
Mechanism and preparation research of binary heteroatom co-doped (X = N, S, P) platinum/carbon black electrocatalysts for an enhanced oxygen reduction reaction via a one-pot pyrolysis method†
Fengning
Bai
ab,
Yantong
Zhang
b,
Dongyu
Hou
ab,
Jian
Chen
ab,
Fanming
Meng
ab,
Michael K. H.
Leung
 c,
Ling
Zhou
d,
Yingjie
Zhang
b,
Chengxu
Zhang
*ab,
Wutao
Wang
*e and
Jue
Hu
*a
c,
Ling
Zhou
d,
Yingjie
Zhang
b,
Chengxu
Zhang
*ab,
Wutao
Wang
*e and
Jue
Hu
*a
aFaculty of Metallurgical and Energy Engineering, Kunming University of Science and Technology, Kunming 650093, China. E-mail: chxzhang@kust.edu.cn
bNational and Local Joint Engineering Research Center for Lithium-ion Batteries and Materials Preparation Technology, China
cAbility R&D Energy Research Centre, School of Energy and Environment, City University of Hong Kong, Hong Kong, China. E-mail: mkh.leung@cityu.edu.hk
dCity College, Kunming University of Science and Technology, Kunming 650093, China
eLuXi KuoBo Precious Metals Co., Ltd, Honghe, 651400, China. E-mail: 642329529@qq.com
First published on 22nd November 2023
Abstract
Due to the depletion of fossil fuels and environmental concerns, hydrogen fuel cells and proton exchange membrane fuel cells emerge as vital alternatives for sustainable energy. Currently, there is a lack of comprehensive understanding and experimental validation regarding the effects of different heteroatom dopants (e.g., N, P, and S) and their combinations on platinum-based catalysts. In this study, a simple one-pot synthesis approach is employed to synthesize Pt-based catalysts supported on heteroatom-doped carbon black. The oxygen reduction performance of these catalysts is evaluated, and the influence of pyrolysis temperature, Pt loading, and the type and amount of heteroatom dopants on the distribution and catalytic activity of Pt in the catalysts is thoroughly investigated. Experimental and theoretical calculations reveal that the incorporation of three types of heteroatoms, namely N, S, and P, exerts varying degrees of impact on the composition and particle size of Pt-based catalysts. The Eo and E1/2 of the Pt/BP-N250S250-900-30% catalyst were found to be 0.962 V vs. RHE and 0.821 V vs. RHE, respectively, surpassing those of other N, S, and P co-doped catalysts, indicating enhanced activity due to N-doping. Additionally, the S-doped Pt/BP-N250S250-900-30% catalyst exhibits superior stability, with only a 40 mV decrease in E1/2 after 5000 cycles of CV testing compared to a 52 mV decrease observed for the commercial Pt/C catalyst. However, the presence of platinum–phosphorus species results in adverse effects on the oxygen reduction process. This facile one-pot synthesis approach for heteroatom-doped carbon supported Pt-based catalysts offers new insights for the scalable production of fuel cell catalysts.
Introduction
With the improvement of living standards and rapid social development, the depletion of Earth's resources, particularly fossil fuels such as coal, oil, and natural gas, has become a pressing issue, leading to unprecedented energy challenges and severe environmental problems.1–4 In response to this problem, the concept of the “dual-carbon” strategy has been proposed by governments, aiming to achieve peak carbon emissions by 2030 and carbon neutrality by 2060. New energy vehicles and hydrogen-powered systems, specifically proton exchange membrane fuel cells (PEMFCs), have emerged as primary alternatives to fossil fuels.3,5 Consequently, hydrogen fuel cells and PEMFCs have witnessed rapid development. PEMFCs are widely recognized as the preferred energy source for electric vehicles and stationary power stations due to their low operating temperature, quick startup, high power density, simple structure, and convenient operation.6,7However, the currently employed PEMFC catalysts predominantly rely on Pt-based catalysts, which are hindered by their high cost and poor stability, severely limiting the commercialization of PEMFCs.8 Therefore, there is an urgent need to develop efficient, stable, and cost-effective catalysts for the oxygen reduction reaction (ORR) at the fuel cell cathode. The ORR is a crucial reaction in fuel cells, but the sluggish kinetics associated with it hampers the improvement of fuel cell conversion efficiency.9 Commercially available PEMFCs typically utilize Pt-based catalysts;10,11 however, their poor stability, coupled with the scarcity and high cost of the precious metal Pt, necessitates research into enhancing the stability of Pt-based catalysts and reducing the content of Pt in the catalyst. This area has received considerable attention in recent years. For instance, Alipour Moghadam Esfahani Reza et al.12 successfully synthesized a Pt/TNTS-Mo electrocatalyst through a modified polyol method. In this catalyst, the sub-oxide species on the carrier surface reduced the OH− adsorption on Pt, thereby improving its ORR activity and stability.
Heteroatom doping has proven to be an effective approach for enhancing the activity and stability of Pt catalysts. EunAe Cho's research group at the Korea Advanced Institute of Science and Technology13 reported the synthesis of a Pt-based catalyst (Pt1/NBP) using nitrogen-doped activated carbon (Black Pearl 2000, NBP) as the support, employing a hydrothermal ethanol reduction method. Through further high-temperature pyrolysis, the researchers successfully introduced melamine as a nitrogen source, altering the coordination structure of Pt and forming single-atom catalysts (Pt1@Pt/NBP). In this catalyst, 47.8% of Pt atoms existed in an isolated form on the carbon support, exhibiting excellent dispersion. Physical characterization and density functional theory (DFT) calculations demonstrated that the doped nitrogen atoms effectively anchored the Pt atoms, promoting their complete dispersion and forming active single-atom sites for efficient four-electron ORR pathways.
A mild autocatalytic reaction method was used to transform NiFe-ANR oxide nanoreactors into efficient OER electrocatalysts.14 The experimental results demonstrate that this catalyst exhibits excellent electrocatalytic activity and stability, with current density and oxygen production rates 2.5 and 1.8 times higher than those of commercial RuO2 electrocatalysts, respectively. This method provides new application prospects in the fields of renewable energy and hydrogen production. Qi Hu's research group15 discovered through density functional theory (DFT) calculations that the d-band center of Ru clusters shifts downward with increasing particle size, leading to reduced adsorption strength for H2O and H*. However, the smallest Ru19 cluster possesses the highest d-band center and hydrolysis ability, with a hydrolysis energy barrier of only 0.03 eV, far lower than that of other Ru clusters and single-atom Ru. Testing the alkaline hydrolysis performance of different Ru clusters and commercial Pt/C in 1 M KOH solution revealed that the Ru-1.0 cluster performed best, requiring only 13 mV of overpotential at 10 mA cm−2, 25 mV lower than that required by Pt/C. Additionally, the Ru-1.0 cluster displayed the smallest Tafel slope (25.3 mV dec−1) and the highest turnover frequency (43.3 s−1), indicating its fast reaction kinetics. This excellent performance is attributed to the strong hydrolysis ability resulting from the high d-band center of the Ru-1.0 cluster. The researchers also used density functional theory (DFT) to calculate the differential charge density of SnCo-HNT, revealing strong electronic transfer and synergistic effects between SnO2 and Co3O4.16 This effect causes electron deficiency in the Sn center and a high electron affinity on the catalyst surface, explaining why SnCo-HNT can effectively adsorb and stabilize key intermediates in CO2 reduction, thereby improving reaction rates and selectivity. Among them, SnO2 was identified as the main active center, while Co3O4 enhances the conductivity and synergistic effect of the composite material. By utilizing a biowaste lychee shell as a carbon source, a carrier with a rich microporous structure and carbon defects was prepared through a carbonization and activation process (DRC).17 Subsequently, subnanometer copper clusters (Cu clusters/DRC) were prepared on DRC through an impregnation–calcination method. The existence of the microporous structure and carbon defects can effectively prevent copper clusters from aggregating at high temperatures, resulting in an average diameter of approximately 1.0 nm. The strong metal–support interaction (SMSI) between Cu clusters and DRC can regulate the electronic structure of Cu clusters, further optimize the adsorption strength of *CO, and enhance the stability and antioxidant capacity of Cu clusters. The experimental results were verified using density functional theory (DFT) calculations.
Doping heteroatoms (N and P atoms) into the carrier material can significantly enhance the chemical reactivity by creating more active sites, facilitating faster electron transfer pathways and thereby improving the electrochemical catalytic activity.18,19 Recent studies by Peng Yin et al. revealed that the adhesion strength between metal nanoclusters and a sulfur-doped carbon support is strengthened due to metal–sulfur interface bonding, which greatly impedes metal atom diffusion and nanocluster migration. During the propane dehydrogenation process at 550 °C, sulfur-doped carbon-supported Pt nanoclusters with interface electronic effects demonstrated higher propylene selectivity and improved durability compared to sulfur-free carbon-supported catalysts.20 However, a comprehensive understanding of the effects of different heteroatoms and their combinations on doped carbon-supported platinum catalysts for the oxygen reduction reaction (ORR) is still lacking in both theoretical and experimental aspects. This study fills this gap and provides the necessary insights into these aspects.
Building upon previous studies, this work proposes a simple one-pot synthesis of heteroatom-doped Pt catalysts and identifies the most promising doping strategy with optimal performance. The article also analyzes the influence of different experimental conditions, such as pyrolysis temperature, Pt loading, types, and doping levels of heteroatoms, on the particle size and catalytic performance of the catalyst. Theoretical calculations confirm that dual-atom doping plays a significant role in performance enhancement and stability improvement. The study aims to investigate the effects of single and dual heteroatom doping on the performance of Pt catalysts, providing new insights for the design and development of high-performance fuel cell electrocatalysts.
