
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning acid extraction of magnesium and calcium from platinum group metal tailings for CO2 conversion and storage†
Caleb M.
Woodall
a,
Katherine
Vaz Gomes
 *b,
Andreas
Voigt
c,
Kai
Sundmacher
d and
Jennifer
Wilcox
b
*b,
Andreas
Voigt
c,
Kai
Sundmacher
d and
Jennifer
Wilcox
b
aIndependent Consultant, Washington, DC, USA
bDepartment of Chemical and Biomolecular Engineering, University of Pennsylvania, Philadelphia, PA, USA. E-mail: katgomes@seas.upenn.edu
cOtto-von-Guericke University Magdeburg, Germany
dDepartment of Process Systems Engineering, Max Planck Institute for Dynamics of Complex Technical Systems, Magdeburg, Germany
First published on 2nd October 2024
Abstract
Avoiding the worst impacts of climate change requires reducing greenhouse gas emissions and removing atmospheric CO2 with permanent storage. The global shift to low- and zero-emission energy sources demands increased metal mining, resulting in substantial mine tailings. Mineral carbonation offers a method to store CO2 in alkaline-rich mine tailings, addressing both waste and excess atmospheric CO2. This study explores the use of a pH-swing process to optimize the extraction of calcium and magnesium from plagioclase feldspar-rich platinum group metal (PGM) mine tailings from the Stillwater Mine in Nye, Montana. Various organic (citric, acetic, oxalic) and mineral (hydrochloric, sulfuric) acids were tested at different concentrations, solid/liquid ratios, and dissolution times. Organic acids, particularly citric and oxalic, were selective for magnesium and calcium, respectively, with citric acid extracting 44% of available magnesium in 72 hours. Sulfuric acid proved most effective in extracting both metals but may be impractical due to corrosion-resistant equipment costs. Carbonation of synthetic leachate indicated precipitation yields above 90% at pressures between 5 and 9 bar, producing carbonate products under 3 μm. Additionally, comparing in situ and ex situ base addition methods suggests that pH pre-swing, i.e., before carbonation is comparable to adding base during the reaction. This study advances the understanding of divalent cation extraction from plagioclase feldspar-rich PGM mine tailings but highlights the need for further research to develop an economic process.
Sustainability spotlightOur research addresses the urgent need to manage mining waste sustainably by utilizing it for carbon storage. Specifically, we focus on a platinum group element mine in Montana, USA, employing a pH swing mechanism involving acid leaching and carbonate precipitation. This technology promises durable carbon storage while reducing mine waste from metal production. Our work supports climate action (SDG12 and 13) by using mine tailings for durable carbon storage, essential for achieving net zero emissions by 2050 and mitigating severe climate impacts. |
Introduction
The most recent IPCC report made abundantly clear that anthropogenic CO2 emissions are causing the warming of our planet's atmosphere, and that it is necessary to develop methods that remove CO2 from the atmosphere and durably store it.1 There are several methods to remove CO2 from the atmosphere, including direct air capture, bioenergy with carbon capture, and reforestation.2,3 Another technology that can be used to both capture and durably store CO2 is mineral carbonation.4Carbon mineralization mimics the natural process of mineral weathering, where alkaline minerals (i.e., those containing Mg or Ca) interact with atmospheric CO2 and form carbonate minerals. This reaction is irreversible at practical conditions, resulting in very durable and nearly permanent CO2 storage. The challenge is to accelerate this reaction from geologic timescales of millennia to climate-relevant timescales of days to years.
Decades of research on carbon mineralization has led to companies using the technology to store CO2 today. In Iceland, Climeworks and CarbFix are constructing the Mammoth direct air capture (DAC) facility: to capture 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tCO2 per year and store underground in deep basaltic rock by 2025.5 In the United States, Heirloom Carbon Technologies won one of the Department of Energy's DAC Hub awards and will be constructing two facilities in Louisiana by 2026 with a combined capture capacity of 320000 tCO2 per year with a mineral looping technology.6 CarbonCure injects CO2 into the concrete curing process, where it reacts and permanently binds with calcium to make a stronger concrete product: the technology won the NRG COSIA XPRIZE and has stored over 150000 tCO2 to date.7 While these companies have seen success with some feedstocks that have relatively reactive mineralogy, other sources exist that require additional research.
000 tCO2 per year and store underground in deep basaltic rock by 2025.5 In the United States, Heirloom Carbon Technologies won one of the Department of Energy's DAC Hub awards and will be constructing two facilities in Louisiana by 2026 with a combined capture capacity of 320000 tCO2 per year with a mineral looping technology.6 CarbonCure injects CO2 into the concrete curing process, where it reacts and permanently binds with calcium to make a stronger concrete product: the technology won the NRG COSIA XPRIZE and has stored over 150000 tCO2 to date.7 While these companies have seen success with some feedstocks that have relatively reactive mineralogy, other sources exist that require additional research.
The Stillwater Mine in Nye, Montana produces platinum, palladium, copper, and nickel. The mine generates about 1 Mt of mine tailings each year, and nearly half of these have been stored in two different tailings storage facilities.8 Indeed, tailings from mines like Stillwater will become ever more relevant in the coming decades, as production of nickel and platinum group metals (PGMs) is expected to grow by a factor of 5 and 10, respectively, due to increased demand through the global transition to clean energy.9 Mineral carbonation could be used to develop an effective waste management practice for tailings generated by the increased mining practices.
A previous study characterized and assessed the reactivity of four tailings samples from Stillwater.10 Characterization results showed that the tailings are heterogeneous, where the primary component is anorthite – the Ca-end member of the plagioclase feldspar group of aluminosilicates. Observation of the tailings storage facilities indicates that older tailings have been slowly carbonating over nearly two decades. However, low reactivity in experiments simulating ambient conditions (atmospheric temperature and pressure) and slightly elevated CO2 concentration (10%) suggests that a process with elevated conditions (e.g., temperature, pressure, time, and/or pH) might be necessary to maximize the CO2 stored in the tailings.
The alternative ÅA route is an indirect carbon mineralization process that uses a thermal solid/solid reaction with ammonium sulfate to extract magnesium.10 A previous study use the alternative ÅA route to increase divalent cation extraction in the Stillwater tailings.11 While this process was optimized to extract up to 41% of the Mg in the Stillwater tailings, the Ca extraction was limited to 13%. This issue stems from the aqueous dissolution step: because CaSO4 is practically insoluble, the extracted calcium is kept with the “waste residue” and does not progress to the carbonation step with the extracted magnesium. This study indicated that to maximize the CO2 storage potential of the Stillwater tailings, a different process is necessary that can extract both calcium and magnesium.
