
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic stabilization of nickel-based oxygen evolution electrocatalysts in the presence of chloride ions using a phosphate additive†
Hiroki
Komiya
 a,
Keisuke
Obata
a,
Tetsuo
Honma
b and
Kazuhiro
Takanabe
*a
a,
Keisuke
Obata
a,
Tetsuo
Honma
b and
Kazuhiro
Takanabe
*a
aDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, Japan. E-mail: takanabe@chemsys.t.u-tokyo.ac.jp
bJapan Synchrotron Radiation Research Institute (JASRI), 1-1-1, Kouto, Sayo-gun, Hyogo, Japan
First published on 12th January 2024
Abstract
Achieving a highly stable and selective oxygen evolution reaction (OER) in the presence of chloride ions (Cl−) is imperative for the future practical application of seawater splitting technology. This approach can streamline complex purification processes and avoid competition with the demand for drinking water. In this report, we present novel electrolyte engineering through mixed buffers – specifically, the utilization of borate/phosphate at a non-extreme pH of 9.2 – to achieve stable OER. Our findings demonstrate that borate serves as the primary buffer, maintaining localized pH stability, while the addition of phosphate acts as the main stabilizer for the Ni component. Electrochemical analysis and ex situ/operando spectroscopic characterization reveal that phosphate selectively forms a stabilized Ni complex. This complex effectively suppresses the corrosion and dissolution of Ni, resulting in an extended operational lifetime without compromising OER performance, even in the presence of Cl−. Furthermore, electrochemical measurements conducted with various redox probes indicate the formation of a passive Ni layer which leads to improved stability rather than the charge repulsion effect proposed in an alkaline pH environment. This study underscores the significant potential arising from the simultaneous advancement of both electrocatalysts and electrolytes.
Introduction
Electrochemical water-splitting technology driven by renewable electricity for hydrogen production has been regarded as an alternative to the current fossil-fuel-based technologies, which inevitably produce CO2. Practical electrolyzers need to purify seawater, which contains various impurities, to produce highly pure reactants of water to avoid the degradation of the whole system (consisting of anodes, cathodes, and gas separators), potentially leading to cost inefficiency. The process of seawater splitting has garnered significant attention due to its potential to curtail hydrogen production expenses. However, the presence of impurities, particularly chloride ions (Cl−) within seawater, instigates the deterioration of electrocatalysts and undermines the selectivity of the oxygen evolution reaction (OER). This phenomenon arises from the competition posed by the Cl− oxidation reaction, generating harmful Cl2 gas and hypochlorite (ClO−) compounds.The NiFeOx OER electrocatalyst has been extensively investigated because of its high intrinsic activity and earth-abundant components.1–6 Recently, some studies have been conducted to improve the stability and activity of NiFe-based electrocatalysts by doping a third element (e.g., S and Nb)7–9 and surface compositional engineering.10,11 However, its stability in the presence of Cl− is lower than that in the absence of Cl− regardless of pH levels.12–14 To prolong the lifetime of catalysts, surface modification techniques are often utilized to protect NiFeOx. For example, we demonstrated that a permselective CeOx layer coating on NiFeOx improved the stability by regulating the diffusion of redox ions in KOH solution even with Cl− while maintaining the intrinsic activity.12 The CeOx nanocoating selectively allowed OH− to reach the active site but not Cl−. As another example, a MnOx layer has been utilized to improve selectivity15,16 and stability.17–19 Wang et al.20 covered NiFeOx with MnOx by an electrochemical deposition method. They claimed that the MnOx overlayer prohibited Cl− from accessing the active site but allowed the permeation of H2O and O2 molecules. Although this technique is powerful, the drawback is that the permselective layers do not allow the diffusion of buffer substances (which are OH− sources) to the active sites underneath, leading to inefficient OER in buffered electrolytes at non-extreme pH levels.12
Another technique to prevent corrosion by Cl− is to introduce anion additives into the electrolyte, which would form a unique solid–liquid interface. Ma et al.21 introduced sulfate (SO42−) into the KOH electrolyte and investigated the OER performance over Ni foam (NF) and NiFeOx/NF. It was reported that SO42− adsorbed on the surface and worked as a negatively charged repulsion layer toward Cl−, preventing significant corrosion. Yu et al.22 added phosphate into the alkaline KOH electrolyte with Cl− and found that NiFeOx was highly stable during long-term stability testing. They claimed that it was because surface-adsorbed phosphate functioned as a charge repulsion layer and as a buffer on the surface, both of which led to low hypochlorite (ClO−) production, suppression of phase segregation, and Fe metal dissolution. The common interpretation of adding the anions under alkaline pH conditions is to utilize ion charge repulsion toward Cl−. This coulomb repulsion capacity is mainly determined by the ionic charge and the ion's radius.22 The other benefit of anion additives is the potential formation of a passive film toward Cl−. In the electrochemical corrosion study, phosphate23 and carbonate24 are often introduced in Cl−-containing solution, and they form a passive metal complex, leading to the high stability of the steel material. These additive salt approaches for electrolysis have not been well explored at non-extreme pH levels. For example, an additive of a fluoride anion in borate buffer is suggested to form BF2(OH)2− enhancing the OER activity of Co(OH)2 through activating a water molecule.25 According to the Pourbaix diagram,26 the desired pH level for electrolysis over NiFeOx is above ca. 8.5 due to the thermodynamic limitation associated with the Ni and Fe metal dissolution. At that pH level, the phosphate can form −2/−3 charged species which have a high enough charge repulsion capacity.22 In addition, the ideal pH for seawater splitting is pH 7–10, where precipitation of metal hydroxide (e.g., Ca(OH)2) can be avoided,27 and a maximal potential gap of 490 mV can be obtained between the OER and hypochlorite formation reaction (HCFR). Considering the thermodynamics mentioned above, the recommended pH for electrolysis over NiFeOx in the presence of Cl− is estimated to be 8.5–10. We demonstrated stable OER even with Cl− using CoFeOxHy on a Ti felt anode in borate buffer solution at pH 9.2 (ref. 14) and borate/carbonate mixed buffer at pH 9.8.28 However, earth-abundant Ni-based electrodes were found to be unstable in the presence of Cl−.
This study demonstrates a new insight into our ongoing efforts at electrolyte engineering by dynamic stabilization of Ni-based OER electrocatalysts in the presence of Cl− by adding phosphate in borate electrolytes at non-extreme pH levels. We tried to clarify the role of phosphate toward OER performance over NiFeOx in terms of stability and selectivity in mixed buffer electrolytes with various redox probes (e.g., not only Cl−, but also other halide ions and alcohols). The main function of the phosphate was found to be a stabilizer in the mixed buffer electrolytes at pH 9.2, which is in contrast to previous reports claiming the charge repulsion effect in alkaline solutions. Electrochemical assessments and ex situ/operando spectroscopic analysis also revealed that the phosphate selectively formed a Ni passive layer, which was not corroded by impurities and suppressed the dissolution, resulting in a greater lifetime. This study highlights the great potential of the concurrent development of electrocatalysts and electrolytes.
