
High-valent iron single-atom catalysts for improved overall water splitting via a reduced energy barrier and stabilization of the active center†
Ayyavu
Shankar‡
,
Sundaramoorthy
Marimuthu‡
and
Govindhan
Maduraiveeran
 *
*
Materials Electrochemistry Laboratory, Department of Chemistry, Faculty of Engineering and Technology, SRM Institute of Science and Technology, Kattankulathur, Chengalpattu, 603 203, Tamil Nadu, India. E-mail: maduraig@srmist.edu.in
First published on 9th November 2023
Abstract
The design of earth-abundant and non-precious transition-metal-based single-atom catalysts (TM-SACs) for promoting the oxygen evolution reaction (OER) and hydrogen evolution reaction (HER) is of great importance for generating green hydrogen (H2). Herein, we demonstrate iron (Fe) single atoms stabilized on carbon–nickel nanosheets (C–Ni) (Fe-SACs|C–Ni NSs) using a facile and single-step metal–organic-framework (MOF)-assisted electrochemical approach. Single-atom iron sites (Fe4+ active center), generated in situ on the C–Ni heterostructure, assist in regulating the binding abilities of hydroxyl ions (OH*) and hydrogen (H*) to accelerate alkaline water splitting reactions. Overpotentials (η) of ∼246 mV and ∼164 mV are required to reach ∼10 mA cm−2 for the OER and HER, respectively, outperforming other recently reported bifunctional catalysts.
1. Introduction
To develop sustainable energy sources, one of the laborious problems for the upcoming decades is how to provide sufficient power for the world. A flourishing alternative for fulfilling the demands for sustainable energy and protecting the environment is electrochemical energy conversion, with technologies including green hydrogen (H2) cycling and beyond.1,2 Hydrogen possesses the highest gravimetric energy density of 120 MJ kg−1 and contains no carbon (C), so is considered a promising renewable candidate to replace fossil fuels.3 Electrochemical water splitting is the most efficient, clean, and environmentally safe strategy for producing green hydrogen among numerous available approaches.4,5 High electrolytic overpotential is a limitation of commercial water electrolysis, which requires a high cell voltage (1.8–2.0 V). The design of efficient electrocatalytic materials is a key to lowering energy consumption.6 To increase the specific activities of the underlying electrocatalysts, one needs to consider improving the surface/interface reactive properties, reducing the size of the electrocatalysts to increase the accessible number of active sites, and stabilization of the active centres.7,8 Despite the substantial advancements made with non-precious catalysts,9,10 platinum (Pt)-, iridium (Ir)- and ruthenium (Ru)-based materials are considered to be the most effective catalysts for electrochemical green-hydrogen generation.11–13 However, the high price and scarcity of state-of-the-art precious-metal-based catalysts hamper their widespread use in electrolyzers for the production of H2.In recent years, numerous alternative non-precious-metal (Fe, Co, Mn, Ni, Cu, etc.)-based catalysts have been investigated and found to have improved catalytic activity and durability.14,15 However, substantial progress in the establishment of bifunctional OER/HER catalysts with enhanced intrinsic activity is still required. In this regard, atomic-scale dispersion of catalysts has drawn remarkable research interest for use in efficient green hydrogen-production systems.16,17 Recent studies have shown that single-atom catalysts (SACs) with high atom utilization efficiency, and tunable coordination and electronic structure deliver boosted bifunctional catalytic OER/HER activity.18–20 It is reported that the metal–support interaction is greater than the metal–metal interaction in the stabilization of SACs with high atomic dispersity.21,22 As a result, it is crucial to build powerful interactions between single metal atoms and the nearby coordination atoms in the support in order to counteract aggregation tendencies.
Generally, the preparation strategies include chemical vapor deposition, atomic layer deposition, etc., for stable SACs.23,24 Nevertheless, these approaches typically have distinct requirements for either the anchored metals or the supports. For example, the pyrolysis approach usually produces carbon-based supports, due to the decomposition of metal–organic complexes.25,26 Such synthesis processes necessitate large amounts of compounds, as well as particular activation techniques (such as solvent extraction and heating), which can take several days.27 In particular, electrically insulating and traditional binders (such as Nafion, poly(vinylidene fluoride), etc.) may impair electrode conductivity and partially block the metal–organic-framework (MOF) particles and active sites, resulting in decreased electrocatalytic performances and increased fabrication costs.28 Strikingly, the coated catalysts may peel off during electrochemical reaction over an extended time period.29,30
In this context, the fabrication of self-standing transition-metal-based active SACs, directly grown on electrodes, delivers encouraging prospects for overall water splitting to attain large current densities over long-term industrial application.31 In general, constructing anion vacancies/cation defects is the strategy typically implemented to prepare SACs with improved OER/HER activity.32 Zhang et al. prepared single Pt atoms immobilized in MXene nanosheets (Mo2TiC2Tx) and onion-shaped carbon-nanosphere supports, which significantly lowered the H-adsorption energy (ΔGH) and allowed the release of the H2 molecule.33 Recently, Zeng and co-workers prepared Fe-SACs on Fe1(OH)x/P–C for improved OER.34 The nucleophilic attack of OH− towards the *O intermediate to create *OOH was encouraged by the high-valent Fe4+ centres with optimum eg orbital filling, decreasing the energy barrier of the rate-determining step.
Despite great advancements in constructing catalysts, it is still challenging to reduce the energy barrier for binding of multiple hydroxyl ions (OH*) and hydrogen (H*) at the active sites of SACs. Consequently, it is essential to discover a single-step strategy for fabricating high-density transition-metal-derived SACs with durable catalytic activity for practical hydrogen generation systems. In this work, we demonstrate earth-abundant iron single-atoms stabilized on a carbon–nickel nanosheet (Fe-SACs|C–Ni NS) electrode using a single-step MOF-assisted electrochemical strategy. It is anticipated that the high intrinsic OER/HER catalytic activity of the Fe-SACs|C–Ni NS electrode would be initiated by the high-valent Fe4+ centres through Fe–C–Ni bonding during the electrocatalytic process, facilitating the water-splitting reaction.
