
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient CO2 electrochemical reduction on dual metal (Co–Ni)–nitrogen sites†
Jianping
Chen
a,
Md Robayet
Ahasan
b,
Jin-Su
Oh
 c,
Jake A.
Tan
d,
Stephen
Hennessey
e,
Mahmoud M.
Kaid
e,
Hani M.
El-Kaderi
e,
Lin
Zhou
cf,
Ka Un
Lao
e,
Ruigang
Wang
b and
Wei-Ning
Wang
*a
c,
Jake A.
Tan
d,
Stephen
Hennessey
e,
Mahmoud M.
Kaid
e,
Hani M.
El-Kaderi
e,
Lin
Zhou
cf,
Ka Un
Lao
e,
Ruigang
Wang
b and
Wei-Ning
Wang
*a
aDepartment of Mechanical and Nuclear Engineering, Virginia Commonwealth University, Richmond, VA 23284, USA. E-mail: wnwang@vcu.edu
bDepartment of Metallurgical and Materials Engineering, The University of Alabama, Tuscaloosa, AL 35487, USA
cDivision of Materials Sciences and Engineering, Ames National Laboratory, Ames, IA 50011, USA
dDepartment of Chemistry, Gottwald Center for the Sciences, University of Richmond, Richmond, VA 23173, USA
eDepartment of Chemistry, Virginia Commonwealth University, Richmond, VA 23284, USA
fDepartment of Materials Science and Engineering, Iowa State University, Ames, IA 50011, USA
First published on 18th January 2024
Abstract
The electrochemical reduction (ECR) of CO2 is a promising approach for CO2 removal and utilization, which is a critical component of the circular carbon economy. However, developing efficient and selective electrocatalysts is still challenging. Single-atom catalysts (SACs) have gained attention because they offer high metal atom utilization and uniform active sites. However, tuning the active metal centres to achieve high activity and selectivity in CO2 reduction remains a significant challenge. This study presents a novel electrocatalyst (Co–Ni–N–C) for CO2 ECR on the diatomic metal–nitrogen sites prepared through ion exchange using a zeolitic imidazolate framework (ZIF) as a precursor. During pyrolysis, nitrogen-doped graphitic carbon serves as the host material, anchoring the diatomic Co–Ni sites. The resulting bimetallic active sites demonstrate exceptional performance, achieving a high CO yield rate of 53.36 mA mgcat.−1 and an impressive CO faradaic efficiency of 94.1% at an overpotential of −0.27 V. Spectroscopic, microscopic, and density functional theory (DFT) analyses collectively unveil the crucial synergistic role of the Co–Ni–N6 moiety in promoting and sustaining exceptional electrocatalytic activities. The successful utilization of bimetallic sites in enhancing catalyst performance highlights the potential of this approach in developing efficient electrocatalysts for various other reactions.
Introduction
The electrochemical reduction of carbon dioxide (CO2 ECR) to value-added fuels and chemicals could enable a shift toward a sustainable energy economy and chemical industry.1 Electrocatalysts are crucial to CO2 ECR research due to their potential to convert CO2 selectively and efficiently, offering both environmental and economic benefits. A plethora of catalysts have been scrutinized for their performance, including metals, alloys, metal oxides, functionalized carbons, and metal complexes, unveiling their remarkable capacity for efficient and selective CO2 conversion. This promising potential, however, coexists with persistent challenges pertaining to energy efficiencies, activity, and undesired off-pathway hydrogen evolution reactions (HER). Therefore, developing high-performance electrocatalysts for CO2 ECR remains an urgent task.2The catalysis field has recently seen a surge in the popularity of embedding active metal atoms in chemically-distinct host materials, a class of materials called single-atom catalysts (SACs),3 for numerous important energy conversion reactions, such as water gas shift reactions,4 hydrogen evolution,5 Fenton reaction,6 nitrate reduction,7,8 oxygen reduction,9 nitrogen reduction,10 methane oxidation,11 and carbon dioxide reduction reactions.12 The research advancements in SACs promise to address CO2 reduction challenges and develop efficient and selective electrocatalysts for sustainable fuel and chemical conversion.13 Such SACs often show the benefits of lower cost and higher performance over conventional metal catalysts due to high metal atom utilization and high uniformity of active sites.14 This is further enhanced by the well-designed and specific structure of SACs, which enables in-depth studies to understand the fundamental mechanisms and active sites critical for CO2 reduction.15 Functionalized carbon is a common host material on which single metal atoms are deposited due to its strong affinity to metal atoms by forming stable chemical bonds, and its high tunability in allowing different dopants, high stability, and good electrical conductivity.16,17
SACs on nitrogenated carbon are excellent model systems for studying catalytic mechanisms at the atomic level.18 Transition metal SACs, such as Ni-SAC on nitrogen-doped graphene, are promising CO2 ECR catalysts with high catalytic activity and CO production efficiency.19 Co and Ni SACs can also be tuned for efficient syngas production.20 However, the catalytic efficiency of SACs is constrained by their sole active centres, inhibiting multi-atom pathways, thus restricting their overall performance.21 In contrast, dual-active site catalysts (DASCs), which utilize two metal species to form a dual-metal single-site catalyst, promise to offer complementary functionalities and synergistic effects.22,23 This is facilitated by electronic interactions between the adjacent heteroatomic metal species, allowing for tailored binding energies with intermediates. This concept is further explored in recent comprehensive reviews, highlighting the advancements and potential of DASCs in various applications.24–29 Nitrogenated carbon-based DASCs have demonstrated advantages in improving CO2 ECR performance.30 For example, the N-bridged Co–N–Ni bimetallic catalyst achieves a CO faradaic efficiency of 96.4%, significantly higher than Co–N4 at 61.5% and Ni–N4 at 45.0%, respectively.31 Despite considerable advancements, the construction of DASCs with superior catalytic performance and multi-functionalities, alongside a comprehensive understanding of their operational mechanisms, remains a formidable challenge in the field.
Herein, we report an efficient CO2 ECR catalyst featuring diatomic metal–nitrogen sites (Co–Ni–N–C) prepared through ion exchange using a zeolitic imidazolate framework (ZIF) as a precursor. During pyrolysis, nitrogen-doped graphitic carbon serves as a host material, anchoring diatomic Co–Ni sites. These bimetallic active sites demonstrate remarkable performance, with a CO yield rate of 53.36 mA mgcat.−1 and an impressive CO faradaic efficiency of 94.1% at an overpotential of −0.27 V. Spectroscopic, microscopic, and density functional theory analyses reveal that the Co–Ni–N3 moiety plays a crucial synergistic role in promoting and sustaining these exceptional ECR activities. This study highlights the viability of utilizing bimetallic sites as an effective approach for enhancing the performance of ECR catalysts and suggests the potential application of this synthetic strategy in the development of various other catalysts.