Results and discussion
Contribution of the phosphate additive to the OER performance in borate buffer solution
Ni–Fe (hydr)oxide (NiFeOx) was deposited on a conductive substrate of Ni foam using the hydrothermal method. The scanning electron microscope (SEM) image of as-made NiFeOx shows flake-like surface morphology which is commonly observed in hydroxide catalysts (Fig. S1†).29,30 NiFeOx/NF showed comparable performance with reported values in an alkaline medium (Fig. S2†).31,32Fig. 1a shows the OER performance in single 1.0 mol kg−1 K–borate, single 1.0 mol kg−1 K–phosphate, and K–borate/phosphate (1.0 mol kg−1 K–borate + 1.0 mol kg−1 K–phosphate) mixed electrolytes at fixed pH of 9.2 and 298 K. The OER performance in single borate was superior to that in single phosphate electrolyte due to its higher buffer capacity than that of the phosphate at specific pH 9.2, indicating that utilizing appropriate pKa is critical for efficient OER which is explained further in the ESI with Fig. S3.† This concentration overpotential trend relative to the identity of electrolyte pKa matched well with previously reported trends over IrOx33 and NiFeOx/NF.34 In mixed buffer electrolytes, in the low current density (j) region (up to 20 mA cm−2), the potential matched well with that under single borate conditions, indicating no significant performance loss even in the presence of phosphate. The double-layer capacitance (Cdl) calculated by cyclic voltammetry (CV) scan shows comparable values even in the presence of phosphate (Fig. S4†). This also insists that phosphate does not have an impact on the OER performance. In the high j region, the potentials reached values lower than that in the single borate system. Fig. 1b shows the monotonical potential decline with increased phosphate molality at fixed j; however, this improvement was not observed in dense (1.5 mol kg−1) borate electrolytes (Fig. S5†). We can conclude that the molality of borate at 1.0 mol kg−1 was not high enough to maintain the local pH shifting to acid during the OER, resulting in the induction of phosphate buffering action (pKa2 = 7.2) to suppress further local pH fluctuation at high current densities. Fig. 1c shows redox features over NiFeOx in two different electrolytes. The observed redox peak can mainly be attributed to the Ni redox species from Ni2+ to Ni3+. Fe-derived peaks cannot be detected, which is in agreement with previous studies.2,35,36 The reduction potential peak shifted to more positive by ca. 40 mV in phosphate-containing electrolyte than in single borate solution, indicating that the Ni2+ formed a more stable complex with phosphate compared to Ni(OH)2. This phenomenon can be explained by the thermodynamic stability of the complex based on the solubility products. Fig. 1d summarizes the redox peak positions of the NiFeOx catalyst and confirms that not borate ions but phosphate ions trigger the reduction shift, indicating the strong interaction between Ni and phosphate. Fig. 1e shows the potential at 10 mA cm−2 over bare Ni foam and NiFeOx/NF as a function of added phosphate molality. Even though potential drastically increased with introducing phosphate over Ni foam by around 200 mV, potentials were not affected over NiFeOx/NF. This result indicates that the phosphate selectively formed the Ni-passive layer, deactivating the Ni hydroxide active site on Ni foam. Meanwhile, the potential did not change significantly on NiFeOx/NF because the Fe site and the Ni site are considered to be active (guest) sites and hosts, respectively, over NiFeOx,37 and Fe hydroxides remain intact based on the reported solubility products.38 The Faradaic efficiency of O2 (FEO2) in the mixed buffer electrolyte was close to unity (Fig. S6†). Fig. S7a† depicts the Pourbaix diagram of Ni in the presence of phosphate calculated using the thermodynamic properties of products (Table S1†) and illustrates that a passive layer can form under mild pH conditions. At alkaline pH levels, the shift of the redox position was not clearly observed in CV profiles with and without phosphate (Fig. S7b†), emphasizing the unique phenomenon at non-extreme pH levels. Fig. 1f exhibits the X-ray photoelectron spectroscopy (XPS) results of Ni 2p over NiFeOx/Ni felt after OER measurements with and without phosphate additives. The peak of Ni 2p3/2 at 856 eV and 857 can be assigned as Ni2+ and Ni3+, respectively.39 When comparing the peak area ratio of two oxidation states, after the measurement in the borate, Ni3+ species existed more than Ni2+ (Ni3+/Ni2+ = 1.9). However, post-sample