
Engineering 3D-printed carbon structures with atomic layer deposition coatings as photoelectrocatalysts for water splitting†
Siowwoon
Ng
 a,
Michela
Sanna
a,
Edurne
Redondo
a and
Martin
Pumera
*abcde
a,
Michela
Sanna
a,
Edurne
Redondo
a and
Martin
Pumera
*abcde
aFuture Energy and Innovation Laboratory, Central European Institute of Technology, Brno University of Technology, Purkyňova 123, 61200 Brno, Czech Republic. E-mail: martin.pumera@ceitec.vutbr.cz
bFaculty of Electrical Engineering and Computer Science, VSB – Technical University of Ostrava, 17. Listopadu 2172/15, Ostrava 70800, Czech Republic
cDepartment of Medical Research, China Medical University Hospital, China Medical University, No. 91 Hsueh-Shih Road, Taichung 40402, Taiwan
dDepartment of Chemical and Biomolecular Engineering, Yonsei University, 50 Yonsei-ro, Seodaemun-gu, Seoul 03722, South Korea
eDepartment of Paediatrics and Inherited Metabolic Disorders, First Faculty of Medicine, Charles University, Ke Karlovu 2, 12808 Prague, Czech Republic
First published on 5th December 2023
Abstract
Three-dimensional (3D)-printing has evolved as a popular technique for producing customized parts and devices. 3D conductive structures made of metals or carbon-based materials are highly preferable in the field of electrochemistry. Compared to their metal counterparts, 3D carbon structures printed by the filament extrusion technique are readily available to end users, with the advantages of reduced electrode mass and broad compatibility with harsh environments that might be required for electrochemical applications. To elevate the applicability of 3D carbon electrodes in sensing, catalysis, energy storage, etc., surface or chemical modifications and coating of functional layers are essential. Atomic layer deposition (ALD) is an ideal deposition tool for creating coatings on geometrically complicated structures, yet the surface chemistry of the inert 3D carbon electrodes critically affects the initial growth. We performed a straightforward surface treatment, also known as ‘activation’, to improve the surface wettability and promote the ALD of TiO2, SiO2, and Al2O3 at low deposition temperatures. We applied the ALD coated electrodes for light-enhanced water splitting hydrogen and oxygen evolution reactions (HER, OER). In addition, we showed that 3D electrodes can be prepared in different geometrical shapes and sizes, as their metal counterparts. This work presents the versatility of ALD coatings on 3D carbon platforms, tunable for many other applications.
Introduction
The rise of additive manufacturing utilizing three-dimensional (3D)-printing technologies is closely associated with the rapid production of functional 3D objects based on individualized designs with minimal waste of materials. In alignment with the direction of Industry 4.0, 3D-printing directly increases the productivity in manufacturing processes, thereby offering an intelligent manufacturing solution across the vast spectrum of materials science and engineering.1 In parallel, it is highly flexible to upscale the printing to the size of buildings2,3 or miniaturize it to wearable electronic devices.4 Altogether, 3D-printing offers absolute freedom from design to manufacturing for functional applications.Transitioning from the traditional machining process to create 3D objects, initially powder bed fusion methods, for instance, electron beam melting (EBM) and selective laser melting (SLM) that fuse fine metal powders with a high-powered electron beam or laser, were employed to develop 3D-printed metal electrodes for electrochemical purposes.5–8 The metal electrodes serve as a conductive platform for directly synthesizing or depositing active materials for electrochemical conversion and storage applications.5,7–11 Several studies have utilized the core advantage of 3D-printing to manufacture tens to hundreds of hollow and solid cones on a flat surface7,8 and centimeter-scaled tubular metal electrodes.12 In recent years, the fused deposition modeling (FDM) technique has emerged as a captivating alternative to produce carbon-composite electrodes.13,14 FDM simplifies the printing process with a benchtop printer or hand-held printing pen that is accessible in every research laboratory. Furthermore, the advantages of the FDM-printed carbon electrodes over SLM- or EBM-printed metal electrodes include using an abundant material, reduction in printing cost, decrease in electrode mass, and compatibility with a wide range of chemical or harsh environments.
To expand the functionalities of the as-printed 3D carbon-based electrode, post-treatments13–17 and post-modifications are carried out to cater to electrochemical applications such as sensing, energy storage and conversion.18–25 Atomic layer deposition (ALD) that features conformal coating is presently the most feasible deposition technique to modify objects with geometrically complicated structures such as high porosity foams,26,27 arrays of spheres,28,29 tubes and channels.30–32 Several studies have employed ALD to deposit an active material on 3D-printed metal10–12 and carbon electrodes.20–23 However, for 3D carbon electrodes, the restriction on reaction temperatures and chemically inert carbon surface critically influence the nucleation and initial growth of ALD processes,20,23 posing limitations to depositing functional materials on carbon electrodes. To address these challenges, surface treatment or functionalization to introduce surface species or to increase defect, anchoring, and nucleation sites on the carbon surfaces are viewed as positive solutions.33
In this work, we provide a direct solution to tune the carbon surface for ALD processes. We demonstrate three universally used materials, TiO2, SiO2, and Al2O3,31,34 coated on 3D-printed carbon electrodes for light-enhanced hydrogen and oxygen evolution reactions (HER, OER). We extend the optimized ALD TiO2 coating on electrodes with different geometrical shapes and sizes up to a 2 inch-diameter disk (analogous to a small silicon wafer), as illustrated in Scheme 1, in contrast to many flat or simple 3D structures in the past. We show that the 3D-printed carbon electrodes can be potentially upscaled for industrial purposes similar to their metal counterparts. The present work demonstrates the versatility of ALD coatings on 3D carbon host structures in design, up-, and down-scaling to cater to the vast applications.
 | ||
| Scheme 1 Schematic representation of the workflow. (Top) Design and 3D-printing of electrodes with various sizes and shapes. (Bottom) ALD of Al2O3, SiO2, or TiO2 on the 3D electrodes. The electrodes are ready to be transferred to the ALD reactor, and the reflective pieces are silicon wafers for reference purposes. | ||
Experimental
3D-printed carbon electrodes
3D carbon electrodes were designed and printed following the settings in our previous work.20 In brief, the graphene/polylactic acid (PLA) composite conductive filament (BlackMagic, Graphene Supermarket) was supplied to a Prusa i3 MK3 printer (Prusa Research) to print the 3D electrodes depicted in Scheme 1. The as-printed 3D electrodes underwent the activation steps following the literature with slight modifications as described below.(i) Electrochemical oxidation + immersion in a reducing agent15 – 2.5 VAg/AgCl for 1000 s in phosphate-buffered saline (PBS, pH 7.2), followed by immersion in 1 M sodium borohydride (NaBH4) for 24 h; denoted as NaBH4.
