
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Machine learning facilitated by microscopic features for discovery of novel magnetic double perovskites†
Shuping
Guo
 *a,
Ryan
Morrow
a,
Jeroen
van den Brink
ab and
Oleg
Janson
*a
*a,
Ryan
Morrow
a,
Jeroen
van den Brink
ab and
Oleg
Janson
*a
aLeibniz Institute for Solid State and Materials Research IFW Dresden, Helmholtzstraße 20, Dresden 01069, Germany. E-mail: shuping.guo@ifw-dresden.de; olegjanson@gmail.com
bDepartment of Physics, Technical University Dresden, Dresden 01069, Germany
First published on 1st February 2024
Abstract
Double perovskites are a growing class of compounds with prospects for realization of novel magnetic behaviors. The rich chemistry of double perovskites calls for high-throughput computational screening that can be followed by or combined with machine-learning techniques. Yet, most approaches neglect the bulk of microscopic information implicitly provided by first-principles calculations, severely reducing the predictive power. In this work, we remedy this drawback by including onsite energies and transfer integrals between the d states of magnetic atoms. These quantities were computed by Wannierization of the relevant energy bands. By combining them with the experimental information on the magnetism of studied materials and applying machine learning, we constructed a model capable of predicting the magnetic properties of the remaining materials whose magnetism has not been addressed experimentally. Our approach combines classification learning to distinguish between double perovskites with dominant ferromagnetic or antiferromagnetic interactions and regression employed to estimate magnetic transition temperatures. In this way, we identified one antiferromagnet and three ferromagnets with a high transition temperature. Another 28 antiferromagnetic candidates were identified as magnetically frustrated compounds. Among them, cubic Ba2LaReO6 shows the highest frustration parameter, which is further validated by a direct first-principles calculation. Our methodology holds promise for eliminating the need for resource-demanding calculations.
1. Introduction
Discovering new materials with desired functionalities and understanding physical mechanisms behind these functionalities are central goals of materials science and engineering. For systematic studies, particularly advantageous are material classes in which the small number of structure types is contrasted by possibly rich chemistry. One such class is double perovskites with the general chemical formula A2BB′O6, where A cations occupy voids of the three-dimensional framework formed by corner-sharing BO6 and B′O6 octahedra. The interplay between electronic correlations and the spin–orbit coupling (SOC) in the d-shells of B and/or B′ can facilitate applications of double perovskites in data storage, energy conversion, contactless sensing, spin-polarized electrical conductivity, superconductivity, and catalysis.1–3The magnetic properties of double perovskites are very diverse. Following the discovery of large magnetoresistance in Sr2FeMoO6,4 several promising candidates with high magnetic transition temperature (Tc) were suggested for spintronic applications, such as Sr2FeReO6 (Tc = 401 K), Sr2CrWO6 (Tc = 450 K), Sr2CrReO6 (Tc = 625 K) and Sr2CrOsO6 (Tc = 725 K).5 On the other hand, long-range magnetic ordering in double perovskites can be suppressed by magnetic frustration, giving rise to the valence bond glass state in Ba2YMoO6.6
This remarkable disparity is driven not only by chemistry, but also by structural degrees of freedom. Alternating BO6 and B′O6 octahedra form a three-dimensional framework; A cations occupy the voids of this framework. In a regular octahedral environment, the d orbitals of B and B′ split into threefold degenerate t2g (dxy, dyz, dxz) and twofold degenerate  states. While the strict degeneracy is often lifted by distortions of octahedra, the crystal field splitting remains significant and largely shapes the electronic properties of double perovskites. In addition to distortions, different sizes of A and B/B′ ions may give rise to octahedral tilts and rotations. As a result, various structural types can be formed, such as cubic Fm
states. While the strict degeneracy is often lifted by distortions of octahedra, the crystal field splitting remains significant and largely shapes the electronic properties of double perovskites. In addition to distortions, different sizes of A and B/B′ ions may give rise to octahedral tilts and rotations. As a result, various structural types can be formed, such as cubic Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, tetragonal I4/m, monoclinic P21/n, I2/m, rhombohedral R and R3. Both distortions and tilts play a major role in the physical properties, as they are strongly intertwined with the charge, orbital, and spin degrees of freedom.7
m, tetragonal I4/m, monoclinic P21/n, I2/m, rhombohedral R and R3. Both distortions and tilts play a major role in the physical properties, as they are strongly intertwined with the charge, orbital, and spin degrees of freedom.7
The magnetic exchange in double perovskites strongly depends on the atomic arrangement and the valence of B and B′ cations. Exchange interactions are contributed by different virtual electron transfer processes that can couple the t2g or eg states of B with the t2g or eg states of B′, giving rise to multiple – and often competing – processes. Two such processes are illustrated in Fig. 1, where the t2g states of B′ are coupled to the t2g states of the B cation on the right, and the eg states of B′ are coupled to the eg states of the B cation on the left. In this case, B and B′ ions in double perovskites interact strongly with each other, and the magnetism is driven predominantly by the B–B′ interaction. Electron transfer processes within each sublattice, B or B′, are also possible. If B′ has an empty d shell, the interaction within the B sublattice determines the magnetic order, despite the large separation between the nearest neighbors.