under the borate/phosphate mixed condition, the existence ratio of the Ni3+ was suppressed (Ni3+/Ni2+ = 1.1). These spectroscopic results highlighted the stabilization of Ni2+ rather than Ni3+ by the formation of a Ni–phosphate complex in the presence of phosphate. The Ni felt was used only in this XPS analysis. Also, the X-ray diffraction patterns shown in Fig. S8† do not present a highly crystalline structure. A cross-sectional SEM image with energy dispersive X-ray (EDX) line scan reveals that phosphorus exists throughout the ca. 10 μm NiFeOx catalyst layer, evenly and close to the Ni plate substrate, as illustrated in Fig. 2. The Ni plate was employed only in this cross-sectional SEM observation because of its flat shape. The transmission electron microscopy (TEM) image was obtained after the OER test in borate/phosphate electrolytes as shown in Fig. S9.† This figure shows the aggregation of the nanoparticles.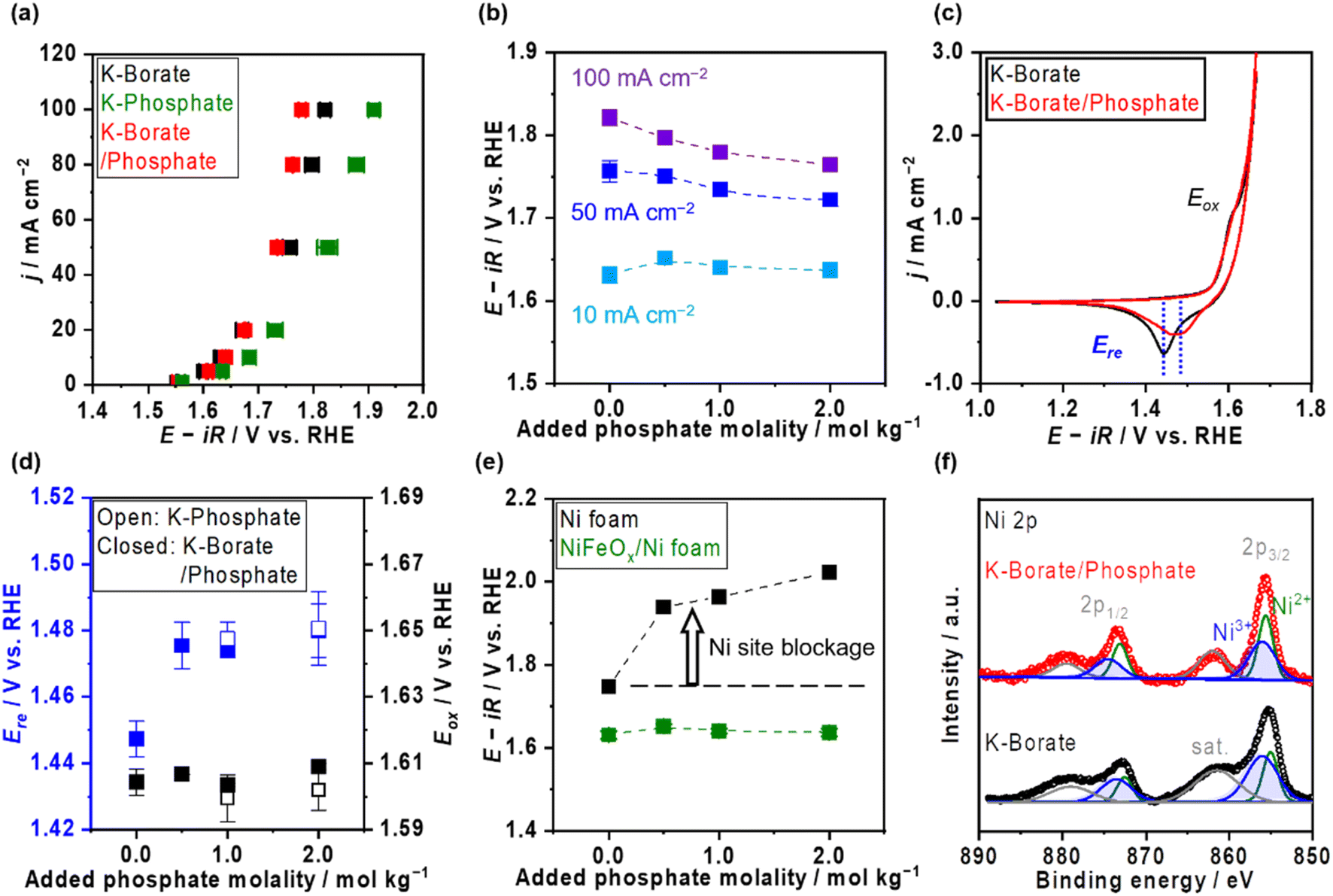 | ||
| Fig. 1 Oxygen evolution reaction (OER) performance over NiFeOx electrocatalyst in K–borate/phosphate mixed buffer electrolytes. (a) Potential and current density (j) relationships in various electrolytes at pH 9.2. Each single buffer molality was 1.0 mol kg−1. The molality of K–borate/phosphate electrolyte was 1.0 mol kg−1 K–borate + 1.0 mol kg−1 K–phosphate. (b) Potentials at different j values as a function of added phosphate molality. The molality of the borate was fixed at 1.0 mol kg−1. Potentials were recorded by using steady state chronopotentiometry (CP). (c) Cyclic voltammetry (CV) profile of NiFeOx in different electrolytes at pH 9.2. The molality of K–borate was 1.0 mol kg−1 and of K–borate/phosphate was 1.0 mol kg−1 K–borate + 1.0 mol kg−1 K–phosphate. CVs were recorded at a scan rate of 1 mV s−1. (d) The redox potential of NiFeOx. Reduction and oxidation potentials (Ere and Eox) were obtained from the CV profiles of each condition. (e) Performance comparison over bare Ni foam and NiFeOx/Ni foam (NF) in phosphate-containing electrolyte with 1.0 mol kg−1 K–borate. The potential values were at 10 mA cm−2. (f) X-ray photoelectron spectroscopy (XPS) spectra of Ni 2p over NiFeOx/Ni felt after OER testing in distinct electrolytes. All OER measurements were conducted with O2 bubbling at 298 K, pH 9.2. | ||
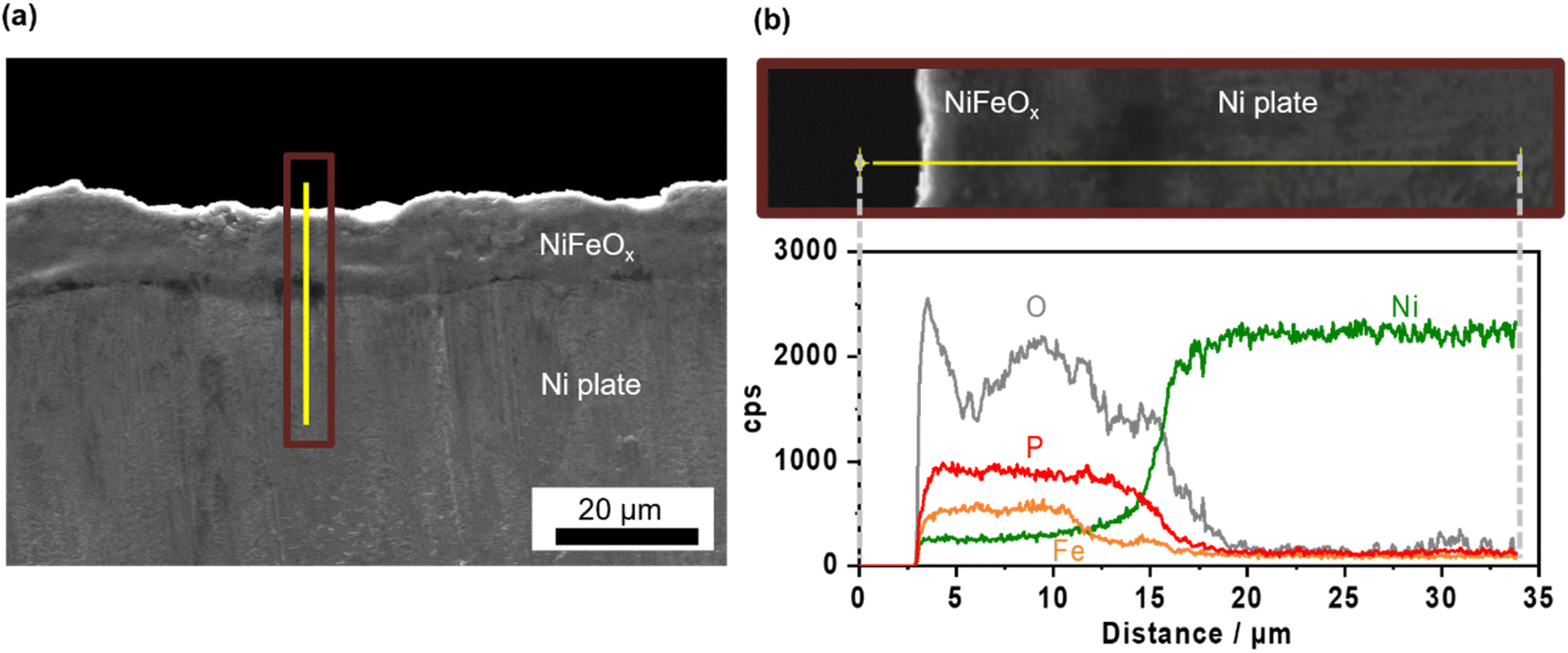 | ||
| Fig. 2 Cross-sectional scanning electron microscope (SEM) images of the NiFeOx/Ni plate. (a) Cross-sectional SEM image of the NiFeOx/Ni plate electrode. (b) Magnified SEM of the NiFeOx/Ni plate and corresponding elemental cps as a function of distance. The yellow line corresponds with the scan line shown in both figures (a) and (b). | ||
Fig. 3a shows the stability testing over NiFeOx/NF in borate and borate/phosphate mixed buffer electrolytes at 50 mA cm−2 for 24 h. The potential increased in the borate electrolyte. In contrast, the potential in the mixed buffer electrolyte was stable for 1 day. CVs after the stability testing are shown in Fig. 3b. In borate electrolytes, the Ni reduction potential shifted to more negative, which can be explained by the phase segregation and the metal dissolution.22 Under alkaline pH conditions, the degradation mechanism of NiFeOx was known to be the overcharging (oxidation) of the Ni hydroxide associated with the formation of phase-segregated FeOOH.40,41 Furthermore, at near-neutral pH levels, the degradation of the NiFeOx catalysts is considered the dissolution of the Ni from the electrode.42 We note that the acquired CV in the anodic scan region does not reflect the pure OER because the measured current was not in the steady state and contained the oxidation of the electrode. Fig. S10a† shows the steady-state potential after stability testing, endorsing deteriorated performance in borate electrolytes. Fig. 3c presents the metal dissolution amount in the electrolytes after stability testing. Ni dissolution was significant and can be attributed to the reason for degradation. In contrast, the reduction potential did not shift as much after continuous CP in the phosphate-containing electrolyte (Fig. 3b). In addition, the Ni dissolution after stability testing was twice lower than that in the single borate electrolyte (Fig. 3c). The phosphate stabilized the Ni and suppressed the dissolution of Ni, which resulted in a stable potential. Fe dissolution is also regarded as a degradation reason. However, the amount of dissolution is one magnitude lower than that of Ni and does not have a prominent relationship, unlikely to be a main factor for the degradation.