2. Results and discussion
A series of nanostructured Fe-SACs catalysts on carbon modified nickel nanosheets (C–Ni NS) were prepared using the electrochemical deposition strategy. Typically, cyclic voltammograms (CVs) were performed for the bare NF in a precursor mixture of different Fe concentrations (0.25(@1), 0.50(@2), 1.0(@3), 1.25(@4), 1.50(@5)) mM in N,N-dimethylformamide (DMF) for five incessant cycles with low scan rate (5.0 mV s−1) in the range of ∼−0.16 to ∼1.63 V against RHE (Scheme S1†). The as-developed Fe-SACs|C–Ni electrodes were represented as Fe-SACs|C–Ni@1, Fe-SACs|C–Ni@2, Fe-SACs|C–Ni@3, Fe-SACs|C–Ni@4 and Fe-SACs|C–Ni@5, respectively (see the ESI†). Fig. 1(a) shows the scanning electron microscopy (SEM) image of the Fe-SACs|C–Ni@3 electrode, presenting a homogeneous dispersion of 3D structures (Fig. S1†). As can be easily seen in Fig. 1(b) and (c), there is a high dispersion of isolated Fe single atoms. The as-grown nanosheet-like C–Ni electrode shows a homogeneous dispersion of Ni, C, and O elements, as displayed in Fig. S2 and S3.† Intriguingly, Fe single atoms have been potentiostatically deposited on the C–Ni electrode for the first-time using a MOF-assisted strategy (Fig. S4†). The pro-base Et3NHCl was primarily reduced to Et3N and H2, which generated a pH buffering pair of Et3NH+/Et3N, allowing the electrode surface pH to be adjusted and facilitating the deprotonation of benzenetricarboxylic acid (H3BTC).35 Meanwhile, Fe3+ was reduced to Fe2+, which could couple with BTC3− to form a MOF, and was deposited as Fe single-atoms on the C–Ni electrode (Scheme S1†). The elemental mapping (Fig. 1(d)) and energy-dispersive X-ray (EDX) studies (Fig. 1(e)) revealed a uniform dispersion of Fe, C, and O elements across the electrode. A high dispersion of Fe single-atoms on the C–Ni support is also revealed. Additionally, the Fe-SACs|C–Ni@3 catalyst exhibited higher Fe loading on the C–Ni support (6.7 μg mL−1) when compared to other catalysts developed in this study, according to inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis, as shown in Table S1.† As depicted in Fig. S5,† the X-ray diffraction (XRD) pattern shows no characteristic Fe peaks, suggesting the absence of Fe clusters and particles in Fe-SACs|C–Ni@3. However, it can been seen that there is a broad XRD peak at ∼26.8° (2θ), corresponding to the (002) plane of carbon.36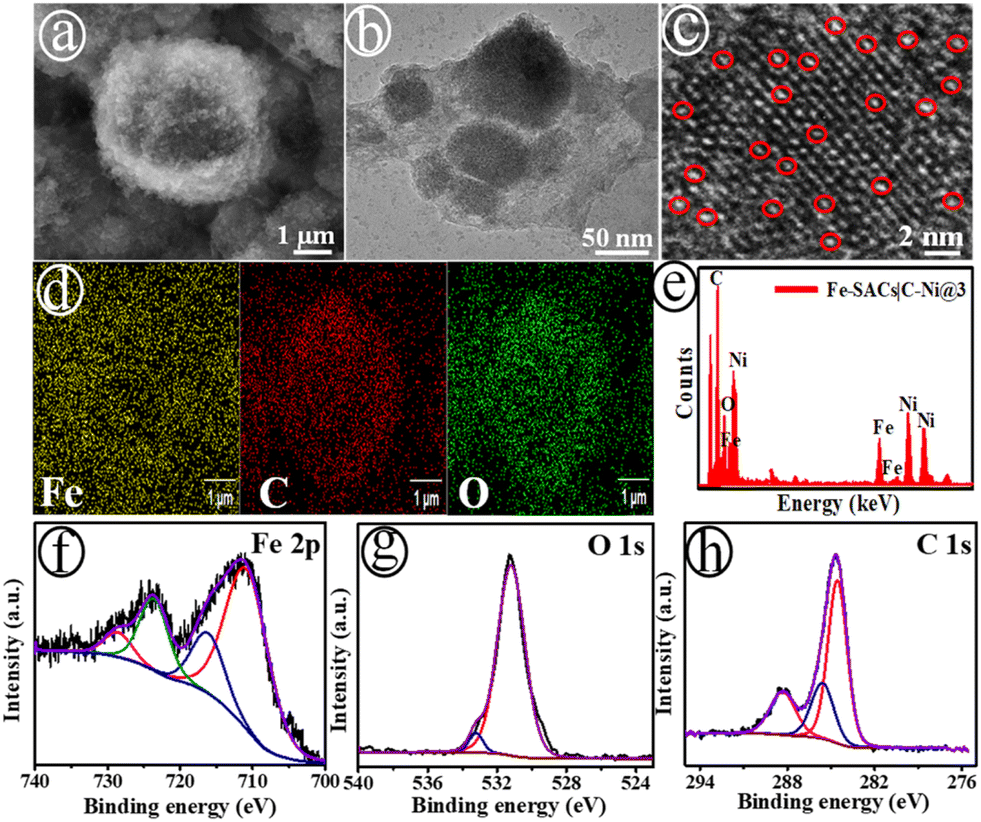 | ||
| Fig. 1 SEM (a), transmission electron microscopy (TEM) (b) and high-resolution TEM (HRTEM) (c) images; inset: the red circles indicates the distribution of the Fe single-atoms over the C–Ni nanosheets, elemental mapping (d), and EDX spectrum (e) of the Fe-SACs|C–Ni@3 electrode; detailed Fe 2p (f), O 1s (g), and C 1s (h) XPS spectra of the Fe-SACs|C–Ni@3 electrode. | ||
In the Fe 2p XPS spectrum (Fig. 1(f)), the major peaks at ∼716.0 eV and ∼728.8 eV are characteristic of Fe 2p3/2 and Fe 2p1/2 core-level electrons, attributed to the Fe4+ octahedral site. The XPS peaks at ∼710.8 eV and ∼723.7 eV confirm the presence of Fe3+ species in the octahedral sites, which is attributed to the carboxylate–iron bond.37,38 Slight peak shifts of 0.8 eV and 0.7 eV towards high binding energy transpired at ∼710.8 eV and ∼723.7 eV, respectively, relative to the peaks for metallic Fe, ascribed to the bonding of Fe with O ions or due to Fe and COO−.39,40 As displayed in Fig. 1(g), O 1s peaks appeared at ∼529.9, ∼531.7 and ∼533.2 eV, corresponding to Fe–O, C–O and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the carboxylate part of the ligand in Fe-SACs|C–Ni@3.40 The deconvoluted C 1s spectrum for the single Fe-sites at the C–Ni heterointerfaces exhibited three major XPS peaks at ∼284.5 eV (C–C), ∼285.4 eV (C–C/C–H), and ∼288.4 eV (O–CO) for the carboxylate group in H3BTC (Fig. 1(h)), suggesting good incorporation of Fe4+.41 The atomic ratio of Fe
O of the carboxylate part of the ligand in Fe-SACs|C–Ni@3.40 The deconvoluted C 1s spectrum for the single Fe-sites at the C–Ni heterointerfaces exhibited three major XPS peaks at ∼284.5 eV (C–C), ∼285.4 eV (C–C/C–H), and ∼288.4 eV (O–CO) for the carboxylate group in H3BTC (Fig. 1(h)), suggesting good incorporation of Fe4+.41 The atomic ratio of Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C:O was found to be 0.1:0.5:0.3 for the Fe-SACs|C–Ni@3 electrode based on the XPS results (Fig. S6†).
:C:O was found to be 0.1:0.5:0.3 for the Fe-SACs|C–Ni@3 electrode based on the XPS results (Fig. S6†).