One process that has been identified is the pH swing using organic acids.12 Weak organic acids such as acetic acid, oxalic acid, and citric acid have been demonstrated to effectively dissolve plagioclase structures.13 Oxalic and citric acids also have displayed selective leaching of Ca, K, Na, and Al from plagioclase.13,14 Because plagioclase feldspar makes up more than half of the Stillwater tailings, use of organic acids could prove effective in divalent cation extraction. There are two different mechanisms at play when studying acidic mineral dissolution. Organic acids aid in dissolution of mineral structures primarily via organic ligands that attack cations at the mineral surface (i.e., ligand-promoted dissolution). Ligands like acetate, citrate, and oxalate adsorb on the mineral surface, forming complexes with metal ions that typically have high solubility.15
Alternatively, mineral acids, such as hydrochloric and sulfuric acids, dissolve minerals via proton-promoted dissolution, where the cations at the mineral surface are protonated. The ability of either of these mechanisms to promote mineral dissolution depends on the nature of the mineral surface and the ligand, and the concentration of surface active sites, ligands, and protons.16 Both mineral and organic acids have been used to extract magnesium and calcium from silicate structures for the purpose of carbon mineralization, with varying degrees of success. The structures of tecto-, ino-,and phyllosilicates possess different susceptibility to proton and ligand attack. The majority of previous studies used mined material that is relatively homogeneous in mineralogy (e.g., serpentine, olivine, pyroxene, plagioclase) and chemistry (i.e., primarily rich in calcium or magnesium). For example, one study used citric and oxalic acids with serpentine samples, finding that citric acid was more effective than oxalic acid at selectively leaching Mg.17 Other studies have shown that mineral acids can leach Mg from phyllosilicates like serpentine at a high rate.18–20 However, these mineral acids have been shown to be less effective than organic acids at dissolving tectosilicate structures, where sulfuric acid can even have a detrimental effect by creating inert surface complexes.14,15,21–23 Meanwhile very few studies consider mineralogically and chemically heterogeneous tailings like those of Stillwater. One study used the pH swing process to extract magnesium and calcium from pyroxene-rich PGM tailings, and found that oxalic acid generally promoted Ca extraction, while Mg was mainly extracted via proton attack from mineral acids.24 These results suggest that both organic and mineral acids might be necessary to maximize the extraction of cations from PGM tailings.
Given the projected production scale of tailings from nickel and PGM mines like the Stillwater Mine, this study aims to develop a method to store CO2 in the tailings, effectively managing mine wastes and providing a sink for our society's excess carbon emissions. Due to the mixed results in the literature surrounding dissolution of chemically and mineralogically heterogeneous tailings like those of Stillwater, the experiments in this study were designed to optimize magnesium and calcium extraction with organic (acetic, citric, oxalic) and mineral (hydrochloric, sulfuric) acids.
To elucidate the prevailing mechanisms in dissolution of PGM tailings, special attention is given to the extent of extraction within each acid. To optimize acidic extraction, new “series” and “parallel” acid extraction processes are introduced. Finally, carbonation experiments were conducted on synthetic extraction fluid to study the precipitation of calcium carbonate and the impact of base addition on the carbonation reaction.
Materials & methods
Materials
In this study were performed using the Stillwater tailings sample Tail 2a. This tailings sample was characterized previously, and a summary of the results is shown in Tables 1 and 2.10| Silicate group | Mineral type | Mineral | Tail 2a | Contributions | |
|---|---|---|---|---|---|
| To Tail 2a | |||||
| Mg | Ca | ||||
| Tecto – (3-D framework) | Plagioclase feldspar | Anorthite – CaAl2SiO8 | 54.3% | 0.0% | 76.6% |
| Albite – NaAlSi3O8 | 3.9% | 0.0% | 0.3% | ||
| Ino – (chain) | Pyroxene | Enstatite – (Mg,Fe)Si2O6 | 11.9% | 35.3% | 0.0% |
| Diopside – CaMgSi2O6 | 9.7% | 13.4% | 18.6% | ||
| Amphibole | Tremolite – Ca2Mg5(Si8O22)(OH)2 | 2.2% | 4.1% | 2.3% | |
| Cummingtonite – Mg7(Si8O22)(OH)2 | 1.3% | 3.4% | 0.0% | ||
| Phyllo – (sheet) | Serpentine | Lizardite – Mg3Si2O5(OH)4 | 8.4% | 27.1% | 0.0% |
| Clay | Talc – Mg3Si4O10(OH)2 | 2.3% | 5.4% | 0.0% | |
| Mica | Clinochlore – (Mg,Fe)5Al2SiO3O10(OH) | 6.0% | 11.2% | 0.0% | |
| TIC (% C) | 0.1% | 0.0% | 2.3% | ||
| d (0.9) (μm) | 51.17 | ||||
| BET reactive surface area (m2 g−1) | 1.29 | ||||
| SiO2 | Al2O3 | Fe2O3 | CaO | MgO | Na2O | K2O | LOI |
|---|---|---|---|---|---|---|---|
| % | % | % | % | % | % | % | % |
| 45.58 | 22.98 | 5.14 | 13.30 | 8.55 | 1.30 | 0.08 | 2.29 |
Five acids were used to facilitate the tailings dissolution in these experiments: acetic, citric, oxalic, sulfuric, and hydrochloric.
Extraction methods
The experiments were run using the pH swing process shown in Fig. 1. Unless otherwise noted, extraction experiments involved dissolving 20 grams of Tail 2a in one liter of acid solution (0.1–1.0 M) for a set time (6–72 hours). Time was extended to 72 hours (and 18 days in two cases) in an attempt to determine the maximum possible extent of cation extraction in each acid. Each dissolution experiment was facilitated in a Schott Duran 1 liter glass volumetric flask, stirred at 500 rpm at room temperature (∼22 °C). After dissolution, solutions were filtered using a round paper filter with pore size ∼ 15 μm. Following filtration, the liquid extraction solution was stored in a 1 liter glass bottle until later use, and the mass of the solid residue was measured after drying overnight.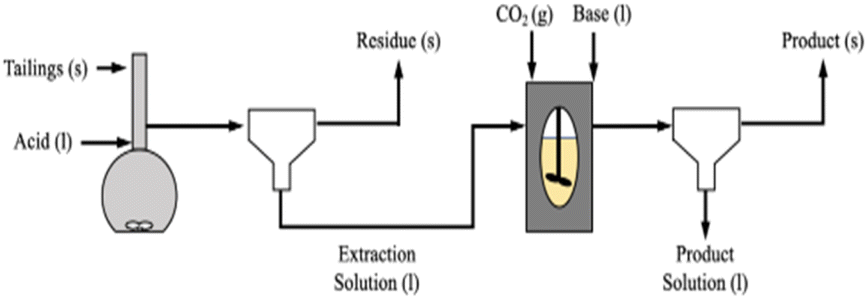 | ||
| Fig. 1 Process flow diagram for pH swing process. | ||
It is difficult to compare all five acids on a consistent basis due to their different dissolution mechanisms, where ligand- and proton-promoted dissolution are primarily affected by the concentration of ligands and protons (pH) in solution, respectively. For this study, it was decided to use constant solution concentration. The approximate associated pH of each acid at each concentration tested is shown in Table 3. The effects of ligand- and proton-promoted dissolution can be directly compared among oxalic, hydrochloric, and sulfuric acids, as these all provide the same solution pH at each concentration.