Experimental section
Synthetic procedures
CO2 ECR evaluation
The CO2 flow was regulated at a flow rate of 30 mL min−1 using a mass flow controller (Aalborg DFC) and introduced into the cathode compartment through a porous glass frit. Each chronoamperometry test had a duration of 30 minutes. The cathode compartment, filled with ca. 50 mL of solution, was continuously stirred at 800 rpm using a magnetic PTFE stirring bar. The effluent of the cathode compartment was connected directly to a gas chromatograph (GC, Agilent 7890B) for online detection of gas-phase products. Liquid-phase products were analyzed after each chronoamperometry test by 1H nuclear magnetic resonance (NMR). Further description of the electrochemistry setup and product analysis can be found in ESI S1.†Catalyst characterization
The particle size and morphology were characterized by scanning electron microscopy (SEM, Su-70, Hitachi), HAADF-STEM imaging (Thermo Fisher Titan Themis), and TEM imaging (Tecnai F20). The structure and crystallinity of the catalyst were determined from powder X-ray diffraction (PXRD, X'Pert PRO, PANalytical). Surface chemistry was characterized by Fourier transform infrared (FT-IR) spectroscopy (Nicolet iS50, Thermo Scientific). Surface active sites and reducible species were respectively characterized by temperature-programmed desorption (TPD) and temperature-programmed reduction (TPR) techniques (Micromeritics AutoChem II 2920). The degree of graphitization of metal–nitrogen-doped carbon materials was analyzed by Raman spectroscopy (LabRAM HR Evolution, Horiba Scientific). The chemical state of the elements was investigated by X-ray photoelectron spectroscopy (XPS, PHI VersaProbe III, ULVAC-PHI). BET surface area and pore size distribution were analyzed using Autosorb iQ (Quantachrome Instruments).Computational procedures
To assess the catalytic activity of the monatomic and diatomic metal–nitrogen sites, DFT calculations were performed at the level of PBE0/def2-SVP.36–39 The RIJCOSX approximation with def2/J and def2-SVP/C as auxiliary basis sets were utilized to speed up the calculations.40 The def2-ECP effective core potential was used for the Ni and Co atoms. Dispersion interactions were considered through the use of Grimme's D3 dispersion correction with Becke-Johnson damping (D3BJ).41 For brevity, we will refer to this calculation method as RI-PBE0-D3BJ/def2-SVP. All electronic structure calculations were performed using the ORCA 5.0.4 package.42–44Results and discussion
Synthesis and characterization of catalysts
Carbon-supported catalysts with dispersed SACs and DASCs and related nanoparticles (NPs) were prepared by pyrolysis of zeolitic imidazolate frameworks (ZIFs) precursors. As shown in Fig. 1, ZIFs have a distinct structural characteristic where metal centres are only coordinated by nitrogen atoms of imidazolate units, resulting in a network of interconnected frameworks.45 The reaction mechanism, Fourier transform infrared (FTIR) spectra, powder X-ray diffraction (PXRD) patterns, and scanning electron microscopy (SEM) images of ZIFs are provided in Fig. S1–S4,† respectively. During thermal activation, such metal coordination with nitrogen can maintain the isolation of metal centres to form SACs, which benefits the in situ formation of uniformly dispersed metal centres.3 In addition to the conventionally fully isolated SACs, the spatial correlation among metal single atom sites can influence the local geometry of the metal centres, demonstrating much higher flexibility in tuning electronic and electrocatalytic properties of correlated SACs with additional metal centres, i.e. DASCs. ZIFs containing abundant metallic elements with low vapourability at high temperatures can lead to phase segregation and induce the formation of metallic NPs. This transformation route (Fig. 1) provides a generalized method for the synthesis of SACs, DASCs, and NPs from ZIFs.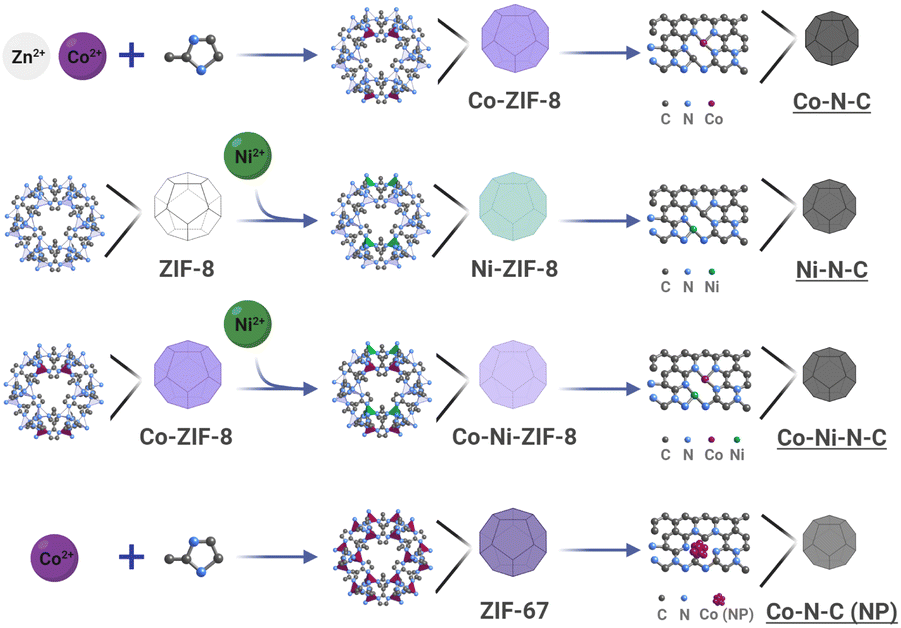 | ||
| Fig. 1 Pyrolysis-induced transformation of ZIFs for the synthesis of nitrogenated carbon-based catalysts: Co–N–C, Ni–N–C, Co–Ni–N–C, and Co–N–C (NP) (NP: nanoparticle). | ||
As confirmed by PXRD in Fig. 2A, the crystalline phases of ZIF precursors underwent a complete transformation in the high-temperature pyrolysis process. The PXRD pattern of Co–N–C (SAC) (Fig. 2A) shows two characteristic peaks at 25 and 44 °C, indexed to the (002) and (101) planes of graphitic carbon, while metal-containing crystal phases were not observed. Likewise, amorphous phases are found in the PXRD patterns of Ni–N–C and Co–Ni–N–C. To make a better comparison, we also synthesized the nanoparticle counterpart Co–N–C (NP) by pyrolyzing ZIF-67 (a type of isostructural ZIFs that only contains Co) at 1100 °C. Without the presence of Zn ions to protect them, the Co nodes tend to form nanoparticles instead of single Co atoms, as a result of the shorter distance between adjacent atoms. In this case, the crystallographic plane (111) of cobalt appeared in Co–N–C (NPs), suggesting the formation of metallic cobalt nanoparticles.46 The pyrolysis of ZIF-8 samples was tracked through thermogravimetric analysis (TGA) (see Fig. S5 and S6†). At temperatures above 900 °C, the weight loss was attributed to the release of zinc metal (bp, 900 °C). Therefore, pyrolysis of ZIF-8 samples at 1100 °C was sufficient to remove the majority of zinc and form SACs. Energy dispersive X-ray spectroscopy (EDS) spectra (Fig. 2B) further confirm the elemental composition in the pyrolyzed samples. Accompanying the major carbon emission peak, a trace amount of metallic element was confirmed by EDS as shown in the inset of Fig. 2B. Zinc evaporated due to its low boiling point, but Co(Kα) and Ni(Kα) peaks were present in the corresponding SACs.47 Due to carbothermal reduction, coordinated metals were easily reduced to metallic states, generating individual metal centres. Analogous to the EDS analysis, the inductive coupled plasma optical emission spectroscopy (ICP-OES, Fig. S7†) results indicate that in Co–Ni-ZIF-8, the zinc content was nearly removed while the Co and Ni content was maintained. Furthermore, Co–Ni–N–C showed similar Co and Ni loadings to those in Co–N–C and Ni–N–C.