(ii) Immersion in polar aprotic solvent + electrochemical oxidation16 – immersion in N,N-dimethylformamide (DMF) for 3 h, followed by 2.5 VAg/AgCl for 300 s; denoted as DMF.
(iii) Saponification14 – immersion in 4 M sodium hydroxide (NaOH) for 4 h, where 4 h was the optimized condition and used for further studies; denoted as NaOH.
All electrodes were rinsed thoroughly with water and dried in air.
Atomic layer deposition of TiO2, Al2O3, and SiO2
After the activation and surface treatment of 3D-printed carbon electrodes, an atomic layer deposition (ALD, Ultratech/CambridgeNanoTech Fiji 200) system was used for material deposition. Tetrakis(dimethylamino)titanium(IV) (TDMATi, Strem Chemicals), trimethylaluminum (TMA, Sigma-Aldrich), and tetrakis(dimethylamido)silane (TDMASi, Sigma-Aldrich) were used as Ti, Al, and Si precursors, respectively. TDMATi was heated to 75 °C to increase the vapor pressure, whereas TMA and TDMASi were not heated. For TiO2 and Al2O3, deionized water (≈18 MΩ) was used as an O2 precursor, and argon (Air Products, purity 99.999%) served as the carrier gas. The plasma for ALD SiO2 was obtained by ionizing O2 gas (Air Products, purity 99.999%). For all substrates temperature was 150 °C. The ALD growth cycle follows the sequence stated below.(i) TiO2 – 0.06 s H2O pulse (Ar 60 sccm), 30 s Ar purge; 0.1 s TDMATi pulse (Ar 200 sccm), 30 s Ar purge.
(ii) Al2O3 – 0.06 s H2O pulse (Ar 30 sccm), 30 s Ar purge; 0.06 s TMA pulse (Ar 100 sccm), 30 s Ar purge.
(iii) SiO2 – 25 s O2 purge (50 sccm), 20 s plasma (300 W), 5 s O2 purge; 0.4 s TDMASi pulse (Ar 100 sccm), 5 s Ar purge.
Materials characterizations
Surface wettability of the activated 3D electrodes was observed using a surface energy evaluation system (See System, Advex Instruments) with proprietary analysis software v7.0. Compositional analyses were carried out by X-ray photoelectron spectroscopy (XPS, Kratos AXIS Supra). The X-ray excitation source was a monochromatic Al Kα (1486.7 eV) with 225 W power. The measured data were analyzed using CasaXPS software. The surface of the electrodes was assessed using a scanning electron microscope (SEM, FEI Verios 460L).Photo- and electrochemical measurements
All electrochemical experiments were performed using a potentiostat (PGSTAT 204, Metrohm Autolab) operated by NOVA software v2.1 in a three-electrode configuration. The blank or ALD TiO2, Al2O3, or SiO2 coated 3D-printed carbon, Ag/AgCl (1 M KCl), and graphite rod, were used as the working, reference, and counter electrodes, respectively. For both the HER and OER, linear sweep voltammetry (LSV) was carried out with a scan rate of 2 mV s−1 in 0.5 M H2SO4 and 1 M NaOH electrolyte, respectively, with and without irradiation. Chronoamperometry photoresponse was measured in the same electrolytes by alternating light on and off with applied potential ≈ −0.4 VRHE for the HER and ≈1.8 VRHE for the OER. The potential vs. Ag/AgCl (VAg/Ag) was converted to the potential vs. the reversible hydrogen electrode (VRHE) according to the literature.35 The blue light irradiation was provided by a customized light-emitting diode setup (λ = 460 nm, LZ4-40B208, LedEngin Inc.).Results and discussion
We printed the 3D carbon electrodes with nanocarbon/PLA filament, one of the most used commercial-based conductive filaments. The as-printed 3D electrodes comprised carbon and PLA in a ratio of ≈1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9.13 We adopted three different activation procedures with minor modifications from the literature to partially remove the insulating PLA, which simultaneously served as the treatment for the inert carbon surface for ALD purposes. The procedures are (i) electrochemical oxidation, followed by immersion in a reducing solution of NaBH4 (denoted as NaBH4),15 (ii) a widely reported physical swelling by DMF, followed by electrochemical oxidation (denoted as DMF),16 and (iii) the less explored saponification by NaOH (as compared to DMF activation, denoted as NaOH).14
:9.13 We adopted three different activation procedures with minor modifications from the literature to partially remove the insulating PLA, which simultaneously served as the treatment for the inert carbon surface for ALD purposes. The procedures are (i) electrochemical oxidation, followed by immersion in a reducing solution of NaBH4 (denoted as NaBH4),15 (ii) a widely reported physical swelling by DMF, followed by electrochemical oxidation (denoted as DMF),16 and (iii) the less explored saponification by NaOH (as compared to DMF activation, denoted as NaOH).14