The FT-IR spectrum of the Fe-SACs|C–Ni@3 electrode showed a strong peak at ∼1612 cm−1, indicating the deprotonation and coordination of the carboxylate groups of BTC (Fig. S7†).42 The Raman spectrum of the Fe-SACs|C–Ni@3 (red curve in Fig. S8†) demonstrated a brand-new double peak at ∼1608 and ∼1417 cm−1, assigned to the in- and out-of-phase stretching modes of the carboxylate group.43 The ultraviolet-visible (UV-vis) adsorption spectra were recorded for revealing the formation process of a single-atom structure. As can be seen in Fig. S9,† a prominent UV-vis peak appeared at ∼288 nm for 1.0 mM FeCl3 (FeCl3 in DMF and H2O), corresponding to Fe3+ ions. In contrast, the peak at ∼288 nm disappeared in 1.0 M KOH containing 1.0 mM FeCl3, while a new peak appeared at ∼358 nm, due to the formation of a hydroxyl–iron species.34
Fig. 2(a) shows the linear sweep voltammetry (LSV) curves of the various Fe-SACs|C–Ni electrodes developed in this study, recorded in 1.0 M KOH. The Fe-SACs|C–Ni@3 electrode exhibited a pair of redox peaks for Fe3+/4+ at ∼1.35 V.44 As displayed in Fig. S10,† the plot of the anodic and cathodic peak current densities (j) against the square roots of the scan rates showed a linear line, suggesting diffusion-controlled electrode processes. Among the other electrodes developed in this study, the Fe-SACs|NiO@3 electrode demonstrated the lowest overpotential (ηOER) of ∼246 mV at 10 mA cm−2 in 1.0 M KOH, as shown in Fig. S11.† In contrast, Fe-SACs|C–Ni@1 (∼285 mV), Fe-SACs|C–Ni@2 (∼311 mV), Fe-SACs|C–Ni@4 (∼320 mV), Fe-SACs|C–Ni@5 (∼349 mV) and commercial RuO2 (∼309 mV) required higher overpotentials (ηOER) to drive ∼10.0 mA cm−2 (Fig. 2(a)). As presented in Fig. 2(b), the Fe-SACs|C–Ni@3 electrode exhibited the smallest Tafel slope of 58.0 mV dec−1, which is lower than those of Fe-SACs|C–Ni@1 (∼71.0 mV dec−1), Fe-SACs|C–Ni@2 (∼94.0 mV dec−1), Fe-SACs|C–Ni@4 (∼70.0 mV dec−1), Fe-SACs|C–Ni@5 (∼91.0 mV dec−1) and commercial RuO2 (∼101.0 mV dec−1) electrodes.
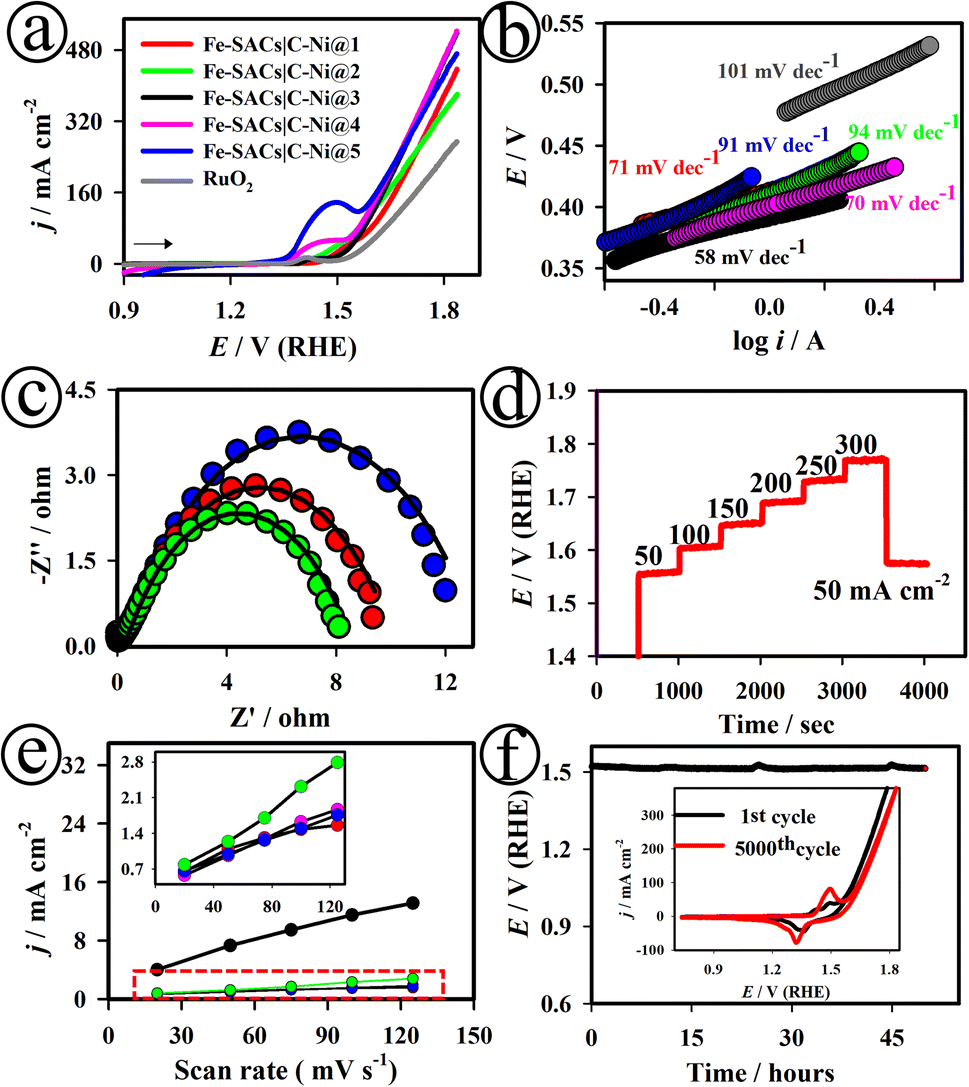 | ||
| Fig. 2 LSV curves (a) and Tafel plots (b) of the various as-developed Fe-SACs|C–Ni electrodes recorded in 1.0 M KOH; Nyquist plots of the Fe-SACs|C–Ni@3 electrode at Eapp values of 1.48 V (blue curve), 1.49 V (red curve) and 1.51 V (green curve) (c); sequential chronopotentiometric measurements of the Fe-SACs|NiO@3 electrode in 1.0 M KOH (d); plots of the anodic current density of Cdlvs. scan rates of the numerous as-developed Fe-SACs|C–Ni electrodes (e); controlled current electrolysis test of the Fe-SACs|C–Ni@3 electrode for the OER at Eapp = 1.48 V (f). Inset in (f): CV curves of Fe-SACs|C–Ni@3 recorded for over 5000 continuous cycles at a scan rate of 50 mV s−1. | ||
Differential pulse voltammetry (DPV) measurements were performed in order to get an insight into the detailed OER at the Fe-SACs|C–Ni@3 electrode (Fig. S12†). Two oxidation peaks were discerned at −0.13 and 0.75 V vs. RHE, which can be assigned to Fe2+/Fe3+ and Fe3+/Fe4+ redox shuttles, respectively, at the Fe-SACs|C–Ni@3 electrode. The as-made Fe-SACs|C–Ni@3 electrode exhibits a high mass loading of Fe and a greater propensity for electron transport. The observed lower positive potential of the Fe2+/Fe3+ and Fe3+/Fe4+ at the Fe-SACs|C–Ni@3 electrode than that of the OER equilibrium potential (1.23 V), revealing that Fe4+ essentially performed as the active center during the OER activity (Fig. S12†). It is predicted that with the in situ formation of electrophilic Fe4+ active centers, the energy barrier for the rate-determining formation of Fe4+–*OOH may be lowered. It is deduced that the transformation of Fe3+ to Fe4+ under an applied potential is largely because of the strong interaction between the Fe center and the Ni–C support through Ni–C–O–Fe bonding (Fig. S12†). The high valent Fe4+ (t2g3eg1) provides more appropriate adsorption energies for reaction intermediates, improving the OER activity. The deprotonation of an OH group coordinated with Fe4+ to form Fe5+–O* was the primary OER step.34 Another OH− ion integrated with O* to form Fe4+–*OOH via nucleophilic attack, followed by the deprotonation of *OOH to release O2 gas (Scheme S2†). The rate determining step (RDS) was the O–O coupling to form *OOH from *O. The present Fe4+ center increased the electrophilicity of the *O intermediate, favouring the formation of *OOH. Thus, the energy barrier of the rate determining conversion of *O to *OOH may be reduced.