 | ||
| Fig. 1 The workflow of the machine-learning part of this work. The training data set comprises 113 experimentally studied double perovskites; the resulting model is used to predict the magnetic properties of the remaining 68 materials. In the first step, a classification model is constructed to distinguish between antiferromagnetic and ferromagnetic compounds. Here, only atomic features are used. In the second step, the regression model is constructed based on the transfer integrals. This latter model predicts the ordering temperature. | ||
Transfer integrals are quantum-mechanical amplitudes describing the virtual electron transfer between a certain pair of orbitals. In the literature,8 the respective terms are called “onsite” if the pertinent orbitals belong to the same atom, and “transfer integrals” or “transfers” otherwise. Unfortunately, the knowledge of all transfer integrals between the magnetically active orbitals is not sufficient to evaluate the respective magnetic exchange: strong electronic correlations in the d shell lead to a many-body quantum problem whose generic solution is unknown. For specific electronic configurations, such as heavy d5 metals in an edge-sharing octahedral environment,9 perturbative treatments or solving the problem on a small cluster can deliver parameterized solutions, provided that interaction parameters and the strength of SOC are known. In all other situations, insights are limited to empirical assessments, such as the Goodenough-Kanamori rules, that are at best qualitative. Therefore, predicting the magnetic ground state is difficult even on a case-by-case basis, let alone in high-throughput calculations.
An alternative, more direct way of obtaining exchange integrals is performing density functional theory (DFT) calculations of different spin configurations. If only collinear arrangements are considered, the DFT total energies can be mapped onto a Heisenberg Hamiltonian and its classical ground state can be addressed by energy minimization. The treatment of noncollinear configurations is even more complicated: the inclusion of a full spin-density matrix and the necessary symmetry reduction leads to a considerably longer computational time.10 Therefore, screening the total energy of various configurations is almost impossible in a high-throughput fashion, especially for systems with metastable solutions that are sensitive to initially set magnetic moments and U values.11
Because no method can be universally applied to derive magnetic interactions, magnetic double perovskites have not been studied much by high-throughput calculations. Machine learning12–14 assisted high-throughput computations show excellent potential for exploring the physical properties particularly when the understanding of underlying mechanisms is elusive. Even though the majority of them don't have a large amount of training data, with experimental inputs or more in-depth features, a machine learning model is capable of accurate predictions for a small dataset.11,14–17 For example, a regression model with 157 experimental known data points proposed 26 new high Curie temperature two-dimensional ferromagnetic materials with a testing root-mean-square error (RMSE) of 174 K.11 Here, we're inclined to explore representative magnetic features which can further improve the predictive power and make up for the limited experimental sample size.
Presently, there are more than 400 ordered double perovskites known experimentally (including doped, high-pressure and high-temperature synthesized phases),18 and this number is sufficient for several machine-learning techniques. But at present, machine learning studies are either based on simple inputs (e.g. radius, valence difference, atomic mass, tolerance factor and so on)13 or total energies obtained for collinear configurations,10 limiting their predictive power. For more accurate assessments, it is crucial to resort to physically relevant inputs and accurate targets which determine the predictive precision of the machine learning.
In this work, we demonstrate that using the microscopic information contained in the transfer integrals in combination with the experimental information can drastically enhance the accuracy, leading to accurate predictions of the magnetically ordered state and the critical temperature. As shown in Fig. 1, by using 113 experimentally known double perovskites as the training dataset, we were able to predict the magnetic properties of 68 further double perovskites. This is done in two steps. First, a classification machine-learning model with atomic features was used to distinguish between antiferromagnets and ferromagnets. In the second step, we constructed a regression model which comprises the onsite energies and the transfer integrals between the first few neighbors; leading transfer integrals in the three channels – t2g–eg, t2g–t2g, and eg–eg – are included separately. Note that these microscopic terms underlie different exchange mechanisms, including direct exchange, superexchange and double exchange. All these terms were calculated by Wannierization of the nonmagnetic band structures of the respective materials. In this way, we obtained a model which predicts the ordering temperature with a RMSE of 18 and 61 K, for antiferromagnets and ferromagnets, respectively. One antiferromagnet and three ferromagnets with a high ordering temperature were identified. We also found 28 antiferromagnets combining a low transition temperature (≤50 K) with sizable transfer integrals (≥100 meV); these materials are likely magnetically frustrated and therefore may harbor exotic magnetic ground states. Since frustration is often assessed as the ratio of the Weiss temperature and the magnetic ordering temperature, we constructed an additional regression model to predict the Weiss temperature (RMSE = 76 K) and identify systems with a large frustration parameter. We obtained the largest ratio of 12 for cubic Ba2LaReO6; the sizable frustration was subsequently confirmed by a direct DFT + U + SOC calculation. This demonstrates that transfer integrals can be efficiently used in machine-learning models that aim at describing magnetic properties.