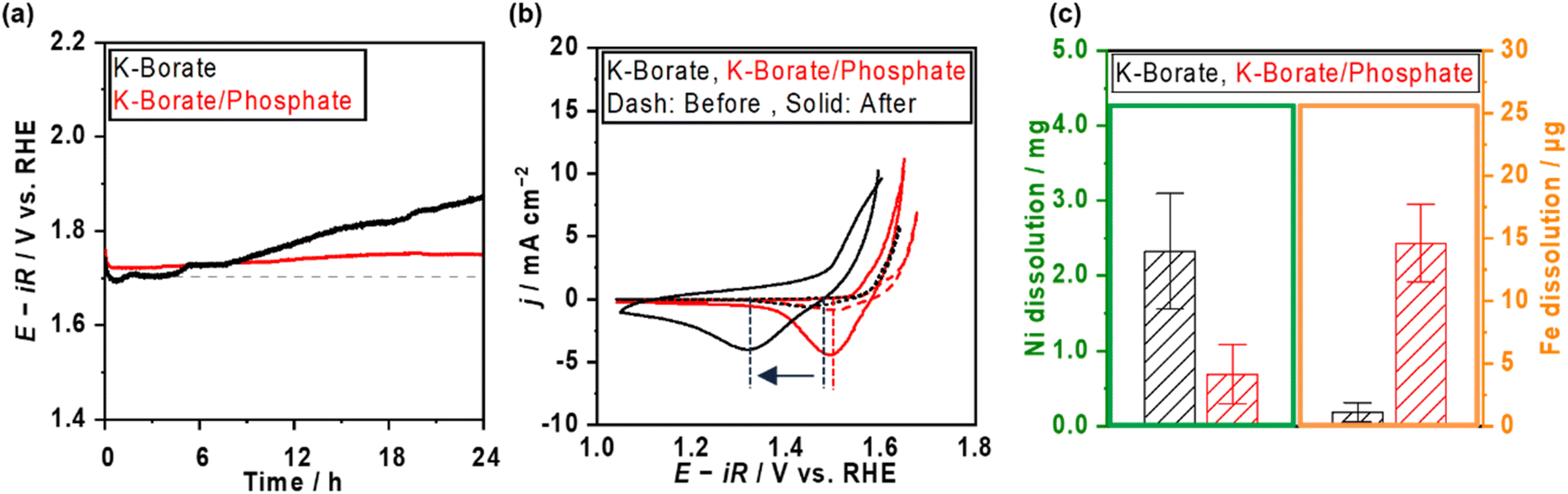 | ||
| Fig. 3 Stability testing over NiFeOx/Ni foam with and without phosphate additives at room temperature. (a) Potential and (b) CV profiles over NiFeOx/Ni foam in 1.5 mol kg−1 K–borate (black) and 1.0 mol kg−1 K–borate + 2.0 mol kg−1 K–phosphate (red) at pH 9.2. The potential profiles were recorded using CP at 50 mA cm−2 for 24 h. CVs were recorded at a scan rate of 1 mV s−1 before and after the stability testing. All electrolytes were saturated with O2 during measurements. (c) Metal dissolution amount of Ni and Fe in the electrolytes after the stability testing. | ||
OER performance in chloride ions containing borate/phosphate mixed buffer solution
The OER performance over NiFeOx/NF was also assessed in the presence of Cl−. Fig. 4a shows the CV profile of NiFeOx in single borate and borate/phosphate mixed electrolytes with and without Cl−. NiFeOx suffered from significant electrochemical corrosion in borate plus Cl− electrolyte (black solid line). Previous studies, including our reports,14,21 have already revealed that this degradation was induced by the Ni corrosion forming Ni(OH)2 green solids. In contrast, interestingly, CV matched well between those with and without Cl− in phosphate-containing electrolytes (red lines). In addition, this stability improvement was observed only when the phosphate was present in the electrolyte. In detail, Fig. S11† shows the CV profiles recorded by replacing the electrolytes. NiFeOx was activated in K–borate/phosphate mixed buffer electrolyte, and CV was depicted after the activation (red). Then, the electrode was washed with water and immersed in a Cl−-containing borate electrolyte without phosphate. Subsequently, the CV was recorded (black). Although the initial anodic current shows negligible Cl− aggression, the CV was unstable from the initial cathodic scan. These results infer the in situ formation of a kind of Ni-passive layer on the surface or that phosphate worked as a charge repulsion layer, highlighting the essential role of phosphate in the electrolyte to protect the electrode. Fig. S12† shows the long-term stability testing over NiFeOx/NF in borate/phosphate + KCl mixed buffer electrolytes at 298 K for 1 day. The potential, CV, and metal dissolution amount matched well with the measurements without KCl, supporting that the Cl− did not corrode the electrode. Considering the formation constant of [Ni(H2O)5Cl]+ and Ni–phosphate at pH 9.2, the Ni ions prefer to form a Ni–phosphate complex;38,43 thus, Cl− cannot corrode the in situ formed Ni-passivated electrode even though Cl− existed on the surface of the electrode confirmed by XPS (Fig. S13†). Our previously reported CoFeOxHy catalyst was unstable when the potential was above the HCFR potential (1.72 V vs. the reversible hydrogen electrode (RHE)),14 while the potential was stable even at 1.73 V vs. RHE in the present system, highlighting the advantage of phosphate additives towards the stability. Fig. S14† shows the OER potentials and FE toward the HCFR (FEHC) as a function of added phosphate molality with 0.5 mol kg−1 of KCl. The potential values almost matched with those under the conditions without Cl−, and FEHC was below 1% even at the potential surpassing the thermodynamic threshold of HCFR potential. The close-to-unity FEO2 during the OER is shown in Fig. S15a,† highlighting the negligible electrode corrosion by Cl−. Also, the OER performance was unaffected when changing the counter cation of the Cl− from K+ to Na+ (Fig. S16†). We also tried a sulfate additive instead of phosphate because it is expected to show a charge repulsion effect to block Cl− similar to HPO42−, which is the major phosphate species in the present study (Fig. S17a†). Fig. S17b† shows the CV profile and photo after CV over NiFeOx/NF in the sulfate and Cl− containing borate electrolytes. A sharp corrosion current was observed, and the electrode formed green solids. This unstable trend can be explained by higher solubility products of Ni–sulfate than phosphate ones implying weaker interaction between Ni and sulfate.38,44 Because the previous literature21 claims that the stability improvement is attributed to the surface-formed charge repulsion layer in sulfate-containing alkaline medium solutions, the charge repulsion theory has been examined at non-extreme pH levels through adding redox probes in the following sections.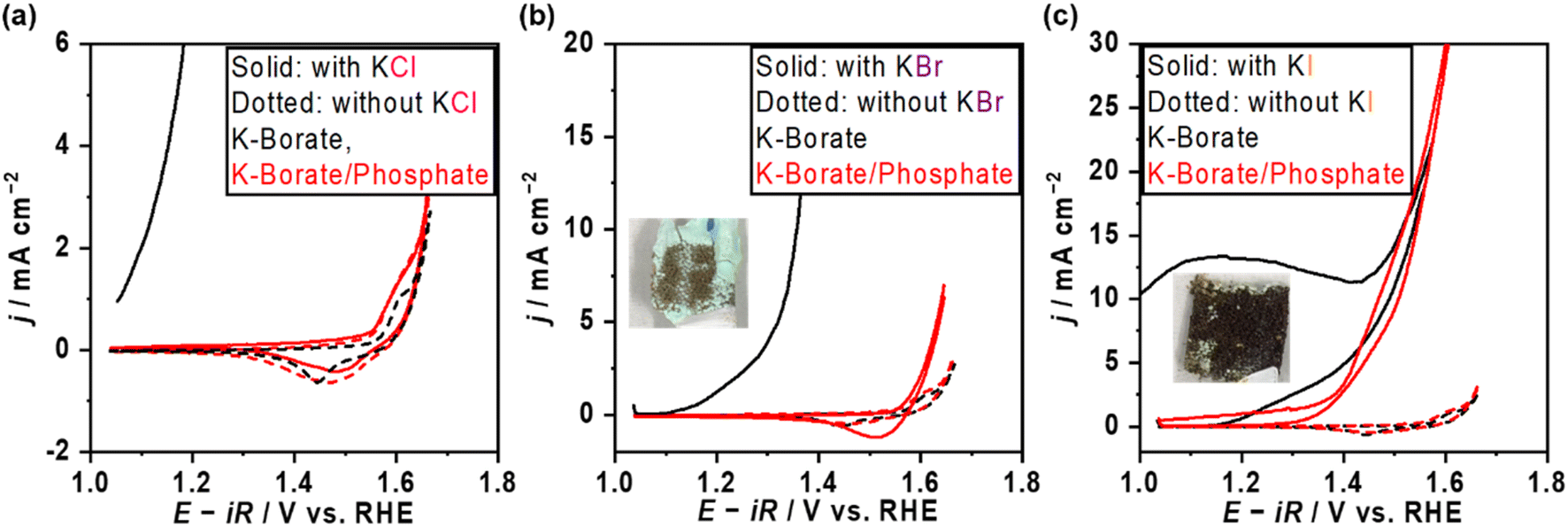 | ||
| Fig. 4 OER performance over NiFeOx in halide ions containing electrolytes. CV profiles in K–borate and K–borate/phosphate electrolytes with (a) KCl, (b) KBr, (c) KI at 298 K, pH 9.2. The molality of K–borate was 1.0 mol kg−1, of K–borate/phosphate was 1.0 mol kg−1 K–borate + 1.0 mol kg−1 K–phosphate, and of the halide salt was 0.5 mol kg−1. All CVs were recorded at a scan rate of 1 mV s−1. | ||
Borate/phosphate mixed buffer electrolyte with various redox probes
The OER performance was also assessed in the presence of various redox probes to describe the versatile impact of the phosphate additive on selectivity and stability. Fig. 4b and c show the CVs in the presence of bromide ions (Br−) and iodine ions (I−). Both ions drastically deactivated NiFeOx/NF with the occurrence of green solids after applying the positive potential in the single borate electrolytes (black). In contrast, stable CVs were observed in mixed borate/phosphate electrolytes, likely due to the formation of a Ni passive layer (red). Table 1 summarizes the FEO2 recorded with various halide ions. Although the potential profiles during FE measurements were stable regardless of the halide ion's identity shown in Fig. S14b,† the FEO2 in the presence of Br− and I− was under the detection limit of gas chromatography, suggesting that almost all the charges were consumed to oxidize the halide