Fig. 1 presents the surface of the as-activated electrodes observed by SEM. The electrode activated in NaBH4 is less fibrous than the other electrodes, as the fibers are covered by the PLA. For reference, an image of the as-printed, non-activated electrode is given in Fig. S1 in the ESI,† where only several fibers are visible on the surface. Both electrodes activated in DMF and NaOH are similar from the microscopic point, where a substantial amount of PLA was removed, consistent with previous works.16,23 However, the surface wettability of these electrodes is completely different, evidenced by the contact angle of a water droplet with the electrode surface in the insets in Fig. 1. Specifically, the contact angle with NaBH4, DMF, and NaOH electrodes is 82.5°, 124.5°, and 17.1°, respectively, showing that the NaOH electrode possesses the highest hydrophilicity among the three electrodes. The NaOH treats the electrodes in two ways. For PLA, the hydroxides contribute to the saponification of the PLA as an aliphatic polyester, eventually breaking down the polymer chain into lactate monomers with oxygen-rich functionalities.14 For carbon, the hydroxides form oxygen-rich functionalities on the carbon surface, altering the wettability of the surface. Transforming the hydrophobic surface to hydrophilic directly improves the accessibility of the electrolyte to the electrode, hence improving the charge transfer,36–38 and lowering the adhesion forces of the produced bubbles on the electrode surfaces. These characteristics strongly influence the OER, HER and other electrochemical reactions.37–40
 | ||
| Fig. 1 3D-printed electrodes activated in different solvents, i.e., NaBH4, DMF, and NaOH. (Top) SEM images of the electrodes after the activation process. The insets present the contact angle of water with the surface of the electrodes. (Bottom) LSV curves show the HER of the blank and 500 ALD cycles TiO2 coated electrodes in 0.5 M H2SO4 electrolyte, without and with blue light (λ = 460 nm) irradiation. | ||
We carried out thermal ALD of TiO2 for 500 cycles at 150 °C on the 3D-printed carbon electrodes activated by different solvents. We evaluated the TiO2 coated electrodes for their HER performance in 0.5 M H2SO4 electrolyte. Comparing the LSV curves in Fig. 1, the TiO2 coated electrodes possess a lower overpotential than the blank counterparts, with further reduced overpotentials by the irradiation of a blue light source. The potential required to drive the HER in descending sequence follows: TiO2 coated NaBH4, DMF, and NaOH electrodes. The nanometer-scale ALD TiO2 coatings are not distinguishable from the fibers of a hundred nanometer diameter by SEM imaging (not shown). The XPS analysis confirms that the surface of the electrodes is composed of C, O, and Ti. The survey spectra of all three TiO2 coated electrodes and atomic concentration of the three elements are given in Fig. S2 in the ESI.† In the high-resolution Ti 2p spectra, the spin–orbit split doublets at ≈458–459 eV and ≈464–465 eV are assigned to Ti 2p3/2 and Ti 2p1/2, respectively,41,42 verifying the formation of Ti4+–O for the ALD TiO2 coatings. Furthermore, the analysis of the survey spectra shows that the atomic concentrations of Ti in TiO2 coated NaBH4, DMF, and NaOH electrodes are ≈0.75, 0.50, and 1.85%, respectively, implying more deposition occurred on the surface of the NaOH electrode under the same ALD conditions. Based on the evaluation from different aspects, we conclude that the surface of NaOH electrodes favors the deposition of ALD coatings. In addition, between the DMF and NaOH electrodes, the latter has a higher mechanical stability, which is preferable for device applications.