2. Methods
2.1 Wannier function calculations
Experimentally reported crystal structures of double perovskites are drawn from the inorganic crystal structure database (ICSD),19 including structures synthesized at high pressure or high temperature. We restrict ourselves to the fully ordered and slightly doped (with <0.1 fractional occupancy of a certain element within an individual Wyckoff position) structures; doping is neglected in the calculations of the latter structures. For all materials, internal atomic coordinates were optimized with respect to the GGA energy by keeping all symmetry operations of the respective space group; the resulting residual forces do not exceed 0.005 eV Å−1. Density functional theory band-structure calculations and Wannier projections are performed using an all-electron full potential local-orbital scheme implemented in the FPLO code;20 the initial projections are done for the d-dominated bands of B and B′ cations in the relevant energy range around the Fermi level. The Brillouin zones were sampled using meshes of 500, 1715 and 1470 k-points per reciprocal atom for cubic Fmm, tetragonal I4/m and monoclinic P21/n space groups, respectively. The Wannier basis, i.e. the presence of t2g and/or eg as well as B and/or B′, and the energy window depend on the material. For instance, in Ba2CoMoO6 we consider the energy range between −2 and 2 eV which contains besides the t2g and eg orbitals of Co also the t2g orbitals of Mo. The 4f electrons of Pr, Nd, Sm, Eu, Gd, Tb, Dy, Er, Tm, and Yb are treated by the open-core approach by putting  electrons of each spin channel into the core.
electrons of each spin channel into the core.
2.2 Machine learning models
Machine learning contains two parts. As mentioned before, the Goldschmidt tolerance factor | (1) |
Then with the computed matrices of the largest t2g–eg, t2g–t2g, eg–eg transfer integral of the short-range connections plus the onsite energies, the AdaBoost regression model was used to fit the magnetic transition temperature (Tc) and Curie–Weiss temperature (Θ). The dataset is randomly split into 80/20 training/testing datasets for validation.
2.3 DFT + U + SOC calculations of Ba2LaRe/RuO6
First-principles DFT calculations of Ba2LaReO6 and Ba2LaRuO6 are performed with the projector-augmented-wave basis set implemented in the Vienna ab initio simulation package.25 The exchange-correlation energy is evaluated within the generalized gradient approximation (GGA) using Perdew, Burke, and Ernzerhof parametrization26 and a roughly constant density of k-points (30 Å3) of a Monkhorst-Pack27 mesh is used to sample the Brillouin zones. Electron correlation effects were treated within DFT + U + SOC with the Coulomb repulsion U = 2 eV (6 eV) and the Hund exchange J = 0.4 eV (0.5 eV) for Re (Ru). For Ba2LaReO6 (Ba2LaRuO6), the total energies of one FM and one (six) AFM spin configurations are mapped onto a Heisenberg model; magnetic exchanges were obtained by a least-squares solution to the system of linear equations. Detailed expressions are provided in the ESI.†3. Results
3.1 Magnetic order: classification machine learning
The classical review by Vasala and Karppinen summarizes the experimental information on more than 400 ordered double perovskites.18 In this work, we first focus on three most populated space groups: cubic Fmm, tetragonal I4/m, and monoclinic P21/n (Fig. 2). These three space groups comprise 181 magnetic and non-magnetic stoichiometric double perovskites with structures available in the Inorganic Crystal Structure Database (ICSD). This set includes, in addition to structures stable under ambient conditions, also high-pressure and high-temperature phases that can be stabilized after quenching.28 For 113 double perovskites the magnetism has been addressed experimentally. We use these experimental observations to train the machine-learning models, which are later applied to the remaining 68 materials. To make the problem tractable, we consider a simplified classification scheme which approximates canted (e.g., Sr2CoOsO6 (ref. 29)) and quasi-two-dimensional (e.g., Sr2YRuO6 (ref. 30)) antiferromagnets as antiferromagnetic (AFM). Similarly, we do not distinguish between ferro- and ferrimagnetic (FM) order. Paramagnetic and spin-glass double perovskites are classified as nonmagnetic (NM). This three-valued classification splits the data set into 19 NM, 23 FM and 71 AFM compounds.