| Acid | Solution concentration | ||
|---|---|---|---|
| 0.1 M | 0.5 M | 1.0 M | |
| Citric | 2.1 | 1.7 | 1.6 |
| Acetic | 2.9 | 2.5 | 2.4 |
| Oxalic | 1.0 | 0.3 | 0.0 |
| Hydrochloric | 1.0 | 0.3 | 0.0 |
| Sulfuric | 1.0 | 0.3 | 0.0 |
Most experimental trials involved optimizing the cation extraction step of the pH swing process, where the liquid extraction solution was analyzed via Ion Chromatography (IC). The IC instrument was a Metrohm 930 Compact IC Flex, with a cation column capable of measuring Ca2+, Mg2+,Na+, K+, NH4+.
In some cases, the solid residue was analyzed for mineralogical changes relative to Tail 2a. This was done via powder X-ray diffraction (XRD) using back-mounting technique. Experimental results were analyzed according to the percentage of cations released from Tail 2a and dissolved into the extraction solution. Aqueous cation concentration results from IC were compared to the initial cation concentration in the alkaline feedstock to yield an extraction efficiency, as in eqn (1):
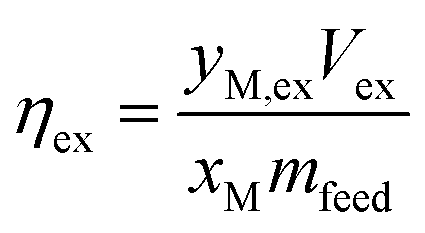 | (1) |
The error in the extraction efficiency values was derived from the IC data used to measure calcium and magnesium concentrations in solution. Multiple measurements of each trial were taken with the IC, and the average of these measurements was used to determine the extraction efficiency. The standard deviation of the measurements from each trial was propagated through the efficiency calculation to represent the error.
Carbonation methods
To carry out carbonation experiments, synthetic leachate was produced using calcium hydroxide or calcium chloride to mimic fluid produced during the extraction step while initially avoiding the presence of other species that may have leached from the silicate phases in the tailings. The level of calcium in these synthetic solutions was intended to simulate the levels of Ca that were obtained from the extraction of step. In cases where calcium chloride was used, sodium hydroxide was added in ex situ and in situ regimes, respectively to the carbonation reaction (Fig. 2). The base was added to achieve a substantially alkaline condition, of a pH of at least 10, to favor carbonate anion formation in solution (pKa = 10.33). Synthetic calcium-rich solutions were selected for carbonation experiments in preference to magnesium-rich solutions due to the propensity of calcium carbonate to form non-hydrated phases. In contrast, magnesium carbonate exhibits multiple hydrated forms, each requiring different stoichiometric amounts of CO2. By opting for calcium-rich solutions, the formation of hydrated carbonates was precluded, allowing for a more precise assessment of product yield and reaction efficiency.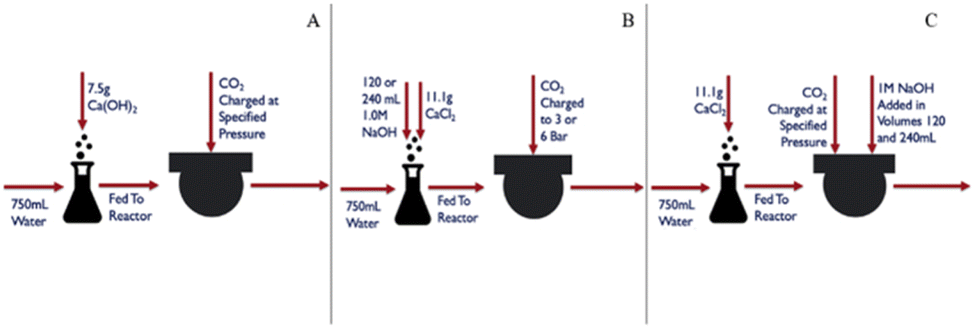 | ||
| Fig. 2 Experimental reaction pathways: (A) pressure variation experiments of calcium hydroxide carbonation; (B) ex situ base addition carbonation of calcium chloride solution; and (C) in situ base addition carbonation of calcium chloride solutions. | ||
The reactor was 1.5 liter stainless steel from Juchheim with a pressure rating of 10 bar. Reaction conditions (temperature, pressure, flow rate) were monitored in real-time using Siemens WinCC for Appelhaus reactor system. The calcium-rich fluid was added to the reactor as a batch addition at the beginning of a carbonation experiment, prior to the introduction of CO2. A CO2 cylinder (Linde AG) was connected to the reactor via stainless steel tubing. CO2 was steadily added to the reaction, at a rate of 1 L min−1, until the reactor reached the desired pressure while the stirring rate was at 500 rpm. During in situ base addition experiments, 1.0 M sodium hydroxide was pumped into the reactor with a ProMinent Beta® BT4a, set at a constant flow rate of 33 mL min−1. The reaction was then allowed to proceed for a residence time of 60 minutes.
After completion, the reaction contents were subject to particle size analysis in a Malvern Mastersizer 3000E with a Hydro M attachment and particle shape analysis by a Sympatec QicPic. The reaction contents were also vacuum filtered using a filter paper with a ∼2.5 μm pore size. The solid product would be analyzed via X-ray diffraction using a Malvern Panalytical Empyrean XRD.
The carbonation results were analyzed by comparing the actual amount of calcium carbonate precipitated to the theoretical yield (eqn (2));
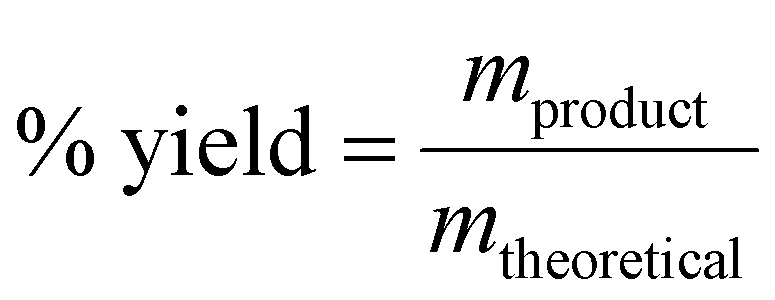 | (2) |
In addition, the carbonation results were also analyzed on the relative amount of carbon mineralized, comparing the total amount of CO2 provided to the reactor to the moles of calcium carbonate product; where mproduct represents the mass of calcium carbonate in the solid phase of the reaction product, MWCaCO3 represents the molar mass of calcium carbonate, PCO2 represents the pressure of CO2 used in the experiment, VCO2 represents the volume of CO2 added to the reactor, R is the universal gas constant, and Trxn is the temperature of the reaction.