With the NaOH electrodes, we moved forward with depositing 1500 cycles of TiO2 and 50 cycles of Al2O3 by thermal ALD and 50 cycles of SiO2 by plasma ALD. The XRD patterns in Fig. S3 in the ESI† present only the diffraction peaks of carbon and PLA,43,44 with no sign of the peaks associated with the respective coatings. All ALD coatings were amorphous because of the 150 °C substrate temperature, consistent with the literature and our previous work.22,23,34,45–47 In addition, the thicknesses of coatings in nanometers are rather thin to confirm the crystallinity.34 We performed XPS analysis to verify the deposition on the 3D-printed carbon electrodes. The survey spectra in Fig. 2 confirm the presence of the expected elements, Al, Si, and Ti in their respective electrodes, along with C from the electrode and O from both the coating and electrode. In addition, Fig. 2 presents the high-resolution Al 2p, Si 2p, and Ti 2p spectra for the electrodes with ALD coatings. The Al 2p spectrum presents a dominant peak at 75.0 eV and a shoulder peak at 72.3 eV, associated with Al–O and Al metal,46,48 respectively. In addition, the small amount of Al metal indicates the incomplete formation of the Al2O3.22,23 The symmetric peak of Si 2p centered at 103.1 eV confirms the deposition of SiO2,49 whereas the spin–orbit split doublet of Ti 2p at 464.2 eV (Ti 2p1/2) and 458.5 eV (Ti 2p3/2) verifies the formation of TiO2.41,42
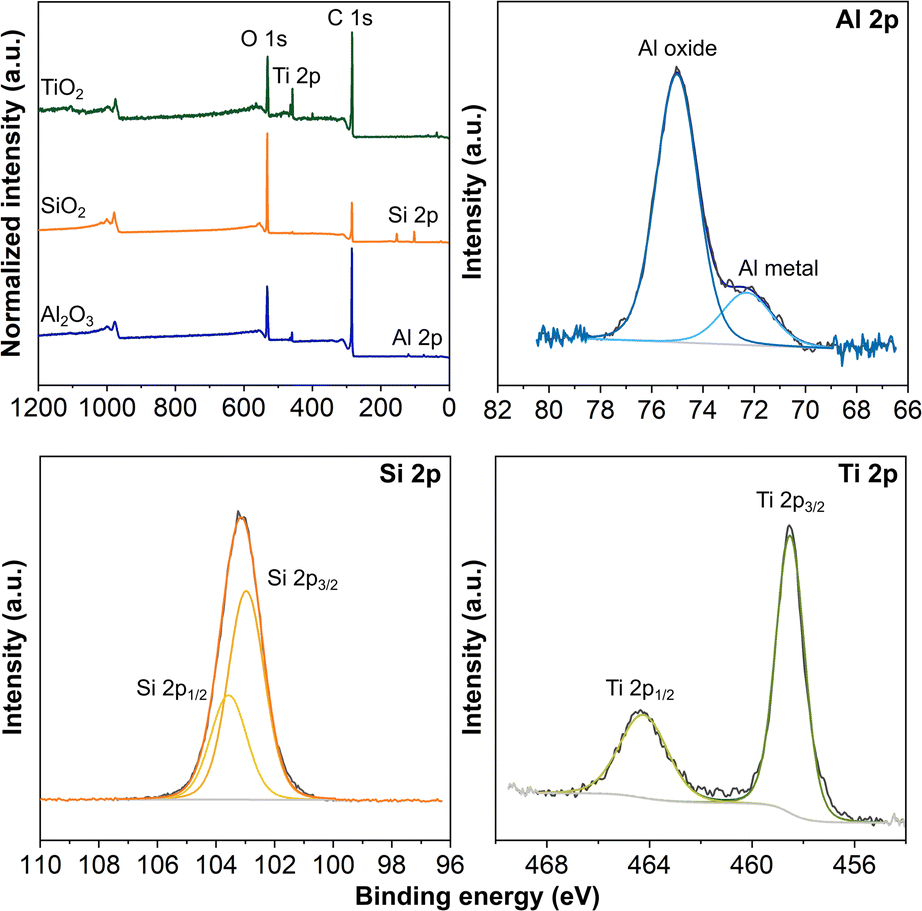 | ||
| Fig. 2 ALD coated 3D-printed carbon electrodes activated by NaOH. XPS survey spectra and high-resolution Al 2p, Si 2p, and Ti 2p spectra for 50 cycles Al2O3, 50 cycles SiO2, and 1500 cycles TiO2 coated electrodes, respectively. | ||
We examined the functionalities of these ALD coated 3D-printed carbon electrodes for the HER. Fig. 3A presents the LSV curves of all Al2O3, SiO2 and TiO2 coated 3D-printed carbon electrodes for the HER with and without irradiation. For reference, the LSV curves comparing the blank and ALD coated electrodes are included in Fig. S4 in the ESI.† The HER was catalyzed by the ALD coatings and further enhanced by the irradiation, as reflected by the lower potential required to drive the reaction. This is particularly obvious for TiO2, which has been widely used as a photocatalytic material. For a better overview of the electrodes' performance, we compiled the overpotential at −10 mA cm−2, as a common comparison current density for electrocatalytic reactions in Fig. 3B. For the HER without irradiation, the overpotential for all three coatings is rather similar, between 0.60 and 0.66 V. With irradiation, the SiO2 coated electrode shows a minor improvement to 0.62 V, followed by Al2O3 coating at 0.47 V, and the most significant improvement by TiO2 coating at 0.31 V. In continuation, we tested the ALD Al2O3, SiO2 and TiO2 coated 3D-printed carbon electrodes for the OER with and without irradiation. Fig. 3D displays the corresponding LSV curves. Similar to the HER, the OER was catalyzed by the added coatings and improved by irradiation. Fig. 3E summarizes OER overpotentials at 10 mA cm−2 for all ALD coated electrodes. Without irradiation, the overpotentials were between 1.83 and 1.98 eV. With irradiation, the overpotentials of Al2O3, SiO2, and TiO2 were reduced to 1.72, 1.71, and 1.47 V, respectively.
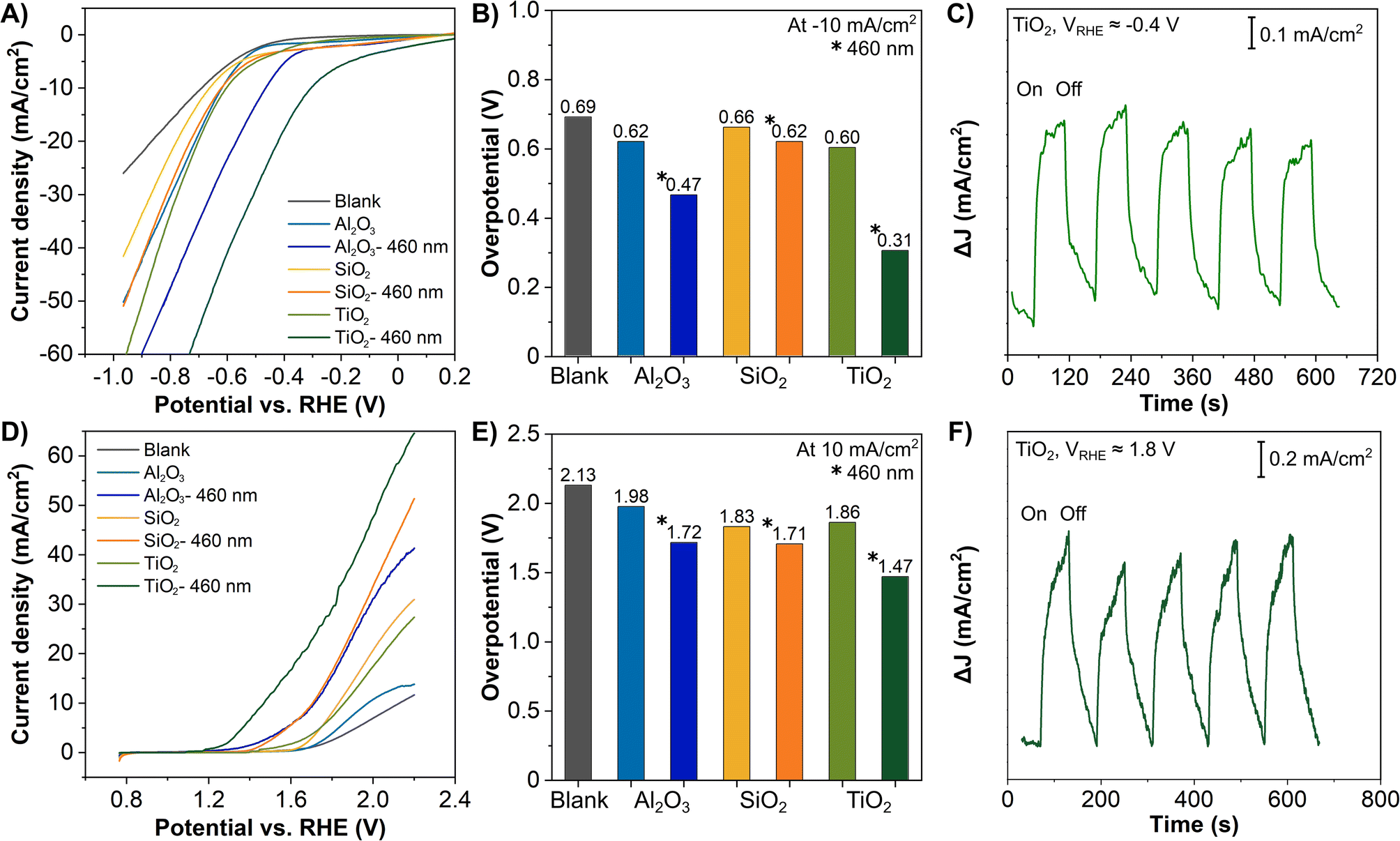 | ||
| Fig. 3 ALD coated 3D-printed carbon electrodes: 50 cycles of Al2O3, 50 cycles of SiO2, and 1500 cycles of TiO2. (A and D) LSV of the HER measured in 0.5 M H2SO4 and OER measured in 1 M NaOH, with and without irradiation by blue light source (λ = 460 nm), (B and E) overpotential of the HER (at −10 mA cm−2) and OER (at 10 mA cm−2). Chronoamperometry of the photoresponses of 1500 ALD cycle TiO2 coated electrodes irradiated by a blue light source with (C) applied potential ≈ −0.4 VRHE in 0.5 M H2SO4 and (F) applied potential of ≈1.8 VRHE in 1.0 M NaOH. | ||
Apparently, TiO2 coatings as a photoactive material have demonstrated the most substantial light enhancement effect in both the HER and OER. While Al2O3 and SiO2 are classified as insulators based on their electronic band structures, both have shown considerable enhancement. In fact, both insulating materials have served as the primary or co-catalytic material in many photo- and electrocatalytic reactions.50–54 Although their contribution and exact role remain disputable, the plausible explanations include the photoactivity of defective Al2O3 phases,50 the effective separation of the electron–hole pairs by the (hydroxylated-) Al2O3 or SiO2,51–53 and the fact that surface states of the insulators (Al2O3 and SiO2) located between the conduction and valence bands act as electron acceptors.54 Moreover, insulator photocatalysts have become increasingly popular in recent years.55