 | ||
| Fig. 2 Crystal structures of A2BB′O6 double perovskites crystallizing in various space groups: (a) Fmm, (b) I4/m, and (c) P21/n. | ||
Our first task is to assess the ground states of 68 double perovskites whose magnetism remains unknown. Following earlier studies that addressed thermodynamic stability,31 we consider geometric atomic features (such as tolerance factor, ionic radius, and atomic number). These are supplemented by electronic atomic features such as the number of electrons in the outermost shell and the oxidation states of B and B′ atoms. To surmount the relatively small size of the data set, leave-one-out validation was used to evaluate the performance. We applied three extensively used multi-class classifiers: MLP, XGBoost and AdaBoost that yielded an accuracy of 63%, 73%, and 78%, respectively. Unfortunately, using microscopic parameters such as transfer integrals (which we do use for predictions of the ordering temperatures, see the next section) as features does not improve the performance of the classification models. We attribute this to the excessive dimensions of such features and the small size of the dataset. The AdaBoost model with the highest accuracy was further used to predict the magnetic order of the 68 remaining double perovskites. In this way, we identified 45 AFM, 7 FM, and 16 NM compounds depicted in Fig. 3 with red, blue, and gray squares.
 | ||
| Fig. 3 Classification ML-model-predicted 16 nonmagnetic (gray cells), 7 ferromagnetic (blue cells) and 45 AFM (red cells) double perovskites with the cubic Fmm (left), monoclinic P21/n (top right), and tetragonal I4/m (bottom right) space groups. Rows and columns denote B and B′ cations, respectively. Atomic symbols in the cell denote A cations; double entries with atomic symbols separated by a slash pertain to both compounds. | ||
3.2 Magnetic ordering temperature: regression machine learning
After applying the classification model to distinguish between potential ferro- and antiferromagnets, we turn to the magnetic ordering temperature Tc. In this case, atomic features are not sufficient. Magnetic ordering is driven by different, sometimes competing magnetic exchanges, that are in turn implicitly (and nontrivially) governed by transfer integrals. While the number of such parameters in each material amounts to several hundreds, the magnetism is mostly determined by the largest transfer integrals between the first, second or third neighbors. Depending on the number of electrons in the d shells and the orbital sector (t2g–t2g, t2g–eg, or eg–eg), such terms contribute to the antiferro- or ferromagnetic exchange; the overall sign and strength depend on the balance between these contributions.To illustrate how such balance is realized in practice, we consider three Co2+-containing double perovskites: the cubic Ba2CoReO6, the tetragonal Sr2CoReO6, and the monoclinic La2CoIrO6. Since Re6+ (Ir4+) has one (five) electron in the 5d shell, exchanges between B and B′ as well as within each sublattice has to be considered. Possible relevant exchanges in these three structure types are schematically illustrated in the left panel of Fig. 4, where the purple, green and orange solid lines represent B–B, B′–B′, and B–B′ exchanges, respectively. Respective directions in the crystal lattice are denoted by superscripts, i.e. is exchange between B and B along the a axis,
is exchange between B and B along the a axis,  is the direction along the body diagonal between B′ and B′, etc. In the right panel of Fig. 4, we show the strongest transfer integrals for a given interatomic separation in each orbital sector; the color map spans the range between −300 (blue) and 300 (red) meV. For the cubic Ba2CoReO6, three first Co–Re nearest-neighbor bonds
is the direction along the body diagonal between B′ and B′, etc. In the right panel of Fig. 4, we show the strongest transfer integrals for a given interatomic separation in each orbital sector; the color map spans the range between −300 (blue) and 300 (red) meV. For the cubic Ba2CoReO6, three first Co–Re nearest-neighbor bonds 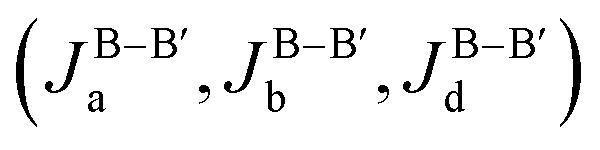 are identical, with three large t2g–t2g transfer integrals (three blue squares in the third row) around −225 meV. Besides, two eg–eg Co–Co transfer integrals
are identical, with three large t2g–t2g transfer integrals (three blue squares in the third row) around −225 meV. Besides, two eg–eg Co–Co transfer integrals 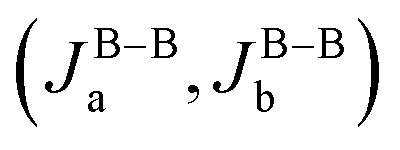 and two t2g–t2g terms of Re–Re
and two t2g–t2g terms of Re–Re  are around −100 meV.
are around −100 meV.