ions. In contrast, the presence of Cl− resulted in almost unity for FEO2. This selectivity difference was explained by their redox potentials. In short, given that if the halide ion oxidation would proceed with the production of the XO− (X = Cl, Br, I), the redox potential (E0) of the Br− oxidation reaction (1.59 VRHE) and iodine oxidation reaction (1.31 VRHE) are below the potentials during FEO2 measurements, leading to their preferential oxidations. On the other hand, only E0 of chloride oxidation (1.72 VRHE) is slightly lower than the measured potential at 50 mA cm−2 during FEO2 measurement, which partially induces the HCFR. The comparison among the halides clarifies the major function of phosphate under the non-extreme pH conditions. If the phosphate would work as a charge repulsion layer on the surface, the Br− and I− would not be oxidized, considering their chemical similarities, such as their Stokes radius (listed in Table 1) and their ion charge (−1). However, the FEO2 was extremely low, indicating that the halides can adsorb on the catalyst surface leading to inner-sphere charge transfer. This highlights that the main role of the phosphate is to stabilize the Ni site, not the charge repulsion layer. Oxidation of other redox probes, such as alcohols (EtOH and i-PrOH) and anions ([Fe(CN)6]4− and formate) is discussed in the ESI (Fig. S18–S21†) to reveal the active site of NiFeOx in the presence of phosphate. During the EtOH oxidation reaction, improved FEO2 and Tafel slope changes were observed in the presence of phosphate. Because Fe hydroxide is not a superior catalyst for the EtOH oxidation reaction to Ni hydroxide,45 we believe that the observed change comes from the switching of active sites from Ni to Fe due to the formation of a Ni-based passive layer with phosphate. Fig. S20a† shows the CV profile in the presence of K4[Fe(CN)6]. Redox behaviors of NiFeOx were considerably different from that in the absence of K4[Fe(CN)6]. The standard redox potential of [Fe(CN)6]4−/3− is 0.37 V vs. the standard hydrogen electrode (0.92 VRHE). Because the oxidation of [Fe(CN)6]4− to [Fe(CN)6]3− proceeds via the facile outer sphere reaction,46 diffusion-limited anodic currents were observed between 1.1 and 1.5 VRHE, which decreased in the presence of phosphate due to the increased viscosity and the reduced diffusion coefficient. FEO2 was not significantly affected by the presence of phosphate (Fig. S20b†), indicating that the highly charged anion can still reach the outer Helmholtz plane. This result strongly suggests that the phosphate does not form the charge repulsion layer at the present non-extreme pH level.| Added halide salts | a/Å | Electrolyte | FEO2/% |
|---|---|---|---|
| KCl | 1.21 | K–borate | Could not be measured due to electrode corrosion |
| K–borate/phosphate | >99 (both 50 and 100 mA cm−2) | ||
| KBr | 1.18 | K–borate | Could not be measured due to electrode corrosion |
| K–borate/phosphate | Under the detection limitation of GC | ||
| KI | 1.20 | K–borate | Could not be measured due to electrode corrosion |
| K–borate/phosphate | Under the detection limitation of GC |
In addition, the OER performance at elevated temperatures and high current density were investigated to clarify the applicability of mixing buffer ions for future large-scale H2 production. Fig. S22a and b† display the E–j relationship and E at various j values as a function of the added molality of phosphate at 353 K in 1.5 mol kg−1 borate containing mixed buffer solutions. In the low j region, the potentials were independent of the added phosphate; however, the potential increased with increased phosphate molality in the high j region (>0.1 A cm−2). When looking at the E–j plots, the difference can be attributed to concentration overpotential induced by the high viscosity of the solution, which caused the low diffusion of the borate. The viscosity of phosphate-containing electrolytes is two times higher than that of the single borate (Fig. S22c†). Optimizing the mixture ratio of borate and phosphate would be important to achieve stable and efficient OER. Furthermore, we compared the OER performances in the presence of the Cl−. The potentials recorded in borate/phosphate mixed buffer electrolyte with 0.5 mol kg−1 KCl overlapped well with all j values below HCFR potentials (Fig. S22b†). Fig. 5a shows the multi-on–off stability testing at 500 mA cm−2 and an elevated temperature of 353 K with and without Cl−. In K–borate with Cl− electrolyte, rapid potential overshooting was observed because of Ni corrosion caused by Cl−. On the other hand, the potential was highly stable in the presence of the phosphate, even in the presence of Cl− additives. Fig. 5b shows the CV after stability testing with and without Cl−. The CVs almost overlapped with each other, confirming that there was no significant corrosion derived from Cl− aggression. These data confirm the great advantage of mixing additional ions into the electrolytes to maintain the OER performance of NiFeOx/NF in the coexistence of Cl−.
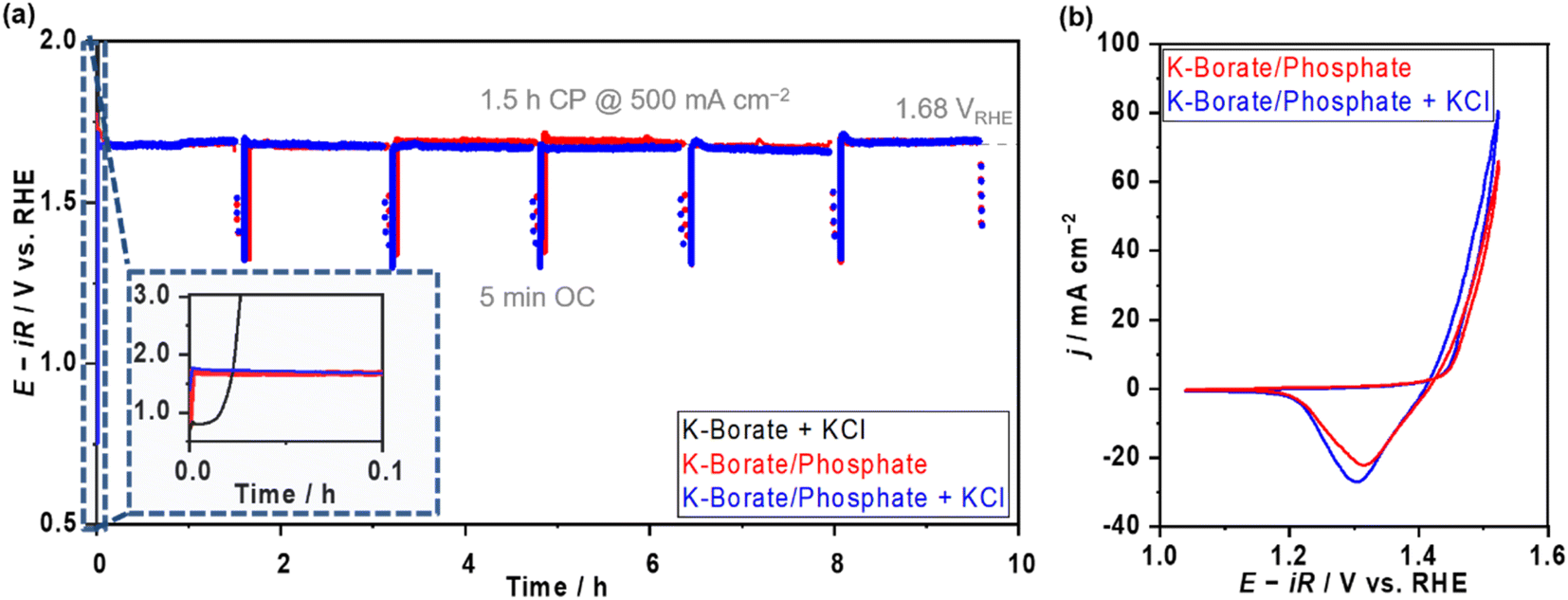 | ||
| Fig. 5 Stability testing over NiFeOx in distinct electrolytes at commercially relevant current density. (a) On–off CP profiles of each electrode at 353 K. The molality of each electrolyte was 1.5 mol kg−1 K–borate + 0.5 mol kg−1 KCl (black) and 1.0 mol kg−1 K–borate + 1.0 mol kg−1 K–phosphate (i.e., K–borate/phosphate (red)) + 0.5 mol kg−1 KCl (blue). The test consists of six on–off cycles (one cycle: 1.5 h CP at 500 mA cm−2 and 5 min open-circuit (OC)). (b) CVs after stability testing. All CVs were recorded at a scan rate of 1 mV s−1. All OER measurements were conducted with O2 bubbling and at pH 9.2. | ||
Operando XAS analysis