For control studies, carbon–nickel (C–Ni) nanosheets were prepared under similar experimental conditions in the absence of Fe single atoms (see details in the Experimental section, ESI†). As displayed in Fig. S13,† the C–Ni|NF electrode exhibited a pair of redox peaks in the range of ∼1.35 to ∼1.45 V (vs. RHE), ascribed to the Ni2+/3+ couple.45 The plot of anodic and cathodic peak current densities (j) against the square roots of the scan rates showed a linear line, suggesting diffusion-controlled electrode processes. The LSV curve of the C–Ni nanosheet electrode exhibited poor OER activity (jOER = 10 mA cm−2, ∼351 mV), and high charge transfer resistance (Rct) in comparison to the Fe-SACs|C–Ni@3 electrode, as shown in Fig. S14 and S15.† Furthermore, electrodes based on various single-metal-atom catalysts, namely Co-SACs|C–Ni, Ni-SACs|C–Ni and Cu-SACs|C–Ni, were prepared under similar experimental conditions, and electrocatalytic OER measurements were conducted (Fig. S16†). As shown in Fig. S16(a),† the as-developed single-metal-atom-derived electrodes exhibited enhanced reversible electrochemical characteristics of the M2+/M3+ couple in the potential range of 1.05–1.55 V (RHE). According to Fig. S16(b),† the OER overpotential values were calculated at 10 mA cm−2 to be ∼336 mV, ∼354 mV, and ∼366 mV for Co-SACs|C–Ni, Ni-SACs|C–Ni and Cu-SACs|C–Ni, respectively.
Fig. 2(c), S17 and S18† show the Nyquist plots of the Fe-SACs|C–Ni@1, Fe-SACs|C–Ni@2, Fe-SACs|C–Ni@3, Fe-SACs|C–Ni@4, and Fe-SACs|C–Ni@5 electrodes at different Eapp values of 1.48, 1.49 and 1.51 V (RHE). As presented in Table S2,† the Fe-SACs|C–Ni@3 electrode exhibited low charge transfer resistance (Rct) values in the range of ∼9.8–84.1 Ω, solution resistance (Rs) values in the range of 3.4–5.4 Ω, and capacitance values in the range of 9.51–175 μF at various Eapp, suggesting the possession of improved electronic conductivity. As presented in Fig. 2(d) and S19,† the multi-step chronopotentiometric curve of the Fe-SACs|C–Ni@3 electrode suggested good mass transport, conductivity, and mechanical robustness. The electrochemically active surface areas (ECSAs) of the developed electrodes were examined by measuring the capacitive current densities at different scan rates, as depicted in Fig. 2(e). The ECSA values were ∼212, ∼487, ∼2160, ∼310, and ∼257 cm2 for Fe-SACs|C–Ni@1, Fe-SACs|C–Ni@2, Fe-SACs|C–Ni@3, Fe-SACs|C–Ni@4, and Fe-SACs|C–Ni@5, respectively. The number of active sites was first examined via CV measurements, as shown in Fig. S20.† The number of active sites on the catalyst Fe-SACs|C–Ni@3 was calculated to be 2.95 × 10−6 mol, which is ∼1.5 fold higher than that on the C–Ni catalyst alone.
The intrinsic catalytic activity of the catalysts is assessed using the turn over frequency (TOF) kinetic index. The TOF values were 0.03 × 10−2 s−1, 0.05 × 10−2 s−1, 0.15 × 10−2 s−1, 0.08 × 10−2 s−1, and 0.001 × 10−2 s−1 for Fe-SACs|C–Ni@1, Fe-SACs|C–Ni@2, Fe-SACs|C–Ni@3, Fe-SACs|C–Ni@4, and Fe-SACs|C–Ni@5, respectively. As depicted in Fig. 2(a), the Fe-SACs|C–Ni@4 and Fe-SACs|C–Ni@5 electrodes exhibited a lesser extent of catalytic OER activity when compared to the Fe-SACs|C–Ni@3 electrode due to the fact of decreased active sites on the aggregated nanostructures of the Fe-SACs|C–Ni@4 and Fe-SACs|C–Ni@5 electrodes (Fig. S21†). Thereby, Fe-SACs|C–Ni@4 and Fe-SACs|C–Ni@5 electrodes showed a decreased electrochemically active surface area, high onset and overpotentials, low charge transfer kinetics, high polarisation resistance and low turn-over frequency value when compared to the as-optimized Fe-SACs|C–Ni@3 electrode, which had a well-defined, uniform and optimal deposition of single Fe active sites. The value of mass activity was measured to be ∼0.50 A g−1 for the Fe-SACs|C–Ni@3 electrode.
In terms of practical use, long-term endurance is particularly significant, especially at ∼10 mA cm−2. After 50 h long-term OER catalytic activity, the electrode potential had stayed at ∼1.49 V (RHE), with no discernible rise, exhibiting the high resistance to corrosion of the Fe-SACs|C–Ni@3 electrode under intensely oxidising circumstances (Fig. 2(f)). As depicted in Fig. 2(f) (inset), the Fe-SACs|C–Ni@3 electrode exhibited an overpotential value that only increased by ∼5 mV at the current density of 10.0 mA cm−2 over 5000 potential cycles. Our electrochemical studies suggested that the Fe-SACs|C–Ni@3 electrode delivered a small overpotential and Tafel slope, high mass activity, large double layer capacitance (Cdl) and large number of active sites due to its synergistic transfer of OH− anions from the as-made Fe-MOF bulk into single Fe sites. It is speculated that the free BTC3− ligand and charged Fe complexes might have proximately escaped, leading to the formation of the metal oxyhydroxides nanosheets. Zhang and coworkers have experimentally proved the conversion of FeCo-MOF bulk to metal oxyhydroxide nanosheets for efficient OER.39 There are huge quantities of free charged molecules, such as the free BTTA2− ligand and potential Fe complexes over the surface and pores of the FeCo-MOF.
