiation than reported in the literature.40 It should be mentioned that in contrast to Foot et al.,38 who detected Li2S above x > 1.6, the final transformation in our experiments is delayed. After de-lithiation, some reflection shifts are visible, indicating re-oxidation of Li2S, but in contrast to Choi et al. no crystalline NiPS3 could be observed.10
A similar multistep decomposition process of NiPS3 with the formation of the Na2S end product was observed during galvanostatic sodium intercalation. The end of the first plateau at x(Na) = 1.8 in the galvanostatic curve coincides with the disappearance of pristine NiPS3 reflections (Fig. 2b). However, in contrast to lithium, no crystalline intermediate products could be detected during structural decomposition. Moreover, the reflections of Na2S are sharper than of its Li counterpart suggesting a higher crystallinity of Na2S. However, the peak evolution occurs at significantly higher x(Na) > 6 values, especially in a constant voltage profile.
3.2 Redox changes in NiPS3 upon alkali metal intercalation
Furthermore, we performed operando XAS measurements at the Ni K-edge upon Li and Na insertion into NiPS3, which provide direct information about the valence state and local surrounding of the absorbing atom.43Fig. 3a shows the evolution of the Ni K-edge spectra recorded operando during electrochemical Li-intercalation, which was limited to 1 Li per NiPS3 formula unit, together with the NiO reference spectrum. The edge positions of the NiO reference material and of NiPS3 are different. The difference arises mostly from various chemical surroundings of Ni and different covalencies of the chemical bonds, which can be qualitatively estimated from Pauling electronegativity values. Although Ni is octahedrally coordinated in both NiO and NiPS3, the more electronegative oxygen atoms (3.44 Pauling units) in ionic Ni–O bonds strongly attract the electron density from Ni, making it more difficult to remove core level electrons and causing a shift to a higher edge energy, in comparison to more covalent Ni–S bonds with S having an electronegativity of 2.589 Pauling units.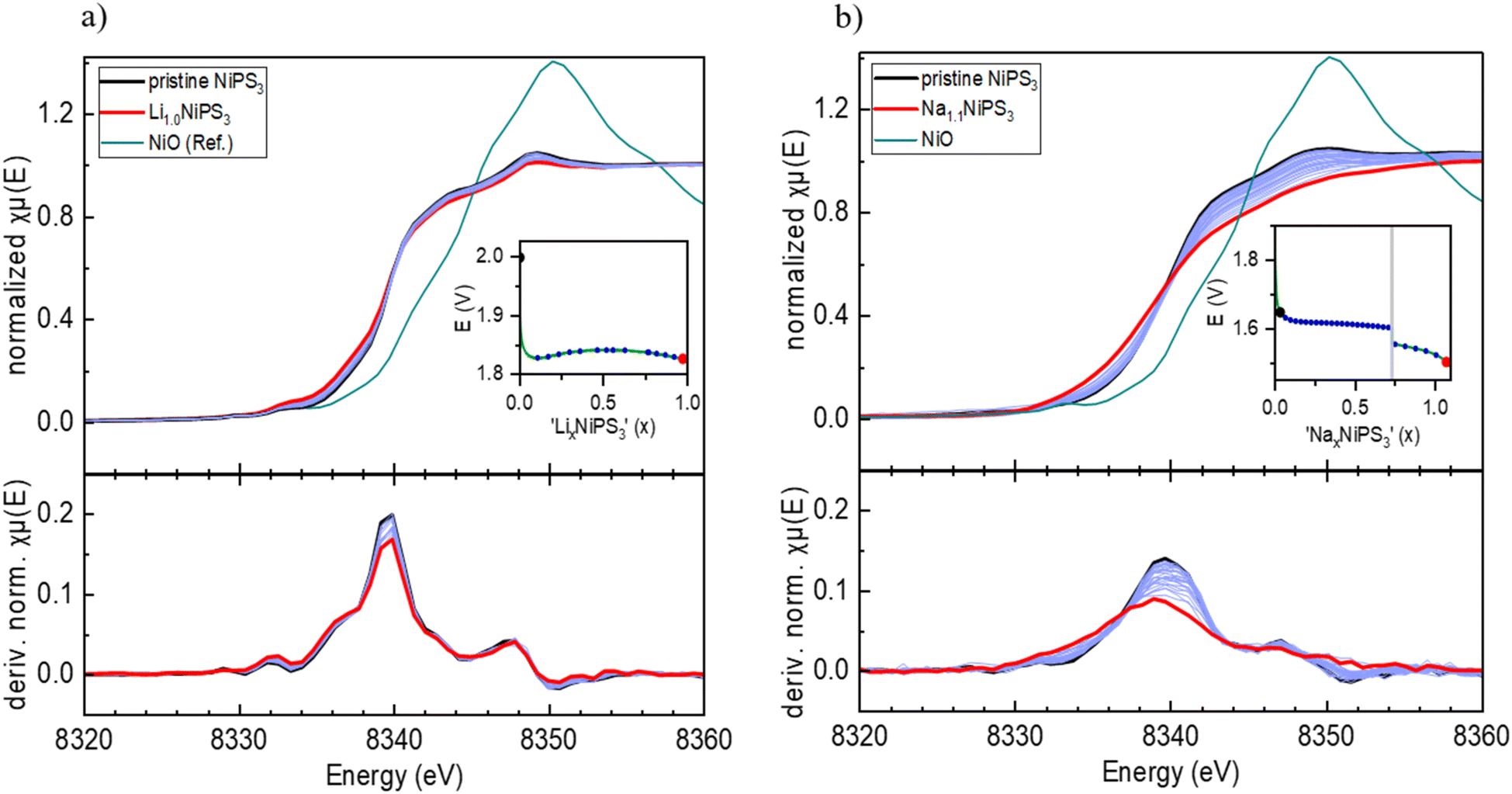 | ||
| Fig. 3 Operando XAS spectra of alkali ion intercalation into NiPS3: (a) Ni K-edge and dμ(E)/dE plots of LixNiPS3 (0 ≤ x ≤ 1). (b) Ni K-edge and dμ(E)/dE plots of NaxNiPS3 (0 ≤ x ≤ 1.1). Insets show the corresponding potential profiles and the specific points of measurement. | ||
According to the work of Brec and Ouvrard,44 the extended X-ray absorption fine structure (EXAFS) analysis of chemically lithiated LixNiPS3 samples points to pronounced migration of Ni cations from the octahedral to tetrahedral sulfur surrounding their simultaneous reduction. Therefore, as per the work of Brec and Ouvrard,44 50% of Ni exists in the zero oxidation state in sulfur tetrahedra in the Li1Ni0.50Ni0.52+PS3 composition. The Ni–S distance for the first Ni coordination sphere is reported to decrease from 2.45 Å (NiS6-octahedra) to 2.30 Å (NiS4-tetrahedra).
However, the reported Ni-reduction and Ni-diffusion in LixNiPS3 are in contradiction to our operando XAS measurements. According to Fig. 3a, showing Ni K-edge spectra of LixNiPS3 also as dμ(E)/dE derivative plots, there are no visible changes in the spectra for compositions in the range of 0 ≤ x(Li) ≤ 1. As is well known, the pre-edge of transition metals in the K-edge spectrum originates from a 1s → 3d electron transition and depends, among other factors, on the symmetry and the coordination number of the central atom.43 In agreement with the dipole selection rules, only a quadrupole transition is allowed, hence showing either zero or a low intensity in the octahedral environment.43 In the case of tetrahedral surrounding, the p- and d-orbitals of Ni overlap and can hybridize, thus enabling a dipole transition from the 1s to a hybridized p-orbital.45 This should strongly enhance the pre-edge intensity with the pronounced Li-insertion. However, such an enhancement could not be detected in Li-containing compositions. There are also no visible shifts in the main edge and post-edge regions of the Ni K-edge spectra, indicating no change in the coordination sphere and Ni oxidation state. Therefore, we conclude that Ni is redox-inactive during lithiation of LixNiPS3.
Similarly, sodium intercalation into NiPS3 (Fig. 3b) was studied up to x(Na) = 1.1. The normalized plots show many more changes upon intercalation in comparison to lithium. Here, the maximum in the derivative plots is shifted from 8340 eV to 8339 eV, showing the beginning of partial Ni reduction, in agreement with the work of van Dinter et al.9 The changes in the post-edge region with increasing sodium content are significant as well. Although no structural changes were observed till x(Na) = 1.5, the larger size of the Na-ion likely leads to local distortions in the structure and the corresponding partial redox.
In order to further explore interlayer modification, intercalation with a larger organic cation was undertaken. In contrast to the literature where organic salts are used in solvent media, an ionic-liquid mediated approach was used without the need for additional component(s). EMIM-TFSI was selected and the corresponding intercalation of an EMIM cation is presented next.
3.3 Intercalation of EMIM and co-intercalation of (EMIM,Li)
Prior to galvanostatic EMIM insertion experiments, the electrochemical stability window of EMIM-TFSI was probed using a symmetric Pt–Pt two-electrode cell setup at room temperature (Fig. 4a). Cyclic voltammetry measurements, performed between −5 V and 1 V at a low scan rate of 0.1 mV s−1, show an irreversible redox process starting below −4.1 V, which lies outside the working potential range of EMIM intercalation between −2.4 V and −3.9 V vs. Pt.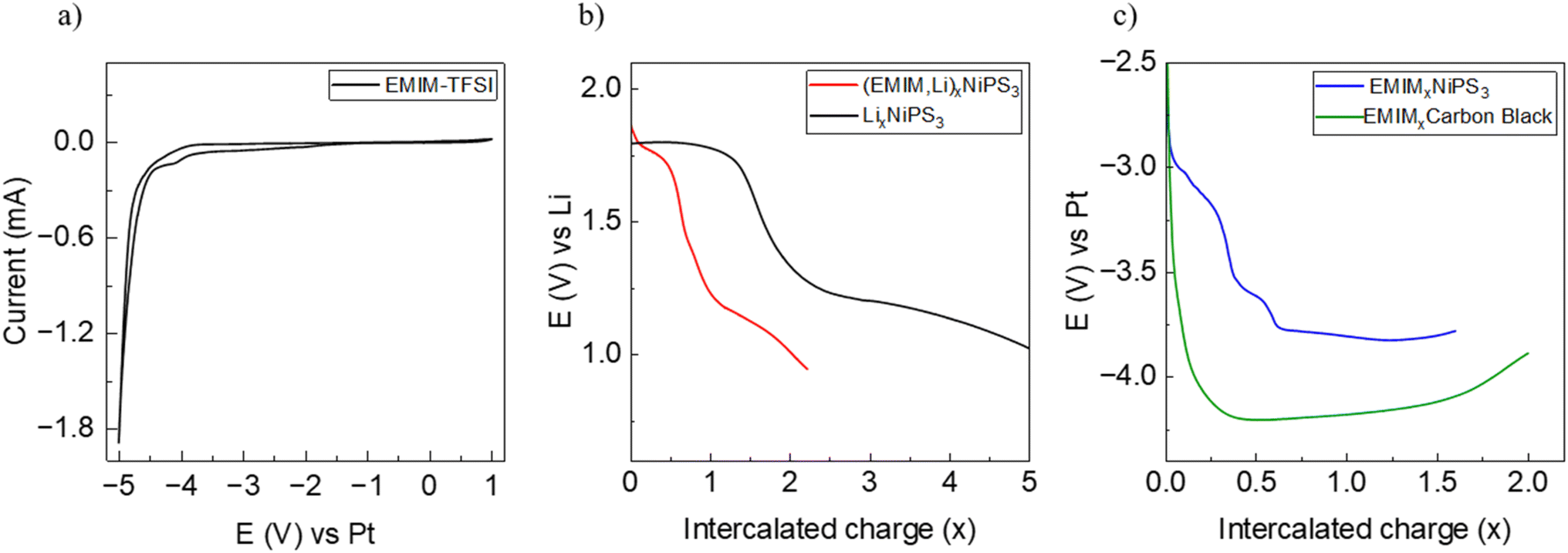 | ||
| Fig. 4 (a) Cyclic voltammetry of EMIM-TFSI in a symmetric Pt–Pt cell, (b) galvanostatic discharge of NiPS3 with Li (black), (EMIM,Li) (red), and (c) EMIM-only (blue), and galvanostatic discharge of a carbon black material in EMIM-TFSI vs. Pt (green). | ||
Insertion of EMIM cations from the EMIM-TFSI electrolyte was performed using either metallic Li or Pt as a counter electrode. In the case of the using a Li-electrode, co-insertion of Li during the long intercalation time is expected and the system is referred to as EMIM-Li. A galvanostatic profile of NiPS3 during insertion, using EMIM-TFSI electrolyte and a lithium electrode is shown in Fig. 4b along with the data for pure Li intercalation for comparison. The potential curve for EMIM-Li shows two distinct plateaus at 1.8 V and 1.2 V, similar to that of Li intercalation. However, the extent of the first plateau is significantly less, ending at around x(Li) ∼0.6.