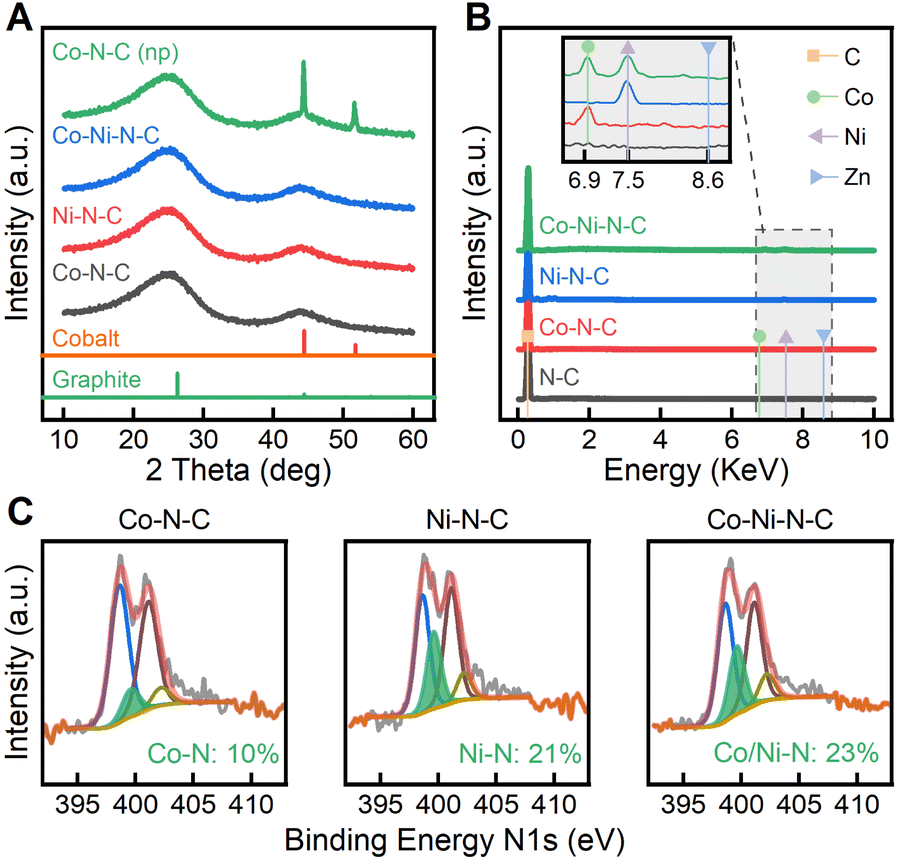 | ||
| Fig. 2 (A) PXRD patterns of the pyrolyzed samples. The crystallographic data used to simulate the powder diffraction pattern of cobalt and graphite were respectively obtained from PDF# 00-015-0806 and PDF# 01-075-1621. (B) EDS spectra of the pyrolyzed samples. The inset, extracted from the dashed box in B, shows the metal emission peaks. Coloured lines show the expected peak positions of Zn (blue), Ni (purple), Co (green), and C (orange). (C) The N1s spectrum changes based on the metal–nitrogen (M–N) peak in the probed sample region. The spectra are linear combinations of pyridinic (blue), pyrrolic (brown), graphitic (gold), and N–Ox (green) contributions. | ||
To understand the coordination status of carbon and nitrogen in anchoring SACs, we investigated the local structure and bonding evolution during SAC synthesis by X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy. The bonding configurations for nitrogen atoms in the samples were characterized with detailed N1s XPS analysis. High-resolution N1s spectra of the samples, as shown in Fig. 2C, proved the existence of various N–C species, including pyridinic Ni (398.6 eV), pyrrolic No (401.1 eV), and graphitic Ng (402.2 eV).48 The pyridinic Ni peak dominates the chemical state of nitrogen. The abundant pyridinic nitrogen provides sufficient coordination sites to active metal centers, which facilitates the formation of metal–nitrogen coordination.49 Consequently, a new nitrogen type Nm originating from nitrogen–metal bonding was observed at 399.6 eV, indicating a transformation of metal–nitrogen tetrahedra within ZIF-8s to active metal–nitrogen moieties embedded in carbon.47 The Ni peak (398.6 eV) stood out in Co–N–C while the percentage of Nm peak was identified to be only 10%, suggesting pyridinic Ni was the dominant defect species. In contrast, 23% of Nm could be found in Co–Ni–N–C, indicating stronger metal coordination with nitrogen.50 From peak area analysis of N1s and Co 2p (Fig. S8†), the ratio of pyridinic N to the correlating Co 2p peaks in Co–Ni–N–C was approximately 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, strongly indicating the formation of porphyrin-like Co–N3 species.51 The Co 2p peak at 780.0 eV obtained from Co–Ni–N–C (Fig. S8†) has a binding energy higher than that for Co(0) species at 778.2 eV, consistent with the accepted chemical shift theory of oxidized cobalt.52 The Ni 2p spectra for Co–Ni–N–C show that the Ni oxidation state is positioned between Ni(0) peak at 852.6 eV and Ni(II) peak at 855.6 eV, indicating the presence of Ni species that are partially oxidized.53 As evident in Fig. S8,† The Co 2p peak for Co–Ni–N–C is found shifted 0.6 eV to lower binding energy in comparison to Co–N–C. Additionally, the Ni 2p peak for Co–Ni–N–C shifts 0.2 eV toward higher binding energy compared to Ni–N–C. It can be interpreted from XPS results that Ni atoms in Co–Ni–N–C serve as the electron donors to the neighbouring Co atoms.54 The presence of electronic interactions between adjacent Co and Ni atoms and metal–N species in Co–Ni–N–C supports the existence of Co–Ni–N coordination. Raman spectra and their fitting results further reveal changes in carbon–carbon (C–C) bonds due to the presence of various defect species (Fig. S9†). The D bands at 1350 cm−1 are intense for all catalysts due to the edge or defect of carbon rings, indicating the existence of strong disorder in the catalysts. The G bands at 1582 cm−1 originating from graphitized carbon–carbon bonds are broad, signifying low crystallinity within the samples. A highly broadened peak at ca. 2700 cm−1 was also observed, which can be assigned to the second order of the D band.55 Based on the integrated area, the ratio of the D band to G band for Co–N–C was calculated to be ID/IG = 0.992, which was similar to the corresponding ID/IG ratio for Ni–N–C (0.995) and Co–Ni–N–C (0.998). This suggests these three samples were comparable in the degree of graphitization. Raman analysis further confirmed the formation of graphitized carbon and the concomitant disorder species, which enhances CO2 ECR activity by serving as an electron transfer medium and hosting the metal–nitrogen active moieties.
:1, strongly indicating the formation of porphyrin-like Co–N3 species.51 The Co 2p peak at 780.0 eV obtained from Co–Ni–N–C (Fig. S8†) has a binding energy higher than that for Co(0) species at 778.2 eV, consistent with the accepted chemical shift theory of oxidized cobalt.52 The Ni 2p spectra for Co–Ni–N–C show that the Ni oxidation state is positioned between Ni(0) peak at 852.6 eV and Ni(II) peak at 855.6 eV, indicating the presence of Ni species that are partially oxidized.53 As evident in Fig. S8,† The Co 2p peak for Co–Ni–N–C is found shifted 0.6 eV to lower binding energy in comparison to Co–N–C. Additionally, the Ni 2p peak for Co–Ni–N–C shifts 0.2 eV toward higher binding energy compared to Ni–N–C. It can be interpreted from XPS results that Ni atoms in Co–Ni–N–C serve as the electron donors to the neighbouring Co atoms.54 The presence of electronic interactions between adjacent Co and Ni atoms and metal–N species in Co–Ni–N–C supports the existence of Co–Ni–N coordination. Raman spectra and their fitting results further reveal changes in carbon–carbon (C–C) bonds due to the presence of various defect species (Fig. S9†). The D bands at 1350 cm−1 are intense for all catalysts due to the edge or defect of carbon rings, indicating the existence of strong disorder in the catalysts. The G bands at 1582 cm−1 originating from graphitized carbon–carbon bonds are broad, signifying low crystallinity within the samples. A highly broadened peak at ca. 2700 cm−1 was also observed, which can be assigned to the second order of the D band.55 Based on the integrated area, the ratio of the D band to G band for Co–N–C was calculated to be ID/IG = 0.992, which was similar to the corresponding ID/IG ratio for Ni–N–C (0.995) and Co–Ni–N–C (0.998). This suggests these three samples were comparable in the degree of graphitization. Raman analysis further confirmed the formation of graphitized carbon and the concomitant disorder species, which enhances CO2 ECR activity by serving as an electron transfer medium and hosting the metal–nitrogen active moieties.