Fig. 3(a) and S22† show the LSV curves of the Fe-SACs|C–Ni@3, commercial PtC (10%) and C–Ni NS electrodes, recorded for the HER in 1.0 M KOH. The Fe-SACs|C–Ni@3 electrode showed a small overpotential (η10) of ∼164 mV at 10 mA cm−2 with an onset potential (ηonset) of ∼152 mV, which is very competitive with the commercial PtC (10%) electrode. Meanwhile, the C–Ni NS electrode showed a high overpotential (η10) of ∼276 mV at 10 mA cm−2 with an onset potential (ηonset) of ∼386 mV. As displayed in Fig. 3(b), the Tafel slope value was found to be ∼147 mV dec−1 for the Fe-SACs|C–Ni@3 electrode, revealing high electrode HER kinetics. Fig. 3(c) displays the Nyquist plots of the Fe-SACs|C–Ni@3 electrode at different applied potentials, which revealed small polarization resistance and high double layer capacitance values. The multi-step chronopotentiometry curve of the Fe-SACs|C–Ni@3 electrode was recorded in 1.0 M KOH with the current increasing from 50 to 300 mA cm−2 and is depicted in Fig. 3(d). This showed good mass transport, conductivity, and mechanical robustness during the HER.
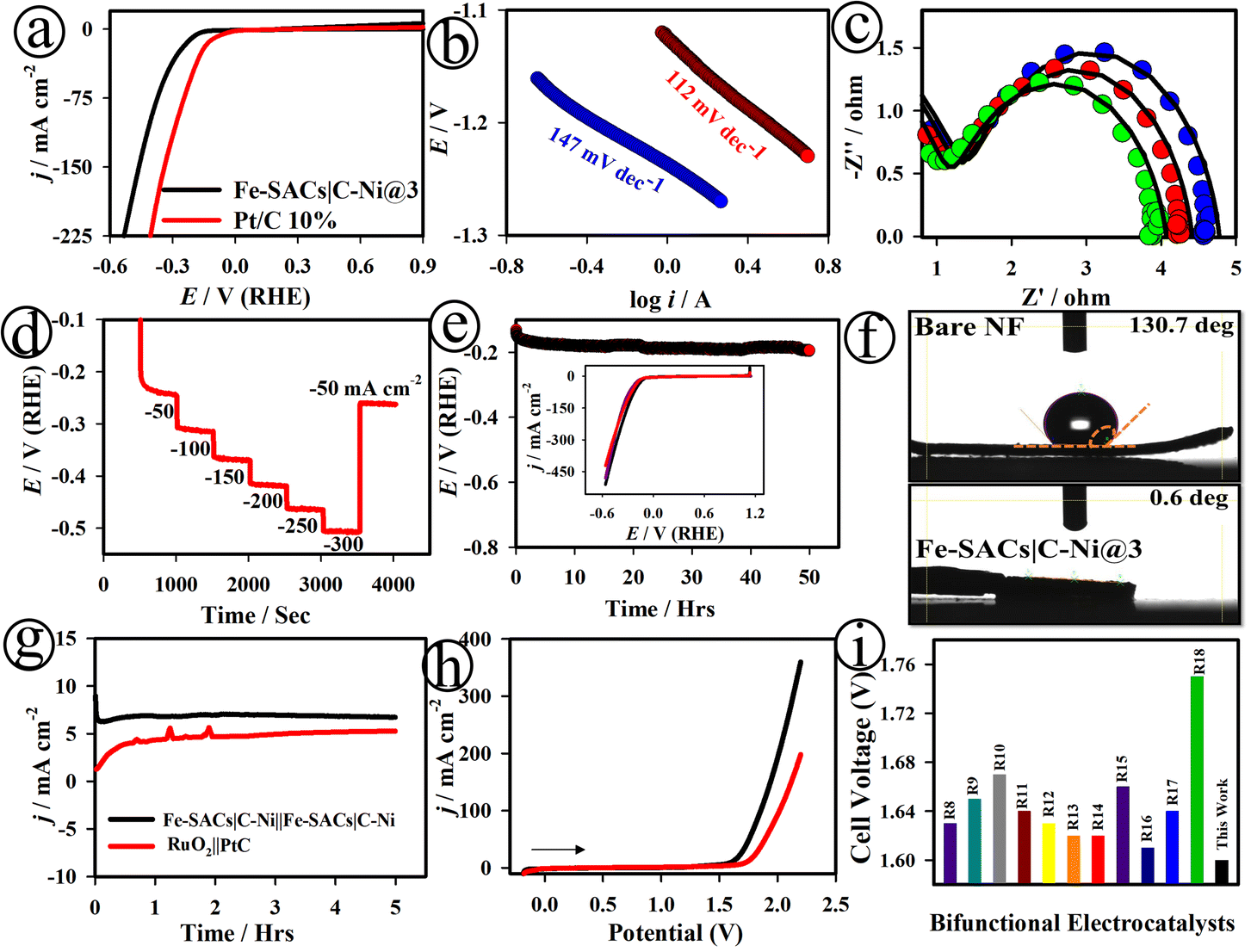 | ||
| Fig. 3 LSV curves (a) and Tafel plots (b) of the Fe-SACs|C–Ni@3 (black) and commercial PtC (10%) (red) electrodes, recorded in 1.0 M KOH. Nyquist plots at Eapp values of 1.48 V (blue curve), 1.49 V (red curve) and 1.51 V (green curve) (c), sequential chronopotentiometric measurements (d), and long-term stability tests (e) for the Fe-SACs|C–Ni@3 electrode; inset in (e): LSV curves of the Fe-SACs|C–Ni@3 nanosheets after the 1st, 2nd, and 4th days of HER tests in 1.0 M KOH solution. Contact angle measurement of the bare NF and Fe-SACs|C–Ni@3 electrodes (f). Chronoamperometry (g) and LSV curves of the Fe-SACs|C–Ni@3‖Fe-SACs|C–Ni@3 (black) and RuO2‖Pt/C (red) couples for overall water splitting (h); comparison of recently reported catalysts (see ESI (Table S3†)) in terms of the cell voltage (Ucell) at 10 mA cm−2 (i). | ||
Based on Fig. S23,† the number of active sites was calculated to be ∼1.67 × 10−6 mol for Fe-SACs|C–Ni@3.46 The TOF value was found to be 0.22 × 10−2 s−1 for the Fe-SACs|C–Ni@3 electrode, which is much greater than that of the other electrodes investigated, Fe-SACs|C–Ni@1 (0.02 × 10−2 s−1), Fe-SACs|C–Ni@2 (0.18 × 10−2 s−1), Fe-SACs|C–Ni@4 (0.12 × 10−2 s−1), and Fe-SACs|C–Ni@5 (0.01 × 10−2 s−1). The Fe-SACs|C–Ni@3 electrode exhibited good long-term electrochemical stability in 1.0 M KOH after undergoing continuous HER measurements (Fig. 3(e)). In addition, HER polarization curves were recorded for the 1st, 2nd, and 4th day of continuous measurements and it was found to maintain its overpotential at 10 mA cm−2 (Fig. 3(e) inset). The interatomic electronic interactions of the Fe–C–Ni species play a vital role in constructing highly active catalytic centers to accelerate the OER/HER reaction kinetics of the Fe-SACs|C–Ni@3 electrode, as depicted in Fig. S13, S14, S16 and S23.† For instance, Friebel et al. have provided evidence that the Fe3+–O bond is shortened during the catalytic process.49 The experimental strengthening of this Fe catalytic centre was shown using in situ Raman characterization with 18O labelling.47 Moreover, studies showed that Fe3+ might eventually develop into high-valent Fe4+ in the Fe4+O motif, adding to the high catalytic activity.48,49 The Fe-SACs|C–Ni@3 electrode promoted the interfacial adsorption of water molecules, and chemisorption of intermediates of *H on the surface of Fe-SACs|C–Ni@3 with the aid of Ni–C–O–Fe bonding. This provides high activity for the dissociation of water moieties and chemisorption of OH−, thereby obviously allowing the Fe-SACs|C–Ni@3 electrode to greatly enhance the rate-determining Volmer step in HER catalysis (Scheme S2†). The as-developed Fe-SACs|C–Ni@3 electrode exhibited a water contact angle of only ∼0.6°, as shown in Fig. 3(f), improving its hydrophilicity.