 | ||
| Fig. 4 Transfer path and corresponding largest t2g–t2g, t2g–eg, and eg–eg transfer integrals of Ba2CoReO6 (Fmm), Sr2CoReO6 (I4/m), and La2CoIrO6 (P21/n), respectively. The purple, green and orange solid lines represent the first nearest neighbors of B–B, B′–B′ and B–B′, respectively. The color from purple to red indicates the value of transfer integrals ranging between −300 and 300 meV. | ||
For Sr2CoReO6 crystallizing in the I4/m space group, leading transfer integrals are three t2g–t2g (one blue and two red squares in the third row) of −167 and 193 meV and two t2g–eg (two red squares in the third row) around 203 meV of the three shortest Co–Re bonds,  and
and 
For the monoclinic La2CoIrO6, the largest components (100 to 200 meV) also pertain to the short-range B–B′ connections. At the same time, both t2g–eg and t2g–t2g channels are active, as evidenced by five blue and one red squares in the third row of Fig. 4 (right). In addition, there are four eg (t2g) transfer integrals operating within the Co (Ir) sublattice. The structure of these matrices determines the sign and the strength of the respective magnetic exchanges.
The leading transfer integrals are complemented by the onsite energies of B and B′ d-orbitals. The resulting model is trained on a dataset of experimental transition temperatures (Tc), which is separated into 21 ferro- and 66 antiferromagnets. To assess the predictive power, we randomly divided each set following the 80/20 rule for the training/testing. As shown in Fig. 5, the training data set of antiferromagnets yields very high accuracy, which allows us to achieve the root mean square error (RMSE) of 18 K for the testing data set. In contrast, RMSE is sizable (around 61 K) for ferromagnets, which can be traced back to the small size of the dataset and the broad distribution of ordering temperatures.
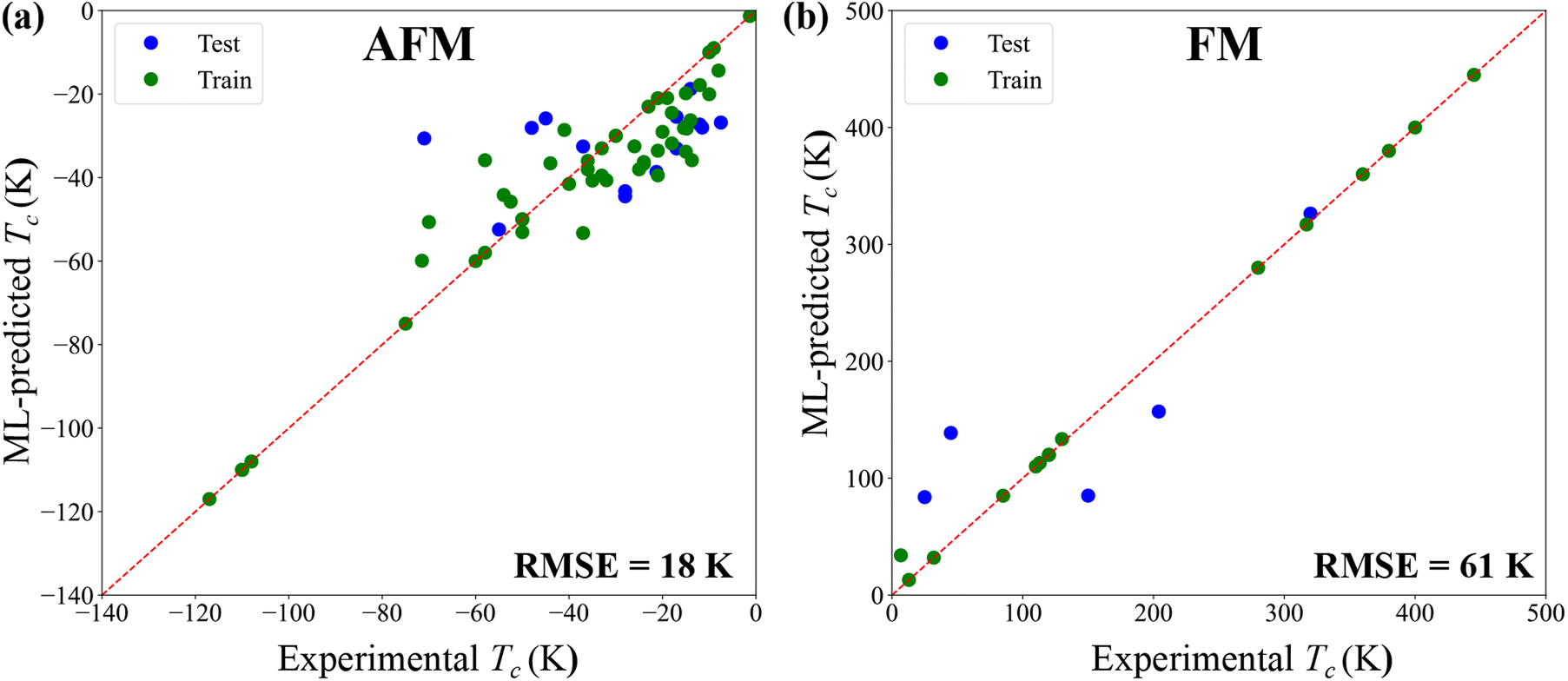 | ||
| Fig. 5 Magnetic transition temperature Tc predicted by the regression models in comparison with the experimental value. Antiferromagnetic (a) and ferromagnetic (b) materials are treated separately. The training and testing data sets are green and blue dots, respectively. | ||
These two pre-trained models are further used to predict the transition temperatures of prospective 45 antiferromagnets and 7 ferromagnets. Among them, we find antiferromagnetic Ba2TbReO6 and three ferromagnets with high predicted critical temperatures: −51 K for Ba2TbReO6 (Fmm), 172 K for Bi2NiMnO6 (P21/n), 209 K for Ba2FeMoO6 (Fmm) and 99 K for Ca2TiMnO6 (I4/m). While the predicted ordering temperature (209 K) of Ba2FeMoO6 is sensibly lower than in the recent experiment (345 K32), our model correctly identifies this material as a high-temperature magnet. Considering this deviation, to test whether the regression models are at risk of overfitting, different validations using resampling are conducted. As shown in Fig. S1,† the robust behavior of the model for antiferromagnets against overfitting gives us hope that the deviations in the model for ferromagnets can be overcome if more samples are added to the training dataset.