In ALD processes, the growth per cycle (GPC) varies for each material. It is largely influenced by various deposition conditions, including substrate temperature, the precursor (as well as co-reactant) dosing and purging time, or briefly, the interaction between the precursor and the substrate. Therefore, the number of cycles applied for different materials does not correspond to the same thickness. TiO2 is an example material with much lower GPC as compared to Al2O3.56–59 For example, Tupala et al. demonstrated that coating thickness of ≈ 5 nm for Al2O3, Ta2O5 and TiO2 required 50, 140, and 376 cycles, respectively.56 In this work, the three ALD coatings served as the catalytic material. For Al2O3 and SiO2 with an insulating nature, overly thick coatings will hinder the charge transfer to the carbon fibers.23 On the other hand, TiO2 is well known as a photoactive material, the thicker coatings hence provide higher photocatalytic and photoelectrochemical activities,10,60 as long as the 1D carbon fibers are not completely clogged, as supported by the comparison in Fig. S5 in the ESI.†
The photoresponse of the ALD TiO2 coated 3D-printed carbon electrodes irradiated by a blue light was exemplified by the chronoamperometry measurement in Fig. 3C and F. The measurement followed the electrolyte used in Fig. 3A and D. The applied potentials of ≈−0.4 VRHE and ≈1.8 VRHE were selected for cathodic and anodic region reactions, respectively, according to the LSV measurements, where the HER and OER were already initiated at these potentials. In both regions, the TiO2 coated 3D electrode showed an immediate response toward irradiation, confirming a photoresponse similar to other TiO2 photoelectrodes, favorable for solar energy harvesting.7,10,61,62
Based on the positive results attained by the ‘regular’ 3D-printed electrodes, we applied the ALD TiO2 coatings to 3D electrodes of different dimensions and shapes, as shown in Fig. 4A. We tested these TiO2 coated electrodes for the HER, where the LSV curves are given in Fig. 4B. Taking into consideration the different geometrical surface areas of the prepared 3D electrodes, at −10 mA cm−2, the overpotential varies in between 0.70 and 0.75 V, with the ‘regular’ 3D electrode showing the lowest overpotential of 0.60 V. The results show that the electrode performance, in other words, its efficiency, does not increase linearly by scaling up the surface area of an electrode. In most cases, electrodes with surface area ≈ 1 cm2 or smaller remain the most optimized photo- and electrocatalytic water splitting electrodes.63–65
 | ||
| Fig. 4 Scaling-up 3D-printed carbon electrodes. (A) Models of 3D electrodes in different shapes and sizes. (B) LSV of the HER of 1500 ALD cycles TiO2 coated on different 3D electrodes. | ||
For large scale electrodes, factors such as electrode homogeneity, distribution of current density, ohmic losses, electrolyte conductivity, and pH gradient might be more pronounced and thus influence the electrode performance, of which some of these factors are also supported by modelling and simulation.63–67 These findings suggest that potential mitigation can be achieved by combining multiple smaller units to minimize the losses described above to achieve a large area device.68 In other words, the scalability is achieved with the continuous repetition of a base unit to form arrays as a large unit. This approach has been demonstrated by many applications,69 for example, photovoltaic cells, solar steam generation for CO2 reduction,70 fluidic batteries,71 and aluminum–air micro-batteries.72 These works show that combining small base units to form a large unit retains the advantages such as a large surface-to-volume ratio, enhanced heat and mass transfer, and precise fluid control, all of which are fundamental aspects for solar and electrochemical applications. Overall, the design and geometry of the electrodes are a major challenge in scaling up photo- and electrocatalytic devices to move from laboratory to industrial scale systems.
To evaluate the performance of the 3D-printed electrodes, we compile Table 1 for the HER and OER performance of other works from the literature. We selected TiO2 as a catalytic material on 3D electrodes printed by other techniques (electrodes were made from other materials, typically metals) and similar carbon-based electrodes printed by the FDM technique with other catalytic materials. Evidently, our 3D electrodes documented comparable HER and OER overpotentials, showing that the carbon-based electrode is a feasible alternative as a 3D conductive platform.