Fig. S23† shows the ex situ X-ray absorption near edge structure (XANES) spectra over post-reacted NiFeOx/carbon paper (CP) electrodes. The white line of Ni K-edge after the OER in phosphate-containing solutions appeared more negative than that after the OER in borate alone and that of the Ni(OH)2 reference, indicating the formation of a kind of Ni–phosphate complex after the OER. The trend of the lower oxidation state of Ni agrees with the previously mentioned XPS results (Fig. 1f). In addition, the Ni oxidation state was unaffected by the Cl−, likely because of the strong interaction between Ni and phosphate. In contrast, the Fe K-edge spectra show almost the same chemical state of Fe3+ under various electrolyte conditions, highlighting that Fe remained stable in the presence of Cl− and that phosphate prefers to interact with Ni. A previous EXAFS study on a NiFe layered double hydroxide catalyst observed a higher oxidation state in the presence of NaCl in KOH solution;47 however, our NiFeOx catalyst studied at non-extreme pH maintained the oxidation state.Operando X-ray absorption spectroscopy (XAS) measurements were conducted at 1.6 VRHE, and the XANES spectra are shown in Fig. 6. In borate electrolytes, the Ni K-edge shifted to higher energy from the ex situ one due to the oxidation of the Ni2+ during the OER. The spectrum of the Fe K-edge did not shift by as much. These results match well with the reported trends in alkaline electrolytes.5 In borate/phosphate electrolytes, although the Ni K-edge at 1.6 VRHE shifted to be more positive than the ex situ spectrum, the spectrum did not overlap with the single borate one, suggesting that further Ni2+ oxidation approaching +3 and +4 was suppressed by the phosphate additive. The formed Ni matrix was partially oxidized at OER potential while partially maintaining the Ni complex, resulting in a unique spectrum. The spectra of the Fe K-edge did not show any noticeable change even under the OER in the presence of phosphate, which supports the strong interaction between Ni and phosphate. In addition, the Ni and Fe K-edge spectra with Cl− represent an almost similar oxidation state to those without Cl− conditions. This Ni oxidation suppression effect confirmed by XANES analysis supports the fact that the major reason for the improved stability under long-term CP (Fig. 3 and S12†) comes from the formation of the Ni-based passive layer.
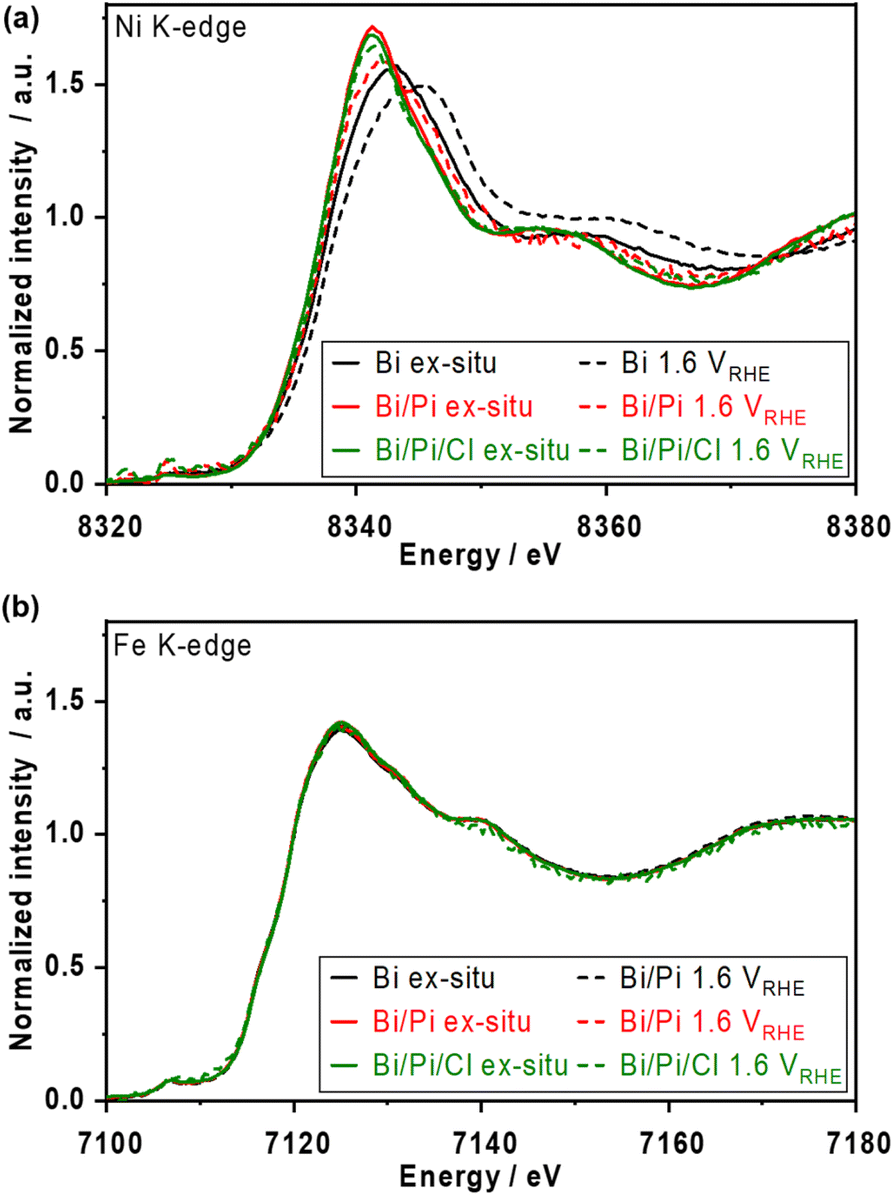 | ||
| Fig. 6 Ex situ and operando X-ray absorption spectroscopy (XAS) analysis over NiFeOx catalysts. X-ray absorption near edge structure (XANES) spectra of (a) Ni K-edge and (b) Fe K-edge. Operando measurements were conducted at 1.6 VRHE in each solution; 1.0 mol kg−1 K–borate (Bi), 1.0 mol kg−1 K–borate + 1.0 mol kg−1 K–phosphate (Bi/Pi), and 1.0 mol kg−1 K–borate + 1.0 mol kg−1 K–phosphate + 0.5 mol kg−1 K–Cl (Bi/Pi/Cl) at pH 9.2, RT. Ex situ represents the samples after CP at 10 mA cm−2 for 1 h in the corresponding electrolytes. | ||
Conclusions
Herein, we introduce the new concept of borate/phosphate mixed buffer electrolyte to stably and selectively electro-catalyze the OER over a NiFeOx/NF electrode in the presence of Cl−. The intensive electrochemical analyses employing various redox probes and ex situ/operando spectroscopic characterizations reveal the role of the borate and phosphate on OER performance. The borate worked as the dominant buffer to maintain the local pH shifting at pH 9.2 due to its closer pKa value. The roles of the phosphate are (mainly) as a stabilizer and (partially) buffer. The function of the charge repulsion layer was not observed under the non-extreme pH conditions investigated even though previous studies endorsed the charge repulsion theory at alkaline pH levels. Phosphate selectively forms a Ni-complex, which stabilized the Ni2+ and suppressed the Ni dissolution and was passive to Cl−, resulting in high electrocatalyst stability at the present non-extreme pH levels. The phosphate functioned as a buffer only when the borate molality was insufficient. The selectivity of redox ion oxidation reactions was dependent on their reaction thermodynamics and kinetics via the Fe site (e.g., the halide ion oxidation reaction) in the presence of the phosphate additive. The mixed buffer approach can be utilized at high j and under elevated temperature conditions in the presence of impurities. The present study demonstrates great potential to explore electrolyte additives and electrocatalysts concurrently for efficient and stable electrocatalysis.Experimental
Materials and chemicals
Ni foam, a Ni plate, and a Pt wire were purchased from Nilaco Corporation. Ni felt was purchased from Nikko Techno Corporation. Carbon paper 060 was purchased from the Fuel Cell Store. KCl-saturated Ag/AgCl and Hg/HgO (1 M NaOH) were purchased from BAS Inc. All chemicals were purchased with the following purities from Sigma-Aldrich unless otherwise specified: NaOH (99.99%), KOH (99.99%), boric acid (≥99.5%), phosphoric acid (≥85 wt%), potassium sulfate (≥99.0%), HCl (35–37%, FUJIFILM-Wako), HNO3 (≈60%, FUJIFILM-Wako), KBr (>99%, PIKE Technologies), KI (>99.5%, TCI), KCl (>99%), NaCl (≥99%), EtOH (≥99.5%), i-PrOH (99.7%, FUJIFILM-Wako), K4[Fe(CN)6]·3H2O (>98.5%), formic acid (≥98.0%), NaClO (effective chlorine > 5%, FUJIFILM-Wako), Ni(NO3)2·6H2O (98%, FUJIFILM-Wako), Fe(NO3)3·9H2O (99.9%, FUJIFILM-Wako), CO(NH2)2 (>99%), Ni standard solution (Ni 1000 ppm, FUJIFILM-Wako), and Fe standard solution (Fe 1000 ppm, FUJIFILM-Wako). The N,N-Diethyl-p-phenylenediamine (DPD) reagent was purchased from Kazusa Corporation. Ar (99.9999%) and O2 (99.99995%) were used. Ultrapure water was used for the preparation of all aqueous solutions.Electrolyte preparation
First, the substances were dissolved in ultrapure water to yield the desired molality (mol kg−1). Then, the pH was adjusted by adding KOH pellets until the solution pH reached the target value.Electrode preparation: NiFeOx/Ni substrates and carbon paper