Fig. 4c shows the electrochemical EMIM intercalation against the Pt counter electrode. The overall potential difference is due to the Pt–Li difference in the galvanic series. The reference measurement (green line) shows the galvanostatic curve of the cell with carbon black as a working electrode. However, since the potential drops below −4.1 V vs. Pt, the observed curve most likely represents decomposition of the electrolyte. An initial plateau is seen at ∼–3.3 V, which gradually shifts to a higher potential during intercalation likely due to a reduction in the overpotential of the material with continued intercalation.
3.4 Crystal and electronic structure changes
Structural changes in NiPS3 during reaction with EMIM-TFSI were monitored by operando XRD measurements using EMIM-TFSI electrolyte. Fig. 5 shows the XRD data where Li and Pt metals were used as counter electrodes. Here, a new crystalline phase is formed during the insertion of EMIM-cations in cells with both Li and Pt counter electrodes. In the case of (EMIM,Li) co-intercalation (Fig. 5b), the conversion represents a two-phase process with subsequent disappearance of the pristine phase, which is characterized by the first galvanostatic plateau up to x = 0.6, and growing reflections of a new phase. The most pronounced change between two phases is related to the (00l) reflections, see for example a clear shift of the (001) reflection from 0.1578 Å−1 in pristine NiPS3 to 0.0885 Å−1 in the new phase (Fig. 5b). The newly formed phase has the (001′) reflection at much smaller reciprocal lattice lengths, signaling an expansion of the NiPS3 interlayers. This phase transformation is completed with the EMIM0.6NiPS3 composition at the end of the first plateau in the potential profile. During further intercalation, no other changes occur in the lattice parameters. Only a fading of the intensity of the (001′) reflection is seen for x > ∼1.4, likely as a result of continued Li intercalation. The intercalation of pure EMIM ions into NiPS3 is similar to the co-intercalation process (Fig. 5a) with the evolution of a new phase. In contrast to the loss of pristine NiPS3 reflections in alkali metal intercalation (Fig. 2) and (EMIM-Li), with pure EMIM intercalation the reflections of the initial compound are merely faded until an effective inserted charge of ∼2.4x. Although a new phase is formed as well, the conversion process is not completed even after reaching a composition of EMIM2.4NiPS3. In addition, the new (001′) reflection is at a slightly higher 1/d position of 0.1010 Å−1, indicating less expansion along the c-axis. Its intensity does not appear to fade and, therefore, one can conclude that there is no visible decomposition here, in contrast to the co-intercalation.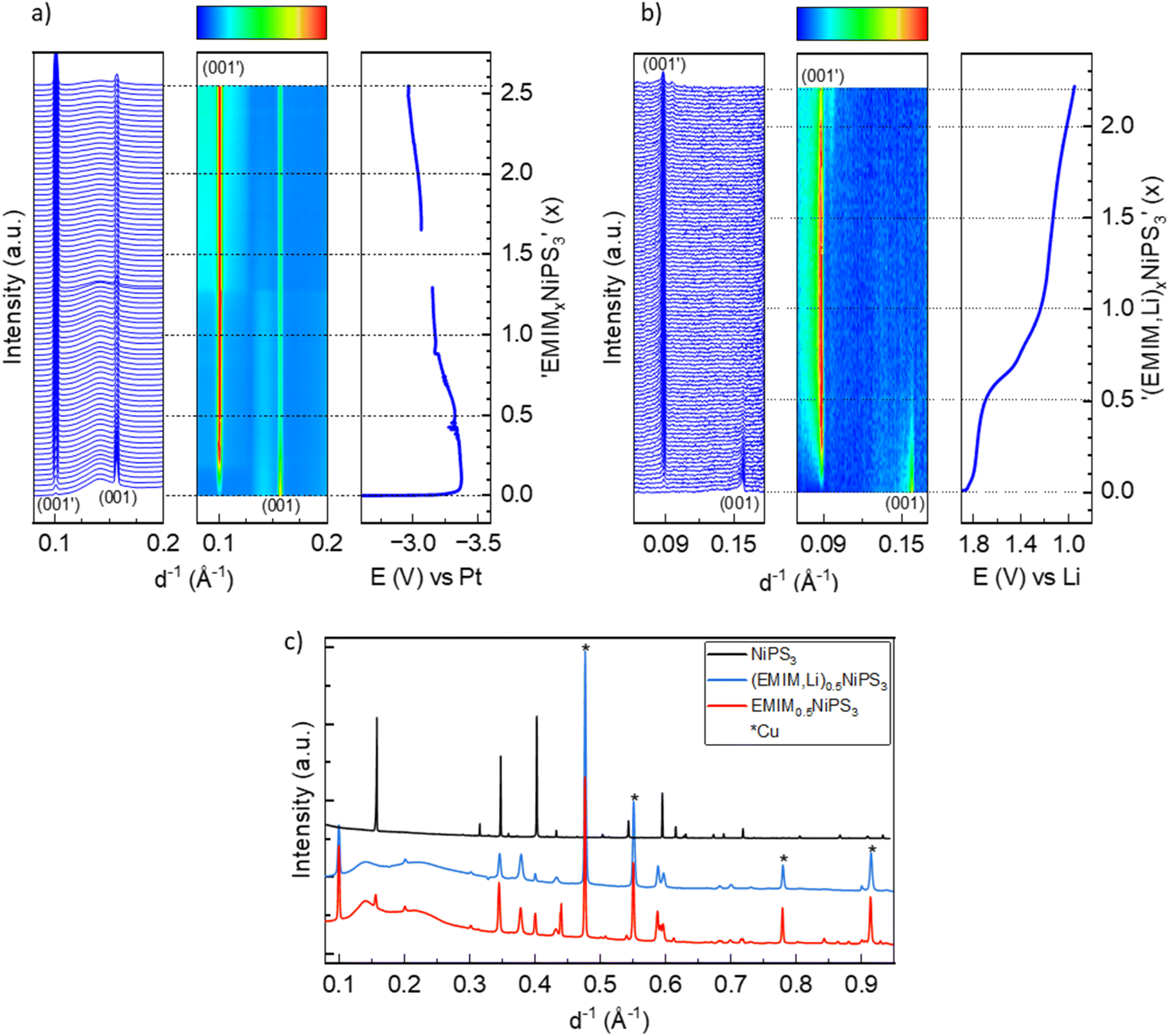 | ||
| Fig. 5 (a) Waterfall plot, contour plot and potential profile from operando XRD of NiPS3 with EMIM-TFSI vs. Pt, and vs. Li (b). (c) XRD of the pristine (black line) and intercalated phases corresponding to the (EMIM,Li)0.5NiPS3 (blue line) and EMIM0.5NiPS3 (red line) composition. | ||
For comparison, Fig. 5c shows the X-ray powder diffractograms of pristine NiPS3 and the materials after intercalation, corresponding to the (EMIM,Li)0.5NiPS3 and EMIM0.5NiPS3 compositions. Additional reflections and an increased background of the intercalated samples originate from operando cell components like the copper current collector and ionic liquid. The increase in the lattice parameter c, which is related to the interlayer distance in the ab-plane, from 6.62 Å to 10.65 Å, defined by using the Le Bail analysis method on the assumption of the same symmetry of the new phase (see Fig. S2 in the ESI†), is in agreement with a common observation for intercalation of organic ions into transition metal phosphorus trisulfides.13,19,21,46 For example, tetrabutylammonium (TBA+) intercalation into NiPS3 was demonstrated by Choi et al. with an increased c-parameter of 14.5–15.3 Å.10 The variation is a result of various crystallographic phases with different orientations of the organic molecule between the layers. In contrast, all EMIM0.5NiPS3 reflections could be assigned to only one crystallographic phase. Interestingly, a similar reaction between TBA+ and NiPS3 was performed by Mi et al., demonstrating an interlayer distance of only 11.46 Å.13 This may be an effect of different amounts of intercalation, leading to the assumption that this is a stepwise process, which is supported by the work of Chen et al.19 They investigated the reaction between FePS3 and 1,10-phenanthroline after certain intercalation intervals with ex situ XRD, and also observed different stages resulting from changes in the intercalant orientation. Changes in oxidation states and the local structure upon EMIM intercalation were monitored with operando Ni K-edge XAS, and ex situ P and S K-edge XAS measurements, as shown in Fig. 6 and 7.