Based on the outstanding OER and HER catalytic activity of the Fe-SACs|C–Ni@3 electrode, it was used as a bifunctional electrode for assembling a two-electrode water electrolyzer (denoted as Fe-SACs|C–Ni@3‖Fe-SACs|C–Ni@3). For comparison, an alkaline water electrolyzer based on the state-of-the-art Pt/C‖RuO2 redox couple was also investigated. The bifunctional Fe-SACs|C–Ni@3‖Fe-SACs|C–Ni@3 pair attained a higher steady-state current density (∼7.9 mA cm−2), as well as a small cell voltage of ∼1.60 V (@10 mA cm−2) compared to the Pt/C‖RuO2 couple (Fig. 3(g) and (h)). The measured overpotential (η10) is ∼374 mV for Fe-SACs|C–Ni@3‖Fe-SACs|C–Ni@3, which was much lower than that of the PtC‖RuO2 couple (502 mV) as well as those of previously reported bifunctional catalysts, as can be seen in Fig. 3(i) and Table S3.†
Fig. 4 displays the SEM (a), TEM (b), and HR-TEM (c) images of the Fe-SACs|NiO@3 electrode after long-term stability tests. The XPS survey (d), Fe 2p (e), and O 1s (f) spectra for the fresh and recovered catalyst of the Fe-SACs|C–Ni@3 electrode are shown in Fig. 4. As shown in Fig. 4(a)–(c), S24 and S25,† the Fe-SACs|C–Ni@3 electrode presented a similar morphological structure, and retained the single-atom Fe sites and high surface wettability. As indicated above, it has been identified that there is a rapid transition of bulk Fe-MOF into single iron sites. Meanwhile, the Fe-MOF contains polar channels that may allow OH− anions to diffuse into its pores and attack its framework from inside. A significant amount of free charged molecules, such as free BTC3− ligand and possible Fe complexes, can be formed in situ on the surface and in the pores of the Fe-MOF during the process of fast hydrolysis.
Moreover, the XPS spectra confirm that the elemental composition did not change during the electric-field assisted hydrolysis, as shown in Fig. 4(d)–(f). In comparison with the spectrum prior to stability testing, the Fe 2p3/2 peak in the XPS spectrum shifted about 0.3 eV after 50 h of continuous testing of Fe-SACs|C–Ni@3 (Fig. 4(e)), revealing the high oxidation state of Fe with the formation of a metal–hydroxide surface. The peaks associated with the carboxylate functional groups in the C 1s spectra suggested that the H3BTC ligand was released from the surfaces of both Fe-MOFs. However, there were only XPS peaks attributed to Fe–O and Fe–OH in the O 1s spectra. The proportions of the oxygen-vacancy (OV) and metal–oxygen (Fe–O) peaks were significantly increased after activation of Fe-SACs|C–Ni@3 (Fig. 4(d) and (f)). Specifically, the proportional area of the OV peak increased from 17% to 22% during activation, while that of the Fe–O peak increased from 11% to 22%. These findings suggest that the activation process led to a substantial increase in the number of oxygen vacancies and Fe–O content at the Fe-SACs|C–Ni@3 electrode, improving water splitting. Furthermore, the oxidation state and electronic environment of the Fe sites also display changes after 50 hours of electrolysis. Based on the XPS tests, it was discovered that the Fe:C:O atomic ratio for the Fe-SACs|C–Ni@3 electrode was 0.08:0.69:0.27, which was similar to the ratio before OER measurements. The attained high surface wettability of the Fe-SACs|C–Ni@3 electrode may be attributed to its surface hydroxyl groups as well as the high roughness of the nanosheets, facilitating the swift adsorption of water and electrolyte infiltration into electrode.