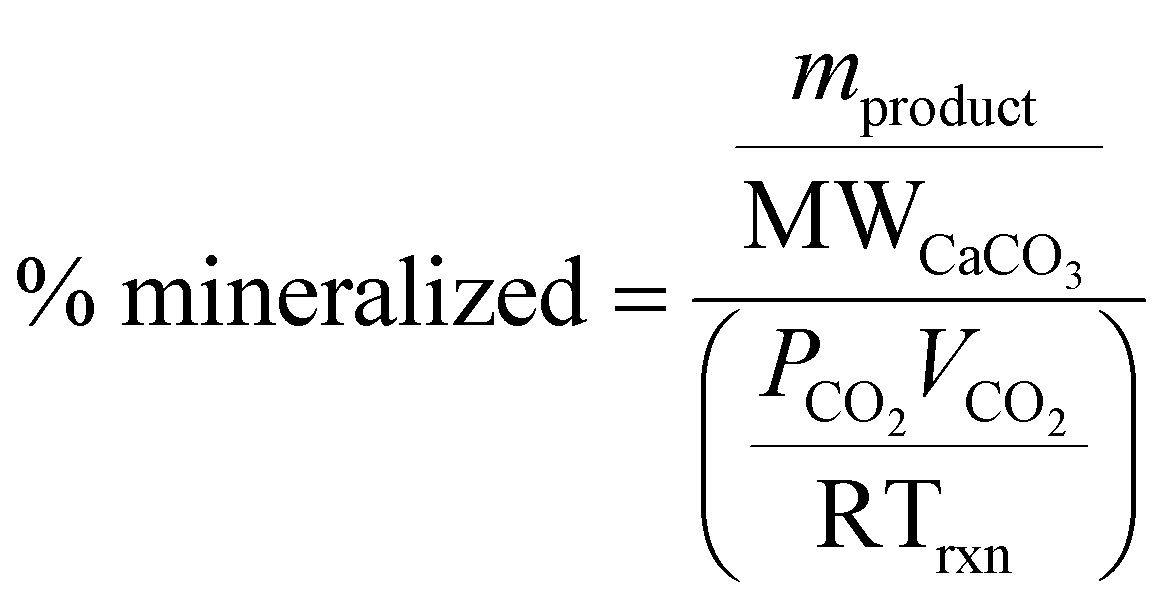 | (3) |
Results & discussion
Comparison of organic and mineral acids
Tail 2a was dissolved overnight (∼18 hours) with separate solutions of five acids at three concentrations (0.1 M, 0.5 M, 1.0 M), with 20 grams of Tail 2a per liter of acid solution. The Ca and Mg extraction efficiencies for each acid solution are displayed in Fig. 3. | ||
| Fig. 3 Extraction efficiencies of all acids at 01 M, 0.5 M, 1.0 M, overnight dissolution. | ||
The mineral acids (i.e., hydrochloric, sulfuric) were much more effective at extracting magnesium than the organic acids (i.e., citric, acetic, oxalic), especially at higher concentrations, indicating that proton-promoted dissolution is more prevalent for the Mg-containing mineral phases in Tail 2a (i.e., phyllosilicates, inosilicates). Meanwhile, the increase in concentration from 0.5 M to 1.0 M had minimal effect on the extraction efficiency for organic acid solutions. The extraction started at low pH of around 1.0 and normally increased slightly during extraction up to 3.5. It is important to note that sulfuric acid was the only diprotic acid employed in the experiments, providing twice the moles of protons compared to hydrochloric acid. This characteristic likely contributed to the observed increase in extraction efficiency when using sulfuric acid.
Based on the relative stability of the ligand–metal complexes (Table 4), the expected performance of each organic acid would be citric > oxalic > sulfuric > acetic for calcium extraction, and oxalic > citric > sulfuric > acetic for magnesium extraction. However, the results in Fig. 3 do not correlate with this predicted order (aside from the low extraction for acetic acid). While sulfuric acid creates relatively weak complexes, it also contributes strongly to dissolution via proton-promoted dissolution, with two protons per sulfate ligand. This likely explains why sulfuric acid extracted the most magnesium (and nearly the most calcium) at all concentrations. Further, the ability of sulfuric acid to contribute to ligand-promoted dissolution could explain the slightly higher extraction relative to hydrochloric acid, which only contributes to proton-promoted dissolution.18,19
K) of relevant ligand–metal complexes at 25 °C, I = 1 M, unless noted
Based on stability constants, oxalic and citric acids would be expected to be more effective in extracting Mg and Ca, respectively, which contradicts the results in Fig. 3. This contradiction is likely due to the difference in the way Ca and Mg are bound to the various silicate structures within Tail 2a, and is investigated further in the following sections.
Effect of time in organic acid dissolution
To further investigate the difference in extraction among citric and oxalic acids, experiments were carried out varying the dissolution time in 0.5 M solutions. 30 grams of Tail 2a was dissolved in 1 liter of solution for dissolution times of 6 hours to 18 days, and results are shown in Fig. 4. While using long dissolution times, like 18 days, helps to demonstrate the maximum cation extraction possible in different acids, it would likely be impractical at scale because it would require an excessively large reactor.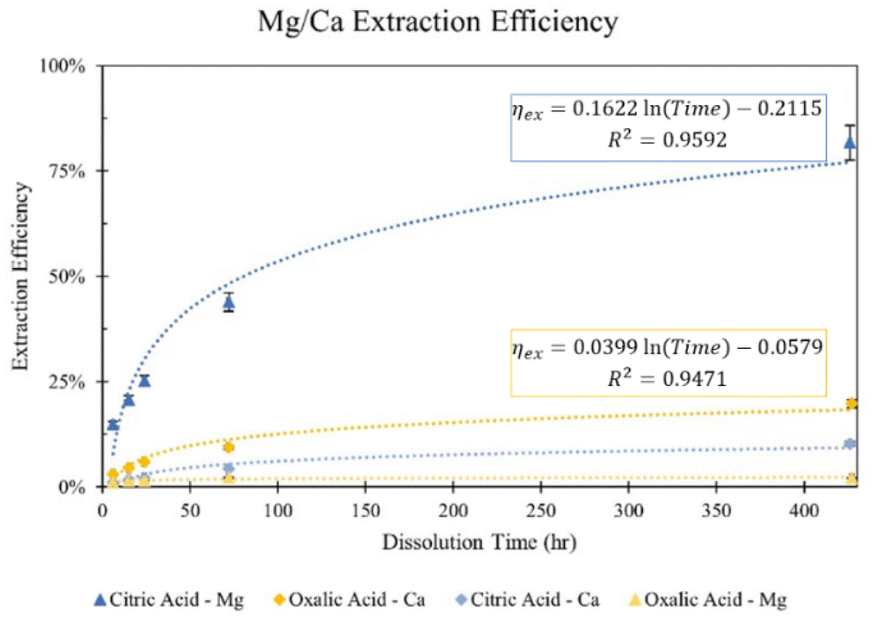 | ||
| Fig. 4 Effect of dissolution time in 0.5 M citric and oxalic acid solutions on Mg and Ca extraction. | ||
Indeed, dissolution at extended periods substantially increases the extraction efficiency of Mg and Ca. The Mg extraction efficiency of citric acid reaches about 44% after dissolving for 72 hours, and even exceeds 80% after 18 days. However, oxalic acid extracts minimal amounts of Mg, only reaching 2% after 18 days. In the case of Ca extraction, oxalic acid nearly extracts 10% in 72 hours, and doubles that in 18 days, while citric acid extracts about half that of oxalic acid at all times.