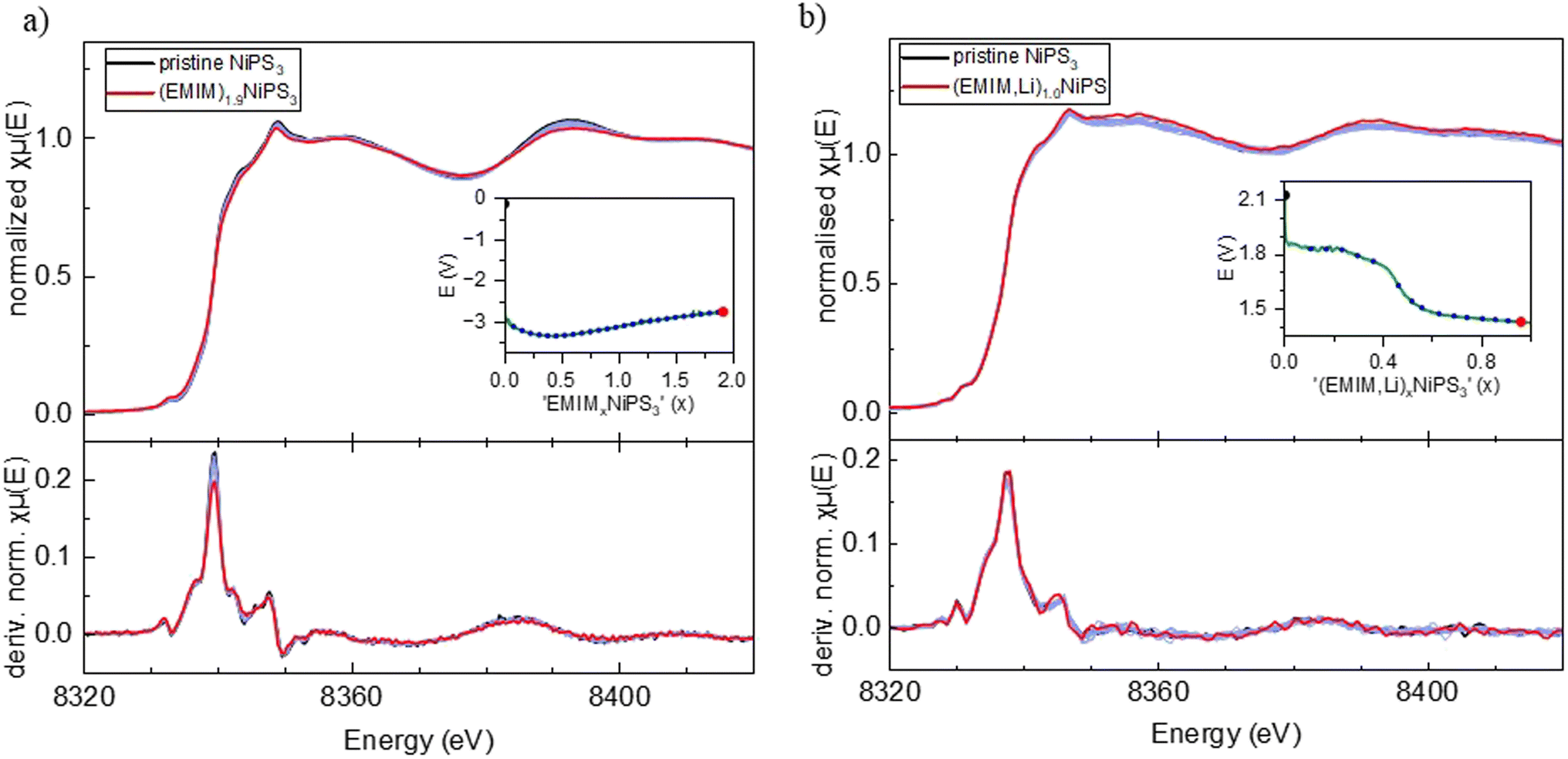 | ||
| Fig. 6 Operando Ni K-edge XAS data. (a) EMIM cation intercalated NiPS3 including the first derivative of the normalized absorption coefficient. (b) EMIM and Li co-intercalated NiPS3 including the first derivative of the normalized absorption coefficient. Insets show the corresponding potential profiles and the specific points of measurement. | ||
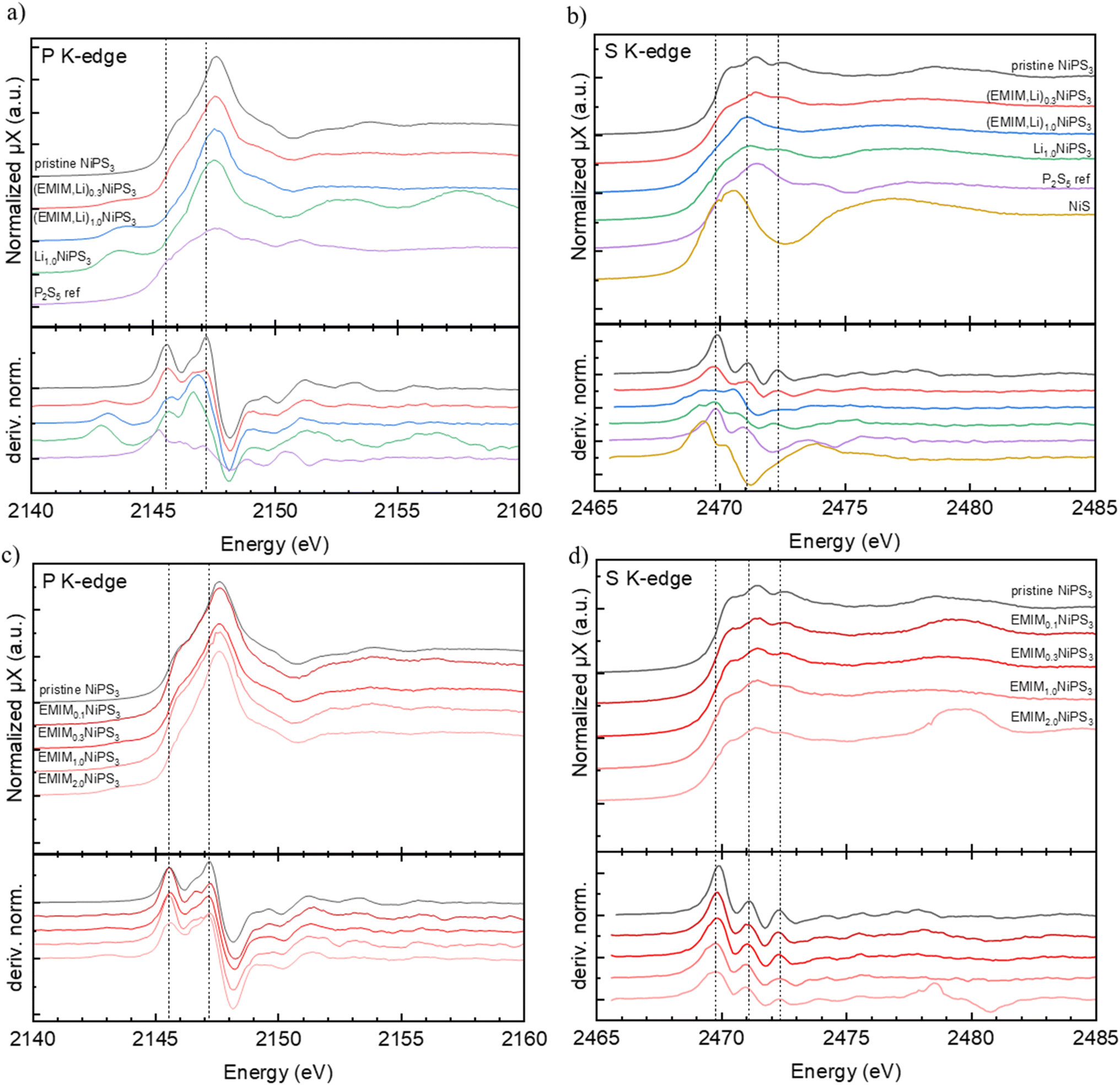 | ||
| Fig. 7 Ex situ XAS data, (a) P K-edge of pristine, EMIM and Li co-intercalated NiPS3 and a P2S5 reference material including the first derivative of the normalized absorption coefficient. (b) S K-edge of pristine, EMIM and Li co-intercalated NiPS3 and a P2S5 reference material including the first derivative of the normalized absorption coefficient. (c) P K-edge of pristine and EMIM intercalated NiPS3. (d) S K-edge of pristine and EMIM intercalated NiPS3. Data has been plotted with y-axis offsets for better visualization. | ||
Similar to Li-insertion into NiPS3, the Ni K-edge spectra of intercalated (EMIM,Li)xNiPS3 and (EMIM)xNiPS3 resemble each other on increasing the x-value, which is clearly seen in the first derivative graphs of the normalized absorption coefficient (Fig. 6).
Here we would like to make a note of caution. As recently reported,47 exposure of battery components such as electrolytes or electrodes to X-ray radiation in XAS experiments can induce some material damage, leading to incorrect interpretation of the obtained results. For example, it was shown that reactivity of the InSb/Al electrode in a Mg-battery with organic electrolytes is significantly retarded during the operando synchrotron X-ray absorption measurement, and the spectra evolution occurred only after a relaxation time, where the beam was powered off.47 To exclude such an effect in our case, we undertook multiple measurements to avoid a misinterpretation of data. First, an operando combined XAS/XRD experiment was performed at the NOTOS beamline (ALBA, Spain) enabling simultaneous monitoring of the crystal and electronic structures of NiPS3 upon EMIM intercalation. The electrochemical cell design including electrode thickness and material density was almost the same as those in the XAS experiments at the P64 Petra III beamline (DESY, Germany). The maximal flux on the sample at NOTOS corresponds to 1 × 1011 Ph s−1 in the energy range of 3d metals, and 1 × 1013 Ph s−1 at the P64 beamline. At NOTOS, the Ni K-edge spectra were recorded with an exposure time of 10 min, which was only half of the exposure time at P64. In addition, ex situ Ni K-edge measurements were performed at HiKE/KMC-1 (BESSY II, Germany) on (EMIM)xNiPS3 samples at various intercalation stages. In the EMIM intercalation experiment at NOTOS, the two-phase reaction mechanism was confirmed via operando XRD while the observed changes in the Ni K-edge spectra were close to zero, in agreement with the operando results from the P64 beamline of PETRA III and ex situ results from the HiKE/KMC-1 beamline of BESSY II. The uniformity of all Ni K-edge spectra for various (EMIM)xNiPS3 samples recorded at different X-ray source facilities, unambiguously confirms Ni-inertness during either Li intercalation, (EMIM-Li) co-intercalation or only EMIM intercalation.
In contrast, changes in the P and S K-edge spectra of pristine and intercalated materials are more noticeable (Fig. 7). The P K-edge spectra of (EMIM,Li)-co-intercalated and (EMIM)-intercalated samples differ from each other, pointing to different electronic processes upon Li and EMIM insertion into NiPS3. The near-edge region of the P K-edge spectrum of NiPS3 consists of two peaks. The energy position of the corresponding adsorption edge was set as the maximum of the first derivative of the normalized absorption coefficient, corresponding to the inflection point.48 Based on the “rigid-band electronic model”,49 the low-energy shoulder at 2145.5 eV might correspond to the transition from the P 1s orbital to the antibonding σ*(P–P) orbital of the (P2S6)4− entity, whereas the maximum at 2147.2 eV probably reflects the transition P 1s → antibonding σ*(P–S) orbital, since the σ*(P–P) level is assumed to be low-lying in energy.50 Upon co-intercalation of EMIM- and Li-cations, the intensity of the first peak noticeably decreases, indicating a decreasing amount of unoccupied states. A natural interpretation of this observation is that these states become occupied by electrons from the intercalated Li or EMIM. This is consistent with the fact that the first shoulder continuously loses intensity upon (EMIM,Li) intercalation. At the same time, the peak at 2147.5 eV shifts slightly to lower energy and becomes significantly broader, pointing to an increased electron density at P (Fig. 7). There are also noticeable changes in the near-edge region at higher energy.
Generally, phosphorus has a narrow range of absorption edge energies for species with various oxidation states, which makes the procedure of conventional determination of oxidation numbers difficult: for example, the energy difference is about 2.5 eV for compounds with the formal oxidation state varying from 0 (elemental phosphorus) to P5+ in Na4P2O7.51 Furthermore, with increasing Li-content in the samples, a new peak at a much lower energy of 2143.2 eV arises for both Li intercalation and (EMIM-Li) co-intercalation, which is in line with the onset of structural decomposition of NiPS3 at the pronounced intercalation confirmed by our operando XRD studies in both.
In the case of pure EMIM intercalation (Fig. 7b), the P K-edge spectra of samples with various amounts of EMIM show less changes. The positions of two main peaks in the derivative plots at 2145.5 eV and 2147.2 eV do not change with the EMIM content. However, the maximum of the white line systematically grows with a higher EMIM content, in contrast to that of the Li-containing samples. The near-edge structures at higher energies also remain almost constant. We conclude that pure EMIM intercalation, in contrast to Li intercalation and (EMIM-Li) intercalation, does not affect the electronic structure and the crystal structure around P.
We also recorded spectra of P2S5 as a reference material for phosphorus and sulfur. In both NiPS3 and P2S5 compounds, P-atoms have a distorted tetrahedral surrounding of three S-atoms with bond lengths varying between 2.02 and 2.10 Å. The fourth bond in the tetrahedron is either shorter like the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S double bond of 1.91 Å in P2S5, or longer like the P–P single bond of 2.15 Å in NiPS3. Despite the similarity in the structural surrounding, the P near-edge spectra of P2S5 and NiPS3 are rather different. The first maximum in the derivative plot of the normalized absorption coefficient for P2S5 is shifted to a lower energy of 2145.05 eV, compared to 2145.5 eV in NiPS3.
S double bond of 1.91 Å in P2S5, or longer like the P–P single bond of 2.15 Å in NiPS3. Despite the similarity in the structural surrounding, the P near-edge spectra of P2S5 and NiPS3 are rather different. The first maximum in the derivative plot of the normalized absorption coefficient for P2S5 is shifted to a lower energy of 2145.05 eV, compared to 2145.5 eV in NiPS3.
Considering the S K-edge spectra, the changes in the case of (EMIM,Li) co-intercalation are again much more pronounced than those after EMIM intercalation (Fig. 7c and d). The near-edge region in NiPS3 is composed of three well-distinguished peaks, which correspond to a transition of S 1s to various possible empty levels such as hybridized S 3p – Ni 3d orbitals and a σ*(P–S) orbital of the (P2S6)4− unit.52 The assignment of peaks to certain transitions in the case of S K-edge spectra is not straightforward without supporting theoretical calculations, since the electronic structure of pristine NiPS3 around the Fermi level has not been well-understood until now.53 The first two peaks in the derivative plots of NiPS3 with energy positions at 2469.8 eV and 2471.1 eV are very close to spectral features of the P2S5 reference.
After (EMIM,Li) co-intercalation (Fig. 7a and b), a new pre-edge feature at 2469.0 eV appears in the S K-edge spectra of (EMIM,Li)xNiPS3 samples. Simultaneously, the derivative peak at 2471.1 eV moved to a lower energy of 2470.5 eV. The third peak at 2472.3 eV remained nearly constant. Similar to the P K-edge, the intensity of the first shoulder at 2470 eV decreases upon (EMIM,Li) intercalation. These states are understood to be occupied by electrons donated by Li. The overall shift to lower energies reflects the increasing electron density. After intercalation of EMIM (Fig. 7c and d), only a broadening of the first maximum in the derivative plots could be detected, and the other peaks remained unchanged. In particular, the intensity of the first shoulder does not decrease.
Therefore, our spectroscopic experiments showed noticeable changes in the electronic structure of S and P in layered NiPS3 upon Li and EMIM co-intercalation, while Ni cations were not affected at all. In particular, our results show that the first unoccupied states of S and P are filled by electrons. Hence, the (P2S6)4− sublattice absorbs the electrons donated by Li-intercalation. The changes are much more pronounced upon insertion of small Li-cations compared to bigger EMIM molecules. EMIM intercalation appears to be completely ineffective in impacting the electronic structure. In the case of Na intercalation, Ni reduction was observed, pointing to a conventional redox reaction mechanism. This is in accordance with the NiPS3 structure decomposition detected by operando XRD measurements.