Results and discussion
Physicochemical characterization of single heteroatom doped catalysts
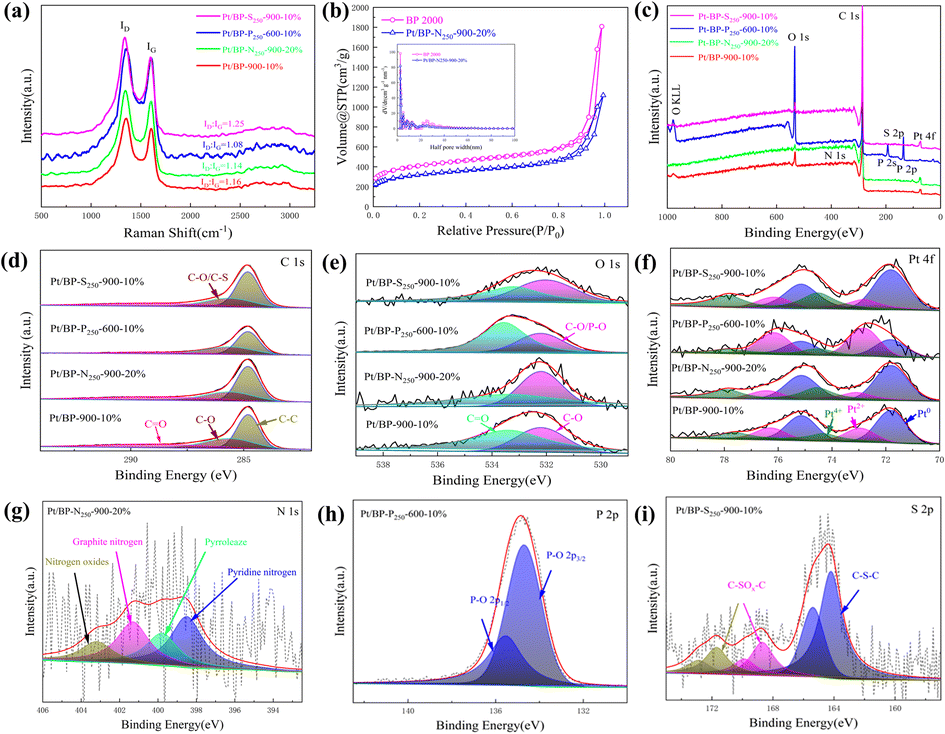
Upon comparing the XRD spectra of the four samples, it was observed that the Pt/BP-P250-600-10% sample exhibited additional diffraction peaks corresponding to elemental P and other impurities, in addition to the characteristic peaks of metallic Pt (Pt PDF#87-0964) (Fig. S1a and b†). This correlated with the inferior ORR performance of Pt/BP-P250-600-10% observed in electrochemical tests. It was found that at lower pyrolysis temperatures, compounds such as PtP2 and Pt–P were formed, which could be attributed to incomplete decomposition of ammonium dihydrogen phosphate, leading to phase changes and decreased conductivity of the catalyst. TEM observations (Fig. S2a and d†) revealed the presence of uniformly dispersed large particles (Fig. S2c†) and small particles (Fig. 1e–h) in the Pt/BP-N250-900-20% catalyst. The average particle size of the large particles was approximately 19.4 ± 0.16 nm, with a lattice spacing of about 0.197 nm corresponding to the Pt (200) crystal plane. The average particle size of the small particles was approximately 3.34 ± 0.02 nm, with a lattice spacing of about 0.226 nm corresponding to the Pt (111) crystal plane. EDS scans confirmed the presence of Pt nanoparticles successfully doped with N on the carbon support. The Pt/BP-900-10% catalyst did not exhibit large particles, and the lattice spacings corresponded to the Pt (111) and (200) crystal planes, with spacings of approximately 0.23 nm and 0.20 nm, respectively. The average particle size was 2.55 ± 0.02 nm (Fig. 4b), and the particles were predominantly composed of metallic Pt (Fig. 1a–d). In the Pt/BP-P250-600-10% catalyst, there were fewer but larger metal particles, with lattice spacings around 0.23 nm corresponding to the Pt (111) crystal plane, and the presence of Pt–P compounds (Fig. 1i–l), as indicated by the XRD peak for Pt–P compounds. The Pt/BP-S250-900-10% catalyst exhibited uniform particle distribution, with lattice spacings corresponding to the Pt (111) crystal plane at approximately 0.23 nm. The average particle size was 3.72 ± 0.24 nm (Fig. 4d), and the particles were mainly composed of metallic Pt, with successful incorporation of S into the support material (Fig. 1m–p). | ||
| Fig. 1 (a)–(d) TEM and EDS mapping of the Pt/BP-900-10% catalyst; (e)–(h) TEM and EDS mapping of the Pt/BP-N250-900-20% catalyst; (i)–(l) TEM and EDS mapping of the Pt/BP-P250-600-10% catalyst; (m)–(p) TEM and EDS mapping of the Pt/BP-S250-900-10% catalyst. | ||
ICP analysis revealed a Pt loading of 13.96% in the Pt/BP-N250-900-20% catalyst, which was lower than the theoretical loading of 20% (Table 1). This could be attributed to the loss of Pt(acac)2 during the pyrolysis process, resulting in incomplete retention of Pt on the carbon support. Raman spectroscopy analysis showed the presence of the D band (at approximately 1340 cm−1) and G band (at approximately 1590 cm−1) in all samples.21 The ID/IG ratio (Fig. 2a and Table 1) was used to evaluate the degree of graphitization and defect density of the carbon materials.22,23 The addition of melamine prior to pyrolysis improved the graphitization and conductivity of the carbon materials, with N atoms filling some of the defect sites on the carbon support. Upon adding ammonium dihydrogen phosphate and subsequent pyrolysis, higher graphitization was achieved; however, the formation of larger PtP2 compound particles occupied more defect sites. The addition of thiourea followed by pyrolysis resulted in the lowest degree of graphitization, leading to decreased conductivity of the catalyst. Nitrogen adsorption–desorption isotherms were used to measure the specific surface area and pore size distribution of the carbon materials in the samples. The specific surface area of Pt/BP-N250-900-20% was 1041.67 m2 g−1, which was lower than that of pure carbon material BP 2000 (1325.27 m2 g−1). The addition of melamine resulted in a decrease in specific surface area as N atoms occupied the active sites (Fig. 2b). Compared to BP 2000, Pt/BP-N250-900-20% lacked mesoporous structures of approximately 26 nm, but both samples exhibited mesoporous structures of approximately 2.6 nm, indicating that smaller mesopores could increase the number of active sites and enhance catalytic activity and stability.
| Samples | Pt content in ICP results (%) | I D/IG | Specific surface area (m2 g−1) |
|---|---|---|---|
| BP 2000 | — | — | 1325.27 |
| Pt/BP-900-10% | 4.99 | 1.16 | |
| Pt/BP-N250-900-20% | 13.96 | 1.14 | 1041.67 |
| Pt/BP-P250-600-10% | 1.94 | 1.08 | — |
| Pt/BP-S250-900-10% | 9.99 | 1.25 | — |
 | ||
| Fig. 2 (a) Raman spectra of catalysts Pt/BP-S250-900-10%, Pt/BP-P250-600-10%, Pt/BP-N250-900-20% and Pt/BP-900-10%; (b) nitrogen adsorption and desorption isotherms of BP 2000 and catalyst Pt/BP-N250-900-20% (the small figure shows the pore size distribution of both); (c) total XPS spectra of Pt/BP-900-10%, Pt/BP-N250-900-20%, Pt/BP-P250-600-10%, and Pt/BP-S250-900-10%; high-resolution spectra of samples Pt/BP-900-10%, Pt/BP-N250-900-20%, Pt/BP-P250-600-10%, and Pt/BP-S250-900-10%: (d) C 1s, (e) O 1s, (f) Pt 4f, (g) N 1s, (h) P 2p, and (i) S 2p. | ||
The elemental composition and oxidation states of the catalysts are shown in Fig. 2c. All four samples contain carbon, oxygen, and platinum elements, while additional specific elements are present in each sample depending on the added species. Semi-quantitative analysis (Table 2) showed that the Pt/BP-N250-900-20% sample had the highest platinum content, which matched the theoretical loading. However, this sample exhibited a lower nitrogen content, possibly due to more complete decomposition of melamine at 900 °C during the pyrolysis process. The successful doping of heteroatoms (N, P, and S) into the support was consistent with the TEM-EDS elemental mapping. Among them, the Pt/BP-P250-600-10% sample contained a small amount of nitrogen, likely a result of incomplete volatilization of residual nitrogen in ammonium dihydrogen phosphate.
| Samples | C 1s (at%) | O 1s (at%) | Pt 4f (at%) | N 1s (at%) | P 2p (at%) | S 2p (at%) |
|---|---|---|---|---|---|---|
| Pt/BP-900-10% | 94.97 | 4.78 | 0.25 | — | — | — |
| Pt/BP-N250-900-20% | 95.10 | 2.43 | 0.39 | 2.08 | — | — |
| Pt/BP-P250-600-10% | 57.54 | 33.53 | 0.11 | — | 8.83 | — |
| Pt/BP-S250-900-10% | 96.19 | 3.05 | 0.34 | — | — | 0.42 |
The C 1s spectra (Fig. 2d) of all four samples showed the presence of C–C (284.8 eV ± 0.3 eV), C–O (285.6 eV ± 0.3 eV), and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (289.0 eV ± 0.3 eV) peaks.24,25 The Pt/BP-S250-900-10% sample also exhibited a C–S bond peak.26,27 The Pt/BP-N250-900-20% sample, with a lower nitrogen content in the catalyst, did not show the presence of a C–N bond. The absence of a C–P bond in the Pt/BP-P250-600-10% sample could be attributed to the strong interaction between phosphorus and metallic Pt, without forming an interaction with the support. The O 1s spectra (Fig. 2e) of all four samples exhibited C–O (532.2 eV ± 0.3 eV) and CO (533.4 eV ± 0.3 eV) peaks.28,29 The Pt/BP-P250-600-10% sample also showed a P–O peak that overlapped with the C–O peak.30
O (289.0 eV ± 0.3 eV) peaks.24,25 The Pt/BP-S250-900-10% sample also exhibited a C–S bond peak.26,27 The Pt/BP-N250-900-20% sample, with a lower nitrogen content in the catalyst, did not show the presence of a C–N bond. The absence of a C–P bond in the Pt/BP-P250-600-10% sample could be attributed to the strong interaction between phosphorus and metallic Pt, without forming an interaction with the support. The O 1s spectra (Fig. 2e) of all four samples exhibited C–O (532.2 eV ± 0.3 eV) and CO (533.4 eV ± 0.3 eV) peaks.28,29 The Pt/BP-P250-600-10% sample also showed a P–O peak that overlapped with the C–O peak.30
The Pt 4f peaks (Fig. 2f) could be fitted into Pt0 (Pt 4f7/2: 71.8 eV ± 0.3 eV; Pt 4f5/2: 75.1 eV), Pt2+ (Pt 4f7/2: 72.8 eV ± 0.3 eV; Pt 4f5/2: 76.1 eV), and Pt4+ (Pt 4f7/2: 74.5 eV ± 0.3 eV; Pt 4f5/2: 77.8 eV) components,31–33 with varying proportions in different samples. The Pt/BP-N250-900-20% sample had the highest Pt0 content, followed by Pt4+, and the lowest Pt2+ content, possibly due to the presence of abundant Pt nanoparticles and the formation of PtN4 structures. The Pt/BP-P250-600-10% sample had the highest Pt2+ content, which could be attributed to the formation of PtP compounds, while Pt0 and Pt4+ were also present, indicating the existence of metallic Pt and PtP2 compounds. The Pt/BP-S250-900-10% sample had the highest Pt0 content, consistent with the XRD results, followed by Pt4+, possibly related to the presence of a small amount of PtS2.