The difference in cation extraction behavior between the two organic acids is unexpected, considering their similar complex stability constants and equal cation concentrations. This behavior cannot be explained by pH alone, as oxalic acid, with its lower pH, provides more protons for dissolution. A possible explanation may lie in the composition of the solid residues remaining after dissolution. In Fig. 5, XRD diffractograms of the solid residues show the time-dependent mineralogical changes of Tail 2a during organic acid dissolution. The largest peak at 28°, associated with anorthite, steadily decreases in both acids. This decrease is more prevalent in oxalic acid, especially after 72 hours, and is coupled with the disappearance of another small anorthite peak at 22°. Another major difference among the two acids is the appearance of a new phase in oxalic acid residues, around 15°. This phase has been identified as whewellite – a highly insoluble calcium oxalate mineral (CaC2O4·H2O), and is not uncommon when Ca-bearing minerals dissolve in oxalic acid.23–25 Precipitation of new phases correlates with the far longer filtration times observed for the oxalic acid solutions following dissolution, which could be caused by increased solid mass in solution.26 These results indicate that while oxalic acid is partially dissolving the anorthite structure, it is forming undesired Ca-precipitates which are not staying in solution for later carbonation.
 | ||
| Fig. 5 Comparison of XRD diffractograms for: (a) raw minerals constituting >5% of mineral phases in Tail 2a, data from RRUFF database; solid residues from dissolution in 0.5 M solutions of (b) citric acid and (c) oxalic acid. In (b) and (c), top diffractogram is unreacted Tail 2a. Letters represent mineral phases: A – anorthite, C – clinochlore, D – diopside, E – enstatite, L – lizardite, W – whewellite. | ||
The precipitation of whewellite during dissolution in oxalic acid could provide some explanation of the extraction efficiency results observed in Fig. 3. First, the precipitation of whewellite from solution removes available oxalate ligands to extract Mg, potentially causing the minimal Mg extraction of oxalic acid.27 Further, if extracted calcium is precipitating as well, then it is not in solution during IC analysis, meaning that the Ca extraction efficiency for oxalic acid is in fact higher than shown in Fig. 4. This finding can be interpreted as good news and bad news – while it is good that oxalic acid actually has extracted Ca and partially dissolved anorthite better than initially suggested, the precipitation of highly insoluble whewellite may in fact render the extracted Ca unusable for the purpose of carbon mineralization. As the particle shape and surface properties strongly influence the extraction of certain ions out of the solid matrix, the observed anorthite dissolution might be influenced by this. The mine tailings were collected in different areas of the mine, how they were treated before storing them in these piles could not be found out. It is clear that milling and breakage of the mine waste particles will lead to changes in the extraction.
Perhaps more impactful is the interaction of oxalate and aluminum, which form strong multiligand complexes with each other.28 Because plagioclase is dissolved at the Al–O sites, every dissolved Ca cation would require formation of a strong complex (e.g., AlOx2−), essentially removing oxalate anions from solution to react with Mg-bearing phases.19 Aluminum leaching data were not collected for these experiments, but future work may consider measuring the concentration of aqueous Al3+ ions to differentiate the effects of anorthite dissolution and whewellite precipitation.
The explanations for extraction in oxalic acid solution do not hold for that in citric acid solution. In Fig. 5, the anorthite peak is steadily dissolved over time in citric acid (albeit at a slower rate than in oxalic acid), but there is no evidence of citrate precipitation to explain a decreased Ca extraction, potentially nullifying the hypothesis that Ca extraction in oxalic acid is actually higher than indicated in IC results. This finding remains an open question that should be further investigated.