To gain a better understanding of the surface sorption and interaction with the reactants, catalyst surface properties were characterized with N2 sorption, diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), and temperature-programmed analyses. Reversible type I isotherms at 77 K (Fig. 3A) are given for the physisorption of N2 on the catalysts.56 At low relative pressures (P/P0 < 0.01), the N2 adsorption isotherm exhibits a rapid increase due to the saturation of micropores (<20 Å). As the relative pressure increased beyond 0.1, hysteresis is observed, and the increase in adsorption is attributed to the filling of mesopores (20 to 500 Å). The non-local density functional theory (NLDFT) was used to calculate the pore size distribution of the catalysts, and the results, shown in Fig. 3B, confirm the presence of both micropores and mesopores.57 A specific range of micropores (4.9 to 9.2 Å) can be found throughout the carbon phase despite minor distribution variations due to surface heterogeneity. Analysis of the N2 sorption isotherms by the Brunauer–Emmett–Teller (BET) method obtained a specific surface area value of 2095.1 m2 g−1 for the Co–N–C catalyst. A notable reduction in BET surface area was observed for Ni–N–C (599.5 m2 g−1) and Co–Ni–N–C (497.4 m2 g−1), possibly due to increased structural defect and disorder.58 The highly microporous structure provides a large adsorption capacity for reactant molecules and enhanced mass-transport properties.59 Furthermore, the extensive microporosity inside the catalysts hosts a large number of defect sites in the vicinity of micropores to facilitate reaction activation.60 We further investigated the adsorption behaviour of CO2 on the catalysts using DRIFTS to provide additional information about CO2 activation. As shown in Fig. 3C, CO2 adsorption produces vibration bands νm at 2340 cm−1 and νf at 2360 cm−1, which can be ascribed to the asymmetric stretching modes of CO2 gas adsorbed on metal sites and functional groups, respectively.61–63 The peak area is closely associated with the amount of adsorbed CO2 molecules and the CO2 adsorption dynamics were obtained by measuring the peak area in the wavenumber range from 2250 to 2400 cm−1 continuously over time. As shown in Fig. S10,† similar CO2 adsorption capacity was observed for the investigated catalysts while Co–N–C had the highest CO2 adsorption rate among all catalysts. We further evaluated the preferential CO2 adsorption site on the catalysts by measuring the ratio of peak area νm to νf. As exhibited in Fig. S10,† a higher νm to νf ratio was found for Co–Ni–N–C, suggesting that a greater proportion of CO2 molecules were adsorbed on the metal sites of Co–Ni–N–C. The dynamic behaviour of CO2 adsorption observed for Co–Ni–N–C implies that dispersed Co and Ni metal sites dominate the adsorption of CO2 over the functional group and potentially promote the CO2 activation process. Temperature-programmed desorption of carbon monoxide (CO-TPD) was performed to investigate the adsorption strength of intermediates. The desorption peak of CO was monitored and found to occur at 356.5 K and 366.3 K for Co–Ni–N–C and N–C catalysts, respectively, as shown in Fig. 3D. The shift of the desorption peak towards a lower temperature indicates that the CO adsorption strength on the Co–Ni–N–C catalyst is weaker than that on the N–C catalyst.64 CO desorption strength on catalysts affects CO2 reduction performance by altering reaction kinetics and surface intermediates.65 Introducing metalation to N–C catalysts offers controlled tuning of CO desorption strength, which directly influences CO2 reduction performance. Hydrogen temperature-programmed reduction (H2-TPR) measurements of Co–Ni–N–C and N–C catalysts, as shown in Fig. 3E, both feature a broad peak at around 500 °C, suggesting no presence of bulk metal oxides, which would otherwise display a reduction temperature peak below 400 °C.66 However, a slightly higher reduction temperature peak was observed for N–C due to a stronger interaction between a higher loading of nitrogen and the carbon matrix. Metalating N–C catalyst with Co and Ni maintains reducibility, ensuring stability in the reduction environment due to unaltered redox properties.
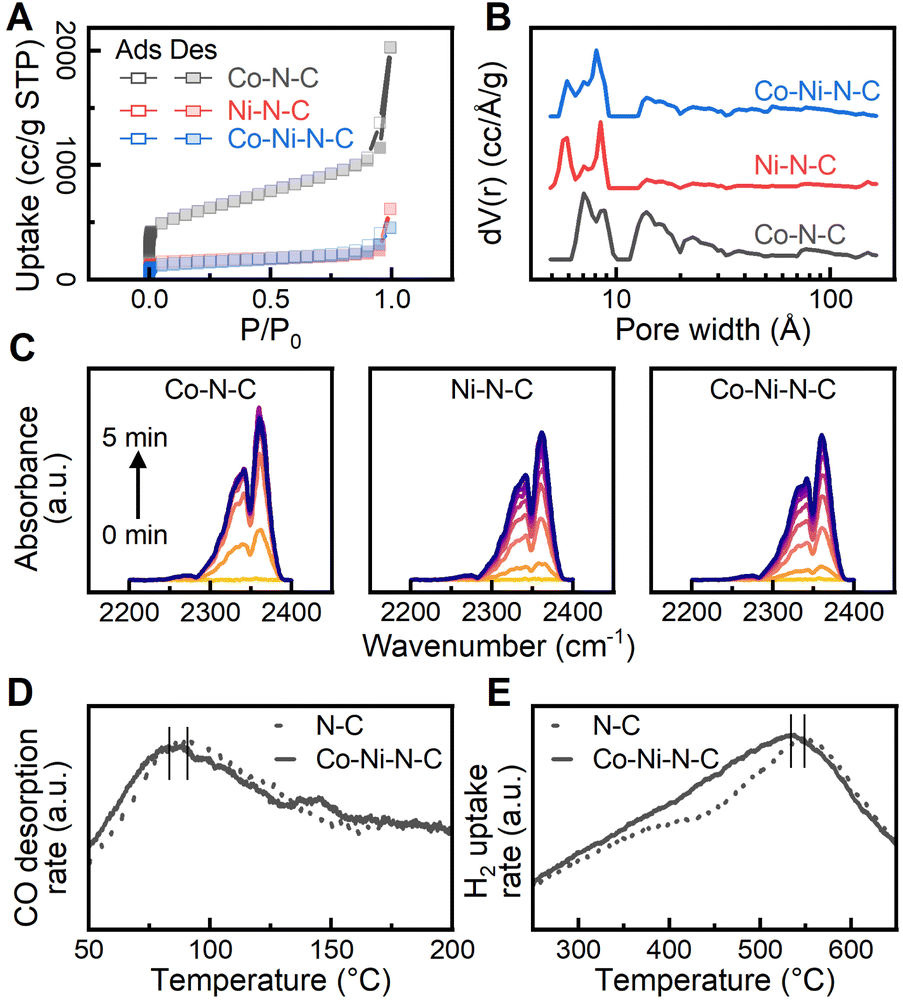 | ||
| Fig. 3 (A) Nitrogen adsorption and desorption isotherms at 77 K. Adsorption capacity is in units of cm3 g−1. Filled symbols correspond to adsorption and open symbols specify desorption. (B) Linear derivative pore size distributions of MNCs calculated based on a NLDFT slit-pore model. (C) Diffuse reflectance infrared spectra of CO2 adsorption on catalysts as a function of time measured at 295 K. CO2 adsorption peaks at 2342 and 2360 cm−1 corresponding to metal sites and functional groups are respectively observed. (D) Temperature-programmed desorption (TPD) measurements for CO desorption on N–C and Co–Ni–N–C. (E) H2 temperature-programmed reduction (TPR) on N–C and Co–Ni–N–C. | ||
The SEM images (Fig. 4A, B) showed decreased particle size after pyrolysis due to thermal shrinkage,67 while the particles still maintained the initial rhombic dodecahedron shape with sharp edges even after undergoing thermal treatment. A 52% reduction in particle size was identified while the uniformity of particle size distribution was retained. The smooth tetragonal facets of the pristine Co-ZIF-8 (Fig. 4A) underwent amorphization and reconstructed into rough and microporous surfaces of Co–N–C (Fig. 4B). Supplementary SEM images (Fig. S4 and S12†) further support observed morphology reconstruction trends attributed to thermal activation effects. As demonstrated in Fig. 4C, the transmission electron microscopy (TEM) image of Co–N–C showed no difference in phase, which further suggests the absence of metal particles. Fig. 4D shows a selected area electron diffraction (SAED) pattern acquired over Co–Ni–N–C. The diffusion halo and the lack of diffraction spots in the SAED pattern suggest the amorphous feature of the Co–Ni–N–C sample. The lack of crystalline phase in the SAED pattern is consistent with the XRD pattern in Fig. 2A, confirming the formation of spatially isolated metal species instead of metal clusters or particles throughout the structure. The homogeneous distribution of Co and Ni atoms in the nitrogen-doped carbon matrix was confirmed by TEM-EDS elemental mapping (Fig. 4E). The morphology and structure of the Co–Ni–N–C catalyst were further characterized through aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM), as illustrated in Fig. 4F. Atomic-resolution HAADF-STEM imaging of Co–Ni–N–C confirms that Co and Ni atoms are isolated and dispersed at the atomic scale on the amorphous carbon support, with no aggregation of metal particles (Fig. 4G). The uniformly distributed bright dots have a size of approximately 0.2nm, which corresponds well to the theoretical size of metal atoms. Additionally, paired metal atoms were clearly observed on carbon support as marked by red boxes in Fig. 4G. Intensity line scan profiles across the numbered regions 1 to 3 in Fig. 4H provide clear evidence for the formation of atomically dispersed Co–Ni pair sites with distances of 2.48 Å, matching well with the size of a hexagonal carbon ring.30 Furthermore, our computational analysis reveals a bond distance of approximately 2.4 Å for Ni–Co in the N3 motif, providing additional support for the formation of Co–Ni pair sites. In light of the above analysis, the as-synthesized Co–Ni–N–C catalysts are hosted in amorphous carbon support rich in defects, and characterized by adjacent atomic metal centres with complementary functionalities, making them suitable candidates for electrocatalytic applications.