The N 1s peaks of the Pt/BP-N250-900-20% sample could be divided into pyridinic nitrogen (398.5 eV ± 0.3 eV), pyrrolic nitrogen (399.8 eV ± 0.3 eV), graphitic nitrogen (401.0 eV ± 0.3 eV), and oxidized nitrogen (403.1 eV ± 0.3 eV) (Fig. 2g).34,35 Among them, pyridinic nitrogen and graphitic nitrogen on the carbon support have been proven to be excellent active sites for the ORR. The P 2p peaks of the Pt/BP-P250-600-10% sample corresponded to P–O bonds (P 2p3/2: 134.7 eV ± 0.3 eV; P 2p1/2: 135.5 eV ± 0.3 eV),36 which could be attributed to the higher content of phosphorus and oxygen in the catalyst (Fig. 2h). The S 2p peaks of the Pt/BP-S250-900-10% sample could be divided into C–S–C (S 2p3/2: 164.2 eV ± 0.3 eV; S 2p1/2: 165.4 eV) and two pairs of peaks corresponding to C–SOx–C (S 2p3/2: 168.7 eV ± 0.3 eV; S 2p1/2: 168.9 eV for C–SO2–C and S 2p3/2: 171.2 eV ± 0.3 eV; S 2p1/2: 172.4 eV for C–SO4–C),30,37 confirming the successful sulfur doping onto the carbon support (Fig. 2i).
Electrochemical testing of single heteroatom doped catalysts
Through electrochemical tests using linear sweep voltammetry (LSV) and cyclic voltammetry (CV), we evaluated the performance of three systems (Pt/BP-N, Pt/BP-P, and Pt/BP-S) as well as a commercial Pt/C (JM 20% wt% Pt) catalyst and included a reference sample, Pt/BP-900-10% (undoped), to validate the experimental methodology. First, by comparing the LSV results of BP, Pt/BP-900-10%, and commercial Pt/C, we found that the reference sample, Pt/BP-900-10%, exhibited superior ORR performance to pure BP (Fig. S7†), confirming the feasibility of the method. We conducted electrochemical tests to compare the performance of the Pt/BP-N, Pt/BP-P, and Pt/BP-S catalyst series. The influence of different pyrolysis temperatures, Pt loading amounts, and heteroatom doping levels on CV and LSV curves was examined. The optimal pyrolysis temperature for all three catalysts was determined to be 900 °C. Different Pt loading amounts were tested for electrochemical performance (Pt/BP-N series: 10%, 20%, 30%, and 40%; Pt/BP-P series and Pt/BP-S series: 5%, 10%, 20%, and 30%). At the optimal pyrolysis temperature and Pt loading amount, the optimization of heteroatom doping was performed. For the Pt/BP-N series, melamine (ammonium dihydrogen phosphate or thiourea) was added at 100 mg, 250 mg, and 500 mg, denoted as Pt/BP-N(SP)100, Pt/BP-N(SP)250, and Pt/BP-N(SP)500, respectively. Among the Pt/BP-N series, the catalyst with the best performance was Pt/BP-N250-900-20%, which exhibited the highest onset potential (Eo), half-wave potential (E1/2), and limiting current density (JL), showing superior ORR performance among the three catalysts in 0.5 M H2SO4 (Fig. S8†). Under the same platinum loading conditions, the half-wave potential of the catalyst Pt/BP-N250-900-20% doped with nitrogen (0.803 V vs. RHE) compared to that of Pt/BP-900-20% (0.722 V vs. RHE) shows a significant advantage, which also indicates that doping with nitrogen enhances the catalyst performance (Fig. S22†). In the Pt/BP-P series, the optimal catalyst was Pt/BP-P250-600-10% (Fig. S9†). In the Pt/BP-S series, the optimal catalyst was Pt/BP-S250-900-10% (Fig. S10†). Further comparison revealed that in the N2-saturated electrolyte, the Pt characteristic peak of the Pt/BP-N250-900-20% catalyst was the strongest, especially the (111) crystal plane peak of Pt. Its ORR performance surpassed that of commercial JM 20% Pt/C, exhibiting higher onset potential (Eo) and half-wave potential (E1/2). The addition of the P source in the Pt/BP-P series led to a decline in performance, while the addition of the S source in the Pt/BP-S series significantly increased the onset potential (Eo) of the catalyst, indicating that the inclusion of S enhanced the intrinsic activity of the catalyst. Moreover, the electron transfer number (n) and hydrogen peroxide yield (H2O2%) are important indicators for evaluating ORR catalysts. In 0.5 M H2SO4, the Pt/BP-N250-900-20% catalyst exhibited an average electron transfer number of 3.95 and an average hydrogen peroxide yield of 2.32%. This suggests that Pt/BP-N250-900-20% displayed an approximate 4-electron process in 0.5 M H2SO4, and its H2O2% was the lowest, further confirming its optimal ORR performance (Fig. 3g).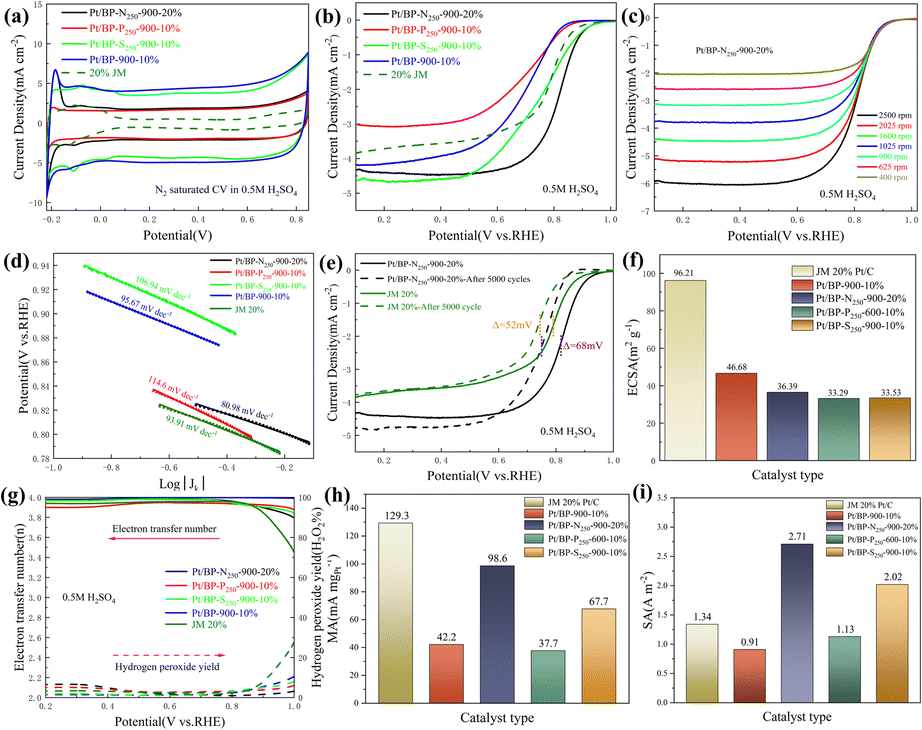 | ||
| Fig. 3 (a) CV diagrams of the obtained catalyst and Pt/C measured at 0.5 M H2SO4 saturated with N2; (b) LSV curves of the obtained catalyst and Pt/C at 0.5 M H2SO4 saturated with O2; (c) LSV curves of sample Pt/BP-N250-900-20% at different speeds; (d) Tafel slope diagram of the catalyst and Pt/C obtained; (e) comparison of the stability of Pt/BP-N250-900-10% and Pt/C; (f) ECSA of the resulting catalyst and Pt/C; (g) the electron transfer number and H2O2% of the obtained catalyst and Pt/C measured using an RRDE; (h) and (i) MA and SA corresponding to the acceleration plan and Pt/C at 0.8 V vs. RHE. | ||
The kinetic current density (Jk) reflects the catalyst's kinetic rate, and the LSV curve data at different rotation speeds can be calculated using the K–L equation. As the rotation speed increases, the ORR current density increases (Fig. 3c and S11a, b†), indicating that the ORR process is diffusion-controlled by O2. The Tafel slope reflects the degree of inhibition the catalyst encounters during the reaction process and can reflect the catalyst's reaction kinetics. From Fig. 3d, it can be observed that the Tafel slope of Pt/BP-N250-900-20% (80.98 mV dec−1) is significantly lower than that of other catalysts and commercial JM 20% Pt/C (93.91 mV dec−1), indicating better reaction kinetics of Pt/BP-N250-900-20%. The addition of melamine as an N dopant in the precursor is beneficial for improving the catalyst's reaction kinetics. In practical applications, catalysts not only require high activity but also high stability. The stability of the catalyst can be evaluated by comparing the LSV curves before and after 5000 cycles of CV testing (Fig. 3e). After 5000 cycles of CV testing, the E1/2 of Pt/BP-N250-900-20% decreased by 68 mV, while that of commercial JM 20% Pt/C decreased by 52 mV, indicating relatively poor stability of Pt/BP-N250-900-20% but still exhibiting good activity.