3.5 Elemental analysis
To address the open question regarding the charge compensation mechanism(s), elemental analysis by ICP-OES was performed before and after intercalation. It is known from other 2D MPS3 materials that the transition metal can leave the host structure during intercalation, for example during intercalation of N-alkylamine in FePS3.54 The mass fractions of Ni and P were determined in both the untreated sample and two intercalated NiPS3 compounds, after EMIM intercalation with Li and Pt counter electrodes for x = 1 charge transfer. The Ni/P molar ratios for the pristine, EMIM-intercalated and (EMIM,Li) co-intercalated samples were found to be rather similar and close to 1, confirming no elemental losses due to dissolution or secondary reactions.Additionally, using ICP-OES the amount of lithium in (EMIM,Li)1NiPS3 (intercalation vs. the Li electrode) was determined to be 0.59 Li per NiPS3 unit. This shows the preferential intercalation of Li ions over EMIM ions, even when the electrolyte did not contain any Li in the beginning. Furthermore, in the case of pure EMIM cation intercalation, the effective EMIM content in the material was evaluated to be 0.74 ± 0.07, based on the mass fraction of Ni and P in EMIM1NiPS3 (1EMIM from the galvanostatic experiment) using ICP-OES. This intercalated amount is less than the calculated introduced charge through GCPL. This difference could be due to the unavoidable washing step to remove excess of ionic liquid, which might also remove surface-adsorbed EMIM ions and/or portions of carbon and the binder. Another possibility may be other redox reactions occurring in parallel to intercalation.
3.6 Cation mobility in intercalated NiPS3
Based on the chemical analysis, a co-intercalation process of Li and EMIM into NiPS3 occurs in the cell with a Li-anode and EMIM-TFSI electrolyte, pointing to unequal rates of Li and EMIM intercalation kinetics. We studied cation mobility in NiPS3 during the intercalation process in the EMIM-TFSI electrolyte with Li or Pt as a counter electrode using the GITT. In the case of (EMIM,Li) co-intercalation, determination of only an average diffusion coefficient for both cations is possible. Starting from 2 × 10−9 cm2 s−1, the average DEMIM,Li diffusion coefficient shows a small maximum during the potential decrease from 1.9 to 1.7 V vs. Li+/Li, followed by a drop of several orders of magnitude, reaching a nearly constant value of 1 × 10−12 cm2 s−1 at a cell potential plateau of 1.7 V vs. Li+/Li (Fig. 8). The diffusion coefficient of a single EMIM cation in NiPS3 measured in cells with a Pt counter electrode shows at the beginning of intercalation a minimum of DEMIM = 1.8 × 10−16 cm2 s−1, and increases again up to 1.7 × 10−13 cm2 s−1. From this value, the coefficient decreases slowly with increasing intercalation. In direct comparison to the average (EMIM,Li) diffusion in (EMIM,Li)xNiP3, the diffusion coefficient of the single EMIM cation in (EMIM)xNIPS3 is always some orders of magnitude lower, since the smaller Li cation has a higher mobility.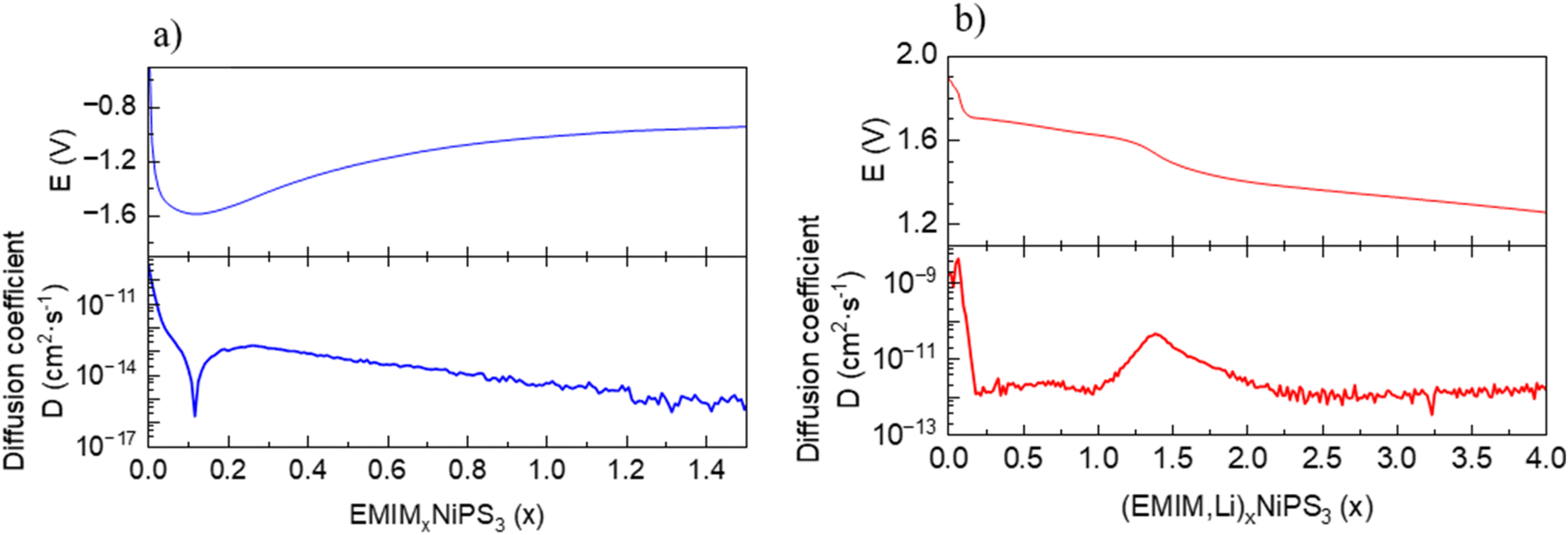 | ||
| Fig. 8 Equilibrium potential and diffusion coefficient dependence on the state of charge for: (a) intercalation of EMIM in NiPS3. (b) Co-intercalation of Li and EMIM in NiPS3. | ||
3.7 Confirmation of cationic intercalation with Raman spectroscopy
Raman spectroscopy was also used to investigate the intercalation of EMIM moieties in the van der Waals interlayers of NiPS3. For pristine NiPS3, 514 nm excitation was used for the Raman measurement due to the lower cut-off filter available with this setup configuration, whereas 405 nm excitation was applied for EMIM-TFSI, pristine NIPS3 (combined with PTFE), and intercalated samples considering an expected enhanced cross-section of the EMIM unit at this wavelength.55 The characteristic peaks of pristine NiPS3 can be seen in the low frequency range (Fig. 9a), matching the values reported in the literature.56 Based on its crystal structure, NiPS3 can have 15 active Raman modes (monoclinic, C2v).56 Due to the layered character of the structure with a small interlayer vibronic interaction (D3d symmetry),57 some vibrations become degenerate, i.e. symmetric and antisymmetric vibrations appear at the same frequency, reducing the number of peaks to 8 fundamental and some higher order modes as presented in Table S1.† Generally, the peaks P1 and P2 can be attributed to translational motions of Ni2+ ions whereas at higher frequencies, P3, P4, P5, and P7 are mostly due to the vibrations from the (P2S6)4− units.58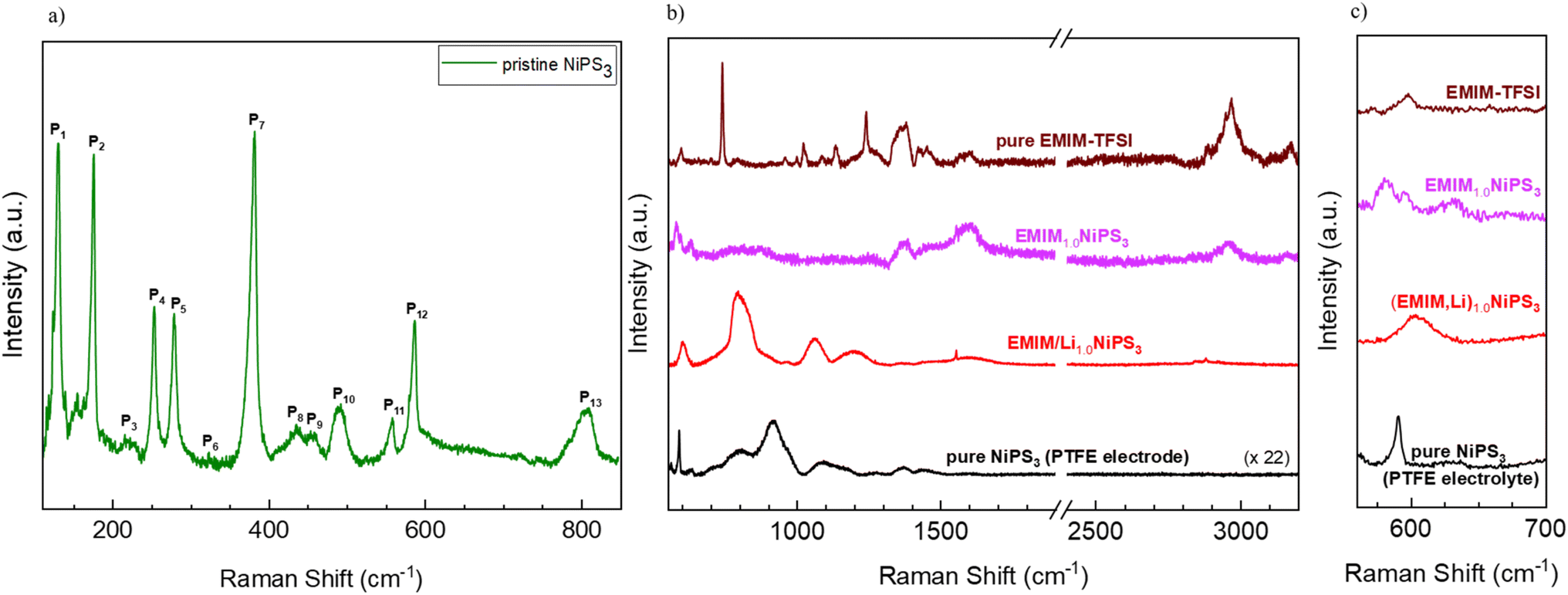 | ||
| Fig. 9 (a) Raman spectrum of pristine NiPS3 in a low-frequency range, using excitation at 514 nm. (b) Raman spectra in the high-frequency range of EMIM-TFSI, EMIM-intercalated NiPS3, (EMIM,Li) co-intercalated NiPS3, and pristine NiPS3 combined with PTFE, using a 405 nm laser. (c) Zoomed-in view of the middle graph. All spectra were obtained using a 2400 grating. In (b) and (c), data are plotted with offset for better visualization. | ||
Characteristic Raman modes of EMIM-TFSI are very pronounced in the high-frequency region (Fig. 9b). The broad peaks between 1350 and 3200 cm−1 belong mostly to vibrations of the EMIM-cation ring and the ethyl- and methyl-groups connected to the ring.59 Two very intense and narrow signals at 760 cm−1 and 1250 cm−1 originate from vibrations in the TFSI-anion.60 The effects of EMIM ion intercalation on the Raman spectrum of NiPS3 are threefold: (i) the normal modes of the NiPS3 host material vanished (for example, P13 at ∼800 cm−1), and some frequency shift occurred (P12 at ∼580 cm−1), (ii) some bands in the spectrum of EMIM1.0NiPS3 can be assigned to EMIM-associated vibrational modes, for example at 1380 cm−1, 1480 cm−1, and 2970 cm−1, and (iii) two new signals at 635 cm−1 and 1600 cm−1 arose (Fig. 9c). Although the signal at 1600 cm−1 is already present in the EMIM-TFSI spectrum, a significant increase in the intensity could imply additional contribution from some processes. Note that the increased intensity in this region also has a contribution from the conductive carbon additive. Nevertheless, the absence of the very intense signals from TFSI− in EMIM1.0NiPS3 confirms the incorporation of only EMIM ions into NiPS3.