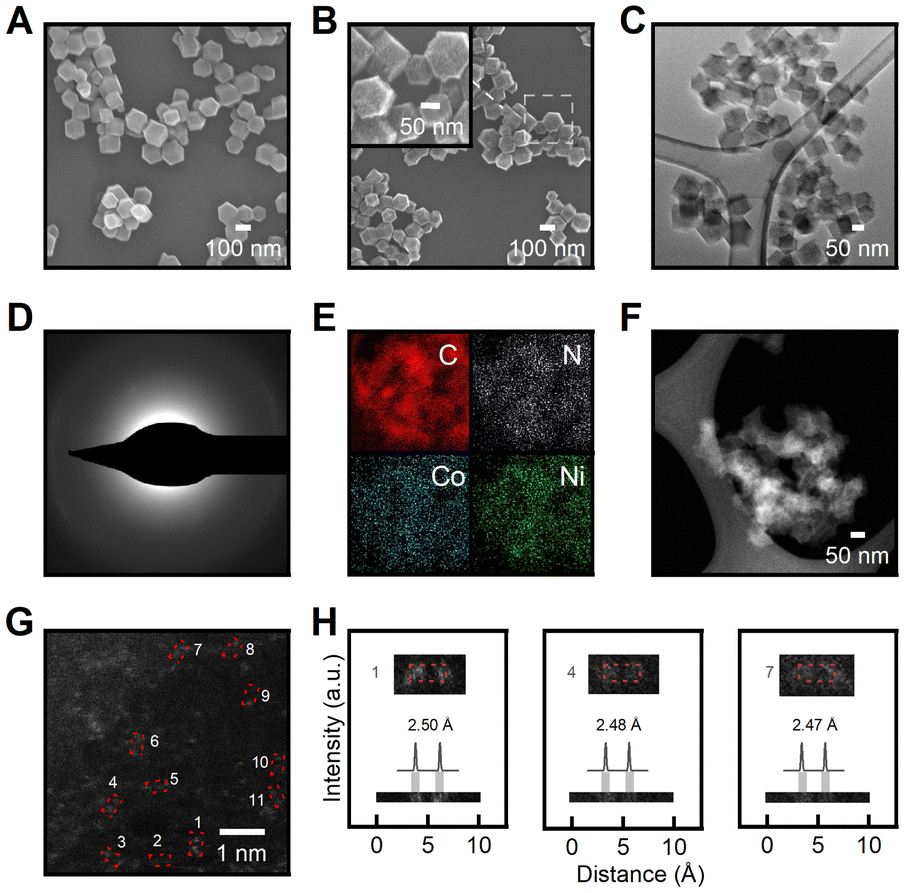 | ||
| Fig. 4 (A and B) SEM images of (A) Co-ZIF-8 and (B) Co–N–C. Inset shows a magnified SEM image of the square region highlighted by the white dashed lines. (C) TEM image of Co–N–C. (D) SAED pattern of Co–Ni–N–C. (E) TEM-EDS elemental map of Co–Ni–N–C. (F) Low-magnification HAADF-STEM image of Co–Ni–N–C. (G) Atomic-resolution HAADF-STEM image of Co–Ni–N–C. (H) Intensity line scan profiles across the numbered regions 1 to 3 in G. | ||
CO2 ECR performance
To evaluate CO2 ECR performance, the catalyst particles were deposited on conductive carbon fabrics to form the working electrode which was then submerged in 0.1 M KHCO3 aqueous electrolyte at room temperature and under atmospheric pressure. We first investigated the overall electrocatalytic performance of the catalysts by linear sweep voltammetry (LSV) in the presence and absence of CO2 (Fig. S14†). The hydrogen evolution reaction (HER) accounted for all the observed faradaic currents in CO2-free electrolytes, while in CO2-saturated electrolytes, the faradaic currents were divided between HER and CO2 ECR. As shown in Fig. S14,† electrochemically active metal species in catalysts enhanced the catalytic activity of both HER and CO2 ECR, as evidenced by substantial current enhancement compared to metal-free N–C. Under CO2-free electrolyte, the voltammogram trace of Co–Ni–N–C exhibited a departure from the baseline at about −0.71 V relative to a reversible hydrogen electrode (RHE). After saturating electrolytes with carbon dioxide, the catalytic onset potential was −0.53 V (RHE), indicating a prominent catalytic effect of Co–Ni–N–C on CO2 ECR over HER. As shown in Fig. 5A, Co–N–C exhibited a relatively low activity, delivering a total current density of −4.3 mA cm−2 at −0.95 V (vs. RHE) whereas Ni–N–C showed a significant increase in total current density. Notably, Co–Ni–N–C delivered a further increased current density of −49.3 mA cm−2 at −0.95 V (vs. RHE), which is 11.5 times higher than that of Co–N–C. As 1H nuclear magnetic resonance (NMR) analysis (Fig. S15†) revealed that no liquid-phase products were detected, our discussion on CO2 ECR performance focuses on gas-phase products. In controlled potential electrolysis conducted in 0.1 M CO2-saturated KHCO3 aqueous electrolyte under applied potential range between −0.5 V and −1.1 V (vs. RHE), CO and H2 were identified as the gas-phase products (Fig. 5B). The product distributions for Co–Ni–N–C in Fig. 5B suggest that CO production was significant at all electrode potentials, with minor H2 production due to the reduction of protons to gaseous hydrogen.68 The Co–Ni–N–C catalyst exhibits a potential-dependent behaviour with maximum faradaic efficiency for CO (FECO) observed at −0.8 V (vs. RHE), equivalent to an overpotential of −0.27 V. Carbon dioxide reduction suffered from sluggish reactivity at more positive potentials, whereas competing proton reduction was promoted at more negative potentials. At a potential of −0.8 V (vs. RHE), Co–Ni–N–C catalyzed CO production at a current