| Hydrogen evolution | ||||
|---|---|---|---|---|
| Catalyst material | 3D electrode | Electrolyte | Performance (overpotential vs. RHE) | Ref. |
| a Abbreviations: EBM = electron-beam melting, SLM = selective laser melting, DIW = direct-ink-writing, FDM = fused deposition modelling. | ||||
| Anodized TiO2 | Ti electrode by EBM | 0.5 M H2SO4 | 0.092 V at −10 mA cm−2 | 73 |
| Electrodeposition of Ni | Carbon electrode by FDM | 1.0 M NaOH | 0.60 V at −50 mA cm−2 | 24 |
| Electrodeposition of NiPt | Carbon electrode by FDM | 1.0 M KOH | 0.27 V at −10 mA cm−2 | 25 |
| ALD MoS2 | Carbon electrode by FDM | 0.5 M H2SO4 | 0.49 V at −10 mA cm−2 | 20 |
| 0.44 V at −10 mA cm−2 (irradiation) | ||||
| ALD TiO2 | Carbon electrode by FDM | 0.5 M H2SO4 | 0.60 V at −10 mA cm−2 | This work |
| 0.31 V at −10 mA cm−2 (irradiation) | ||||
| Oxygen evolution | ||||
|---|---|---|---|---|
| Catalyst material | 3D electrode | Electrolyte | Performance (overpotential vs. RHE or current density) | Ref. |
| Anodized and hydrogenated TiO2 | Ti electrode by SLM | 1.0 M NaOH | ≈0.3 mA cm−2 at 1.2 V (irradiation) | 7 |
| ALD TiO2 | Stainless steel electrode by SLM | 1.0 M NaOH | ≈0.9 mA cm−2 at 1.23 V (irradiation) | 10 |
| ALD Ir coating within anodized TiO2 | Ti6Al4V electrode by selective EBM | 1.0 M H2SO4 at 60 °C | 0.29 V at 10 mA cm−2 or 65.2 mA cm−2 at 0.5 V | 12 |
| Electrodeposition of NiFe-layered double hydroxide | Graphene electrode by DIW | 1.0 M KOH | 1.50 V at 10 mA cm−2 | 74 |
| Electrodeposition of Ni–Fe (oxy)hydroxide | Graphene electrode by FDM | 0.1 M KOH | 0.52 V at 10 mA cm−2 | 75 |
| Electrodeposition of Ni | Carbon electrode by FDM | 1.0 M NaOH | 1.80 V at 50 mA cm−2 | 24 |
| Electrodeposition ReS2 | Carbon electrode by FDM | 1.0 M NaOH | ≈7 μA cm−2 at 1.23 V (irradiation) | 18 |
| ALD TiO2 | Carbon electrode by FDM | 1.0 M NaOH | 1.86 V at 10 mA cm−2 | This work |
| 1.47 V at 10 mA cm−2 (irradiation) | ||||
Conclusions
We demonstrated a simple treatment step in aqueous NaOH to prepare the surface of 3D-printed carbon electrodes for subsequent low-temperature ALD processes. The surface wettability of the carbon electrodes is entirely different after treatment in different solvents. We showed three commonly used ALD coatings, TiO2, SiO2, and Al2O3 on 3D-printed carbon electrodes for light-enhanced HER and OER. All ALD coated electrodes presented improved performance with blue light irradiation. TiO2 coated electrodes recorded the lowest HER overpotentials at −10 mA cm−2 at 0.60 V (dark) and 0.31 V (irradiation), and OER overpotentials at 10 mA cm−2 at 1.86 V (dark) and 1.47 V (irradiation). We applied the ALD TiO2 coating on 3D electrodes with different shapes and sizes, such as multiple rod structures and 2 inch-diameter disks, to show the freedom of 3D design and scalability by FDM-based printing. Similar to other upscaled electrodes, the HER performance of the larger 3D electrodes suffered a slight degradation. Thus, engineering and designing efficient electrodes will be essential for large scale electrochemical energy conversion in the near future. The present work reports an improved carbon surface ready for diverse ALD coatings to increase the functionalities of 3D electrodes with absolute freedom in design and scaling for catalytic applications and beyond.Author contributions
SN, MS, and ER prepared the electrodes. SN and MS carried out ALD, surface treatment, and electrochemical experiments. MS conducted XRD measurement. SN performed SEM and XPS measurements, and wrote the manuscript. MP acquired funding and supervised the work. All authors reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MP acknowledges the support from ERDF/ESF project TECHSCALE (No. CZ.02.01.01/00/22_008/0004587). MS is thankful for the Brno PhD Talent Scholarship funded by the Brno City Municipality. This research was co-funded by the European Union under the REFRESH-Research Excellence For REgion Sustainability and High-tech Industries project number CZ.10.03.01/00/22_003/0000048 via the Operational Programme Just Transition. This work was supported by the Ministry of Health of Czech Republic, grant no. NU21-08-00407. Material deposition and characterizations were carried out at the CEITEC Nano Research Infrastructure supported by CzechNanoLab project LM2023051, MEYS CR. We thank Dr Josef Polčák and Dr Marek Eliáš for technical assistance for XPS and ALD, respectively.References
- S. Vaidya, P. Ambad and S. Bhosle, Procedia Manuf., 2018, 20, 233–238 CrossRef.
- J. V. Vaghasiya, C. C. Mayorga-Martinez and M. Pumera, Adv. Funct. Mater., 2021, 31, 2106990 CrossRef CAS.
- 3D-printed Houses, https://all3dp.com/2/best-companies-building-3d-printed-houses/, accessed 27.12.2022.
- Y. Du, R. Wang, M. Zeng, S. Xu, M. Saeidi-Javash, W. Wu and Y. Zhang, Nano Energy, 2021, 90, 106522 CrossRef CAS.
- Z.-S. Wu, A. Winter, L. Chen, Y. Sun, A. Turchanin, X. Feng and K. Müllen, Adv. Mater., 2012, 24, 5130–5135 CrossRef CAS PubMed.
- A. Ambrosi and M. Pumera, Adv. Funct. Mater., 2018, 28, 1700655 CrossRef.
- C. Y. Lee, A. C. Taylor, S. Beirne and G. G. Wallace, Adv. Energy Mater., 2017, 7, 1701060 CrossRef.
- S. Chang, X. Huang, C. Y. A. Ong, L. Zhao, L. Li, X. Wang and J. Ding, J. Mater. Chem. A, 2019, 7, 18338–18347 RSC.
- A. Ambrosi, J. G. S. Moo and M. Pumera, Adv. Funct. Mater., 2016, 26, 698–703 CrossRef CAS.
- M. P. Browne, J. Plutnar, A. M. Pourrahimi, Z. Sofer and M. Pumera, Adv. Energy Mater., 2019, 9, 1900994 CrossRef.
- V. Urbanová, J. Plutnar and M. Pumera, Appl. Mater. Today, 2021, 24, 101131 CrossRef.
- A. Hofer, S. Wachter, D. Döhler, A. Laube, B. S. Batalla, Z. Fu, C. Weidlich, T. Struckmann, C. Körner and J. Bachmann, Electrochim. Acta, 2022, 417, 140308 CrossRef CAS.
- C. W. Foster, M. P. Down, Y. Zhang, X. Ji, S. J. Rowley-Neale, G. C. Smith, P. J. Kelly and C. E. Banks, Sci. Rep., 2017, 7, 1–11 CrossRef PubMed.