It is known from the literature that at sufficiently low potentials, EMIM cations can be electrochemically reduced on a Ag-electrode.61 Two additional signals at 1330 cm−1 and 1607 cm−1 were registered amongst others in the Raman spectra of reduction products under 638 nm laser excitation, which were ascribed to free radicals, carbenes, and different dimers.61 A reductive decomposition of EMIM cations might occur in contact with NiPS3 as well. We assume that the EMIM cations are first intercalated into the van der Waals interlayers, as also evidenced by the layer expansion determined via XRD. Then, during the reduction process, dimers are possibly formed. On the other hand, reduction could also occur on the NiPS3-surface followed by desorption or release of generated neutral molecules. However, other spectroscopic techniques such as proton nuclear magnetic resonance spectroscopy (H-NMR), infrared spectroscopy (IR) and electron paramagnetic resonance (EPR) spectroscopy have to be employed for further detailed investigations. Interestingly, (EMIM,Li) co-insertion results in a quite different Raman response, with a strong shift of signals of the NiPS3 host (Fig. 9b). Unfortunately, in the literature, Raman studies of Li-intercalated NiPS3 are limited to the low-frequency region,53 pointing to a need for deeper Raman studies in a future work.
3.8 Magnetization studies
As is known, NiPS3 represents a quasi-two-dimensional antiferromagnet with a Néel temperature of 155 K.62 Above this temperature, there is a broad maximum in magnetization reflecting strong short-range magnetic interactions up to around 300 K.40 It was also reported that intercalated LixNiPS3 compounds exhibit a paramagnetic region above 300 K but decompose below 350 K.63 Changes in magnetic properties of NiPS3 after intercalation of Li and EMIM cations were evaluated using a SQUID magnetometer. Fig. 10 depicts the temperature dependence of susceptibility for the pristine compound and samples at different intercalation levels. The susceptibility curve of the initial NiPS3 shows a broad maximum at 280 K, in accordance with the literature.35,57 Observations of a Curie–Weiss paramagnetic behaviour up to the upper temperature range of 330 K was not possible. A small peak at 55 K, observed in all measurements, came from oxygen traces in the system, which becomes solid at that point.64 At temperatures below 10 K, there is another significant increase in magnetization, arising from defects in crystalline NiPS3.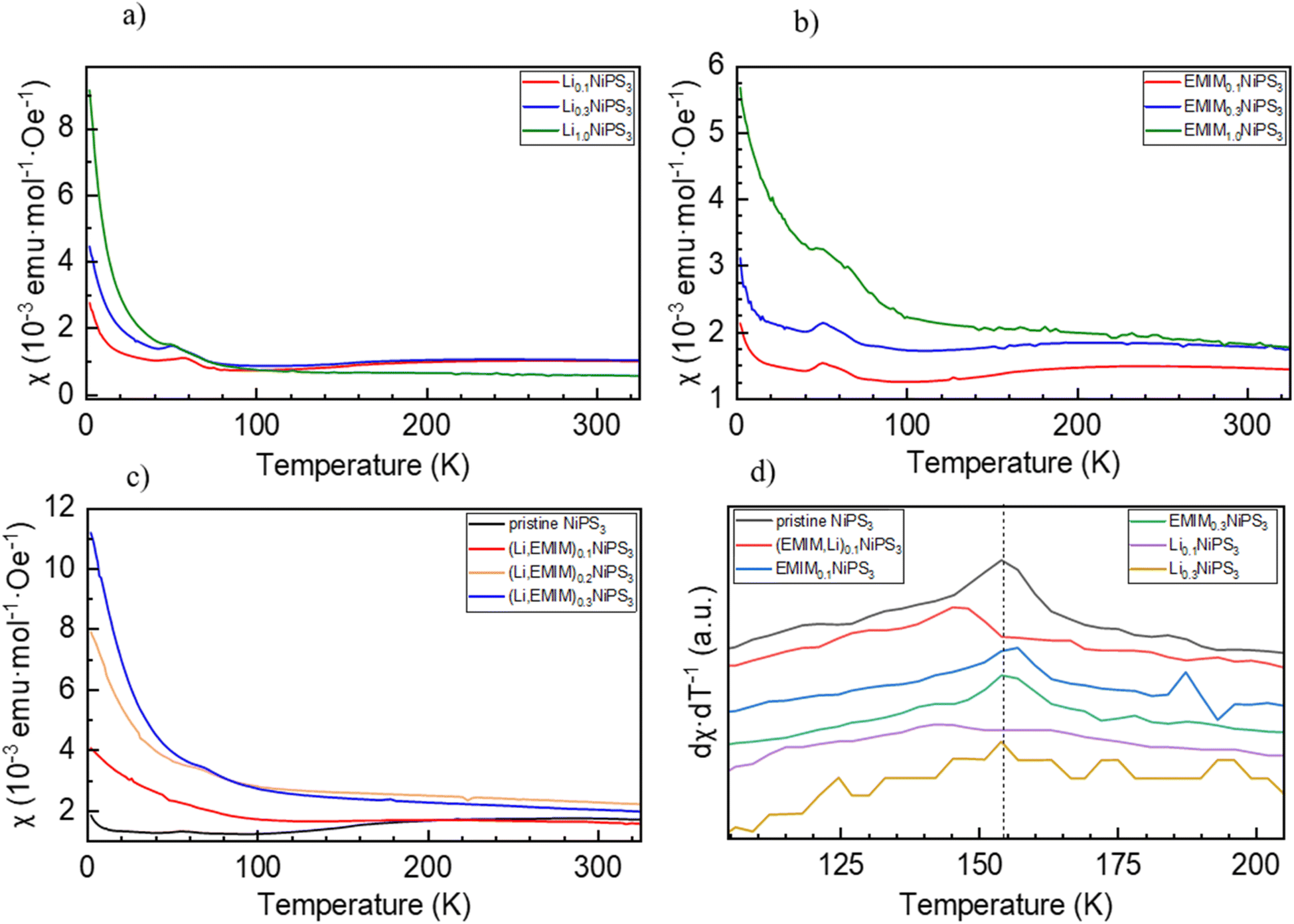 | ||
| Fig. 10 Susceptibility (zero-field cooling) versus temperature plot for intercalated NiPS3 powder samples. (a) Exclusively lithium ion intercalation, (b) exclusively EMIM intercalation, (c) Li and EMIM cation co-intercalation, and (d) first derivative of the temperature dependent susceptibility curves. The maximum corresponds to the Néel temperature of the compound. | ||
For all intercalated samples, the susceptibility increases in comparison to that of pristine NiPS3, especially at low temperatures, while the broad maximum at 280 K vanishes. The Néel temperature of intercalated materials was determined as a maximum of the first derivative of the susceptibility versus temperature plot. As one can see from Fig. 10d, all materials show nearly the same transition temperature of 154 K. Therefore, we conclude that the susceptibility curves emerge from a combination of the antiferromagnetic pristine phase and a paramagnetic-like intercalated phase. By comparing the magnetic behaviour of the three different intercalation systems (Li+, EMIM ion and co-intercalation), it is notable that co-intercalation leads to the highest susceptibilities, both in low and high temperature ranges. Applying the modified Curie–Weiss law (1) with a temperature-independent 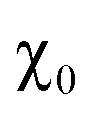 contribution from core diamagnetism and Van Vleck paramagnetism in the temperature range of 300–330 K gives values of the Curie constant C of 0.07 emu K mol−1 Oe−1 for Li1.0NiPS3, 0.10 for EMIM1.0NiPS3 and 0.13 for (EMIM,Li)0.3NiPS3.
contribution from core diamagnetism and Van Vleck paramagnetism in the temperature range of 300–330 K gives values of the Curie constant C of 0.07 emu K mol−1 Oe−1 for Li1.0NiPS3, 0.10 for EMIM1.0NiPS3 and 0.13 for (EMIM,Li)0.3NiPS3.
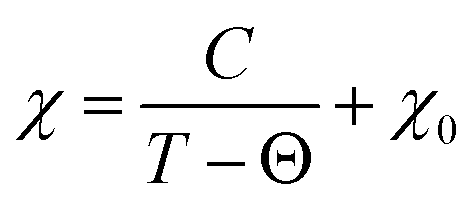 | (1) |
These values correspond to nearly similar paramagnetic moments, calculated as 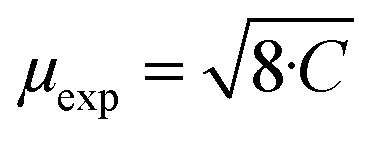 , of 0.75 μB for Li1.0NiPS3, 0.93 μB for EMIM1.0NiPS3 and 1.0 μB for (EMIM,Li)0.3NiPS3, which are significantly lower than the spin-only theoretical paramagnetic moment of 2.83 μB for Ni2+ with a d8 electron configuration (S = 1). This can indicate that there are still very strong magnetic interactions between Ni cations in this narrow temperature range of 300–330 K, which decrease the calculated paramagnetic moment. The more paramagnetic-like magnetization curve for intercalated materials points to decreased interaction between NiPS3-layers due to the interlayer expansion. From these results, it is not possible to conclude whether Ni is reduced during intercalation. Formation of Ni+ or a mixture of 0.5Ni2+/0.5Ni0 would reduce the total paramagnetic moment of the system to 1.73 μB or 2.0 μB, respectively, which is still much higher than the experimental value.
, of 0.75 μB for Li1.0NiPS3, 0.93 μB for EMIM1.0NiPS3 and 1.0 μB for (EMIM,Li)0.3NiPS3, which are significantly lower than the spin-only theoretical paramagnetic moment of 2.83 μB for Ni2+ with a d8 electron configuration (S = 1). This can indicate that there are still very strong magnetic interactions between Ni cations in this narrow temperature range of 300–330 K, which decrease the calculated paramagnetic moment. The more paramagnetic-like magnetization curve for intercalated materials points to decreased interaction between NiPS3-layers due to the interlayer expansion. From these results, it is not possible to conclude whether Ni is reduced during intercalation. Formation of Ni+ or a mixture of 0.5Ni2+/0.5Ni0 would reduce the total paramagnetic moment of the system to 1.73 μB or 2.0 μB, respectively, which is still much higher than the experimental value.