density of 13.34 mA cm−2 (corresponding to 53.36 mA per milligram of catalyst), demonstrating a remarkable level of CO selectivity over H2 production (FECO = 94.1%). This result represents a substantial improvement over Co–N–C or Ni–N–C, for which the FECO values were merely 24.6% and 48.3% respectively under the same applied potentials. Comparing the current density for CO (jCO) (Fig. 5C), Co–Ni–N–C outperformed its counterparts with a CO production rate 24.7 and 2.9 times greater than that of Co–N–C and Ni–N–C respectively at an applied potential of −0.8 V (vs. RHE).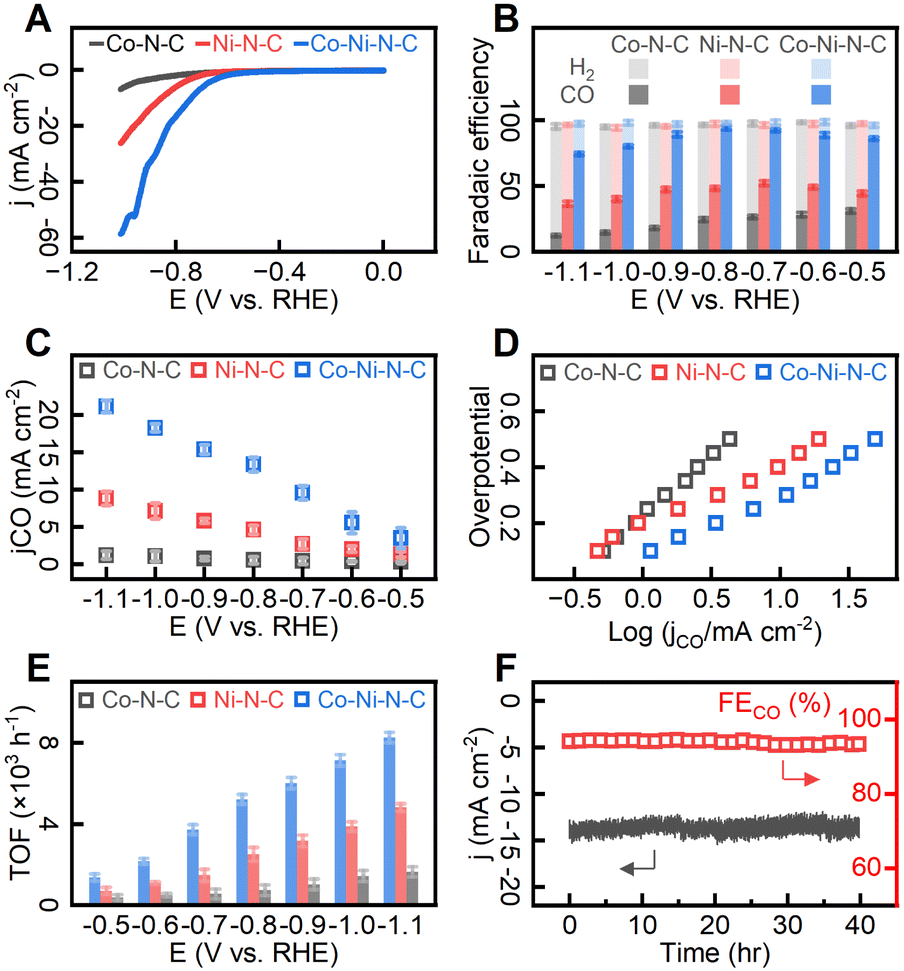 | ||
| Fig. 5 (A) The LSV plots of the tested catalysts for CO2 ECR. (B) Faradaic efficiency (FE) for CO2 ECR product CO and H2 for tested catalysts at corrected potentials ranging from −0.5 to −1.1 V (RHE). The highest FE for CO reaches 94.1%. (C) Partial current density towards CO production over the catalysts. (D) Tafel plots of different samples. (E) Comparison of the TOFs of the catalysts as a function of potential. (F) Stability tests of Co–Ni–N–C catalyst at −0.8 V (RHE). All measurements were performed at 30 sccm CO2 and 0.1 M KHCO3 using an FAS-50 membrane in an H-cell. Error bars are the standard deviation calculated from three independent measurements. | ||
To gain insight into the high activity of the Co–Ni–N–C catalyst, the electrochemically active surface area (ECSA, see S1†) determined from double-layer capacitance (CDL) and Nyquist plots are given in Fig. S16.† The double-layer capacitance measured on all samples fell within the range of 10.3 to 12.8 mF cm−2. The charge transfer resistance was also measured to be similar (ranging from 8.4 to 10.2 Ω) across the investigated catalysts. Further, the kinetics and mechanism of the electrochemical reactions on different sites were investigated by the Tafel analysis. Fig. 5D shows that the Tafel slopes for Co–N–C, Ni–N–C, and Co–Ni–N–C are 0.42, 0.22, and 0.23 mV dec−1, respectively. The Tafel slope of Co–Ni–N–C (0.23 mV dec−1) is slower than that of Co–N–C (0.42 mV dec−1), indicating enhanced initial electron transfer after the incorporation of Ni atoms.69 As shown in Fig. 5E, the calculated turnover frequency (TOF) values (see ESI S1†) per active site for CO production at −0.8 V (vs. RHE) in Co–Ni–N–C catalyst (5.22 × 103 h−1) showed a remarkable improvement over Co–N–C (0.74 × 103 h−1) and Ni–N–C (2.51 × 103 h−1). For a duration of 40 hours, the Co–Ni–N–C catalyst exhibited consistent catalytic activity (Fig. 5F) and generated over 489.6 mL CO gas (standard temperature and pressure, equivalent to 21.86 mmol) per milligram of catalyst. The FECO (ca. 94%) and jCO (ca. −13.5 mA cm−2). remained virtually unchanged for 40 hours of reaction, indicative of relatively high durability. As demonstrated in Table S1,† our Co–Ni–N–C catalyst exhibits performance on par with selected CO2 ECR catalysts. Based on the above analyses, the Co–Ni–N–C catalyst with paired metal sites demonstrates excellent electrocatalytic activity, selectivity, and stability toward CO2 ECR.