- D. M. Wirth, M. J. Sheaff, J. V. Waldman, M. P. Symcox, H. D. Whitehead, J. D. Sharp, J. R. Doerfler, A. A. Lamar and G. LeBlanc, Anal. Chem., 2019, 91, 5553–5557 CrossRef CAS PubMed.
- E. Redondo, J. Muñoz and M. Pumera, Carbon, 2021, 175, 413–419 CrossRef CAS.
- M. P. Browne, F. Novotný, Z. Sofer and M. Pumera, ACS Appl. Mater. Interfaces, 2018, 10, 40294–40301 CrossRef CAS PubMed.
- D. P. Rocha, R. G. Rocha, S. V. F. Castro, M. A. G. Trindade, R. A. A. Munoz, E. M. Richter and L. Angnes, Electrochem. Sci. Adv., 2022, 2, 202100136 Search PubMed.
- S. Ng, C. Iffelsberger, Z. Sofer and M. Pumera, Adv. Funct. Mater., 2020, 30, 1910193 CrossRef CAS.
- K. A. Novčić, C. Iffelsberger, S. Ng and M. Pumera, Nanoscale, 2021, 13, 5324–5332 RSC.
- S. Ng, R. Zazpe, J. Rodriguez-Pereira, J. Michalička, J. M. Macak and M. Pumera, J. Mater. Chem. A, 2021, 9, 11405–11414 RSC.
- W. Gao, J. Michalička and M. Pumera, Small, 2022, 18, 1–13 Search PubMed.
- L. Wang, S. Ng, Jyoti and M. Pumera, ACS Appl. Nano Mater., 2022, 5, 9719–9727 CrossRef CAS.
- S. Ng, C. Iffelsberger, J. Michalička and M. Pumera, ACS Nano, 2021, 15, 686–697 CrossRef CAS PubMed.
- J. C. Bui, J. T. Davis and D. V. Esposito, Sustainable Energy Fuels, 2020, 4, 213–225 RSC.
- B. Hüner, N. Demir and M. F. Kaya, Fuel, 2023, 331, 125971 CrossRef.
- I. Levchuk, C. Guillard, F. Dappozze, S. Parola, D. Leonard and M. Sillanpää, J. Photochem. Photobiol., A, 2016, 328, 16–23 CrossRef CAS.
- L. Tian, A. Soum-Glaude, F. Volpi, L. Salvo, G. Berthomé, S. Coindeau, A. Mantoux, R. Boichot, S. Lay, V. Brizé, E. Blanquet, G. Giusti and D. Bellet, J. Vac. Sci. Technol., A, 2015, 33, 01A141 CrossRef.
- J. S. King, E. Graugnard, O. M. Roche, D. N. Sharp, J. Scrimgeour, R. G. Denning, A. J. Turberfield and C. J. Summers, Adv. Mater., 2006, 18, 1561–1565 CrossRef CAS.
- J. R. Oh, J. H. Moon, H. K. Park, J. H. Park, H. Chung, J. Jeong, W. Kim and Y. R. Do, J. Mater. Chem., 2010, 20, 5025–5029 RSC.
- F. Dvorak, R. Zazpe, M. Krbal, H. Sopha, J. Prikryl, S. Ng, L. Hromadko, F. Bures and J. M. Macak, Appl. Mater. Today, 2019, 14, 1–20 CrossRef.
- A. Spende, N. Sobel, M. Lukas, R. Zierold, J. C. Riedl, L. Gura, I. Schubert, J. M. M. Moreno, K. Nielsch, B. Stühn, C. Hess, C. Trautmann and M. E. Toimil-Molares, Nanotechnology, 2015, 26, 335301 CrossRef PubMed.
- S. Ng, J. Prášek, R. Zazpe, Z. Pytlíček, Z. Spotz, J. R. Pereira, J. Michalička, J. Přikryl, M. Krbal, H. Sopha, J. Hubálek and J. M. Macák, ACS Appl. Mater. Interfaces, 2020, 12, 33386–33396 CrossRef CAS PubMed.
- C. Marichy and N. Pinna, Coord. Chem. Rev., 2013, 257, 3232–3253 CrossRef CAS.
- J. Tupala, M. Kemell, E. Härkönen, M. Ritala and M. Leskelä, Nanotechnology, 2012, 23, 125707 CrossRef PubMed.
- R. van de Krol and M. Graetzel, Photoelectrochemical Hydrogen Production, Springer US, Boston, MA, 2012, vol. 102 Search PubMed.
- D. Mattia, M. P. Rossi, B. M. Kim, G. Korneva, H. H. Bau and Y. Gogotsi, J. Phys. Chem. B, 2006, 110, 9850–9855 CrossRef CAS PubMed.
- C. Zhang, D. Long, B. Xing, W. Qiao, R. Zhang, L. Zhan, X. Liang and L. Ling, Electrochem. Commun., 2008, 10, 1809–1811 CrossRef CAS.
- Z. Zhang, J. Xi, H. Zhou and X. Qiu, Electrochim. Acta, 2016, 218, 15–23 CrossRef CAS.
- Q. Zhang, T. Li, J. Liang, N. Wang, X. Kong, J. Wang, H. Qian, Y. Zhou, F. Liu, C. Wei, Y. Zhao and X. Zhang, J. Mater. Chem. A, 2018, 6, 7509–7516 RSC.
- C. Meng, B. Wang, Z. Gao, Z. Liu, Q. Zhang and J. Zhai, Sci. Rep., 2017, 7, 41825 CrossRef CAS PubMed.
- U. Diebold and T. E. Madey, Surf. Sci. Spectra, 1996, 4, 227 CrossRef CAS.
- A. C. Bronneberg, C. Höhn and R. van de Krol, J. Phys. Chem. C, 2017, 121, 5531–5538 CrossRef CAS.
- M. Kalani and R. Yunus, Int. J. Nanomed., 2012, 2165–2172 CrossRef CAS PubMed.
- K. Ghosh and M. Pumera, Nanoscale, 2021, 13, 5744–5756 RSC.
- S. Ng, H. Sopha, R. Zazpe, Z. Spotz, V. Bijalwan, F. Dvorak, L. Hromadko, J. Prikryl and J. M. Macak, Front. Chem., 2019, 7, 38 CrossRef CAS PubMed.
- L. Zheng, X. Cheng and G. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 7014–7019 CrossRef CAS PubMed.