3.9 Single crystal X-ray diffraction (SCXRD) and TEM characterization of intercalated NiPS3
A combination of electron diffraction, TEM, and X-ray diffraction was employed in order to analyse the local structure of (EMIM,Li)xNiPS3 (Fig. 11). 3D electron diffraction taken from (EMIM,Li)xNiPS3 powder exhibits characteristic in-plane d-spacings, d(200) = 2.88 Å and d(020) = 1.69 Å. We can correlate the d-spacing values with the values obtained for the parent NiPS3 and, hence, assign the (hkl) values. However, in the direction orthogonal to the sample plane, only lines with continuous intensity distribution are seen instead of spots due to a thin film diffraction effect. A d-spacing for the longest axis of about 10 Å was taken from powder XRD data, in agreement with the above-discussed statement.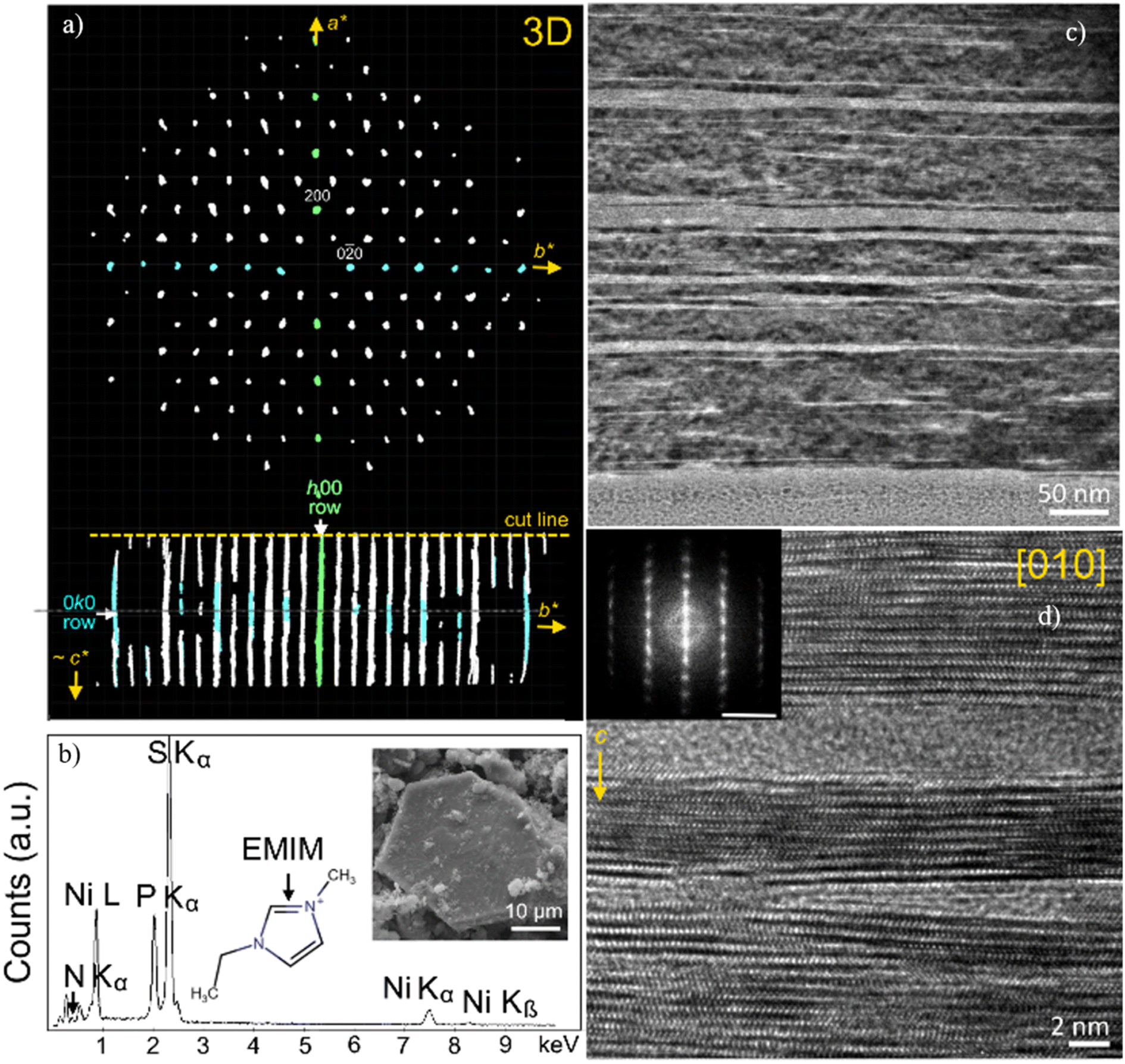 | ||
| Fig. 11 (a) 3D ED pattern of (EMIM)xNiPS3 visualized by REDp software.65 Two spots in the basal plane are indexed as (200) and (020) according to the corresponding diffraction pattern of the parent NiPS3. In the c-axis direction, only lines with a continuous intensity distribution are seen due to a thin film diffraction effect. (b) SEM EDX spectra of (EMIM)xNiPS3. The inset shows the crystal used for data collection. (c and d) HRTEM images of the specimen after EMIM intercalation with regions of crystalline NiPS3 and amorphous regions. The inset shows the FFT of the corresponding HRTEM image. The scale bar is 5 nm−1. | ||
SCXRD reflections collected from a (EMIM,Li)xNiPS3 crystal can be indexed either in a monoclinic space group C2/m with the cell parameters a = 5.867(3) Å, b = 3.391(2) Å, c = 10.075(6) Å, β = 101.36(2)°, and V = 196.5(2) Å3, or in a higher symmetry trigonal space group R-3 with the cell parameters a = b = 3.394(2) Å, c = 29.72(3) Å, β = 101.36(2)°, and V = 296.6(5) Å3. The latter is consistent with electron diffraction and powder XRD data and reflects a known behavior of intercalates when, due to a massive stacking disorder in the out-of-plane dimension, the space group symmetry for the average structure downgrades into a layer group symmetry. A trigonal lattice was used for further full matrix least-squares refinement, and the final cycle was based on 604 reflections measured (4.11° ≤ 2Θ ≤ 56.178°); of them 168 were unique (Rint = 0.0445, Rsigma = 0.0463) and converged to final R1 = 0.1321 (I > 2σ(I)) and wR2 = 0.3223 (all data). The largest peak/hole in the difference Fourier map (Fo − Fc) was 1.77 and −1.45/e Å−1 respectively. Thus, the EMIM-NiPS3 intercalate preserves the hexagonal lattice of Ni atoms bonded with six S atoms to form edge-sharing NiS6 octahedra. The S atoms are connected to two P atoms located above and below the Ni plane, forming a (P2S6)4− anion with a pyramidal structure. While the monoclinic symmetry of the parent NiPS3 structure exhibits the ABC stacking of the atomic layers along the c-axis, the intercalate structure reveals a significant degree of stacking disorder. The guest contribution to the structure factors, estimated by back-Fourier transformation of the electron density found in the solvent-accessible region (in the interlayer space) of a phase-optimized difference electron-density map,66 is about 26 e−/unit cell, or approximately 1/3 of the EMIM cation per unit cell. It is worth noting that for this estimation, the EMIM cation was assumed to be the only guest molecule in the interlayer space. This finding supports the results of ICP-OES, revealing the EMIM0.41Li0.59NiPS3 composition after cation intercalation in the cell with the Li anode and EMIM-TFSI electrolyte.
Cross sections of a larger (EMIM,Li)xNiPS3 crystal and pristine NiPS3 were prepared and analyzed by TEM on a nanometer scale. The TEM images highlight significant morphological changes after electrochemical intercalation with regions of crystalline NiPS3 and amorphous regions (see Fig. 11c). The nanoscale inhomogeneity is seen at the nm-level as increased distances between atomic layers, especially in the vicinity of amorphous regions and numerous stacking defects (Fig. 11d). The characteristic fast Fourier transform of the HRTEM image in Fig. 11d corresponds to ED of the {010} crystal plane in the NiPS3 crystal structure. The calculated d-spacings varies between 6.31 Å, corresponding to d(001) of pristine NiPS3 (ICSD entry 259148), 6.35 Å and 11.2 Å. Very minor changes in the d-spacings from 6.31 Å to 6.35 Å may indicate that the crystalline regions are not pristine NiPS3, but that they additionally host some Li as a co-intercalate, in line with the literature.40 Regions with a large distance of 11.2 Å between layers, caused by intercalation of EMIM molecules, border on some amorphous regions. This can reflect a weaker stability of (EMIM)xNiPS3 domains upon electron beam irradiation, or a partial crystallinity loss associated with EMIM insertion.
4. Discussion
Intercalation of organic species into layered inorganic structures represents an interesting approach for creating ordered organic–inorganic nanocomposites with novel properties, which are distinct from the parent compounds. Upon cation insertion into layered MPS3 with transition metal cations M, three possibilities for electronic transfer are usually discussed in the literature: (i) to a discrete atomic level, for example a 3d level of the transition metal, (ii) to a molecular level of a discrete polyatomic (P2S6)4− unit within the host structure, or (iii) to a part of a host conduction band.67 The last case is common for layered transition metal chalcogenides such as FeS2.67As we realized for NiPS3, electrochemical insertion of Li and Na occurs via different charge compensation mechanisms, resulting most probably from the difference in ionic radii of alkali cations and different thermodynamic stabilities of the products. Upon insertion of Li cations, the electron density on the (P2S6)4− unit increases. It is somewhat surprising, since NiPS3 is a charge-transfer insulator, and the first unoccupied density of states at ∼1.3 eV above Fermi energy represents a narrow Ni 3d band hybridized with S 3p, with a dominant Ni character.52 Probably, intercalated Li cations changed the crystal structure and also exerted an electrostatic potential. In contrast, insertion of larger Na cations increases the electron density on Ni. Intercalation of much bigger EMIM cations leads, however, to very little changes in electronic states of the elements. The redox nature of EMIM intercalation is confirmed by the electrochemical process. However, direct contact of NiPS3 and EMIM-TFSI in a beaker within an Ar-filled glove box for 24 h does not result in (EMIM)xNiPS3 formation. Currently, two different reaction mechanisms of intercalation of big organic molecules into NiPS3 are discussed in the literature. First, an increase in the electron density on Ni, P and S was observed after intercalation of cobaltocene,68 tetrabutylammonium69 or tetraheptylammonium cations.70 The increase in the electron density on S, being in the formal oxidation state −2 in the pristine compound, is reasonable, since a partial charge transfer from S to Ni with a d9L dominant character was calculated for NiPS3.52 The second reaction mechanism is displacive in nature, including creation of metal cation vacancies and intercalation of an appropriate number of organic cations for charge balancing without any change in the oxidation state of elements.18 A replacement mechanism in the case of EMIM insertion can be omitted, according to the chemical analysis results, which show the same Ni:P:S ratio in pristine and intercalated compounds. Reduction of Ni, P and S in NiPS3 can be excluded as well, as confirmed by us with numerous spectroscopic studies. Therefore, reduction of EMIM cations upon electrochemical intercalation into NiPS3 must be the only possible redox mechanism. As is known, imidazolium cations with a hydrogen atom in the 2-position can be considered as a protonated N-heterocyclic carbene, which can easily be de-protonated. A de-protonating agent can be the anion of the ionic liquid (for example, the acetate anion [OAc]−),71 chemical reduction (with alkali metal hydrides NaH or KH),72 or electrochemical reduction.73 The stability of such imidazolium-derived carbenes depends on steric and electronic effects,74 including electronic stabilization via a π-donation into the carbene out-of-plane p-orbital by the electron-rich π-system (N–CC–N) and the σ-electronegativity effect of nitrogen atoms.
Some formally uncharged carbenes can be isolated as products of a redox reaction, such as, for example, 1,3,4,5-tetramethylimidazol-2-ylidene from 1,3,4,5-tetramethyl-imidazolium chloride, with a cationic composition resembling 1-ethyl-3-methylimidazolium (EMIM).75 Electrochemical decomposition of 1-butyl-3-methylimidazolium tetrafluoroborate ionic liquid led to the formation of 1-butyl-3-methylimidazolium radicals that react with each other in a radical–radical coupling reaction, and in a disproportionation reaction.76 Formation of CC double bond dimers through dimerization of carbenes, and C–C single bond dimers through the reaction of single-electron radicals was considered in their work as possible products of the cathodic process of EMIM-TFSI on Ag electrodes.61 Similarities in the Raman spectra of (EMIM)xNiPS3 in our work, and the reaction product spectra in the work of Lu et al.61 point to the possibility of the EMIM cations here undergoing a similar reduction mechanism as presented there. Their reported onset potential for electrochemical EMIM reduction is also very close to the reaction plateau in the cell with a Li-anode during (EMIM,Li) co-intercalation that we present here. Chemical analysis of (EMIM)1NiPS3 yielded a smaller amount of inserted EMIM than that from galvanostatic measurements. Electrochemical reduction processes in EMIM can be the reason for this difference.
It is fascinating that EMIM reduction occurs between NiPS3 layers and does not take place at the same potential on the surface of a Pt-electrode in symmetric Pt–Pt cells. A co-intercalation of Li and EMIM cations shows a reduction of the (P2S6)4− unit, similar to pure Li-intercalation.
SQUID measurements of (EMIM)xNiPS3 and (EMIM,Li)xNiPS3 materials show an increase in the absolute magnetization values with an increased amount of intercalated species. In both cases, the antiferromagnetic ordering vanished, reflecting the importance of a Ni–Ni interlayer interaction and its destruction upon insertion.