Density functional theory (DFT) calculations
To elucidate the influence of the coordination configuration of metal sites on CO2 ECR behaviour, we calculated the free energy diagram of the lowest-energy pathways for CO2 reduction into CO using density functional theory (DFT) (refer to the Computational Procedures section above and the Section S16† for more calculation details). All DFT calculations were carried out using the ORCA 5.0.4 suite of programs.42 In a simple CO2–H2O system, once adsorbed on the catalyst's active site, the CO2 molecule reacts with a proton and an electron to form COOH*, which is usually considered the initial intermediate for CO formation.70 As shown in Fig. 6A, the free energy change to generate COOH* from CO2 is endergonic, and the Co–Ni–N–C site emerges as the most favorable site for COOH* formation (0.227 eV). This validates the efficiency of a dual Co–Ni site in activating CO2. Additionally, Fig. S19 and S20† delve into the free energies associated with the hydrogen evolution reaction (HER). DFT calculations show that H* formation on dual Co–Ni site is energetically unfavourable, presenting an energy barrier of 1.00 eV. As elucidated by DFT calculations for COOH* and H* formation, dual Co–Ni metal sites not only facilitate CO2 activation but also impede hydrogen formation, thereby enhancing the overall efficiency of CO2 ECR. The conversion of COOH* to CO* through hydrogenation is exothermic on all three catalysts (Fig. 6A). However, the desorption process of CO* from Co–N–C and Co–Ni–N–C to produce CO is endergonic, with binding energies of 0.504 and 0.380 eV, respectively. In contrast, the free energy change for CO* desorption from Ni–N–C sites is negative, indicating spontaneous CO desorption and a binding energy of −0.193 eV.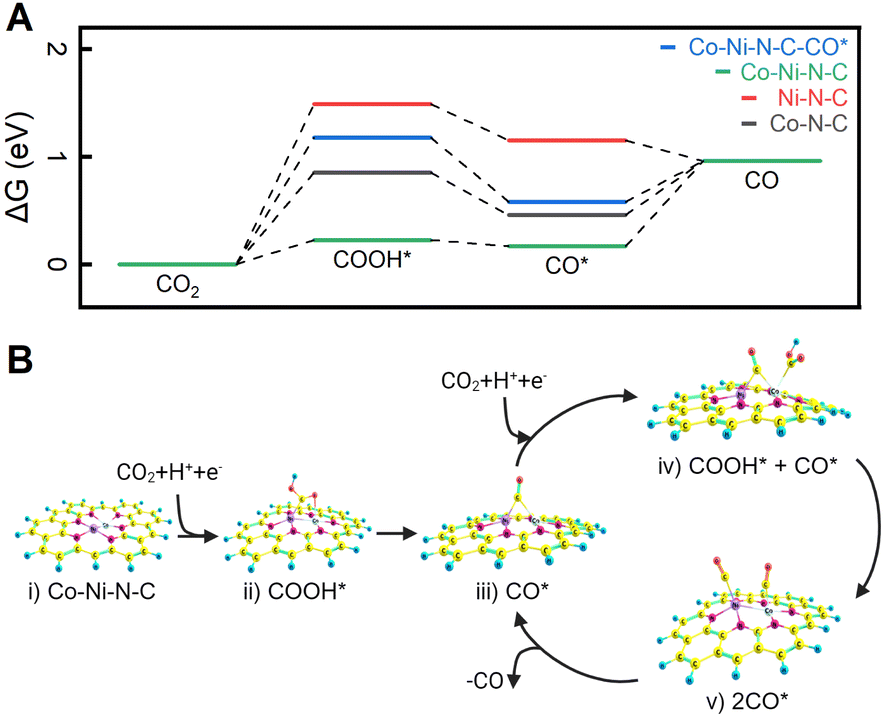 | ||
| Fig. 6 (A) Calculated free energy diagrams illustrating the lowest-energy pathways to CO. (B) Catalytic pathway on the dual Co–Ni site based on the optimized structures of adsorbed intermediates COOH* and CO*. | ||
There are two possible pathways for CO formation over a dual Co–Ni site. In the first pathway, CO* is desorbed from the Co–Ni site, resulting in the release of CO and the regeneration of the catalyst. The free energy diagram for this pathway is illustrated by the green plot in Fig. 6A. For the second possible reaction pathway, a dual Co–Ni site can reduce another CO2 while a CO is bound to this site, thereby forming COOH* + CO* intermediates. The free energy landscape for the second pathway is shown in Fig. 6A (blue plot). This possibility is further investigated in the mechanism for the Co–Ni–N–C site in Fig. 6B. Starting from the bare surface (Fig. 6B-i), the electroreduction of CO2 occurs to form the COOH* intermediate (Fig. 6B-ii). A further reduction to CO* leads to CO serving as a bridging ligand between the two metals (Fig. 6B-iii). Reduction of another CO2 to COOH* can occur either at the Co or Ni sites. When the reduction occurs at the Co atom, ΔG is 0.950 eV, as shown in Fig. 6B-iv. However, CO2 reduction to COOH* on the Ni atom requires a ΔG of 0.989 eV, slightly higher than the reduction on the Co atom. Further reduction of the COOH* moiety on the Co atom leads to a doubly adsorbed CO (Fig. 6B-v). One of the CO can then be desorbed, forming back the CO* intermediate (Fig. 6B-iii). In the second reaction pathway, DFT calculations show that free energies in the formation of COOH* and the desorption of CO* are optimized simultaneously. This is consistent with the experimental results that CO2 ECR is more active on Co–Ni–N–C than on Co–N–C and Ni–N–C.
Conclusions
In summary, we have successfully synthesized carbon-supported SACs, DASCs, and NPs from ZIFs with abundant metallic centres. The formation mechanism and the properties of the catalysts have been characterized by various analytical techniques. The Co–Ni–N–C catalyst with adjacent atomic Co–Ni pairs has been demonstrated to effectively activate CO2 ECR and selectively produce CO with a high FE of 95% at −0.8 V. The highly microporous structure of the catalysts provides a large adsorption capacity for reactant molecules and enhances mass-transport properties. The extensive microporosity inside the catalysts hosts a large number of defect sites in the vicinity of micropores to facilitate reaction activation. DFT studies also discuss the potential of a dual-metal nitrogen site for recycling the catalytic site, offering a strategy to create dual active site catalysts with complementary functionalities for electrocatalytic applications.Author contributions
The project was conceptualized by J. C. and W.-N. W. and was under the supervision of W.-N. W. J. C. prepared the catalysts and conducted the catalytic tests. Materials characterization and analysis were performed by J. C. with the assistance of H. E., L. Z., and R. W. The TPD, TPR, TEM, TEM-EDS, and SAED analyses were contributed by M. R. A., and AC-HAADF-STEM analysis was contributed by J. O. M. K. contributed to TGA and ICP-OES analyses, while DFT calculations were done by J. A. T. and S. H., under the supervision of K. U. L. The paper was written by J. C. with input from all authors. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial supports from the National Science Foundation (CMMI-1727553) and Department of Defence (W911NF-23-1-0196) are gratefully acknowledged. Md Robayet Ahasan and Ruigang Wang acknowledge the financial support from the National Science Foundation (CBET-1856729). AC-HAADF-STEM was performed in the Sensitive Instrument Facility at Ames Laboratory, which is operated for the U.S. DOE by Iowa State University under Contract No. DE-AC02-07CH11358. The DFT computational part used resources from the National Energy Research Scientific Computing Center, a DOE Office of Science User Facility supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 using NERSC award BES-ERCAP0020838. High-Performance Computing resources provided by the High-Performance Research Computing (HPRC) Core Facility at Virginia Commonwealth University (https://chipc.vcu.edu) were also used for conducting the research reported in this work.References
- D.-H. Nam, P. De Luna, A. Rosas-Hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- J. Jiao, R. Lin, S. Liu, W.-C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S.-F. Hung, H. M. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222–228 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem, 2018, 2, 65–81 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- H. Fei, J. Dong, M. J. Arellano-Jiménez, G. Ye, N. Dong Kim, E. L. G. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. J. Yacaman, P. M. Ajayan, D. Chen and J. M. Tour, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- G. Li, Y. Zheng, G. Hu, B. Chen, Y. Gu, J. Yang, H. Yang, F. Hu, C. Li and C. Guo, Small, 2023, 19, 2301540 CrossRef CAS PubMed.
- H. Liu, J. Li, F. Du, L. Yang, S. Huang, J. Gao, C. Li and C. Guo, Green Energy Environ., 2023, 8, 1619–1629 CrossRef CAS.
- C. Wang, Z. Liu, L. Dong, F. Du, J. Li, C. Chen, R. Ma, C. Li and C. Guo, J. Power Sources, 2023, 556, 232523 CrossRef CAS.
- K. Liu, J. Fu, Y. Lin, T. Luo, G. Ni, H. Li, Z. Lin and M. Liu, Nat. Commun., 2022, 13, 2075 CrossRef CAS PubMed.
- H. Zou, W. Rong, S. Wei, Y. Ji and L. Duan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 29462–29468 CrossRef CAS PubMed.
- L. Luo, J. Luo, H. Li, F. Ren, Y. Zhang, A. Liu, W.-X. Li and J. Zeng, Nat. Commun., 2021, 12, 1218 CrossRef CAS.
- Z. Yin, J. Yu, Z. Xie, S.-W. Yu, L. Zhang, T. Akauola, J. G. Chen, W. Huang, L. Qi and S. Zhang, J. Am. Chem. Soc., 2022, 144, 20931–20938 CrossRef CAS PubMed.
- Z. Luo, Z. Yin, J. Yu, Y. Yan, B. Hu, R. Nie, A. F. Kolln, X. Wu, R. K. Behera, M. Chen, L. Zhou, F. Liu, B. Wang, W. Huang, S. Zhang and L. Qi, Small, 2022, 18, 2107799 CrossRef CAS PubMed.
- S. K. Kaiser, Z. Chen, D. Faust Akl, S. Mitchell and J. Pérez-Ramírez, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS.
- S. Wang, L. Wang, D. Wang and Y. Li, Energy Environ. Sci., 2023, 16, 2759–2803 RSC.
- S. Mitchell and J. Pérez-Ramírez, Nat. Commun., 2020, 11, 4302 CrossRef CAS PubMed.