- M. Putkonen, M. Bosund, O. M. E. Ylivaara, R. L. Puurunen, L. Kilpi, H. Ronkainen, S. Sintonen, S. Ali, H. Lipsanen, X. Liu, E. Haimi, S.-P. Hannula, T. Sajavaara, I. Buchanan, E. Karwacki and M. Vähä-Nissi, Thin Solid Films, 2014, 558, 93–98 CrossRef CAS.
- B. R. Strohmeier, Surf. Interface Anal., 1990, 15, 51–56 CrossRef CAS.
- N. Koshizaki, H. Umehara and T. Oyama, Thin Solid Films, 1998, 325, 130–136 CrossRef CAS.
- F. Li, S. Liu, Y. Xue, X. Wang, Y. Hao, J. Zhao, R. Liu and D. Zhao, Chem.–Eur. J., 2015, 21, 10149–10159 CrossRef CAS PubMed.
- C.-S. Yang, Y.-J. Wang, M.-S. Shih, Y.-T. Chang and C.-C. Hon, Appl. Catal., A, 2009, 364, 182–190 CrossRef CAS.
- F. Tzompantzi, Y. Piña, A. Mantilla, O. Aguilar-Martínez, F. Galindo-Hernández, X. Bokhimi and A. Barrera, Catal. Today, 2014, 220–222, 49–55 CrossRef CAS.
- C. Iffelsberger, D. Rojas and M. Pumera, J. Phys. Chem. C, 2022, 126, 9016–9026 CrossRef CAS.
- R. Li, X. Wang, S. Jin, X. Zhou, Z. Feng, Z. Li, J. Shi, Q. Zhang and C. Li, Sci. Rep., 2015, 5, 13475 CrossRef CAS PubMed.
- K. Li, S. Zhang, Q. Tan, X. Wu, Y. Li, Q. Li, J. Fan and K. Lv, Chem. Eng. J., 2021, 426, 130772 CrossRef CAS.
- J. Tupala, M. Kemell, E. Härkönen, M. Ritala, M. Leskelä, V. Pore, T. T. Isimjan, A. El Ruby, S. Rohani, J. Tupala, M. Kemell and H. Emma, Nanotechnology, 2012, 23, 125707 CrossRef PubMed.
- A. Szeghalmi, M. Helgert, R. Brunner, F. Heyroth, U. Gösele and M. Knez, Appl. Opt., 2009, 48, 1727–1732 CrossRef CAS PubMed.
- J. A. Kropp, Y. Cai, Z. Yao, W. Zhu and T. Gougousi, J. Vac. Sci. Technol., A, 2018, 36, 06A101 CrossRef.
- L.-S. Gao, Q.-Y. Cai, E.-T. Hu, Q.-Y. Zhang, Y.-T. Yang, Y.-B. Xiong, B.-J. Liu, W.-B. Duan, T.-Y. Yu and D.-Q. Liu, Opt. Express, 2023, 31, 13503 CrossRef CAS PubMed.
- H. Sopha, M. Krbal, S. Ng, J. Prikryl, R. Zazpe, F. K. Yam and J. M. Macak, Appl. Mater. Today, 2017, 9, 104–110 CrossRef.
- S. Ng, F. K. Yam, S. N. Sohimee, K. P. Beh, S. S. Tneh, Y. L. Cheong and Z. Hassan, Sens. Actuators, A, 2018, 279, 263–271 CrossRef CAS.
- C. Iffelsberger, S. Ng and M. Pumera, Chem. Eng. J., 2022, 446, 136995 CrossRef CAS.
- I. Y. Ahmet, Y. Ma, J.-W. W. Jang, T. Henschel, B. Stannowski, T. Lopes, A. Vilanova, A. Mendes, F. F. Abdi and R. van de Krol, Sustainable Energy Fuels, 2019, 3, 2366–2379 RSC.
- S. Dilger, M. Trottmann and S. Pokrant, ChemSusChem, 2019, 12, 1931–1938 CrossRef CAS PubMed.
- H. Lu, V. Andrei, K. J. Jenkinson, A. Regoutz, N. Li, C. E. Creissen, A. E. H. Wheatley, H. Hao, E. Reisner, D. S. Wright and S. D. Pike, Adv. Mater., 2018, 30, 1804033 CrossRef PubMed.
- S. Haussener, C. Xiang, J. M. Spurgeon, S. Ardo, N. S. Lewis and A. Z. Weber, Energy Environ. Sci., 2012, 5, 9922 RSC.
- A. Hankin, F. E. Bedoya-Lora, C. K. Ong, J. C. Alexander, F. Petter and G. H. Kelsall, Energy Environ. Sci., 2017, 10, 346–360 RSC.
- B. Turan, J.-P. Becker, F. Urbain, F. Finger, U. Rau and S. Haas, Nat. Commun., 2016, 7, 12681 CrossRef PubMed.
- S. K. S. Cheng, T. Li, S. S. Meena, Q. Cao, B. Li, B. K. Kosgei, T. Cheng, P. Luo, Q. Liu, G. Zhu, Q. Liu and R. P. S. Han, Adv. Energy Sustainability Res., 2022, 3, 2200060 CrossRef.
- H. Liu, H. Ye, M. Gao, Q. Li, Z. Liu, A. Xie, L. Zhu, G. W. Ho and S. Chen, Adv. Sci., 2021, 8, 2101232 CrossRef CAS PubMed.
- N. K. Thom, K. Yeung, M. B. Pillion and S. T. Phillips, Lab Chip, 2012, 12, 1768 RSC.
- L.-L. Shen, G.-R. Zhang, M. Biesalski and B. J. M. Etzold, Lab Chip, 2019, 19, 3438–3447 RSC.
- X. Li, Y. Xue, R. Dehoff, C. Tsouris and P. Taboada-Serrano, Journal of Energy and Power Technology, 2020, 2, 1–16 CrossRef.
- J. Ahn, Y. S. Park, S. Lee, J. Yang, J. Pyo, J. Lee, G. H. Kim, S. M. Choi and S. K. Seol, Sci. Rep., 2022, 12, 346 CrossRef CAS PubMed.
- P. L. Santos, S. J. Rowley-Neale, A. G. -M. Ferrari, J. A. Bonacin and C. E. Banks, ChemElectroChem, 2019, 6, 5633–5641 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional SEM image, XPS and XRD analyses, and LSV curves for the HER. See DOI: https://doi.org/10.1039/d3ta04460b |
| This journal is © The Royal Society of Chemistry 2024 |