5. Conclusion
We investigated electrochemical cationic intercalation of Li, Na and EMIM into the 2D NiPS3 structure. Three different mechanisms could be recognized based on comprehensive operando XRD, XAS (Ni K-edge) and ex situ XAS (P K-edge and S K-edge) as well as elemental analysis studies. Intercalation of small Li cations up to x(Li) = 0.5 in LixNiPS3 occurs into free octahedral spaces between layers without any structural changes, according to the synchrotron XRD measurements. Operando XAS investigations did not show any changes in the Ni K-edge spectra, implying inertness of Ni upon Li insertion. In contrast, noticeable changes in the electronic structure of S and P were detected, revealing the role of the (P2S6)4− unit in the redox process. Intercalation of more than 0.5 Li per NiPS3 formula unit results in the formation of Li4P2S6 as an intermediate, and Li2S as the final product of lithiation. Intercalation of Na into NiPS3 is accompanied by a Ni redox process visible in changes in the Ni K-edge spectra. Insertion of the much larger organic EMIM cations leads to a significant expansion in interlayer distance but, surprisingly, does not result in any changes in electronic states of Ni, S, and P. The underlying process here is associated with the reduction of EMIM cations through their de-protonation, and a further possible dimerization. It is understood that the EMIM cations first enter between layers of the 2D material followed by the redox reaction of the organic cations. Li- and EMIM-insertion leads to vanishing of the initial antiferromagnetic ordering of NiPS3 and to strengthening of its paramagnetic behavior.A detailed understanding of the structural changes and element-specific redox and electronic modifications occurring during intercalation of alkali and larger organic cation species shall be of significance in the development of hybrid systems based on 2D materials towards desired applications in opto-electronics and catalysis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the IFW Dresden excellence program, German Federal Ministry of Education and Research (BMBF) in the projects ExcellBattMat-KaSiLi (03XP0254D) and HeNa (03XP0390C) and by the German Research Foundation (DFG) in the project KIBSS (448719339) and AS 523/4-1. The authors thank Andrea Voss and Anne Voidel (IFW Dresden) for performing ICP-OES measurements. This research has benefitted from beamtime allocation at beamlines P02.01 and P64 at the PETRA III synchrotron (DESY, Hamburg, Germany), NOTOS and MSPD beamlines at the ALBA synchrotron (Barcelona, Spain), and the HiKE/KMC-1 beamline at the BESSY II synchrotron (Berlin, Germany). We are very grateful to Prof. I. Weidinger (TU Dresden) for providing access to her laboratory to carry out Raman measurements.References
- S. Selter, Y. Shemerliuk, M.-I. Sturza, A. U. B. Wolter, B. Büchner and S. Aswartham, Crystal Growth and Anisotropic Magnetic Properties of Quasi-Two-Dimensional (Fe1−xNix)2P2S6, Phys. Rev. Mater., 2021, 5(7), 073401, DOI:10.1103/PhysRevMaterials.5.073401.
- N. Sivadas, M. W. Daniels, R. H. Swendsen, S. Okamoto and D. Xiao, Magnetic Ground State of Semiconducting Transition-Metal Trichalcogenide Monolayers, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91(23), 235425, DOI:10.1103/PhysRevB.91.235425.
- P. A. Joy and S. Vasudevan, Magnetism in the Layered Transition-Metal Thiophosphates MPS3 (M = Mn, Fe, and Ni), Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46(9), 5425–5433, DOI:10.1103/PhysRevB.46.5425.
- J. Wang, X. Li, B. Wei, R. Sun, W. Yu, H. Y. Hoh, H. Xu, J. Li, X. Ge, Z. Chen, C. Su and Z. Wang, Activating Basal Planes of NiPS3 for Hydrogen Evolution by Nonmetal Heteroatom Doping, Adv. Funct. Mater., 2020, 30(12), 1908708, DOI:10.1002/adfm.201908708.
- Ye. V. Kuz’minskii, B. M. Voronin, I. M. Petrushina, N. N. Redin and G. P. Prikhodko, Nickel Phosphorus Trisulfide: An Electroactive Material for Medium-Temperature Lithium Batteries, J. Power Sources, 1995, 55(1), 1–6, DOI:10.1016/0378-7753(94)01932-L.
- R. Gusmão, Z. Sofer and M. Pumera, Metal Phosphorous Trichalcogenides (MPCh3): From Synthesis to Contemporary Energy Challenges, Angew. Chem., Int. Ed., 2019, 58(28), 9326–9337, DOI:10.1002/anie.201810309.
- R. Samal, G. Sanyal, B. Chakraborty and C. S. Rout, Two-Dimensional Transition Metal Phosphorous Trichalcogenides (MPX3): A Review on Emerging Trends, Current State and Future Perspectives, J. Mater. Chem. A, 2021, 9(5), 2560–2591, 10.1039/D0TA09752G.
- L. Silipigni, C. Calareso, G. M. Curró, F. Neri, V. Grasso, H. Berger, G. Margaritondo and R. Ponterio, Effects of Lithium Intercalation on the Electronic Properties of FePS3 Single Crystals, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53(20), 13928–13933, DOI:10.1103/PhysRevB.53.13928.
- J. van Dinter, K. Synnatschke, T. A. Engesser, S. Indris, N. Wolff, O. Gronenberg, M. Etter, G. Cibin, L. Kienle, C. Backes and W. Bensch, What Happens Structurally and Chemically during Sodium Uptake and Release by Ni2P2S6: A Combined X-Ray Diffraction, X-Ray Absorption, Pair Distribution Function and MAS NMR Analysis, J. Mater. Chem. A, 2020, 8(42), 22401–22415, 10.1039/D0TA07889A.
- C. Choi, D. Ashby, Y. Rao, E. Anber, J. L. Hart, D. Butts, C. Wilson, E. Levin, M. Taheri, M. Ghazisaeidi, B. Dunn and V. Doan-Nguyen, Mechanistic Insight and Local Structure Evolution of NiPS3 upon Electrochemical Lithiation, ACS Appl. Mater. Interfaces, 2022, 14(3), 3980–3990, DOI:10.1021/acsami.1c19963.
- M. Barj, Infrared Studies of Lithium Intercalation in the FePS3 and NiPS3 Layer-Type Compounds, Solid State Ionics, 1983, 11(2), 179–183, DOI:10.1016/0167-2738(83)90054-1.
- G. M. Currò, V. Grasso, F. Neri and L. Silipigni, The Effects of the Lithium Intercalation on the X-Ray Photoelectron Spectra of NiPS3, Nuovo Cimento Soc. Ital. Fis., D, 1995, 17(1), 37–52, DOI:10.1007/bf02451601.
- M. Mi, X. Zheng, S. Wang, Y. Zhou, L. Yu, H. Xiao, H. Song, B. Shen, F. Li, L. Bai, Y. Chen, S. Wang, X. Liu and Y. Wang, Variation between Antiferromagnetism and Ferrimagnetism in NiPS3 by Electron Doping, Chem, 2021 DOI:10.26434/chemrxiv-2021-mk8zv.
- D. Tezze, J. M. Pereira, Y. Asensio, M. Ipatov, F. Calavalle, F. Casanova, A. M. Bittner, M. Ormaza, B. Martín-García, L. E. Hueso and M. Gobbi, Tuning the Magnetic Properties of NiPS3 through Organic-Ion Intercalation, Nanoscale, 2022, 14(4), 1165–1173, 10.1039/d1nr07281a.
- I. M. Diaz Mesa, P. J. S. Foot and R. A. Kresinski, Synthesis, Structure and Properties of Crystalline and Nanocrystalline MnPS3-Poly(Phenylene Vinylene) Intercalates, Mater. Res. Bull., 2016, 84, 403–413, DOI:10.1016/j.materresbull.2016.08.037.
- M. Hangyo, S. Nakashima, A. Mitsuishi, K. Kurosawa and S. Saito, Raman Spectra of MnPS3 Intercalated with Pyridine, Solid State Commun., 1988, 65(5), 419–423, DOI:10.1016/0038-1098(88)90729-6.
- J. Luxa, Š. Cintl, L. Spejchalová, J.-Y. Lin and Z. Sofer, Potential Dependent Electrochemical Exfoliation of NiPS3 and Implications for Hydrogen Evolution Reaction, ACS Appl. Energy Mater., 2020, 3(12), 11992–11999, DOI:10.1021/acsaem.0c02168.
- X. Ma, L. Zhang, C. Xu, Q. Dong, R. I. Walton, Z. Li, H. Shi, G. Chen, J. Hu, J. Li and H. Yang, The Intercalation of 1,10-Phenanthroline into Layered NiPS3via Iron Dopant Seeding, Chem. Commun., 2020, 56(33), 4603–4606, 10.1039/d0cc00636j.
- X. Chen, C. Yang, J. Qin, K. Yakushi, Y. Nakazawa and K. Ichimura, The Intercalation Reaction of 1,10-Phenanthroline with Layered Compound FePS3, J. Solid State Chem., 2000, 150(2), 258–265, DOI:10.1006/jssc.1999.8584.
- T. Placke, O. Fromm, S. F. Lux, P. Bieker, S. Rothermel, H.-W. Meyer, S. Passerini and M. Winter, Reversible Intercalation of Bis(Trifluoromethanesulfonyl)Imide Anions from an Ionic Liquid Electrolyte into Graphite for High Performance Dual-Ion Cells, J. Electrochem. Soc., 2012, 159(11), A1755–A1765, DOI:10.1149/2.011211jes.
- T. Miyazaki, K. Ichimura, S. Matsuzaki and M. Sano, Pyridine-Intercalated MnPS3 Single Crystals, J. Phys. Chem. Solids, 1993, 54(9), 1023–1026, DOI:10.1016/0022-3697(93)90008-F.
- P. A. Joy and S. Vasudevan, The Intercalation Reaction of Pyridine with Manganese Thiophosphate, MnPS3, J. Am. Chem. Soc., 1992, 114(20), 7792–7801, DOI:10.1021/ja00046a027.
- E. Manova and I. Mitov, Catalytic Sulphide ions oxidation OVER MPS3 (M = Ni, Fe), Ni1-XFexPS3 (x = 0.4, 0.7) layered compounds and some related intercalation compounds, Bulg. Chem. Commun., 1998, 30(1–4), 227–240 CAS.
- F. Endres and S. Z. E. Abedin, Air and Water Stable Ionic Liquids in Physical Chemistry, Phys. Chem. Chem. Phys., 2006, 8(18), 2101–2116, 10.1039/B600519p.
- D. Monti, E. Jónsson, M. R. Palacín and P. Johansson, Ionic Liquid Based Electrolytes for Sodium-Ion Batteries: Na+ Solvation and Ionic Conductivity, J. Power Sources, 2014, 245, 630–636, DOI:10.1016/j.jpowsour.2013.06.153.
- C. A. Bridges, M. L. Martins, C. J. Jafta, X. G. Sun, M. P. Paranthaman, J. Liu, S. Dai and E. Mamontov, Dynamics of Emim + in [Emim][TFSI]/LiTFSI Solutions as Bulk and under Confinement in a Quasi-Liquid Solid Electrolyte, J. Phys. Chem. B, 2021, 125(20), 5443–5450, DOI:10.1021/acs.jpcb.1c02383.
- L. J. Hardwick, M. Holzapfel, A. Wokaun and P. Novák, Raman Study of Lithium Coordination in EMI-TFSI Additive Systems as Lithium-Ion Battery Ionic Liquid Electrolytes, J. Raman Spectrosc., 2007, 38(1), 110–112, DOI:10.1002/jrs.1632.
- M. P. S. Mousavi, B. E. Wilson, S. Kashefolgheta, E. L. Anderson, S. He, P. Bühlmann and A. Stein, Ionic Liquids as Electrolytes for Electrochemical Double-Layer Capacitors: Structures That Optimize Specific Energy, ACS Appl. Mater. Interfaces, 2016, 8(5), 3396–3406, DOI:10.1021/acsami.5b11353.
- M. Herklotz, J. Weiss, E. Ahrens, M. Yavuz, L. Mereacre, N. Kiziltas-Yavuz, C. Dräger, H. Ehrenberg, J. Eckert, F. Fauth, L. Giebeler and M. Knapp, A Novel High-Throughput Setup for in Situ Powder Diffraction on Coin Cell Batteries, J. Appl. Crystallogr., 2016, 49(1), 340–345, DOI:10.1107/S1600576715022165.
- V. Petříček, M. Dušek and L. Palatinus, Crystallographic Computing System JANA2006: General features, Z. Kristallogr. Cryst. Mater., 2014, 229(5), 345–352, DOI:10.1515/zkri-2014-1737.
- M. Gorgoi, S. Svensson, F. Schäfers, G. Öhrwall, M. Mertin, P. Bressler, O. Karis, H. Siegbahn, A. Sandell, H. Rensmo, W. Doherty, C. Jung, W. Braun and W. Eberhardt, The High Kinetic Energy Photoelectron Spectroscopy Facility at BESSY Progress and First Results, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 601(1), 48–53, DOI:10.1016/j.nima.2008.12.244.
- F. Schaefers, M. Mertin and M. Gorgoi, KMC-1: A High Resolution and High Flux Soft x-Ray Beamline at BESSY, Rev. Sci. Instrum., 2007, 78(12), 123102, DOI:10.1063/1.2808334.
- B. Ravel and M. Newville, ATHENA and ARTEMIS Interactive Graphical Data Analysis using IFEFFIT, Phys. Scr., 2005, 1007 DOI:10.1238/Physica.Topical.115a01007.
- APEX3, SAINT-Plus and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2016.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination, J Appl Cryst, 2015, 48(1), 3–10, DOI:10.1107/S1600576714022985.
- G. M. Sheldrick, SHELXT – Integrated Space-Group and Crystal-Structure Determination, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71(1), 3–8, DOI:10.1107/S2053273314026370.