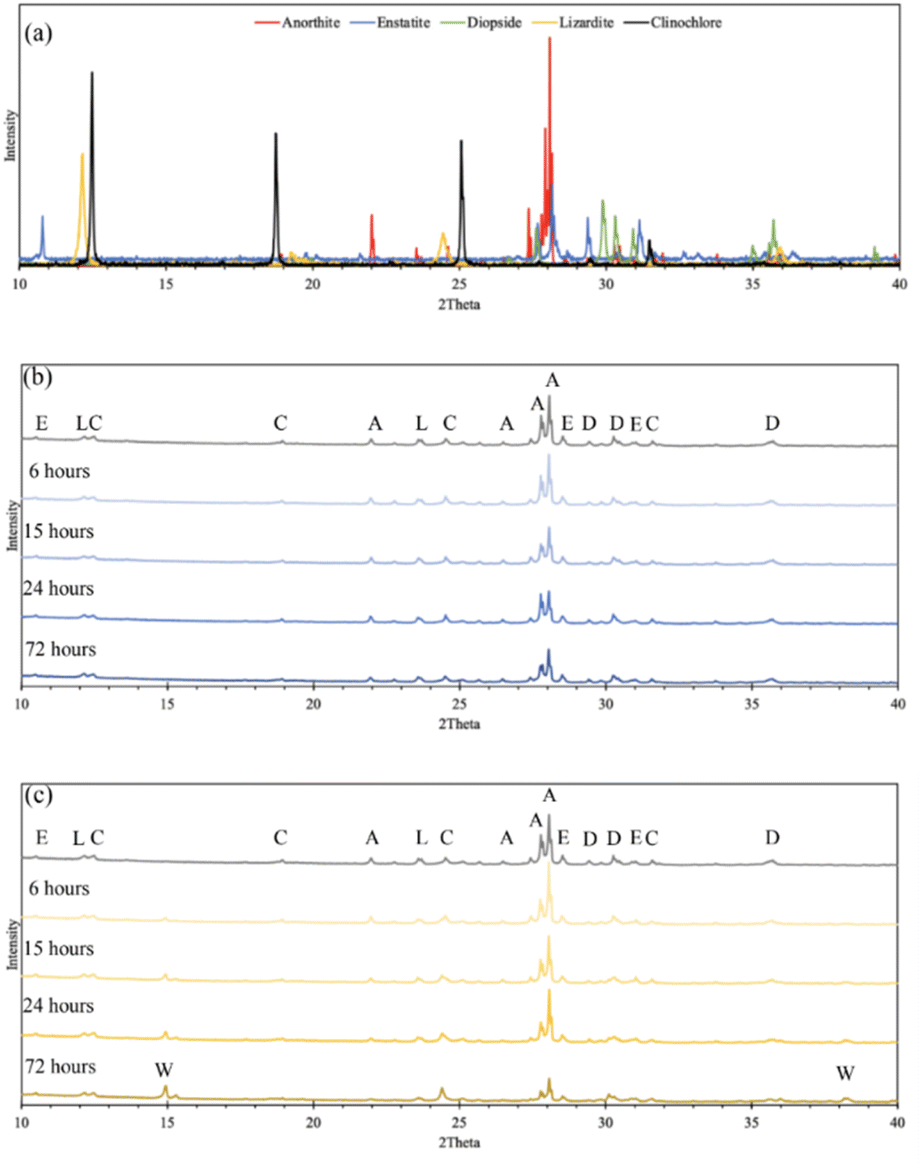
Effect of solid/liquid ratio
In an attempt to minimize the volume of acid required by the dissolution process, the ratio of solid Tail 2a mass to acid solution volume was increased from the 20 g L−1 ratio used in the initial studies. First, the ratio was increased to 30 g L−1 in an overnight (∼18 hour) dissolution period. Minimizing the volume of acid used in the extraction step is key to tuning a scalable process by limiting the reactor volume needed to conduct dissolution at scale, therefore minimizing the capital cost associated with this part of pH-swing indirect carbonation. Fig. 6a shows that during overnight dissolution, the extraction efficiency of Mg2+ and Ca2+ in both acids decreased due to an increase in the solid/liquid ratio. This would be expected, as less ligands are available to extract each cation. However, this same effect was not observed using elevated solid/liquid ratios during 72 hour dissolution (Fig. 6b). In this case, the decreases in efficiency due to increased solid/liquid ratios were minimized, and even some marginal increases in efficiency were observed for oxalic acid. These observations have practical implications for later scale-up, indicating that if dissolution time is long enough, less acid is necessary for similar extraction efficiency results.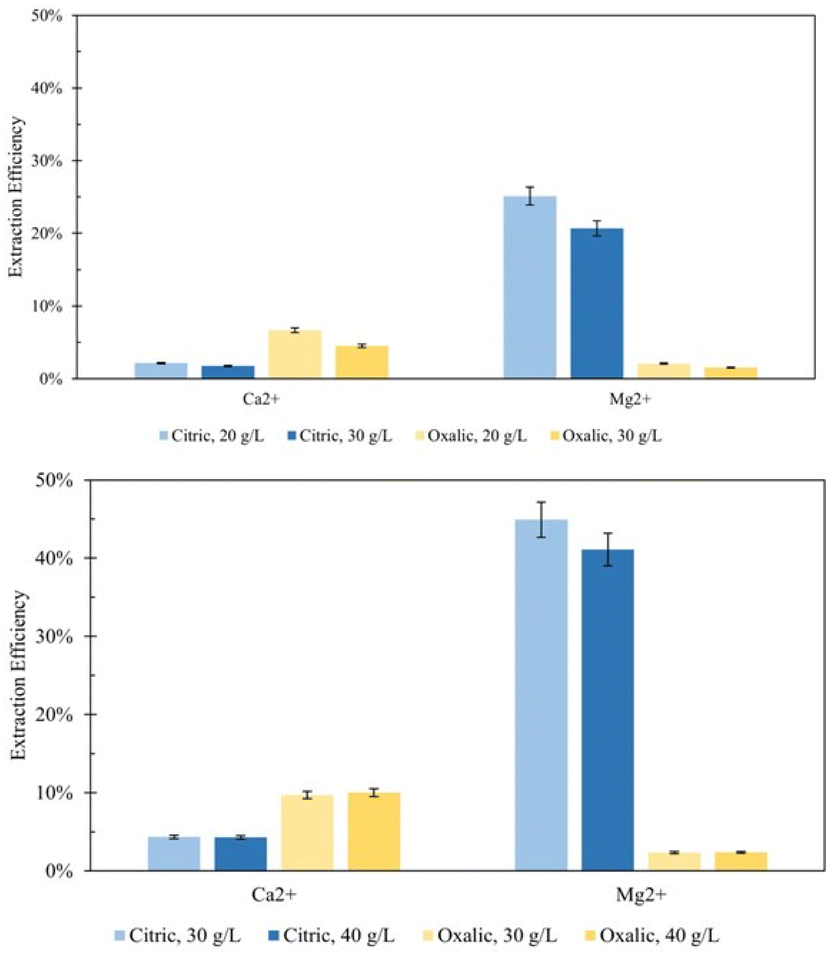 | ||
| Fig. 6 Effect of solid/liquid ratio on calcium and magnesium extraction in 0.5 M citric and oxalic acid solutions dissolving for (top) 18 hours and (bottom) 72 hours. Percentage differences are displayed above each set of bars. | ||
The effect of solid/liquid ratio was also investigated on extraction in sulfuric acid solutions. Two experiments were performed with 1 liter 0.5 M and 1.0 M sulfuric acid solutions, each dissolving 30 g Tail 2a for 72 hours. In Fig. 7, these results are compared to results of sulfuric acid solutions from Fig. 3. When comparing solutions of the same concentration, the Ca2+ and Mg2+ extraction efficiency results differ drastically.