- C. Xia, Y. Qiu, Y. Xia, P. Zhu, G. King, X. Zhang, Z. Wu, J. Y. Kim, D. A. Cullen, D. Zheng, P. Li, M. Shakouri, E. Heredia, P. Cui, H. N. Alshareef, Y. Hu and H. Wang, Nat. Chem., 2021, 13, 887–894 CrossRef CAS PubMed.
- S. Liang, L. Huang, Y. Gao, Q. Wang and B. Liu, Adv. Sci., 2021, 8, 2102886 CrossRef CAS PubMed.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- Q. He, D. Liu, J. H. Lee, Y. Liu, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Angew. Chem., Int. Ed., 2020, 59, 3033–3037 CrossRef CAS.
- S. Zhang, M. Hou, Y. Zhai, H. Liu, D. Zhai, Y. Zhu, L. Ma, B. Wei and J. Huang, Small, 2023, 2302739 CrossRef CAS.
- C. Hu, Y. Wang, J. Chen, H.-F. Wang, K. Shen, K. Tang, L. Chen and Y. Li, Small, 2022, 18, 2201391 CrossRef CAS PubMed.
- W. H. Ren, X. Tan, W. F. Yang, C. Jia, S. M. Xu, K. X. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS.
- T. Cui, Q. Liu and S. Chen, Energy Technol., 2023, 11, 2201456 CrossRef CAS.
- Y. Gao, B. Liu and D. Wang, Adv. Mater., 2023, 35, 2209654 CrossRef CAS PubMed.
- M. Jafarzadeh and K. Daasbjerg, ACS Appl. Energy Mater., 2023, 6, 6851–6882 CrossRef CAS.
- K. Liu, J. Li, Y. Liu, M. Wang and H. Cui, J. Energy Chem., 2023, 79, 515–534 CrossRef CAS.
- L. Liu, X. Wu, F. Wang, L. Zhang, X. Wang, S. Song and H. Zhang, Chem.–Eur. J., 2023, 29, e202300583 CrossRef CAS PubMed.
- M. Zhou, W. Kong, M. Xue, H. Li, M. A. Khan, B. Liu, F. Lu and X. Zeng, Catal. Sci. Technol., 2023, 13, 4615–4634 RSC.
- R. Li and D. Wang, Adv. Energy Mater., 2022, 12, 2103564 CrossRef CAS.
- J. Pei, T. Wang, R. Sui, X. Zhang, D. Zhou, F. Qin, X. Zhao, Q. Liu, W. Yan, J. Dong, L. Zheng, A. Li, J. Mao, W. Zhu, W. Chen and Z. Zhuang, Energy Environ. Sci., 2021, 14, 3019–3028 RSC.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed.
- T. Li, Y. Yao, Z. Huang, P. Xie, Z. Liu, M. Yang, J. Gao, K. Zeng, A. H. Brozena, G. Pastel, M. Jiao, Q. Dong, J. Dai, S. Li, H. Zong, M. Chi, J. Luo, Y. Mo, G. Wang, C. Wang, R. Shahbazian-Yassar and L. Hu, Nat. Catal., 2021, 4, 62–70 CrossRef CAS.
- H. Zhou, Y. Zhao, J. Gan, J. Xu, Y. Wang, H. Lv, S. Fang, Z. Wang, Z. Deng, X. Wang, P. Liu, W. Guo, B. Mao, H. Wang, T. Yao, X. Hong, S. Wei, X. Duan, J. Luo and Y. Wu, J. Am. Chem. Soc., 2020, 142, 12643–12650 CrossRef CAS PubMed.
- X. Li, A.-E. Surkus, J. Rabeah, M. Anwar, S. Dastigir, H. Junge, A. Brückner and M. Beller, Angew. Chem., Int. Ed., 2020, 59, 15849–15854 CrossRef CAS PubMed.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12, e1606 CrossRef.
- F. Neese, J. Comput. Chem., 2023, 44, 381–396 CrossRef CAS PubMed.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- Y. Gao, Z. Han, S. Hong, T. Wu, X. Li, J. Qiu and Z. Sun, ACS Appl. Energy Mater., 2019, 2, 6071–6077 CrossRef CAS.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- R. Zhang, Z. Liu, S. Zheng, L. Wang, L. Zhang and Z.-A. Qiao, Adv. Mater., 2023, 2209635 CrossRef CAS PubMed.
- M. Ferrandon, A. J. Kropf, D. J. Myers, K. Artyushkova, U. Kramm, P. Bogdanoff, G. Wu, C. M. Johnston and P. Zelenay, J. Phys. Chem. C, 2012, 116, 16001–16013 CrossRef CAS.
- D. H. Lee, W. J. Lee, W. J. Lee, S. O. Kim and Y.-H. Kim, Phys. Rev. Lett., 2011, 106, 175502 CrossRef PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- L. Qiu, F. Liu, L. Zhao, W. Yang and J. Yao, Langmuir, 2006, 22, 4480–4482 CrossRef CAS PubMed.
- J. S. Park, A. Reina, R. Saito, J. Kong, G. Dresselhaus and M. S. Dresselhaus, Carbon, 2009, 47, 1303–1310 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- G. Kupgan, T. P. Liyana-Arachchi and C. M. Colina, Langmuir, 2017, 33, 11138–11145 CrossRef CAS PubMed.
- R. Yuan, Y. Dong, R. Hou, L. Shang, J. Zhang, S. Zhang, X. Chen and H. Song, Chem. Eng. J., 2023, 454, 140418 CrossRef CAS.
- P. Sazama, J. Pastvova, C. Rizescu, A. Tirsoaga, V. I. Parvulescu, H. Garcia, L. Kobera, J. Seidel, J. Rathousky, P. Klein, I. Jirka, J. Moravkova and V. Blechta, ACS Catal., 2018, 8, 1779–1789 CrossRef CAS.
- B. Chen, D. Wang, B. Zhang, X. Zhong, Y. Liu, J. Sheng, Q. Zhang, X. Zou, G. Zhou and H.-M. Cheng, ACS Nano, 2021, 15, 9841–9850 CrossRef CAS.
- R. Roque-Malherbe, R. Polanco-Estrella and F. Marquez-Linares, J. Phys. Chem. C, 2010, 114, 17773–17787 CrossRef CAS.
- T. Iwasita and F. C. Nart, Prog. Surf. Sci., 1997, 55, 271–340 CrossRef CAS.
- N. J. Firet and W. A. Smith, ACS Catal., 2017, 7, 606–612 CrossRef CAS.
- L. Lin, H. Li, Y. Wang, H. Li, P. Wei, B. Nan, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2021, 60, 26582–26586 CrossRef CAS PubMed.
- R. Reske, H. Mistry, F. Behafarid, B. Roldan Cuenya and P. Strasser, J. Am. Chem. Soc., 2014, 136, 6978–6986 CrossRef CAS PubMed.
- R. L. Oliveira, M. C. Ben Ghorbel, S. Praetz, D. Meiling, C. Schlesiger, R. Schomäcker and A. Thomas, ACS Sustain. Chem. Eng., 2020, 8, 11171–11182 CrossRef CAS.
- X. Wan, X. Liu, Y. Li, R. Yu, L. Zheng, W. Yan, H. Wang, M. Xu and J. Shui, Nat. Catal., 2019, 2, 259–268 CrossRef CAS.
- Z. Yan, J. L. Hitt, Z. Zeng, M. A. Hickner and T. E. Mallouk, Nat. Chem., 2021, 13, 33–40 CrossRef CAS PubMed.
- Z. Zeng, L. Y. Gan, H. Bin Yang, X. Su, J. Gao, W. Liu, H. Matsumoto, J. Gong, J. Zhang, W. Cai, Z. Zhang, Y. Yan, B. Liu and P. Chen, Nat. Commun., 2021, 12, 4088 CrossRef CAS PubMed.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta05654f |
| This journal is © The Royal Society of Chemistry 2024 |