Furthermore, the ECSA, MA, and SA of the catalysts were compared. Commercial Pt/C had the highest ECSA, while Pt/BP-N250-900-20% has only 36.39 m2 g−1 (Fig. 3f), indicating good intrinsic activity of Pt/BP-N250-900-20%. At the same time, comparing the MA (Jk = 0.8 V vs. RHE) and SA (Jk = 0.8 V vs. RHE), it was found that the MA of Pt/BP-N250-900-20% was lower than that of commercial Pt/C, but its SA was the largest at 2.71 A m−2 (Fig. 3i), indicating that Pt/BP-N250-900-20% exhibited the highest current on a unit electrochemical active surface area and the maximum activity per unit area, further demonstrating its good catalytic activity. In summary, the Pt/BP-N250-900-20% catalyst exhibits good kinetic rate, reaction kinetics, and catalytic activity. Despite relatively poor stability, it still shows potential as a highly active oxygen reduction catalyst (Table 3).
| Samples | E o | E 1/2 | J L | n | H2O2% | ECSA | MA | SA |
|---|---|---|---|---|---|---|---|---|
| Pt/BP-N250-900-20% | 0.931 | 0.803 | 3.76 | 3.95 | 2.32 | 36.39 | 98.6 | 2.71 |
| Pt/BP-P250-900-10% | 0.868 | 0.705 | 3.04 | 3.92 | 3.76 | 33.29 | 37.7 | 1.13 |
| Pt/BP-S250-900-10% | 0.951 | 0.744 | 4.66 | 3.95 | 2.35 | 33.53 | 67.7 | 2.02 |
| Pt/BP-900-10% | 0.85 | 0.695 | 4.14 | 3.94 | 3.05 | 46.68 | 42.2 | 0.91 |
| JM 20% Pt/C | 0.924 | 0.785 | 3.63 | 3.91 | 4.58 | 96.21 | 129.3 | 1.34 |
Physicochemical characterization of double heteroatom doped catalysts
Based on the conclusions of the previous sections, we have inferred that a catalyst doped with both N and S atoms simultaneously may yield the best performance among dual heteroatom-doped catalysts. Furthermore, we will examine whether there exists a phenomenon similar to that in Section 1, i.e., whether doping P atoms will also affect the performance of platinum–phosphorus compounds. With the experience gained from Section 2, we can easily explore the approximate range of temperature, loading, and doping level suitable for screening catalysts with excellent performance. Additionally, we will also examine the influence of different factors on the performance of catalysts to further optimize experimental conditions. In this section, we will continue to delve deeper into the performance optimization of dual heteroatom-doped catalysts and its associated influencing factors.The phase compositions of the three samples were examined through XRD analysis (Fig. S5†), revealing significant differences. The XRD pattern of the Pt/BP-N250S250-900-30% catalyst only displayed diffraction peaks corresponding to metallic Pt (PDF#04-0802), while no diffraction peaks of metallic Pt were detected in the Pt/BP-N250P250-900-10% and Pt/BP-S250P250-900-30% catalysts. Conversely, the analysis indicated the presence of diffraction peaks attributed to PtP2 (PDF#03-1204) in these two catalysts, which was consistent with the experimental data from the previous section. However, there was a significant disparity in the intensity of the PtP2 peaks between the Pt/BP-N250P250-900-10% and Pt/BP-S250P250-900-30% catalysts. This disparity can be attributed to the difference in the theoretical Pt loading during catalyst synthesis. The Pt/BP-N250P250-900-10% catalyst had a theoretical Pt loading of 10%, resulting in a lower concentration of PtP2 and weaker peak intensity, making some faint diffraction peaks indistinct. On the other hand, the Pt/BP-S250P250-900-30% catalyst had a theoretical Pt loading of 30%, leading to a higher concentration of PtP2 and stronger peak intensity, making the diffraction peaks more prominent. Furthermore, it was observed that the addition of ammonium dihydrogen phosphate and other P sources to the catalyst precursors resulted in the transformation of metallic Pt to PtP compounds during the high-temperature pyrolysis process, thereby affecting its catalytic performance.
Through transmission electron microscopy (TEM) imaging (Fig. 4a and S21†), a significant number of metal nanoparticles were observed on the carbon support of the Pt/BP-N250S250-900-30% catalyst. Further magnification revealed that the metal nanoparticles in the catalyst did not exhibit significant aggregation. Statistical analysis determined an average particle size of approximately 4.33 nm for these metal nanoparticles (Fig. 4b and S21†). In high-resolution images, clear lattice fringes were observed, with variations in interplanar spacing. Measurements confirmed interplanar spacings of 0.23 nm, 0.20 nm, and 0.14 nm, corresponding to the (111), (200), and (220) crystallographic planes of metallic Pt, respectively (Fig. S6f, g and i†). This indicates that the metal nanoparticles in the catalyst consist of metallic Pt, and no lattice fringes corresponding to other metals were observed, consistent with the X-ray diffraction (XRD) analysis results (Fig. S5†). Additionally, a region was selected for energy-dispersive X-ray spectroscopy (EDS) mapping (Fig. 4d–i), which confirmed the presence of Pt metal particles in the catalyst. Furthermore, the carbon support of the catalyst exhibited uniform distribution of elements such as N, O, and S, demonstrating the successful doping of N and S heteroatoms onto the carbon support.
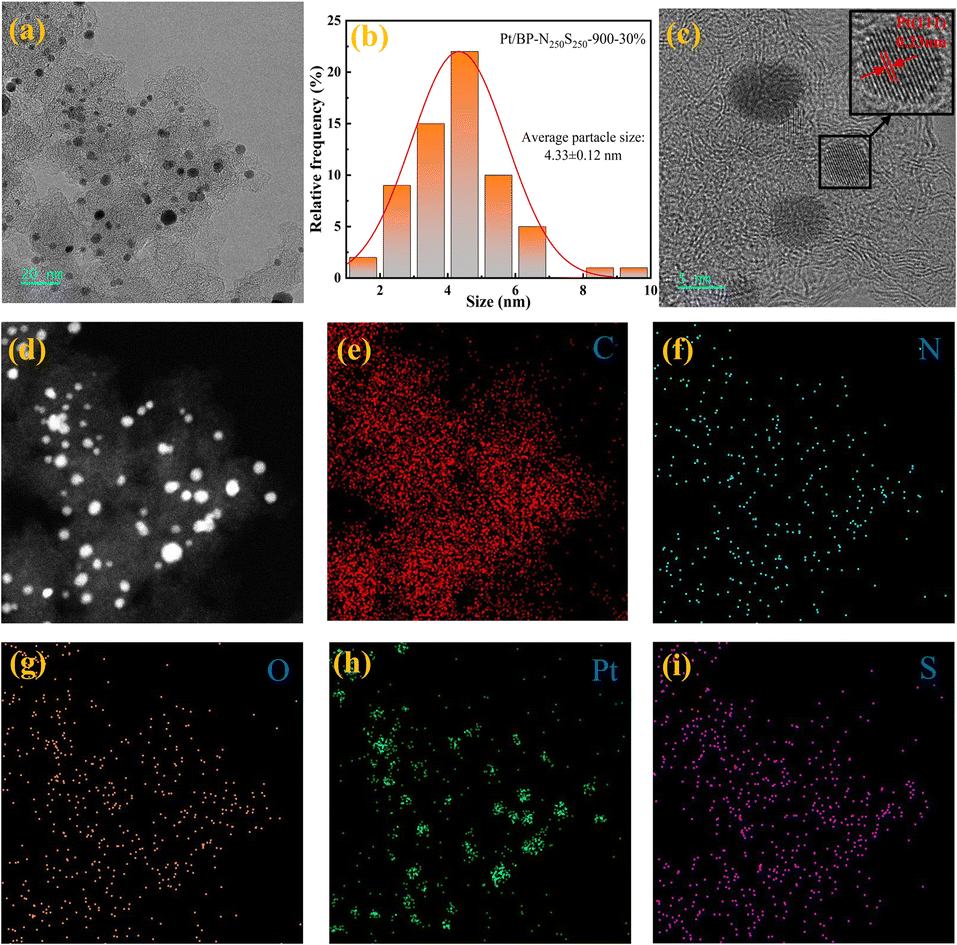 | ||
| Fig. 4 (a) TEM image of the Pt/BP-N250S250-900-30% catalyst at different magnifications; (b) particle size distribution of the Pt/BP-N250S250-900-30% catalyst; (c) HAADF-STEM images; (d)–(i) EDS elemental mapping of Pt/BP-N250S250-900-30%. | ||
Raman spectroscopy was employed to further analyze the graphitization degree and carbon defect density of the carbon materials in the catalyst. In all the catalysts prepared in our study, the D peak and G peak of graphitic carbon were detected at 1346 cm−1 and 1599 cm−1, respectively. The D peak represents the amorphous structure and defect level of the carbon materials in the catalyst, while the G peak represents the degree of graphitization. When comparing the ID/IG ratios of the catalysts (Fig. 5a), it was found that the Pt/BP-N250S250-900-30% catalyst had the smallest ID/IG ratio (ID/IG = 1.11), indicating a higher degree of graphitization in the carbon materials of this catalyst. Compared to Pt/BP-N250-900-20% from the previous section (ID/IG = 1.14), this demonstrates that the addition of thiourea (as a sulfur source) can increase the graphitization degree of the carbon materials, which has a favorable promoting effect on the catalytic ORR. Nitrogen adsorption–desorption isotherm measurements were performed to characterize the specific surface area and pore size distribution of the catalysts (Fig. 5b). The specific surface area of the carbon support used was 1325.27 m2 g−1, while the specific surface area of the prepared Pt/BP-N250S250-900-30% catalyst was 744.58 m2 g−1. This indicates that after the doping of N and S, the catalyst was successfully loaded onto the surface of the carbon support, resulting in a significant reduction in specific surface area. Meanwhile, no significant changes were observed in the pore structure, which maintained a mesoporous size of approximately 2.6 nm. The N and S elements occupied a substantial portion of the mesoporous structure in the carbon support, and no significant macropores were found in the catalyst (Table 4).