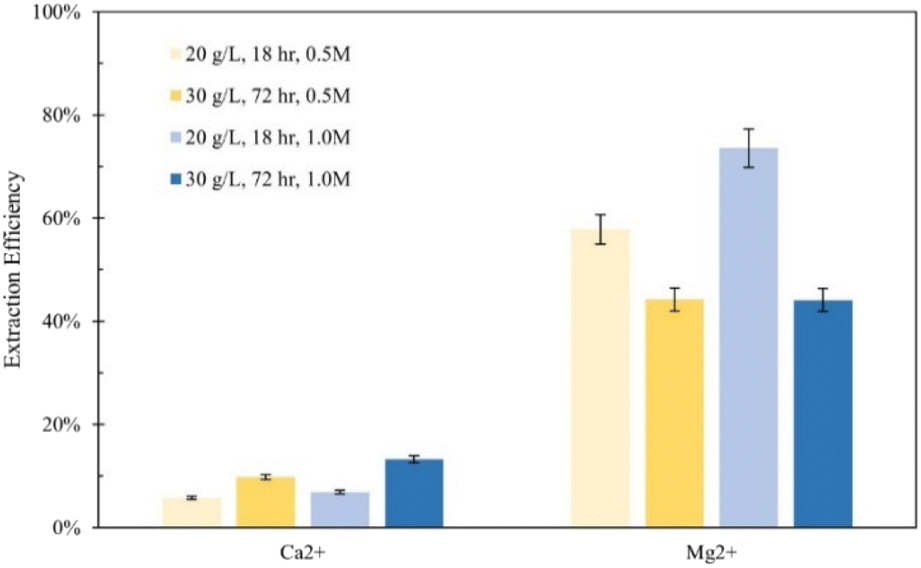 | ||
| Fig. 7 Effect of solid/liquid ratio on calcium and magnesium extraction in 0.5 M and 1.0 M sulfuric acid solutions dissolving for 18 hours and 72 hours. Percentage differences are displayed above each set of bars. | ||
In both solution concentrations, the extraction efficiency for Ca2+ increased, while it decreased for Mg2+. There might be a mechanism occurring at longer dissolution times that is preventing Mg2+ from being released with sulfuric acid. Oxoanions, like sulfates, can inhibit dissolution by forming multinuclear dissolution-inert surface complexes, occupying otherwise reactive sites.16,27 However, it is difficult to elucidate what exactly caused these observations because two parameters were changed between the two sulfuric acid trials. More studies should be done to further investigate dissolution with sulfuric acid at various solid/liquid ratios and dissolution times.
Fig. 7 demonstrates the tendency of sulfuric acid to extract a higher amount of magnesium according to its extraction efficiency. Despite that the Stillwater tailings contain more calcium by mass, a larger mass of magnesium was extracted overall: the 0.5 M and 1.0 M sulfuric acid treatments extracted only 113–390 mg Ca2+ per L and 613–780 mg Mg2+ per L.
Series and parallel dissolution for increased efficiency
To further elucidate the effect of different acids on dissolution of the varying mineral phases in Tail 2a, and potentially further increase cation extraction, multiple acids were combined in one process. As illustrated in Fig. 8, this was be performed in two different ways: (1) “parallel,” where both citric and oxalic acids are combined in one reaction solution, and (2) “series,” where one acid is used to produce a liquid extraction solution and solid residue, and a different acid is used to dissolve the solid residue.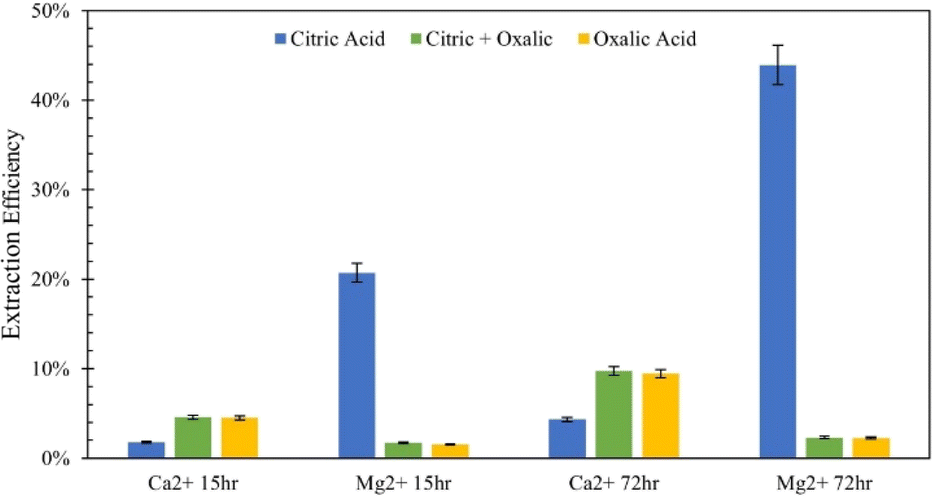 | ||
| Fig. 8 Comparison of extraction efficiency through parallel dissolution (green) and single acid dissolution (blue/yellow) at 15 and 72 hours. In all cases, each acid was 0.5 M in solution. | ||
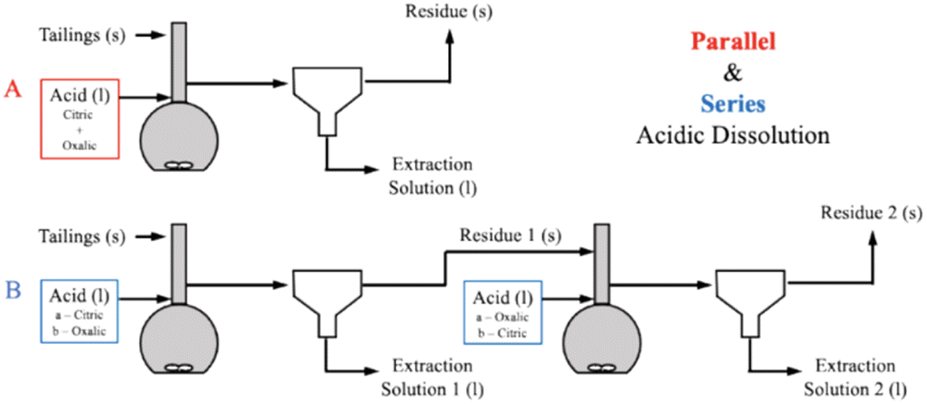 | ||
| Fig. 9 Process flow diagrams of (A) parallel and (B) series acidic calcium and magnesium extraction process configurations. | ||
Parallel dissolution with organic acids
Parallel acid dissolution was performed for 15 hours and 72 hours, where citric and oxalic acid were added to one 1 liter solution, each with concentration of 0.5 M and 30 g Tail 2a. In Fig. 8, extraction results from parallel dissolution are compared with results from single acid dissolution, which were shown in Fig. 5.Instead of enhancing the extraction of both cations, the parallel acid dissolution produced results nearly identical to those of oxalic acid alone. Evidently, when citric and oxalic acids are combined at the same concentration with Tail 2a, oxalic acid exhibits a dominance over citric acid. As noted in Table 4, oxalate forms stronger Mg complexes than citrate. This could be evidence that oxalate is irreversibly adhering to the surface of Mg-containing mineral phases in Tail 2a, essentially rendering them inert. If this is true, it could also explain the limited Mg extraction in oxalic acid observed in Fig. 4.