A precursor solution was prepared from 569.9 mg of Ni(NO3)2·6H2O, 759.5 mg of Fe(NO3)3·9H2O, and 570.6 mg of CO(NH2)2 mixed in 152 mL of Milli-Q water with stirring at room temperature.14 Then, the solution was transferred to a 190 mL Teflon-lined stainless-steel autoclave, where nickel substrates were cleaned with EtOH. To deposit on the Ni plate, the plate was washed with undiluted HCl until bubbles evolved from the surface. To deposit on the carbon paper, the substrate was washed with undiluted HNO3 for more than 1 hour. A sealed autoclave was moved to an oven and heated at 393 K for 12 h. After hydrothermal synthesis, the prepared electrode was rinsed with plenty of water and dried in the atmosphere. The double-layer capacitances (Cdl) values calculated by CV of NiFeOx/Ni foam, NiFeOx/Ni felt, and the NiFeOx/Ni plate were 0.876, 0.166, and 0.091 mF cm−2, respectively (see Fig. S4 and S24†). The NiFeOx/Ni foam was used to compare the OER performances in various electrolytes. The others were used for specific characterization (e.g., XPS and cross-sectional SEM).Electrochemical measurement
Electrochemical measurements were conducted by using three-electrode systems. A Pt wire was employed as a counter electrode. Ag/AgCl (saturated KCl) and Hg/HgO (1 M NaOH) reference electrodes were used as reference electrodes at mild pH and alkaline pH, respectively. The specified gas was supplied to the cell during measurements: O2 for the OER and Ar for Faradaic efficiency and Cdl measurements. All electrochemical measurements were performed using a 16-channel research-grade potentiostat system (VMP3; BioLogic Science Instruments). The reference electrodes were kept at 25 °C by using the long jacket (BAS Inc.), and water-jacketed glass cells (BAS Inc.) were heated with hot water for elevated temperature experiments. All working electrodes were sealed with PTFE tape to secure the effective geometric electrode area. All current–potential relationships described in this paper, except the specified explanation, were iR-corrected using the measured impedance values (≥100 kHz, amplitude 10 mV).Faradaic efficiency measurements
For the Faradaic efficiency of O2: we employed a one-chamber sealed cell. Ar gas was fed in one-way as a carrier gas. The cell was connected to a gas chromatograph (GC-8A; Shimadzu Co. Ltd.) equipped with a thermal conductivity detector (TCD) and a molecular sieve 5A column. The Faradaic efficiency of O2 was calculated based on the measured O2 generation rate in mol.For the Faradaic efficiency of hypochlorite: spent electrolytes were taken at 0.1 mL and diluted with water until 10 mL. Then, N,N-diethyl-p-phenylenediamine (DPD) powder was introduced. UV-Vis spectra were recorded within 3 min. The amount of hypochlorite was calculated based on the recorded intensity referencing the calibration line.
XAS measurements
All XAS measurements were conducted in a fluorescence mode with a 19-element Ge solid-state detector. First, for the ex situ measurements, the electrodes were fixed in a cell with a Kapton window. The electrolyte was then introduced into the cell, and operando measurements were performed. The cell is a plastic box with a wall thickness of 1 mm and a hole (window) of 1.5 × 1.0 cm (width × height). One or two pieces of Kapton tape (0.1 mm thick) were laminated together to create a transmissive window through which X-rays could reach the sample. The auxiliary and reference electrodes were fixed in appropriate positions with Kapton tape.Characterization
X-ray photoelectron spectroscopy (XPS) was employed with a JEOL JPS-9030 using Mg anode 300 W, 12 kV, 25 mA. The C 1s at 284.6 eV was used as an internal standard to calibrate all acquired data. Inductively coupled plasma (ICP) measurements were conducted with the Thermo iCAP PRO series. Ultraviolet-visible absorption spectroscopy (UV-Vis) spectra were recorded with a JASCO V-770. An Anton Paar SVM 2001 was employed to evaluate the solution viscosity. Scanning electron microscopy (SEM) and elemental line scan images were taken with a JEOL JSM IT-800 equipped with an ULTIM MAX. XAS measurements were conducted at the beamline BL14B2 at SPring-8, Hyogo, Japan. The energy was calibrated using a fraction of the K-edge step of the metal plate. E0 of Fe and Ni were set at 7112 and 8333 eV, respectively. The acquired data were analyzed using the software of Athena. The transmission electron microscope (TEM) image was captured with a JEM-2010HC JEOL.Author contributions
H. K. conducted most of the experiments and characterizations and wrote the first draft of the manuscript. K. O. supported the XAS measurements and wrote the manuscript with H. K. T. H. supervised the XAS measurements. K. T. obtained the funding and supervised the project. All the authors contributed to reviewing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the support from MEXT Program: Data Creation and Utilization-Type Material Research and Development Project Grant Number JPMXP1122712807. XAS measurements were approved by the Japan Synchrotron Radiation Research Institute under proposal number 2023A1885. This work was also supported by the Mohammed bin Salman Center for Future Science and Technology for Saudi-Japan Vision 2030 at The University of Tokyo (MbSC2030), the Science and Technology Research Partnership for Sustainable Development (SATREPS) in collaboration between the Japan Science and Technology Agency (JST, JPMJSA2104) and Japan International Cooperation Agency (JICA), and JSPS KAKENHI Grant Number 19KK0126.References
- M. Görlin, P. Chernev, J. Ferreira de Araújo, T. Reier, S. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- Y.-F. Li and A. Selloni, ACS Catal., 2014, 4, 1148–1153 CrossRef CAS.