- G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71(1), 3–8, DOI:10.1107/S2053229614024218.
- P. Foot and B. Nevett, Lithium Ion Diffusion in LixNiPS3 Single Crystals, Solid State Ionics, 1983, 8(2), 169–172, DOI:10.1016/0167-2738(83)90081-4.
- R. Brec, G. Ouvrard, A. Louisy, J. Rouxel and A. Lemehaute, The Influence, on Lithium Electrochemical Intercalation, of Bond Ionicity in Layered Chalcogenophosphates of Transition Metals, Solid State Ionics, 1982, 6(2), 185–190, DOI:10.1016/0167-2738(82)90086-8.
- R. Brec, Review on Structural and Chemical Properties of Transition Metal Phosphorous Trisulfides MPS3, Solid State Ionics, 1986, 22(1), 3–30, DOI:10.1016/0167-2738(86)90055-X.
- Ye. V. Kuz’minskii, B. M. Voronin, I. M. Petrushina, N. N. Redin and G. P. Prikhodko, Nickel Phosphorus Trisulfide: An Electroactive Material for Medium-Temperature Lithium Batteries, J. Power Sources, 1995, 55(1), 1–6, DOI:10.1016/0378-7753(94)01932-L.
- C. Dietrich, M. Sadowski, S. Sicolo, D. A. Weber, S. J. Sedlmaier, K. S. Weldert, S. Indris, K. Albe, J. Janek and W. G. Zeier, Local Structural Investigations, Defect Formation, and Ionic Conductivity of the Lithium Ionic Conductor Li4P2S6, Chem. Mater., 2016, 28(23), 8764–8773, DOI:10.1021/acs.chemmater.6b04175.
- M. H. Nilsen, C. Nordhei, A. L. Ramstad, D. G. Nicholson, M. Poliakoff and A. Cabañas, XAS (XANES and EXAFS) Investigations of Nanoparticulate Ferrites Synthesized Continuously in Near Critical and Supercritical Water, J. Phys. Chem. C, 2007, 111(17), 6252–6262, DOI:10.1021/jp0626723.
- G. Ouvrard, E. Prouzet, R. Brec, S. Benazeth and H. Dexpert, X-Ray Absorption Study of Lithium Intercalated Thiophosphate NiPS3, J. Solid State Chem., 1990, 86(2), 238–248, DOI:10.1016/0022-4596(90)90139-O.
- T. Yamamoto, Assignment of Pre-Edge Peaks in K-Edge x-Ray Absorption Spectra of 3d Transition Metal Compounds: Electric Dipole or Quadrupole?, X-Ray Spectrom., 2008, 37(6), 572–584, DOI:10.1002/xrs.1103.
- P. A. Joy and S. Vasudevan, Intercalation of N-Alkylamines in Iron Thiohypophosphate (FePS3), Chem. Mater., 1993, 5(8), 1182–1191, DOI:10.1021/cm00032a024.
- L. Blondeau, S. Surblé, E. Foy, H. Khodja, S. Belin and M. Gauthier, Are Operando Measurements of Rechargeable Batteries Always Reliable? An Example of Beam Effect with a Mg Battery, Anal. Chem., 2022, 94(27), 9683–9689, DOI:10.1021/acs.analchem.2c01056.
- M. Newville, Fundamentals of XAFS. Consortium for Advanced Radiation Sources, University of Chicago, USA, http://xafs.org, 2004, vol. 78, DOI:10.2138/rmg.2014.78.2.
- C. M. Julien, Lithium Intercalated Compounds: Charge Transfer and Related Properties, Mater. Sci. Eng., R, 2003, 40(2), 47–102, DOI:10.1016/S0927-796X(02)00104-3.
- M. H. Whangbo, R. Brec, G. Ouvrard and J. Rouxel, Reduction Sites of Transition-Metal Phosphorus Trichalcogenides MPX3, Inorg. Chem., 1985, 24(15), 2459–2461, DOI:10.1021/ic00209a028.
- I. Persson, W. Klysubun and D. Lundberg, A K-Edge P XANES Study of Phosphorus Compounds in Solution, J. Mol. Struct., 2019, 1179, 608–611, DOI:10.1016/j.molstruc.2018.11.059.
- S. Y. Kim, T. Y. Kim, L. J. Sandilands, S. Sinn, M.-C. Lee, J. Son, S. Lee, K.-Y. Choi, W. Kim, B.-G. Park, C. Jeon, H.-D. Kim, C.-H. Park, J.-G. Park, S. J. Moon and T. W. Noh, Charge-Spin Correlation in van Der Waals Antiferromagnet NiPS3, Phys. Rev. Lett., 2018, 120(13), 136402, DOI:10.1103/PhysRevLett.120.136402.
- I. Kerrache, Raman Scattering Studies of Lithium-Intercalated NiPS3, Solid State Ionics, 1996, 92(1–2), 37–43, DOI:10.1016/S0167-2738(96)00466-3.
- P. A. Joy and S. Vasudevan, Intercalation of N-Alkylamines in Iron Thiohypophosphate (FePS3), Chem. Mater., 1993, 5(8), 1182–1191, DOI:10.1021/cm00032a024.
- D. M. AL-Aqmar, H. A. S. Al-Shamiri, J. M. AL-Shareef, M. T. H. Abou Kana and H. M. Kandel, Spectroscopic and Photo-Physical Properties of Near-IR Laser Dye in Novel Benign Green Solvents, J. Fluoresc., 2020, 30(5), 1095–1103, DOI:10.1007/s10895-020-02576-1.
- K. Kim, S. Y. Lim, J.-U. Lee, S. Lee, T. Y. Kim, K. Park, G. S. Jeon, C.-H. Park, J.-G. Park and H. Cheong, Suppression of Magnetic Ordering in XXZ-Type Antiferromagnetic Monolayer NiPS3, Nat. Commun., 2019, 10(1), 345, DOI:10.1038/s41467-018-08284-6.
- C.-T. Kuo, M. Neumann, K. Balamurugan, H. J. Park, S. Kang, H. W. Shiu, J. H. Kang, B. H. Hong, M. Han, T. W. Noh and J.-G. Park, Exfoliation and Raman Spectroscopic Fingerprint of Few-Layer NiPS3 van Der Waals Crystals, Sci. Rep., 2016, 6(1), 20904, DOI:10.1038/srep20904.
- K. Kim, J.-U. Lee and H. Cheong, Raman Spectroscopy of Two-Dimensional Magnetic van Der Waals Materials, Nanotechnology, 2019, 30(45), 452001, DOI:10.1088/1361-6528/ab37a4.
- E. R. Talaty, S. Raja, V. J. Storhaug, A. Dölle and W. R. Carper, Raman and Infrared Spectra and Ab Initio Calculations of C2-4MIM Imidazolium Hexafluorophosphate Ionic Liquids, J. Phys. Chem. B, 2004, 108(35), 13177–13184, DOI:10.1021/jp040199s.
- J. Kiefer, J. Fries and A. Leipertz, Experimental Vibrational Study of Imidazolium-Based Ionic Liquids: Raman and Infrared Spectra of 1-Ethyl-3-Methylimidazolium Bis(Trifluoromethylsulfonyl)Imide and 1-Ethyl-3-Methylimidazolium Ethylsulfate, Appl. Spectrosc., 2007, 61(12), 1306–1311, DOI:10.1366/000370207783292000.
- W. Lu, Y. Gu, X.-Y. Hu, S. Tang, X. Li, D.-Y. Wu, J.-W. Yan, B.-W. Mao and Z.-Q. Tian, An In Situ Raman Spectroscopic Study on the Cathodic Process of EMITFSI Ionic Liquid on Ag Electrodes, J. Electroanal. Chem., 2018, 819, 435–441, DOI:10.1016/j.jelechem.2017.12.002.
- A. R. Wildes, V. Simonet, E. Ressouche, G. J. McIntyre, M. Avdeev, E. Suard, S. A. J. Kimber, D. Lançon, G. Pepe, B. Moubaraki and T. J. Hicks, Magnetic Structure of the Quasi-Two-Dimensional Antiferromagnet NiPS3, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92(22), 224408, DOI:10.1103/PhysRevB.92.224408.
- R. Brec, D. M. Schleich, G. Ouvrard, A. Louisy and J. Rouxel, Physical Properties of Lithium Intercalation Compounds of the Layered Transition-Metal Chalcogenophosphites, Inorg. Chem., 1979, 18(7), 1814–1818, DOI:10.1021/ic50197a018.
- T. Nomura, Y. H. Matsuda and T. C. Kobayashi, Solid and Liquid Oxygen under Ultrahigh Magnetic Fields, Oxygen, 2022, 2(2), 152–163, DOI:10.3390/oxygen2020013.
- W. Wan, J. Sun, J. Su, S. Hovmoller and X. Zou, Three-Dimensional Rotation Electron Diffraction: Software RED for Automated Data Collection and Data Processing, J. Appl. Crystallogr., 2013, 46, 1863–1873, DOI:10.1107/s0021889813027714.
- A. L. Spek, Platon Squeeze: A Tool for the Calculation of the Disordered Solvent Contribution to the Calculated Structure Factors, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71(1), 9–18, DOI:10.1107/s2053229614024929.
- G. Giunta, V. Grasso, F. Neri and L. Silipigni, Electrical Conductivity of Lithium-Intercalated Thiophosphate NiPS3 Single Crystals, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(12), 8189–8194, DOI:10.1103/PhysRevB.50.8189.
- E. Manova, A. Leaustic, I. Mitov, D. Gonbeau and R. Clement, The NiPS3-Cobaltocene Intercalation Compound: A New Ferromagnet, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1998, 311(1), 155–160, DOI:10.1080/10587259808042381.
- D. Tezze, J. M. Pereira, Y. Asensio, M. Ipatov, F. Calavalle, F. Casanova, A. M. Bittner, M. Ormaza, B. Martín-García, L. E. Hueso and M. Gobbi, Tuning the Magnetic Properties of NiPS3 through Organic-Ion Intercalation, Nanoscale, 2022, 14(4), 1165–1173, 10.1039/d1nr07281a.
- M. Mi, X. Zheng, S. Wang, Y. Zhou, L. Yu, H. Xiao, H. Song, B. Shen, F. Li, L. Bai, Y. Chen, S. Wang, X. Liu and Y. Wang, Variation between Antiferromagnetism and Ferrimagnetism in NiPS3 by Electron Doping, Adv. Funct. Mater., 2022, 32(29), 2112750, DOI:10.1002/adfm.202112750.
- N. M. A. N. Daud, E. Bakis, J. P. Hallett, C. C. Weber and T. Welton, Evidence for the Spontaneous Formation of N-Heterocyclic Carbenes in Imidazolium Based Ionic Liquids, Chem. Commun., 2017, 53(81), 11154–11156, 10.1039/c7cc06112a.
- S. T. Kerrick and P. Beak, Asymmetric Deprotonations: Enantioselective Syntheses of 2-Substituted Tert-(Butoxycarbonyl)Pyrrolidines, J. Am. Chem. Soc., 1991, 113(25), 9708–9710, DOI:10.1021/ja00025a066.
- B. Gorodetsky, T. Ramnial, N. R. Branda and J. A. C. Clyburne, Electrochemical Reduction of an Imidazolium Cation : A Convenient Preparation of Imidazole-2-Ylidenes and Their Observation in an Ionic Liquid, Chem. Commun., 2004,(17), 1972–1973, 10.1039/b407386j.
- A. J. I. Arduengo, R. L. Harlow and M. Kline, A Stable Crystalline Carbene, J. Am. Chem. Soc., 1991, 113(1), 361–363, DOI:10.1021/ja00001a054.
- A. J. I. Arduengo, H. V. R. Dias, R. L. Harlow and M. Kline, Electronic Stabilization of Nucleophilic Carbenes, J. Am. Chem. Soc., 1992, 114(14), 5530–5534, DOI:10.1021/ja00040a007.
- M. C. Kroon, W. Buijs, C. J. Peters and G.-J. Witkamp, Decomposition of Ionic Liquids in Electrochemical Processing, Green Chem., 2006, 8(3), 241–245, 10.1039/b512724f.
Footnote |
| † Electronic supplementary information (ESI) available: Operando synchrotron measurement data from NOTOS (ALBA, Spain), crystallographic data of EMIMxNiPS3, diffusion coefficient determination by the GITT, and Raman data of pristine NiPS3. See DOI: https://doi.org/10.1039/d3ta06196e |
| This journal is © The Royal Society of Chemistry 2024 |