3.3 Identification and validation of prospective frustrated magnets
Except for the handful of prospective high-temperature antiferromagnets, all remaining antiferromagnets are predicted to have low Tc below 50 K. Among these 44 compounds, 28 feature sizable transfer integrals (≥100 meV) that can underlie strong antiferromagnetic exchange. To test this assumption, we turn to another characteristic temperature – the Weiss temperature Θ. This quantity represents a sum of all magnetic exchanges (multiplied with respective multiplicities) and can be estimated by fits to the magnetic susceptibility at high temperatures. Since strong antiferromagnetic exchanges give rise to sizable Θ, the magnetic frustration is often characterized by the ratio f = |Θ|/Tc, where Tc is the magnetic ordering temperature. Values of f exceeding ∼5 indicate sizable magnetic frustration, which gives rise to an exotic spin-liquid regime between Θ and Tc.30To identify relevant candidates, we collect experimentally reported the Curie–Weiss temperatures (Θ) of 63 known antiferromagnetic double perovskites, and then used the transfer integrals to train the regression ML model (leaving as before 20% for testing). In this way, we obtain a RMSE of 76 K for the testing data set (Fig. 6). Next, we calculate Θ for each of the 45 prospective antiferromagnets. As shown in Fig. 7, most monoclinic double perovskites like Ba2LaRuO6 (Tc = −30 K and Θ = −127 K) lie near the f = 5 isoline. At the same time, many cubic double perovskites lie in a more frustrated regime. In particular, three Re-based compounds are predicted to have f > 10: Ba2LuReO66 (Tc = −43 K and Θ = −485 K), Ba2EuReO6 (Tc = −50 K and Θ = −572 K) and Ba2LaReO6 (Tc = −42 K and Θ = −521 K). We note that the predicted temperatures for Ba2LuReO6 are consistent with recent experimental work33 reporting Tc = −31 K and Θ = −678 K.
 | ||
| Fig. 6 Regression ML model predicted as well as experimentally reported Weiss temperature Θ of AFM candidates. The training and testing dataset are green and blue dots, respectively. | ||
 | ||
| Fig. 7 Magnetic transition temperature Tc and the Weiss temperature Θ for 45 prospective antiferromagnets as predicted by the regression model. Light yellow shading marks the strongly frustrated regime (f > 5). The red rhombus, green square and purple pentagon represent materials crystallizing in the cubic Fmm, tetragonal I4/m and monoclinic P21/n space groups, respectively. | ||
For an independent assessment of the accuracy, we consider the cubic Ba2LaReO6 and perform DFT + U + SOC calculations for the commonly observed AFM configuration in experiment34 as well as the ferromagnetic configuration. The energy of the antiferromagnetic configuration is lower by −151.7 meV/f.u., indicating that  and
and  are exchanges that are sizable (110 K assuming S = 1) and antiferromagnetic, which is in line with large (−154 meV, see Fig. 8) transfer integrals in the t2g channel. We note that in the absence of magnetic moments on B atoms, B′ moments (localized on Re) form a face-centered cubic lattice, which is geometrically frustrated.34
are exchanges that are sizable (110 K assuming S = 1) and antiferromagnetic, which is in line with large (−154 meV, see Fig. 8) transfer integrals in the t2g channel. We note that in the absence of magnetic moments on B atoms, B′ moments (localized on Re) form a face-centered cubic lattice, which is geometrically frustrated.34
 | ||
| Fig. 8 Transfer paths and transfer integrals of Ba2LaReO6 and Ba2LaRuO6. To be consistent with the visual representation of Fig. 4, we show B–B and B–B′ parts despite the absence of a partly filled electronic shell in La3+. Parameters whose absolute value is around 100 meV or larger are labeled; other terms are denoted by color. | ||
For comparison, we perform a similar DFT + U + SOC calculation for the monoclinic Ba2LaRuO6. Since the low symmetry generates several independent terms, more supercells with different magnetic configurations (ferromagnetic and six antiferromagnetic) are needed. Total energies for all magnetic configurations are listed in the ESI.† By mapping these energies onto a Heisenberg model, we estimate the short-range exchange integrals Ja, Jb, Jd and Jd′. For simplicity, the difference between Jd and Jd′ is neglected; this is justified by the similarity of crystalline environments pertaining to both exchange pathways.35 The resulting Ja, Jb, and Jd = Jd′ are 20, 11 and 11 K, respectively, with an accuracy of 0.5 K. These antiferromagnetic exchanges mainly stem from electron transfer in the t2g channel; due to the smaller spatial extent of 4d orbitals compared to 5d, these transfer integrals are much weaker than in Ba2LaReO6.