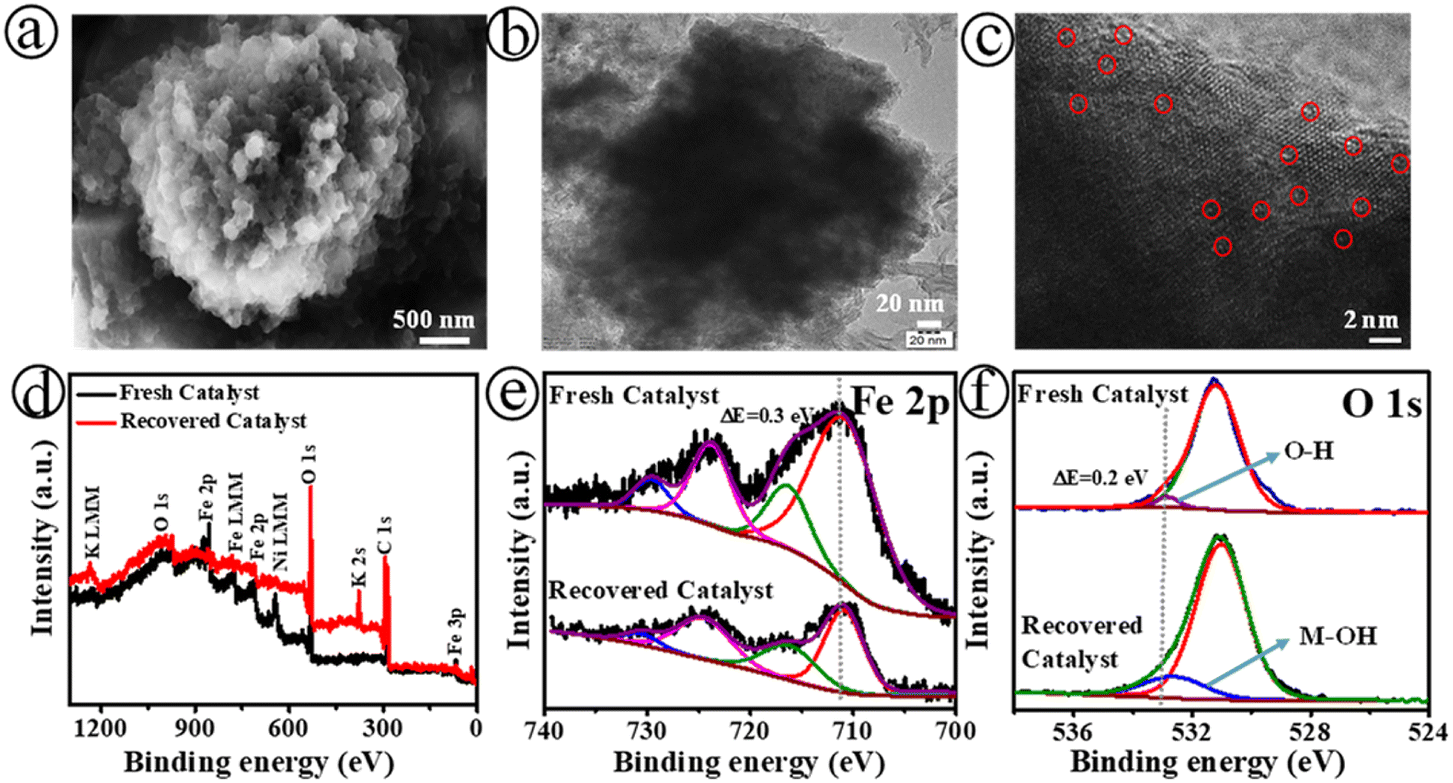 | ||
| Fig. 4 SEM (a), TEM (b), and HR-TEM (c) images of the Fe-SAC|NiO@3 electrode after a long-term stability test, (inset): the red circles indicates the distribution of the Fe single-atoms over the C–Ni nanosheets. XPS survey (d), Fe 2p (e) and O 1s (f) spectra for fresh and recovered catalyst of the Fe-SACs|C–Ni@3 electrode. | ||
3. Conclusions
Improving the utilization efficiency of metal atoms is an emerging approach for reducing the consumption of precious metals, which is of critical importance for decreasing the catalyst cost and promoting sustainability. Herein, we report a simple MOF-assisted electrodeposition method for the preparation of Fe single-atom catalysts over C–Ni heterostructure nanosheets that have effective electrocatalytic OER and HER activity, with low overpotentials of ∼246 and ∼164 mV at 10 mA cm−2, respectively. The electrocatalytic performance of the Fe-SACs|C–Ni@3 electrode was mainly ascribed to the synergistic characteristics of Fe single-atom sites on the C–Ni matrix, with a large volume of active sites, rapid charge transport properties, high-valent Fe4+ centers, electrophilicity, and a low energy barrier for the rate-limiting *OOH formation. The powerful carbon–nickel (C–Ni) support interaction with the Fe-SACs can be used to change the valence state of the metal, which may be useful for synthesis of other high-valent metal centers. Regulating the coordination environment of the metal atoms and optimizing the metal loading of SACs are highly desirable to attain a high-performance hydrogen production system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Central Power Research Institute (CPRI), Bangalore (Ref. CPRI/R&D/TC/GDEC/2022). The authors acknowledge the SRM Institute of Science and Technology (SRM IST) for providing all the research facilities, including the SRM-SCIF and NRC for SEM, TEM, and XPS measurements.Notes and references
- T. Gong, K. K. Rudman, B. Guo and A. S. Hall, Acc. Chem. Res., 2023, 56, 1373–1383 CrossRef CAS PubMed.
- G. Maduraiveeran, M. Sasidharan and W. Jin, Prog. Mater. Sci., 2019, 106, 100574 CrossRef CAS.
- H. Khani, A. R. P. Santiago and T. He, Angew. Chem., Int. Ed., 2023, 62, e202306103 CrossRef CAS PubMed.
- Y. Lv, K. Wang, W. Sun, P. Peng and S. Zang, Adv. Sci., 2023, 2304656, DOI:10.1002/advs.202304656.
- A. Shankar, S. Marimuthu and G. Maduraiveeran, Int. J. Hydrogen Energy, 2023, 48, 7683–7697 CrossRef CAS.
- Y. Yu, T. Wang, Y. Zhang, J. You, F. Hu and H. Zhang, Chem. Rec., 2023, 202300109, DOI:10.1002/tcr.202300109.
- C. Wang, Y. Kong, M. Soldemo, Z. Wu, H. Tissot, B. Karagoz, K. Marks, J. H. Stenlid, A. Shavorskiy, E. Kokkonen, S. Kaya, D. J. Stacchiola and J. Weissenrieder, Chem. Mater., 2022, 34, 2313–2320 CrossRef CAS.
- S. Marimuthu, A. Shankar and G. Maduraiveeran, Chem. Commun., 2023, 59, 2600–2603 RSC.
- C. Zhu, Q. Shi, S. Feng, D. Du and Y. Lin, ACS Energy Lett., 2018, 3, 1713–1721 CrossRef CAS.
- C. Zhu, Q. Shi, B. Z. Xu, S. Fu, G. Wan, C. Yang, S. Yao, J. Song, H. Zhou, D. Du, S. P. Beckman, D. Su and Y. Lin, Adv. Energy Mater., 2018, 8, 1801956 CrossRef.
- L. Liu, Y. Wang, Y. Zhao, Y. Wang, Z. Zhang, T. Wu, W. Qin, S. Liu, B. Jia, H. Wu, D. Zhang, X. Qu, M. Chhowalla and M. Qin, Adv. Funct. Mater., 2022, 32, 2112207 CrossRef CAS.
- Q. Wang, X. Huang, Z. L. Zhao, M. Wang, B. Xiang, J. Li, Z. Feng, H. Xu and M. Gu, J. Am. Chem. Soc., 2020, 142, 7425–7433 CrossRef CAS PubMed.
- P. Zhai, M. Xia, Y. Wu, G. Zhang, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, Z. Fan, C. Wang, X. Zhang, J. T. Miller, L. Sun and J. Hou, Nat. Commun., 2021, 12, 4587 CrossRef CAS PubMed.
- F. Abdelghafar, X. Xu, S. P. Jiang and Z. Shao, Mater. Rep.: Energy, 2022, 2, 100144 CAS.
- Y. Zhu, J. Wang and J. Ma, Small Sci., 2023, 3, 2300010 CrossRef CAS.