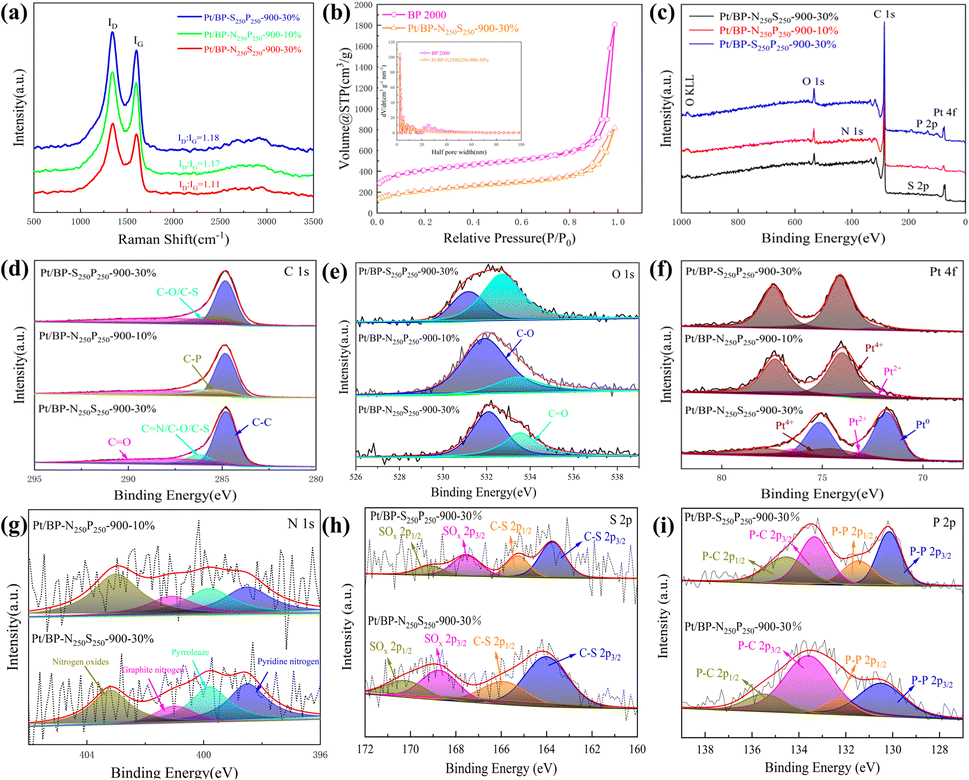 | ||
| Fig. 5 (a) Raman spectra of Pt/BP-N250S250-900-30%, Pt/BP-N250P250-900-10% and Pt/BP-S250P250-900-30% catalysts; (b) nitrogen adsorption and desorption isotherms of BP 2000 and catalyst Pt/BP-N250S250-900-30% (small figure shows pore size distribution of the two samples) (c)–(i) XPS spectrogram analysis of catalysts Pt/BP-N250S250-900-30%, Pt/BP-N250P250-900-10% and Pt/BP-S250P250-900-30%: (c) XPS total spectrum; (d) C 1s fine spectrum; (e) O 1s fine spectrum; (f) Pt 4f fine spectrum; (g) N 1s fine spectrum; (h) S 2p fine spectrum; (i) P 2p fine spectrum. | ||
| Samples | Pt content in ICP results (%) | I D/IG | Specific surface area (m2 g−1) |
|---|---|---|---|
| BP 2000 | — | — | 1325.27 |
| Pt/BP-900-10% | 4.99 | 1.16 | |
| Pt/BP-N250-900-20% | 13.96 | 1.14 | 1041.67 |
| Pt/BP-P250-600-10% | 1.94 | 1.08 | — |
| Pt/BP-S250-900-10% | 9.99 | 1.25 | — |
| Pt/BP-N250S250-900-30% | 20.45 | 1.11 | 744.58 |
| Pt/BP-N250P250-900-10% | 8.76 | 1.17 | 811.57 |
| Pt/BP-S250P250-900-30% | 20.76 | 1.18 | 225.97 |
Among them, two catalysts doped with nitrogen (Pt/BP-N250S250-900-30% and Pt/BP-N250P250-900-10%) showed distinct N 1s peaks that could be fitted into four smaller peaks (Fig. 4g). These peaks corresponded to pyridinic N (398.5 eV ± 0.3 eV), pyrrolic N (399.8 eV ± 0.3 eV), graphitic N (401.1 eV ± 0.3 eV), and oxidized N (403.2 eV ± 0.3 eV).34,35 Previous studies have shown that pyrrolic N has a promoting effect on the 4-electron process of the ORR, which indirectly indicates the superior ORR catalytic performance of this catalyst. In the two sulfur doping cases (Pt/BP-N250S250-900-30% and Pt/BP-S250P250-900-30%), the S 2p spectrum can be fitted with four peak shapes, corresponding to C–S (164.3 eV ± 0.3 eV) and SOx (167.5 eV ± 0.3 eV) respectively.38,39 A comparison (Table 5) revealed that the presence of nitrogen could effectively preserve the presence of sulfur, indirectly indicating that oxidized sulfur species did not have a positive effect on the ORR. For the two phosphorus doping cases (Pt/BP-N250P250-900-10% and Pt/BP-S250P250-900-30%), the P 2p spectrum can be fitted with four peak shapes, corresponding to P 2p3/2 (129.5 eV ± 0.3 eV), P 2p1/2 (130.3 eV ± 0.3 eV), and P–C (133.9 eV ± 0.3 eV).40 This may be attributed to the presence of a small amount of elemental phosphorus in the catalyst, which was present in trace amounts and could not be detected in XRD analysis.
| Samples | C 1s (at%) | O 1s (at%) | Pt 4f (at%) | N 1s (at%) | P 2p (at%) | S 2p (at%) |
|---|---|---|---|---|---|---|
| Pt/BP-900-10% | 94.97 | 4.78 | 0.25 | — | — | — |
| Pt/BP-N250-900-20% | 95.10 | 2.43 | 0.39 | 2.08 | — | — |
| Pt/BP-P250-600-10% | 57.54 | 33.53 | 0.11 | — | 8.83 | — |
| Pt/BP-S250-900-10% | 96.19 | 3.05 | 0.34 | — | — | 0.42 |
| Pt/BP-N250S250-900-30% | 94.10 | 3.80 | 1.00 | 0.22 | — | 0.88 |
| Pt/BP-N250P250-900-10% | 92.07 | 5.72 | 0.33 | 0.29 | 1.58 | — |
| Pt/BP-S250P250-900-30% | 90.71 | 6.04 | 0.79 | — | 2.26 | 0.19 |
By evaluating the performance of Pt catalysts supported on carbon black doped with three different single heteroatoms (N, P, and S), we identified the catalyst with the best performance. Among these catalysts, the N-doped carbon black-supported Pt catalyst exhibited excellent ORR catalytic performance in 0.5 M H2SO4. The outstanding performance of the Pt/BP-N250-900-20% catalyst can be attributed to several factors: first, the catalyst had a high porosity and large specific surface area. Although heteroatom doping led to a slight reduction in specific surface area, it still possessed a larger surface area compared to other carbon black catalysts, providing more active sites. Among the catalysts doped with two heteroatoms, Pt/BP-N250S250-900-30% demonstrated superior catalytic activity and stability due to the following reasons: firstly, Pt nanoparticles were uniformly distributed with an average particle size of approximately 3.26 nm and were present on three crystal facets of the catalyst. Secondly, the catalyst had a large specific surface area and a small mesoporous structure, providing more attachment sites for heteroatoms, increasing the number of active sites and enhancing catalytic performance. Lastly, N and S heteroatoms were successfully incorporated. The increased proportion of pyridinic and graphitic nitrogen may synergistically interact with Pt, forming more active Pt–N species. Additionally, the inclusion of sulfur and the formation of C–S bonds significantly enhanced the stability of the catalyst. Electronic paramagnetic resonance (EPR) further characterized the defect structure of the material (Fig. S22†). The three structures had the same Lorenz line width, indicating a similarity in the dopant radical species. The g-values of all three structures were 2.005, which corresponds to the unpaired electron on the π-conjugated carbon atom, confirming the formation of carbon vacancies. The approximate peak intensity suggested a similar concentration of carbon vacancies, providing strong evidence for our subsequent lateral comparison.