4. Conclusions
We develop a machine learning model to discover new magnetic double perovskites. Our two-step procedure includes classification learning based on atomic inputs and regression learning based on microscopic inputs – transfer integrals and onsite energies. The goal of the first step is to distinguish between prospective antiferromagnets and ferromagnets; this model is reasonably reliable, particularly for antiferromagnets (78%). The second model aims at predicting the magnetic transition temperature. Here, we achieve a root mean square error (RMSE) of 18 and 61 K for antiferro- and ferromagnets, respectively.We identify Ba2TbReO6 as an antiferromagnet with a high magnetic ordering transition temperature of about 50 K, and three prospective ferromagnets – Bi2NiMnO6, Ba2FeMoO6, and Ca2TiMnO6 – with the ordering temperatures of 172, 209, and 99 K, respectively. We additionally identified 28 prospective frustrated antiferromagnets that combine sizable transfer integrals (100 meV and larger) with a low ordering temperature (below 50 K). Detailed total-energy calculations of magnetic supercells support this conjecture for Ba2LaReO6.
In addition to providing new candidate double perovskites with high magnetic transition temperature or sizable magnetic frustration, our work opens new insights for machine learning assisted high-throughput calculations. Estimation of transfer integrals is becoming a routine task that can be done in a high-throughput fashion. These microscopic parameters underlie the electronic and magnetic properties, but quantitative information on the magnetic properties can be obtained using analytical or perturbative expressions that are available for several specific cases only.9,36,37 Machine learning methods can be an appealing alternative to such analytical approaches, because of its potential to capture the elusive, yet inherent link between the transfer integrals and physical observables.
Author contributions
S. G. and O. J. devised the research. S. G. conducted the research and drafted the manuscript. S. G., R. M., J. v. d. B. and O. J. analyzed the results and edited the manuscript. J. v. d. B. and O. J. supervised the research.Conflicts of interest
All authors declare no competing interests.Acknowledgements
S. G., R. M. and O. J. acknowledge financial support from the IFW Excellence Program. R. M., J. v. d. B. and O. J. were supported by the Collaborative Research Center SFB 1143 (Project No. 247310070). J. v. d. B. thanks the Deutsche Forschungsgemeinschaft (DFG) for support through the Würzburg-Dresden Cluster of Excellence on Complexity and Topology in Quantum Matter ct.qmat (EXC 2147, Project No. 39085490). S. G. thanks fruitful discussions with Prof G. Jeffrey Snyder (Northwestern University), Denys Kononenko (IFW Dresden) and Dmitry Chernyavsky (IFW Dresden) as well as technical assistance of Ulrike Nitzsche (IFW Dresden). S. G. acknowledges computing resources provided by HPC-TU Dresden.References
- C. Jia, S. Onoda, N. Nagaosa and J. H. Han, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 144424 CrossRef.
- A. Rüegg and G. A. Fiete, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 201103 CrossRef.
- A. Erickson, S. Misra, G. J. Miller, R. Gupta, Z. Schlesinger, W. Harrison, J. Kim and I. Fisher, Phys. Rev. Lett., 2007, 99, 016404 CrossRef CAS PubMed.
- K.-I. Kobayashi, T. Kimura, H. Sawada, K. Terakura and Y. Tokura, Nature, 1998, 395, 677–680 CrossRef CAS.
- A. E. Taylor, R. Morrow, M. D. Lumsden, S. Calder, M. H. Upton, A. I. Kolesnikov, M. B. Stone, R. S. Fishman, A. Paramekanti, P. M. Woodward and A. D. Christianson, Phys. Rev. B, 2018, 98, 214422 CrossRef CAS.