- H. Yang, L. Shang, Q. Zhang, R. Shi, G. I. N. Waterhouse, L. Gu and T. Zhang, Nat. Commun., 2019, 10, 4585 CrossRef PubMed.
- Y. Shi, W.-M. Huang, J. Li, Y. Zhou, Z.-Q. Li, Y.-C. Yin and X.-H. Xia, Nat. Commun., 2020, 11, 4558 CrossRef CAS PubMed.
- Z. Jin and A. J. Bard, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 12651–12656 CrossRef CAS PubMed.
- M. Zhou, J. E. Dick and A. J. Bard, J. Am. Chem. Soc., 2017, 139, 17677–17682 CrossRef CAS PubMed.
- N. Xuan, J. Chen, J. Shi, Y. Yue, P. Zhuang, K. Ba, Y. Sun, J. Shen, Y. Liu, B. Ge and Z. Sun, Chem. Mater., 2019, 31, 429–435 CrossRef CAS.
- F. Gong, Y. Liu, Y. Zhao, W. Liu, G. Zeng, G. Wang, Y. Zhang, L. Gong and J. Liu, Angew. Chem., Int. Ed., 2023, 62, e202308091 CrossRef CAS PubMed.
- D. Cao, Z. Zhang, Y. Cui, R. Zhang, L. Zhang, J. Zeng and D. Cheng, Angew. Chem., Int. Ed., 2023, 62, e202214259 CrossRef CAS PubMed.
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chem. Rev., 2020, 120, 11900–11955 CrossRef CAS PubMed.
- X. Mu, X. Gu, S. Dai, J. Chen, Y. Cui, Q. Chen, M. Yu, C. Chen, S. Liu and S. Mu, Energy Environ. Sci., 2022, 15, 4048–4057 RSC.
- A. Kumar, V. Q. Bui, J. Lee, L. Wang, A. R. Jadhav, X. Liu, X. Shao, Y. Liu, J. Yu, Y. Hwang, H. T. D. Bui, S. Ajmal, M. G. Kim, S.-G. Kim, G.-S. Park, Y. Kawazoe and H. Lee, Nat. Commun., 2021, 12, 6766 CrossRef CAS PubMed.
- T. Sun, L. Xu, D. Wang and Y. Li, Nano Res., 2019, 12, 2067–2080 CrossRef CAS.
- W. Wan, Y. Zhao, S. Wei, C. A. Triana, J. Li, A. Arcifa, C. S. Allen, R. Cao and G. R. Patzke, Nat. Commun., 2021, 12, 5589 CrossRef CAS PubMed.
- F. Xiao, G. L. Xu, C. J. Sun, M. Xu, W. Wen, Q. Wang, M. Gu, S. Zhu, Y. Li, Z. Wei, X. Pan, J. Wang, K. Amine and M. Shao, Nano Energy, 2019, 61, 60–68 CrossRef CAS.
- L. Yu, Y. Li and Y. Ruan, Angew. Chem., Int. Ed., 2021, 60, 25296–25301 CrossRef CAS PubMed.
- A. Zhang, M. Zhou, S. Liu, M. Chai and S. Jiang, Front. Catal., 2021, 1 DOI:10.3389/fctls.2021.754167.
- Z. Zhang, C. Feng, C. Liu, M. Zuo, L. Qin, X. Yan, Y. Xing, H. Li, R. Si, S. Zhou and J. Zeng, Nat. Commun., 2020, 11, 1215 CrossRef CAS PubMed.
- J. Zhang, X. Wu, W.-C. Cheong, W. Chen, R. Lin, J. Li, L. Zheng, W. Yan, L. Gu, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 1002 CrossRef PubMed.
- J. Zhang, Y. Zhao, X. Guo, C. Chen, C.-L. Dong, R.-S. Liu, C.-P. Han, Y. Li, Y. Gogotsi and G. Wang, Nat. Catal., 2018, 1, 985–992 CrossRef CAS.
- Z. Zhang, C. Feng, X. Li, C. Liu, D. Wang, R. Si, J. Yang, S. Zhou and J. Zeng, Nano Lett., 2021, 21, 4795–4801 CrossRef CAS PubMed.
- L. Ji, J. Wang, K. Wu and N. Yang, Adv. Funct. Mater., 2018, 28, 1706961 CrossRef.
- E. S. Agudosi, E. C. Abdullah, A. Numan, N. M. Mubarak, S. R. Aid, R. Benages-Vilau, P. Gómez-Romero, M. Khalid and N. Omar, Sci. Rep., 2020, 10, 11214 CrossRef CAS PubMed.
- R. Rajagopalan, B. Chen, Z. Zhang, X.-L. Wu, Y. Du, Y. Huang, B. Li, Y. Zong, J. Wang, G.-H. Nam, M. Sindoro, S. X. Dou, H. K. Liu and H. Zhang, Adv. Mater., 2017, 29, 1605694 CrossRef PubMed.
- S. Zhou, Y. Yang, H. Chen and Y. Ling, Ceram. Int., 2020, 46, 18331–18338 CrossRef CAS.
- J. Tian, F. Jiang, D. Yuan, L. Zhang, Q. Chen and M. Hong, Angew. Chem., Int. Ed., 2020, 59, 13101–13108 CrossRef CAS PubMed.
- B. Zhang, P. Huang, J. Chen, X. Dang, Y. Hu, Y. Ai, D. Zheng and H. Chen, Appl. Surf. Sci., 2020, 504, 144504 CrossRef CAS.
- Y. Xu, M. Xie, X. Li, F. Shao, S. Li, S. Li, Y. Xu, J. Chen, F. Zeng and Y. Jiao, J. Colloid Interface Sci., 2022, 626, 426–434 CrossRef CAS PubMed.
- L. Wang, Y. Wu, R. Cao, L. Ren, M. Chen, X. Feng, J. Zhou and B. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 16736–16743 CrossRef CAS PubMed.
- Y. Wang, S. Chen, S. Zhao, Q. Chen and J. Zhang, J. Mater. Chem. A, 2020, 8, 15845–15852 RSC.
- S. Tomyn, S. I. Shylin, D. Bykov, V. Ksenofontov, E. Gumienna-Kontecka, V. Bon and I. O. Fritsky, Nat. Commun., 2017, 8, 14099 CrossRef CAS PubMed.
- N. Zhang, X. Feng, D. Rao, X. Deng, L. Cai, B. Qiu, R. Long, Y. Xiong, Y. Lu and Y. Chai, Nat. Commun., 2020, 11, 4066 CrossRef CAS PubMed.
- J. Yin, P. Zhou, L. An, L. Huang, C. Shao, J. Wang, H. Liu and P. Xi, Nanoscale, 2016, 8, 1390–1400 RSC.
- S. Lee, K. Banjac, M. Lingenfelder and X. Hu, Angew. Chem., Int. Ed., 2019, 58, 10295–10299 CrossRef CAS PubMed.
- J. Y. C. Chen, L. Dang, H. Liang, W. Bi, J. B. Gerken, S. Jin, E. E. Alp and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 15090–15093 CrossRef CAS PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05863h |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2024 |