Electrochemical testing of double heteroatom doped catalysts
In the previous section, this section investigates the effects of doping amounts of two dopants (melamine, thiourea, and ammonium dihydrogen phosphate) in various combinations (each doped at 250 mg) and the loading amount of Pt (10% loading) on the electrochemical performance of the catalyst (Fig. 6a). Subsequently, the influence of the Pt loading amount and the influence of dual-atom doping amounts were explored (Fig. 6b). By examining the CV curves under N2 saturation, the CV curves of catalysts under N2 saturation at different loading amounts (Fig. 6c), LSV under O2 saturation, and comprehensive evaluation of kinetic rates, three samples with excellent ORR catalytic performance in N2-saturated 0.5 M H2SO4 were identified: Pt/BP-N250S250-900-30%, Pt/BP-N250P250-900-10%, and Pt/BP-S250P250-900-30% (Fig. S12–S14† and Table 6). By comparing their cyclic voltammetry (CV) and linear sweep voltammetry (LSV) curves under N2-saturated and O2-saturated conditions, it was found that in the CV curve of Pt/BP-N250S250-900-30%, the characteristic peak of the Pt (111) crystal plane was the most prominent, while Pt/BP-N250P250-900-10% had the largest surface area. This is consistent with the previous analysis which found that increasing the loading amount reduces the catalyst's surface area. On the LSV curve, Pt/BP-N250S250-900-30% exhibited the best catalytic performance (Eo = 0.962 V vs. RHE and E1/2 = 0.821 V vs. RHE), significantly outperforming Pt/BP-N250P250-900-10% (Eo = 0.862 V vs. RHE and E1/2 = 0.753 V vs. RHE), Pt/BP-S250P250-900-30% (Eo = 0.814 V vs. RHE and E1/2 = 0.664 V vs. RHE), and commercial JM 20% Pt/C (Eo = 0.924 V vs. RHE and E1/2 = 0.785 V vs. RHE). Moreover, the catalytic activity of Pt/BP-N250S250-900-30% was superior to that of Pt/BP-N250-900-20% in the previous section (Eo = 0.931 V vs. RHE and E1/2 = 0.803 V vs. RHE), further confirming the effectiveness of S-doping in enhancing catalytic activity. We also compared the catalytic performance of the three catalysts under the same conditions (Fig. S20†), which is clearly consistent with our conclusions.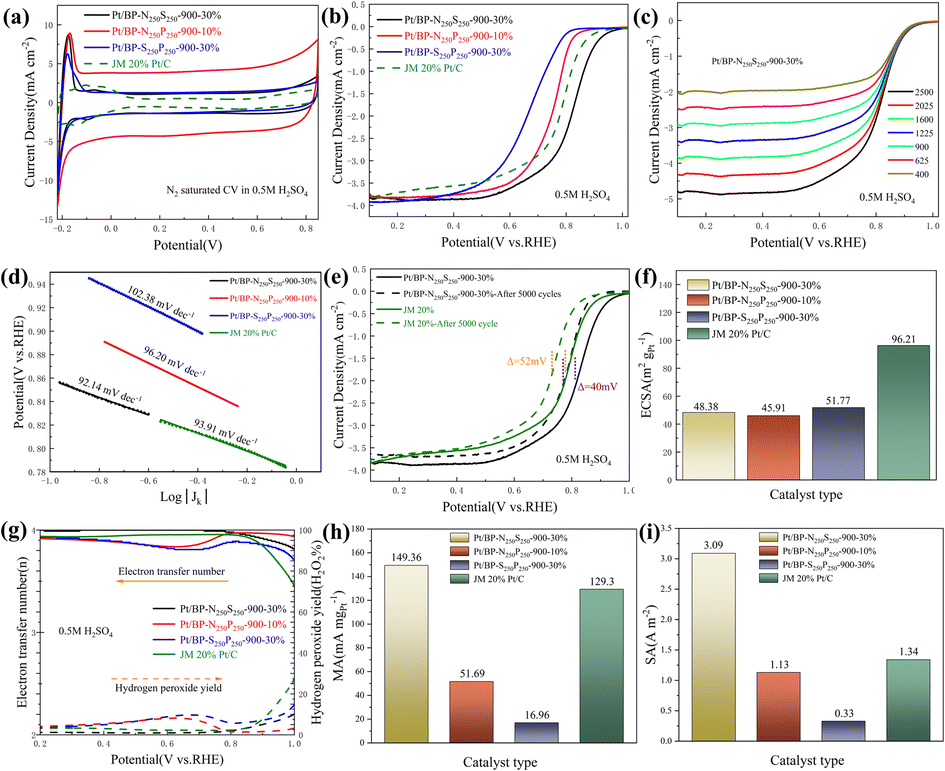 | ||
| Fig. 6 Electrochemical properties of catalysts Pt/BP-N250S250-900-30%, Pt/BP-N250P250-900-10%, Pt/BP-S250P250-900-30% and commercial Pt/C at 0.5 M H2SO4: (a) CV curves of the catalysts at N2 saturation; (b) LSV curves of catalysts under O2 saturation; (c) LSV curves of Pt/BP-N250S250-900-30% at different speeds; (d) Tafel slope of catalysts; (e) Pt/BP-N250S250-900-30% stability test; (f) comparison of ECSA; (g) the electron transfer number and H2O2% of the obtained catalyst and Pt/C measured using the RRDE; (h) and (i) MA and SA corresponding to the acceleration plan and Pt/C at 0.8 V vs. RHE. | ||
| Samples | E o | E 1/2 | J L | n | H2O2% | ECSA | MA | SA |
|---|---|---|---|---|---|---|---|---|
| Pt/BP-N250S250-900-30% | 0.962 | 0.821 | 3.86 | 3.98 | 2.16 | 48.38 | 149.36 | 3.09 |
| Pt/BP-N250P250-900-10% | 0.862 | 0.753 | 3.82 | 3.90 | 4.80 | 45.91 | 51.69 | 1.13 |
| Pt/BP-S250P250-900-30% | 0.814 | 0.664 | 3.9 | 3.86 | 6.86 | 51.77 | 16.96 | 0.33 |
| Pt/BP-N250-900-20% | 0.931 | 0.803 | 3.76 | 3.95 | 2.32 | 36.39 | 98.6 | 2.71 |
| Pt/BP-P250-900-10% | 0.868 | 0.705 | 3.04 | 3.92 | 3.76 | 33.29 | 37.7 | 1.13 |
| Pt/BP-S250-900-10% | 0.951 | 0.744 | 4.66 | 3.95 | 2.35 | 33.53 | 67.7 | 2.02 |
| Pt/BP-900-10% | 0.85 | 0.695 | 4.14 | 3.94 | 3.05 | 46.68 | 42.2 | 0.91 |
| JM 20% Pt/C | 0.924 | 0.785 | 3.63 | 3.91 | 4.58 | 96.21 | 129.3 | 1.34 |
We have considered the experimental results of single doping and double doping of heteroatoms and inferred that the catalyst obtained by co-doping three heteroatoms may be contaminated by platinum–phosphide compounds, making it unsuitable for obtaining an excellent catalyst. We conducted electrochemical testing to support our hypothesis (Fig. S19†). The testing results were in line with our expected outcome, as the synthesized catalyst contained platinum–phosphide compounds that had a negative impact on the experimental results.
To further validate whether these catalysts conform to the 4-electron reaction mechanism, rotating ring-disk electrode (RRDE) tests were conducted on the three catalysts. According to the results shown in ESI Fig. S6g,† the average number of transferred electrons for Pt/BP-N250S250-900-30%, Pt/BP-N250P250-900-10%, and Pt/BP-S250P250-900-30% were 3.98, 3.90, and 3.86, respectively, with corresponding hydrogen peroxide yields of 2.16%, 4.8%, and 6.86%. Among them, Pt/BP-N250S250-900-30% had the closest number of transferred electrons to 4 and the lowest hydrogen peroxide yield, indicating its effectiveness as a good ORR catalyst.
In addition, Pt/BP-N250S250-900-30% and Pt/BP-S250P250-900-30% were tested at different scan rates. According to the LSV curves in Fig. 6c and ESI Fig. S15a, b,† as well as the kinetic current density calculated using the K–L equation, it can be observed that their current density increases with an increasing scan rate. The Tafel slope of a catalyst reflects the degree of hindrance in the reaction process and can indicate the reaction kinetics of the catalyst. Meanwhile, according to the results in Fig. 6d, Pt/BP-N250S250-900-30% has the lowest Tafel slope (92.14 mV dec−1), indicating faster reaction kinetics, which is similar to the commercial platinum carbon catalyst, demonstrating its excellent catalytic performance. Compared to commercial Pt/C, the Tafel slope of Pt/BP-N250S250-900-30% was close, indicating similar reaction kinetics to commercial Pt/C, and considering the previous comparisons, it exhibited the best catalytic activity. Stability is also an important indicator for evaluating catalyst quality. From Fig. 6e, it can be seen that after 5000 cycles of CV, the E1/2 of Pt/BP-N250S250-900-30% only decreased by 40 mV, while that of commercial Pt/C decreased by 52 mV. Compared to the previous data, the stability of Pt/BP-N250S250-900-30% significantly improved after the incorporation of the S element, surpassing commercial Pt/C.
To further demonstrate that the catalyst Pt/BP-N250S250-900-30% is the best ORR catalyst among the three series, calculations and comparisons were conducted for ECSA (electrochemical surface area), MA (mass activity), and SA (specific activity). As shown in Fig. 6f, h and i, the ECSA of Pt/BP-N250S250-900-30% was 48.38 m2 gPt−1, lower than that of commercial Pt/C, indicating better intrinsic activity of Pt/BP-N250S250-900-30%. Furthermore, the calculated results for MA and SA showed that Pt/BP-N250S250-900-30% had the highest MA and SA, indicating better activity in terms of per unit area and per unit electrochemical active surface area, thereby confirming its superior catalytic activity. Pt/BP-N250S250-900-30% is the catalyst with the best ORR catalytic performance in N2-saturated 0.5 M H2SO4. It exhibits prominent crystal plane characteristic peaks, a larger surface area, excellent LSV performance, a transferred electron number close to that of a 4-electron reaction, and a lower hydrogen peroxide yield. Additionally, Pt/BP-N250S250-900-30% demonstrates increased current density at different scan rates and a Tafel slope closer to that of commercial Pt/C. Its stability surpasses that of commercial Pt/C, and it outperforms commercial Pt/C in terms of ECSA, MA, and SA, indicating that Pt/BP-N250S250-900-30% is the most outstanding ORR catalyst.
Electrochemical impedance spectroscopy (EIS) analysis was carried out on dual-doped catalysts Pt/BP-N250S250-900-30%, Pt/BP-S250P250-900-30%, and Pt/BP-N250P250-900-30% and undoped catalyst Pt/BP-900-30%. Among them, the main catalyst Pt/BP-N250S250-900-30% had the smallest Rct value of 0.271 Ω and the best catalytic activity. The undoped catalyst Pt/BP-900-30% had an Rct value of 2.01 Ω, which was the highest among those of the four catalysts. This confirms that the introduction of dopants reduces impedance and increases conductivity. We found that the impedance value of Pt/BP-N250S250-900-30% (0.271 Ω) was like that of Pt/BP-S250P250-900-30% (0.272 Ω), while the latter's impedance value was much lower than that of Pt/BP-N250P250-900-30% (1.723 Ω). This led us to infer that the platinum phosphine compound might increase impedance. We inferred that the platinum phosphine compound may increase impedance. In the case of the dual-doped catalysts, Pt/BP-N250S250-900-30% doped with N and S atoms simultaneously had an Rs value of 3.302 Ω. Pt/BP-N250P250-900-30%, incorporating N and P atoms simultaneously, had an Rs value of 3.26 Ω. Similarly, Pt/BP-S250P250-900-30%, involving P and S atoms simultaneously, had an Rs value of 3.278 Ω, while the undoped catalyst Pt/BP-900-30% had an Rs value of 4.981 Ω. Doping heteroatoms can reduce the solution resistance of the catalyst, thereby increasing the conductivity, which is helpful for improving performance (Fig. S16†).