- H. Ali-Löytty, M. W. Louie, M. R. Singh, L. Li, H. G. Sanchez Casalongue, H. Ogasawara, E. J. Crumlin, Z. Liu, A. T. Bell, A. Nilsson and D. Friebel, J. Phys. Chem. C, 2016, 120, 2247–2253 CrossRef.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- J. Y. C. Chen, L. Dang, H. Liang, W. Bi, J. B. Gerken, S. Jin, E. E. Alp and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 15090–15093 CrossRef CAS PubMed.
- H.-J. Liu, R.-N. Luan, L.-Y. Li, R.-Q. Lv, Y.-M. Chai and B. Dong, Chem. Eng. J., 2023, 461, 141714 CrossRef CAS.
- Y.-N. Zhou, F.-L. Wang, S.-Y. Dou, Z.-N. Shi, B. Dong, W.-L. Yu, H.-Y. Zhao, F.-G. Wang, J.-F. Yu and Y.-M. Chai, Chem. Eng. J., 2022, 427, 131643 CrossRef CAS.
- Y.-N. Zhou, W.-L. Yu, Y.-N. Cao, J. Zhao, B. Dong, Y. Ma, F.-L. Wang, R.-Y. Fan, Y.-L. Zhou and Y.-M. Chai, Appl. Catal., B, 2021, 292, 120150 CrossRef CAS.
- R.-Y. Fan, H.-Y. Zhao, Z.-Y. Zhao, W.-H. Hu, X. Liu, J.-F. Yu, H. Hu, Y.-M. Chai and B. Dong, Nano Res., 2023, 16, 12026–12034 CrossRef CAS.
- R.-Y. Fan, J.-Y. Xie, H.-J. Liu, H.-Y. Wang, M.-X. Li, N. Yu, R.-N. Luan, Y.-M. Chai and B. Dong, Chem. Eng. J., 2022, 431, 134040 CrossRef CAS.
- K. Obata and K. Takanabe, Angew. Chem., Int. Ed., 2018, 57, 1616–1620 CrossRef CAS PubMed.
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS PubMed.
- H. Komiya, T. Shinagawa and K. Takanabe, ChemSusChem, 2022, 15, e202201088 CrossRef CAS PubMed.
- J. G. Vos, T. A. Wezendonk, A. W. Jeremiasse and M. T. Koper, J. Am. Chem. Soc., 2018, 140, 10270–10281 CrossRef CAS PubMed.
- T. Okada, H. Abe, A. Murakami, T. Shimizu, K. Fujii, T. Wakabayashi and M. Nakayama, Langmuir, 2020, 36, 5227–5235 CrossRef CAS PubMed.
- K. Izumiya, E. Akiyama, H. Habazaki, N. Kumagai, A. Kawashima and K. Hashimoto, Electrochim. Acta, 1998, 43, 3303–3312 CrossRef CAS.
- K. Fujimura, K. Izumiya, A. Kawashima, E. Akiyama, H. Habazaki, N. Kumagai and K. Hashimoto, J. Appl. Electrochem., 1999, 29, 765–771 CrossRef CAS.
- N. A. Abdel Ghany, N. Kumagai, S. Meguro, K. Asami and K. Hashimoto, Electrochim. Acta, 2002, 48, 21–28 CrossRef CAS.
- Z. Wang, C. Wang, L. Ye, X. Liu, L. Xin, Y. Yang, L. Wang, W. Hou, Y. Wen and T. Zhan, Inorg. Chem., 2022, 61, 15256–15265 CrossRef CAS PubMed.
- T. Ma, W. Xu, B. Li, X. Chen, J. Zhao, S. Wan, K. Jiang, S. Zhang, Z. Wang, Z. Tian, Z. Lu and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 22740–22744 CrossRef CAS PubMed.
- M. Yu, J. Li, F. Liu, J. Liu, W. Xu, H. Hu, X. Chen, W. Wang and F. Cheng, J. Energy Chem., 2022, 72, 361–369 CrossRef CAS.
- S. Mandal, J. K. Singh, D.-E. Lee and T. Park, Materials, 2020, 13, 3642 CrossRef CAS PubMed.
- V. A. Alves and C. M. A. Brett, Electrochim. Acta, 2002, 47, 2081–2091 CrossRef CAS.
- K. Zhao, Y. Tao, L. Fu, C. Li and B. Xu, Angew. Chem., Int. Ed., 2023, 62, e202308335 CrossRef CAS PubMed.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, National Association of Corrosion Engineers, Houston, Texas, 2nd edn, 1974 Search PubMed.
- E. M. Kapp, Biol. Bull., 1928, 55, 453–458 CrossRef CAS.
- H. Komiya, K. Obata, M. Wada, T. Nishimoto and K. Takanabe, ACS Sustain. Chem. Eng., 2023, 11, 12614–12622 CrossRef CAS.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- E. Nurlaela, T. Shinagawa, M. Qureshi, D. S. Dhawale and K. Takanabe, ACS Catal., 2016, 6, 1713–1722 CrossRef CAS.
- T. Shinagawa, M. T. Ng and K. Takanabe, Angew. Chem., Int. Ed., 2017, 56, 5061–5065 CrossRef CAS PubMed.
- T. Nishimoto, T. Shinagawa, T. Naito and K. Takanabe, ChemSusChem, 2021, 14, 1554–1564 CrossRef CAS PubMed.
- T. Nishimoto, T. Shinagawa, T. Naito, K. Harada, M. Yoshida and K. Takanabe, ChemSusChem, 2023, 16, e202201808 CrossRef CAS PubMed.
- F. Dionigi, Z. Zeng, I. Sinev, T. Merzdorf, S. Deshpande, M. B. Lopez, S. Kunze, I. Zegkinoglou, H. Sarodnik, D. Fan, A. Bergmann, J. Drnec, J. F. de Araujo, M. Gliech, D. Teschner, J. Zhu, W.-X. Li, J. Greeley, B. R. Cuenya and P. Strasser, Nat. Commun., 2020, 11, 2522 CrossRef CAS PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- C. D. Young, P. P. Lopes, F. B. D. M. Pedro, H. He, K. Tomoya, P. Zapol, Y. Hoydoo, T. Dusan, S. Dusan, Z. Yisi, S. Soenke, S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 222–230 CrossRef.
- W. M. Haynes and D. R. Lide, Handbook of Chemistry and Physics, CRC Press, 92nd edn, 2011 Search PubMed.
- NIST X-ray Photoelectron Spectroscopy Database, Version 4.1, National Institute of Standards and Technology, Gaithersburg, 2012, https://srdata.nist.gov/xps/, accessed April 3, 2022 Search PubMed.
- F. Bao, E. Kemppainen, I. Dorbandt, F. Xi, R. Bors, N. Maticiuc, R. Wenisch, R. Bagacki, C. Schary, U. Michalczik, P. Bogdanoff, I. Lauermann, R. van de Krol, R. Schlatmann and S. Calnan, ACS Catal., 2021, 11, 10537–10552 CrossRef CAS.
- C. Kuai, Z. Xu, C. Xi, A. Hu, Z. Yang, Y. Zhang, C.-J. Sun, L. Li, D. Sokaras, C. Dong, S.-Z. Qiao, X.-W. Du and F. Lin, Nat. Catal., 2020, 3, 743–753 CrossRef CAS.
- M. Görlin, M. Gliech, J. F. de Araújo, S. Dresp, A. Bergmann and P. Strasser, Catal. Today, 2016, 262, 65–73 CrossRef.
- W. Liu, A. Migdisov and A. Williams-Jones, Geochim. Cosmochim. Acta, 2012, 94, 276–290 CrossRef CAS.
- C. CHRISTOV, J. Chem. Thermodyn., 2003, 35, 1775–1792 CrossRef CAS.
- D. Martín-Yerga, G. Henriksson and A. Cornell, Electrocatalysis, 2019, 10, 489–498 CrossRef.
- A. Zahl, R. van Eldik and T. W. Swaddle, Inorg. Chem., 2002, 41, 757–764 CrossRef CAS PubMed.
- S. Dresp, T. Ngo Thanh, M. Klingenhof, S. Brückner, P. Hauke and P. Strasser, Energy Environ. Sci., 2020, 13, 1725–1729 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05566c |
| This journal is © The Royal Society of Chemistry 2024 |