- M. A. de Vries, A. Mclaughlin and J.-W. Bos, Phys. Rev. Lett., 2010, 104, 177202 CrossRef CAS PubMed.
- R. Morrow, J. W. Freeland and P. M. Woodward, Inorg. Chem., 2014, 53, 7983–7992 CrossRef CAS PubMed.
- K. Koepernik, O. Janson, Y. Sun and J. van den Brink, Phys. Rev. B, 2023, 107, 235135 CrossRef CAS.
- S. M. Winter, Y. Li, H. O. Jeschke and R. Valentí, Phys. Rev. B, 2016, 93, 214431 CrossRef.
- M. K. Horton, J. H. Montoya, M. Liu and K. A. Persson, NPJ Comput. Mater., 2019, 5, 64 CrossRef.
- A. Kabiraj, M. Kumar and S. Mahapatra, NPJ Comput. Mater., 2020, 6, 35 CrossRef.
- A. Talapatra, B. P. Uberuaga, C. R. Stanek and G. Pilania, Chem. Mater., 2021, 33, 845–858 CrossRef CAS.
- A. Halder, A. Ghosh and T. S. Dasgupta, Phys. Rev. Mater., 2019, 3, 084418 CrossRef CAS.
- P. V. Balachandran, B. Kowalski, A. Sehirlioglu and T. Lookman, Nat. Commun., 2018, 9, 1668 CrossRef PubMed.
- Z.-X. Shen, C. Su and L. He, NPJ Comput. Mater., 2022, 8, 132 CrossRef CAS.
- W. Liu, Y. Lu, D. Wei, X. Huo, X. Huang, Y. Li, J. Meng, S. Zhao, B. Qiao and Z. Liang, et al. , J. Mater. Chem. A, 2022, 10, 17782–17789 RSC.
- Y. Hu, X. Hu, L. Zhang, T. Zheng, J. You, B. Jia, Y. Ma, X. Du, L. Zhang and J. Wang, et al. , Adv. Energy Mater., 2022, 12, 2201463 CrossRef CAS.
- S. Vasala and M. Karppinen, Prog. Solid State Chem., 2015, 43, 1–36 CrossRef CAS.
- A. Belsky, M. Hellenbrandt, V. L. Karen and P. Luksch, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2002, 58, 364–369 CrossRef PubMed.
- K. Koepernik and H. Eschrig, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1743 CrossRef CAS.
- X. Glorot and Y. Bengio, J. Mach. Learn. Res., 2010, 9, 249–256 Search PubMed.
- J. H. Friedman, Ann. Stat., 2001, 1189–1232 Search PubMed.
- T. Hastie, S. Rosset, J. Zhu and H. Zou, Statist. Interface, 2009, 2, 349–360 CrossRef.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and E. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Condens. Matter Mater. Phys., 1976, 13, 5188 CrossRef.
- P. Kayser, S. Injac, B. Ranjbar, B. J. Kennedy, M. Avdeev and K. Yamaura, Inorg. Chem., 2017, 56, 9009–9018 CrossRef CAS PubMed.
- B. Yan, A. K. Paul, S. Kanungo, M. Reehuis, A. Hoser, D. M. Többens, W. Schnelle, R. C. Williams, T. Lancaster and F. Xiao, et al. , Phys. Rev. Lett., 2014, 112, 147202 CrossRef PubMed.
- E. Granado, J. W. Lynn, R. F. Jardim and M. S. Torikachvili, Phys. Rev. Lett., 2013, 110, 017202 CrossRef CAS PubMed.
- A. Talapatra, B. P. Uberuaga, C. R. Stanek and G. Pilania, Chem. Mater., 2021, 33, 845–858 CrossRef CAS.
- S. B. Kim, E. J. Hahn and C. S. Kim, J. Korean Phys. Soc., 2019, 75, 466–470 CrossRef CAS.
- J. Xiong, J. Yan, A. A. Aczel and P. M. Woodward, J. Solid State Chem., 2018, 258, 762–767 CrossRef CAS.
- C. Wang, Y. Xu, W. Hao, R. Wang, X. Zhang, K. Sun and X. Hao, Phys. Rev. B, 2019, 99, 035126 CrossRef CAS.
- H. Wang, S. Zhu, X. Ou and H. Wu, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 054406 CrossRef CAS.
- V. Mazurenko, S. Skornyakov, A. Kozhevnikov, F. Mila and V. Anisimov, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 224408 CrossRef.
- S. M. Winter, J. Phys. Mater., 2022, 5, 045003 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Full lists of experimental and machine learning predicted magnetic transition temperature, Weiss temperature and frustration parameters; detailed formulae of DFT + U + SOC calculations. See DOI: https://doi.org/10.1039/d3ta05679a |
| This journal is © The Royal Society of Chemistry 2024 |