Theoretical calculations
To gain a deeper understanding of the synergistic effects between Pt clusters and doped heteroatoms or intrinsic activity of Pt cluster active sites towards the oxygen reduction reaction (ORR), extensive theoretical investigations were carried out using relativistic density functional theory (DFT). Pyridinic nitrogen modifications have been identified as strong anchoring points for metal or heteroatom atoms,38,39,41,42 and we focused solely on pyridinic nitrogen-based active sites (Fig. 7a and b).40 Additionally, the ORR free energy diagram was computed following the methodology developed by Nørskov et al. to elucidate the reaction pathways in an acidic medium (Fig. 7c and d).43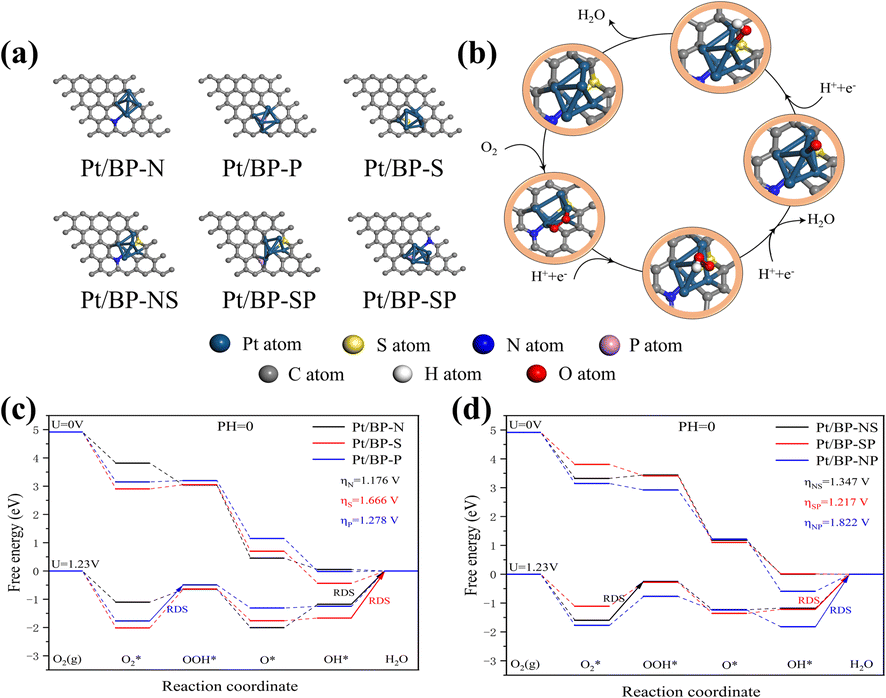 | ||
| Fig. 7 Different heteroatom-doped carbon black supported platinum catalyst configurations and the ORR step diagram, (a) six catalyst configurations, (b) ORR reaction process diagram (royal blue globule represents the Pt atom, the yellow globule represents the S atom, the blue globule represents the N atom, the pink globule represents the P atom, the gray globule represents the C atom, the white globule represents the H atom, and the red globule represents the O atom) and (c) and (d) single heteroatom doping and double heteroatom doping step diagram. | ||
To reduce computational burden while improving the reaction system and simplifying the number of clusters, we increased the surface area of the substrate to minimize periodicity effects. The energy comparison between the top adsorbed oxygen structure and the bridge oxygen structure facilitated the selection of stable structures (Fig. S16 and Table S2†). As the overpotential is a crucial indicator of the catalytic properties of a catalyst,43,44 we calculated the ORR overpotential at each catalytic site. From the obtained free energy diagrams, it was revealed that the ORR overpotential for the Pt/BP-N sites was 1.176 V, consistently lower than that of comparable catalysts, confirming that N atom doping enhances the activity of the active sites, aligning with the previous experimental results, wherein OH desorption was the rate-limiting step at all potentials.
It is worth noting that although Pt/BP-NS exhibits a higher energy barrier compared to Pt/BP-SP, its stability surpasses that of Pt/BP-SP (Tables S4 and S5†). Furthermore, during the actual process, platinum phosphide compounds are formed, influencing the oxygen reduction process (as indicated by XRD). Therefore, based on comprehensive evaluations, the Pt/BP-NS catalyst outperforms Pt/BP-SP. In comparison to conventional carbon-supported heteroatom-based platinum catalysts, both experimentally and theoretically known to have an overpotential of 1.68 V,43,45 the performance of Pt/BP-NS and Pt/BP-N catalysts significantly exceeds that of conventional catalysts.
The support material elucidates the ORR active sites in two catalysts, Pt/BP-N and Pt/BP-NS. During the high-temperature annealing process, the Pt clusters supported on Pt/BP-N and Pt/BP-NS undergo reconstruction in the nitrogen–carbon coordination environment. The Pt clusters coordinate with pyridinic nitrogen groups in a non-planar structure (Fig. 2g and 4g). Compared to Pt/BP-N catalysts, Pt/BP-NS catalysts capture a platinum atom from the Pt clusters to form a new stable structure, leading to better anchoring of Pt clusters on the active sites.46
The O–O bond length of the adsorbed O2 on the catalyst's active site is a key factor influencing the electrocatalytic ORR selectivity. A longer O–O bond length facilitates O–O bond breaking, leading to the four-electron pathway for H2O production.42 Conversely, a shorter O–O bond length favors the two-electron pathway for H2O2 production. Traditional Pt nanoparticle-based ORR active sites involve O2 molecules adsorbed on two or three adjacent Pt atoms in a lateral configuration, resulting in an O–O bond length of 1.37 Å, accommodating both the four-electron and two-electron ORR pathways.42,47
To examine the superior ORR activity of Pt/BP-NS and Pt/BP-N catalysts, we used density functional theory (DFT) calculations to determine the O–O bond lengths. Based on the experimental results, model structures were established (as shown in ESI Fig. S19†). When O2 molecules adsorb in a lateral configuration on the Pt clusters at the active sites of Pt/BP-NS and Pt/BP-N, the computed O–O bond lengths are 1.422 Å and 1.480 Å (Fig. S17†), respectively, longer than the traditional Pt nanoparticle-based length of 1.37 Å. Consequently, the ORR is dominated by the four-electron pathway, indicating that Pt clusters coordinated with one pyridinic nitrogen and two carbon atoms in Pt/BP-NS and Pt/BP-N catalysts can lead to the four-electron ORR pathway and higher ORR performance.42
The above discussions also suggest that high-temperature annealing triggers the reconstruction of the coordination environment around Pt clusters prepared on the doped support, resulting in more stable and efficient four-electron pathway active sites. This is of significant importance for the rational design of ORR electrocatalysts aimed at efficient four-electron processes.
To understand the origin of high stability of the catalyst, the interaction energy (Eint) between the doped active sites and Pt clusters was investigated (Table S8, ESI†).48 Among the single heteroatomdoped catalysts, the Eint value for sulfur (S) atom doping in Pt/BP-S was the highest among the three catalysts in the series, indicating that S atom doping in Pt/BP-S is more stable than other types of doping. Similarly, in the double heteroatom doped series, both Pt/BP-NS and Pt/BP-SP doped catalysts were more stable than the Pt/BP-NP doped catalyst, demonstrating that S atom doping indeed enhances the structure's stability. It is noteworthy that the Eint value for Pt/BP-NS is greater than that of the same type, indicating that Pt/BP-NS exhibits the most exceptional stability within this category.
Conclusions
This paper proposes a straightforward method for the synthesis of heteroatom-doped Pt-based catalysts with high activity and stability, using melamine, ammonium dihydrogen phosphate, and thiourea as dopants. These catalysts present a promising alternative to commercially expensive Pt/C catalysts. The study investigates the effects of pyrolysis temperature, Pt loading, and the type and amount of heteroatom dopants on the distribution and catalytic performance of Pt in the catalysts, as well as the influence of dual heteroatom doping on the metal particle size and catalytic activity. Experimental and theoretical calculations reveal that S atom doping enhances structural stability, while N and P atom doping improves the oxygen reduction process's activity. However, there are challenges in practical applications, such as platinum–phosphorus compounds occupying active sites, making N atom doping a more preferable option in overall evaluations. The results show that the Pt/BP-N250S250-900-30% catalyst has a higher initial potential than RHE's 0.962 V and a higher half-wave potential than RHE's half-wave potential. Due to N doping, its activity is higher than that of the undoped platinum carbon catalyst. Pt/BP-N250S250-900-30% has an ECSA of 48.38 m2 gPt−1, which is lower than that of commercial Pt/C, indicating that Pt/BP-N250S250-900-30% has better intrinsic activity. At the same time, it exhibits the highest MA (149.36 mA mgpt−1) and SA (3.09 A m−2), indicating better activity per unit area and per unit of the electrochemical active surface area, thereby confirming its excellent catalytic activity. In addition, the S-doped Pt/BP-N250S250-900-30% catalyst exhibited excellent stability. After 5000 cycles of CV testing, the half-wave potential only decreased by 40 mV, while that of the commercial Pt/C catalyst decreased by 52 mV. At the same time, compared with other catalysts, Pt/BP-N250S250-900-30% shows a good performance (Table S6†). Future research could explore the impact of heteroatom doping on the formation mechanism of Pt species in catalysts, aiming to reduce costs and enhance the catalyst's practicality and scalability. Additionally, investigations into the application of heteroatom-doped Pt-carbon catalysts in other fields, such as batteries and photocatalysis, could also be pursued.Author contributions
N. F. B. and T. Y. Z. contributed equally to this work. C. X. Z., W. T. W. and J. H. conceived and designed the study. Y. J. Z. provided laboratory supplies for the study. Y. T. Z., F. N. B., D. Y. H., J. C. and F. M. M. performed experiments and analysed the data. L. Z. contributed to the graphical abstract. C. X. Z. provided the method. F. N. B. and M. H. L. contributed to the quantum chemical calculation. F. N. B., C. X. Z. and J. H. revised the manuscript. All authors contributed to the general discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Nature Science Foundation of China (No. 52364041). This research was supported by the Fund of the State Key Laboratory of Advanced Technology for Comprehensive Utilization of Platinum Metals (CXHZ-2021-0101) and the Kunming University of Science and Technology (No. KKKP201707010 and KKKP201752011).Notes and references
- K. R. Abbasi, M. Shahbaz, J. Zhang, M. Irfan and R. Alvarado, Renewable Energy, 2022, 187, 390–402 CrossRef CAS.
- J. Han and H. Chang, Appl. Sci., 2022, 12, 4783 CrossRef CAS.
- Nature, 2022, 601, 7 Search PubMed.
- D. Hou, F. Bai, P. Dong, J. Chen, Y. Zhang, F. Meng, Z. Zhang, C. Zhang, Y. Zhang and J. Hu, J. Power Sources, 2023, 584, 233599 CrossRef CAS.
- Z. Zhang and C. Hu, Int. J. Hydrogen Energy, 2014, 39, 12973–12979 CrossRef CAS.
- R. E. Rosli, A. B. Sulong, W. R. W. Daud, M. A. Zulkifley, T. Husaini, M. I. Rosli, E. H. Majlan and M. A. Haque, Int. J. Hydrogen Energy, 2017, 42, 9293–9314 CrossRef CAS.
- C. Bernay, M. Marchand and M. Cassir, J. Power Sources, 2002, 108, 139–152 CrossRef CAS.
- Y. Qiu, Z. Hu, H. Li, Q. Ren, Y. Chen and S. Hu, Chem. Eng. J., 2022, 430, 132769 CrossRef CAS.
- S. Dey, B. Mondal, S. Chatterjee, A. Rana, S. Amanullah and A. Dey, Nat. Rev. Chem, 2017, 1, 0098 CrossRef CAS.
- D. Banham, S. Ye, K. Pei, J. Ozaki, T. Kishimoto and Y. Imashiro, J. Power Sources, 2015, 285, 334–348 CrossRef CAS.
- Y.-J. Wang, W. Long, L. Wang, R. Yuan, A. Ignaszak, B. Fang and D. P. Wilkinson, Energy Environ. Sci., 2018, 11, 258–275 RSC.
- R. Alipour Moghadam Esfahani, L. M. Rivera Gavidia, G. García, E. Pastor and S. Specchia, Renewable Energy, 2018, 120, 209–219 CrossRef CAS.
- J. Liu, J. Bak, J. Roh, K.-S. Lee, A. Cho, J. W. Han and E. Cho, ACS Catal., 2021, 11, 466–475 CrossRef CAS.
- X. Li, H. Zhang, Q. Hu, W. Zhou, J. Shao, X. Jiang, C. Feng, H. Yang and C. He, Angew. Chem., Int. Ed., 2023, 62, e202300478 CrossRef CAS PubMed.
- Q. Hu, K. Gao, X. Wang, H. Zheng, J. Cao, L. Mi, Q. Huo, H. Yang, J. Liu and C. He, Nat. Commun., 2022, 13, 3958 CrossRef CAS PubMed.
- X. Jiang, X. Li, Y. Kong, C. Deng, X. Li, Q. Hu, H. Yang and C. He, J. Energy Chem., 2023, 76, 462–469 CrossRef CAS.
- Q. Hu, Z. Han, X. Wang, G. Li, Z. Wang, X. Huang, H. Yang, X. Ren, Q. Zhang, J. Liu and C. He, Angew. Chem., Int. Ed., 2020, 59, 19054–19059 CrossRef CAS PubMed.
- M. S. Çögenli and A. Bayrakçeken Yurtcan, Int. J. Energy Res., 2022, 46, 24130–24147 CrossRef.
- C. Lu, D. Yu, Z. Yan, X. Yang, W. Yang, Z. Lv, Z. Tian and Q. Tian, Int. J. Electrochem. Sci., 2021, 16, 210555 CrossRef CAS.
- P. Yin, X. Luo, Y. Ma, S.-Q. Chu, S. Chen, X. Zheng, J. Lu, X.-J. Wu and H.-W. Liang, Nat. Commun., 2021, 12, 3135 CrossRef CAS PubMed.
- G. de Falco, M. Florent and T. J. Bandosz, Carbon, 2022, 189, 230–239 CrossRef CAS.
- Y. Chen, C. Fan, X. Li, J. Ren, G. Zhang, H. Xie, B. Liu and G. Zhou, J. Chem. Technol. Biotechnol., 2023, 98, 117–128 CrossRef CAS.
- L. Wang, Y. Wang, M. Wu, Z. Wei, C. Cui, M. Mao, J. Zhang, X. Han, Q. Liu and J. Ma, Small, 2018, 14, 1800737 CrossRef PubMed.
- R. Samadder, N. Akter, A. C. Roy, M. M. Uddin, M. J. Hossen and M. S. Azam, RSC Adv., 2020, 10, 11945–11956 RSC.
- M. R. Pallavolu, S. Prabhu, R. R. Nallapureddy, A. S. Kumar, A. N. Banerjee and S. W. Joo, Carbon, 2023, 202, 93–102 CrossRef CAS.
- J. Xu, Q. Liang, Z. Li, V. Y. Osipov, Y. Lin, B. Ge, Q. Xu, J. Zhu and H. Bi, Adv. Mater., 2022, 34, 2200011 CrossRef CAS PubMed.
- L. Zhang, J. Niu, M. Li and Z. Xia, J. Phys. Chem. C, 2014, 118, 3545–3553 CrossRef CAS.
- F. Gao, Y. Xie, Y. Zang, G. Zhou, J. Qu and M. Wu, New Carbon Mater., 2022, 37, 752–763 CrossRef CAS.
- H. Da, S. Pan, J. Li, J. Huang, X. Yuan, H. Dong, J. Liu and H. Zhang, Energy Storage Mater., 2023, 56, 457–467 CrossRef.
- M. A. Patel, F. Luo, M. R. Khoshi, E. Rabie, Q. Zhang, C. R. Flach, R. Mendelsohn, E. Garfunkel, M. Szostak and H. He, ACS Nano, 2016, 10, 2305–2315 CrossRef CAS PubMed.
- Y. Tan, R. Xie, S. Zhao, X. Lu, L. Liu, F. Zhao, C. Li, H. Jiang, G. Chai, D. J. L. Brett, P. R. Shearing, G. He and I. P. Parkin, Adv. Funct. Mater., 2021, 31, 2105579 CrossRef CAS.
- C. Wu, J. Zhang, J. Guo, L. Sun, J. Ming, H. Dong, Y. Zhao, J. Tian and X. Yang, ACS Sustain. Chem. Eng., 2018, 6, 7451–7457 CrossRef CAS.
- K. Wu, L. Zhou, C.-J. Jia, L.-D. Sun and C.-H. Yan, Mater. Chem. Front., 2017, 1, 1754–1763 RSC.
- Y. Lv, L. Yang and D. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 32859–32867 CrossRef CAS PubMed.
- Z. Chen, R. Liu, S. Liu, J. Huang, L. Chen, R. Nadimicherla, D. Wu and R. Fu, Chem. Commun., 2020, 56, 12921–12924 RSC.
- J.-S. Li, S. Zhang, J.-Q. Sha, H. Wang, M.-Z. Liu, L.-X. Kong and G.-D. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 17140–17146 CrossRef CAS PubMed.
- H.-Y. Lü, X.-H. Zhang, F. Wan, D.-S. Liu, C.-Y. Fan, H.-M. Xu, G. Wang and X.-L. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 12518–12527 CrossRef PubMed.
- L.-S. Zhang, X.-Q. Liang, W.-G. Song and Z.-Y. Wu, Phys. Chem. Chem. Phys., 2010, 12, 12055 RSC.
- S. Zhang, H. Wang, N. Zhang, F. Kong, H. Liu and G. Yin, J. Power Sources, 2012, 197, 44–49 CrossRef CAS.
- H. Wei, X. Liu, A. Wang, L. Zhang, B. Qiao, X. Yang, Y. Huang, S. Miao, J. Liu and T. Zhang, Nat. Commun., 2014, 5, 5634 CrossRef CAS PubMed.
- B. Xiong, Y. Zhou, Y. Zhao, J. Wang, X. Chen, R. O'Hayre and Z. Shao, Carbon, 2013, 52, 181–192 CrossRef CAS.
- J. Liu, M. Jiao, L. Lu, H. M. Barkholtz, Y. Li, Y. Wang, L. Jiang, Z. Wu, D. Liu, L. Zhuang, C. Ma, J. Zeng, B. Zhang, D. Su, P. Song, W. Xing, W. Xu, Y. Wang, Z. Jiang and G. Sun, Nat. Commun., 2017, 8, 15938 CrossRef CAS PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. Zhang, Z. Zhao, Z. Xia and L. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- D.-H. Lim and J. Wilcox, J. Phys. Chem. C, 2012, 116, 3653–3660 CrossRef CAS.
- H. Kim, I. H. Lee, J. Cho, S. Shin, H. C. Ham, J. Y. Kim and H. Lee, ChemElectroChem, 2019, 6, 4757–4764 CrossRef CAS.
- X.-G. Wang and G. B. Fisher, Phys. Rev. Lett., 2007, 99, 066101 CrossRef PubMed.
- T. Zhang, B. Zhang, Q. Peng, J. Zhou and Z. Sun, J. Mater. Chem. A, 2021, 9, 433–441 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta04599d |
| This journal is © The Royal Society of Chemistry 2024 |