Series dissolution with organic acids
Extraction of Mg for oxalic/citric improves upon that of oxalic alone, but this improvement decreases as with increasing time of initial oxalic acid dissolution. This might be further evidence of irreversible oxalate adsorption onto mineral surfaces, where longer dissolution times allow more oxalate adsorption.Although citric/oxalic series dissolution improves upon the Ca extraction of citric acid alone, it remains relatively constant all four times. As proposed earlier, the Ca extraction from anorthite could in fact be enhanced with citric/oxalic dissolution, but the Ca could have precipitated as whewellite. Some evidence shows that Mg can inhibit Ca-oxalate aggregation.28 In citric/oxalic, Mg has already been removed from Tail 2a via citric acid dissolution, providing less Mg to inhibit whewellite precipitation during oxalic acid dissolution (Fig. 10).
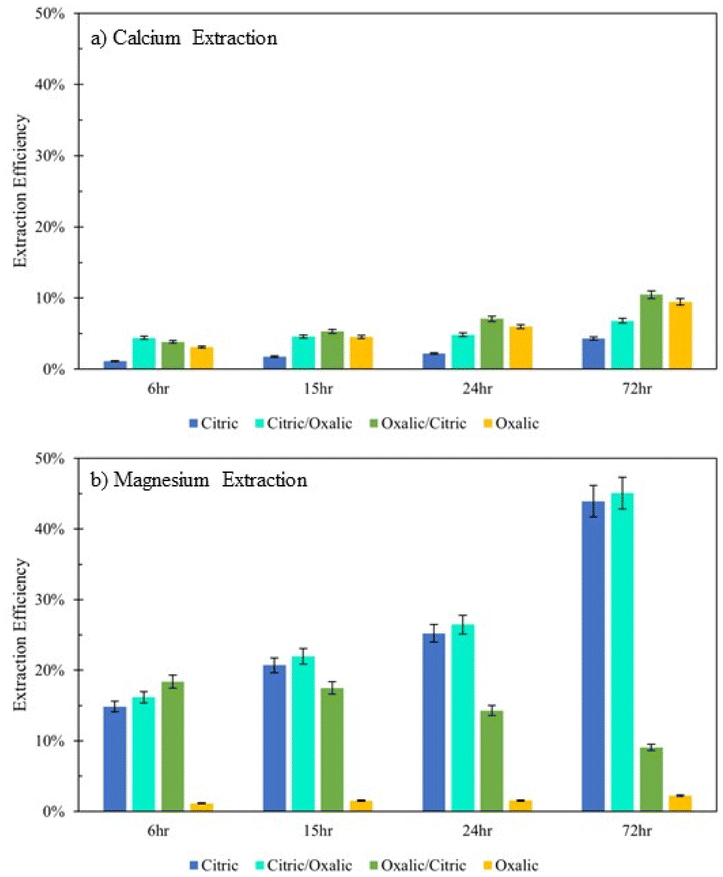 | ||
| Fig. 10 Extraction efficiency of (a) Ca and (b) Mg in organic acid series dissolution, compared to single acid dissolution. | ||
Viewing the total calcium and magnesium extracted on a molar basis gives a sense of how much CO2 can be stored with each dissolution method. As illustrated in Fig. 11, while Mg extraction is already higher than Ca extraction on a mass basis, the disparity is exacerbated in overall calcium and magnesium extraction on a molar basis due to the lower molecular weight of magnesium.
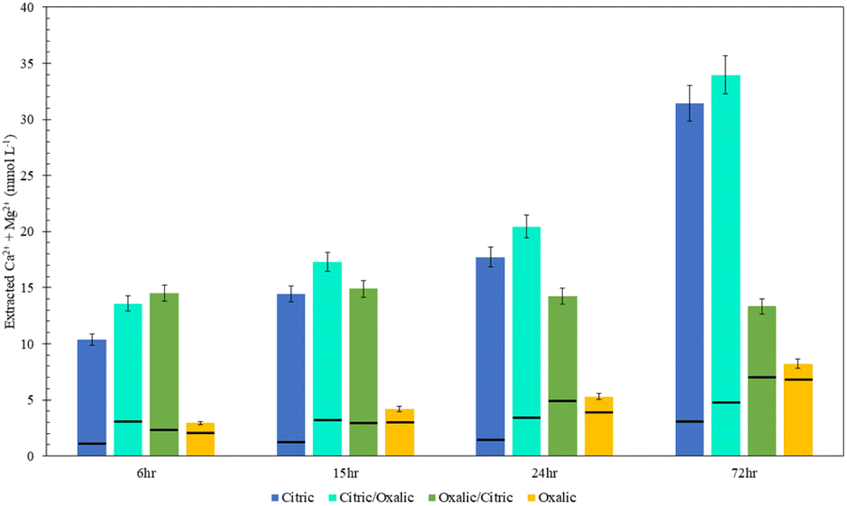 | ||
| Fig. 11 Combined calcium and magnesium extraction from acid series dissolution, compared to single acid dissolution, where the black bar represents the split between calcium (bottom) and magnesium (top). | ||
The citric/oxalic series extracted 13–33 mmol Ca2+ and Mg2+ per liter, with the large majority of that (10–29 mmol L−1) comprised of Mg2+. Similarly, the oxalic/citric series extracted a total of 13–14 mmol L−1, where Mg2+ comprised 5–11 mmol L−1. Although oxalic acid was effective in releasing Ca, its poor Mg extraction causes it to have the lowest overall extraction. Meanwhile, the reduced Ca extraction of citric acid is compensated by its high Mg extraction. While citric/oxalic has the highest overall extraction at all times tested, the marginal increase in extraction over that of citric is likely not worth double the acid consumption. Hence, the citric acid is the optimal organic acid for dissolving Tail 2a among those tested.
Series dissolution with mineral acids
Series dissolution experiments were also performed with the residues produced from the mineral acid experiments displayed in Fig. 3. As noted by Table 3, the three acids tested here provide the same pH at 0.5 M concentrations, so ligand – and proton – promoted dissolution can be directly compared in these experiments. Here, 10 grams of solid residue were placed in a 0.5 liter, 0.5 M solution of oxalic acid for 72 hours (20 g L−1). Citric acid was not used in series with either mineral acid because ins ingle acid trials, all three demonstrated a proclivity to extraction magnesium over calcium. Oxalic acid was used in series with mineral acids to understand if opposing extraction effects could be coupled to extract more magnesium and calcium overall. Results are displayed in Fig. 12, where data are organized by the solution concentration for the first step of the series, and the naming convention refers to the order of acid used (e.g., hydrochloric/oxalic means hydrochloric acid in the first step, followed by oxalic acid in the second step). The hydrochloric/oxalic treatment extracted 160–170 mg Ca2+ per L and 334–659 mg Mg2+ per L. The sulfuric/oxalic treatment extracted 167–207 mg Ca2+ per L and 373–789 mg Mg2+ per L.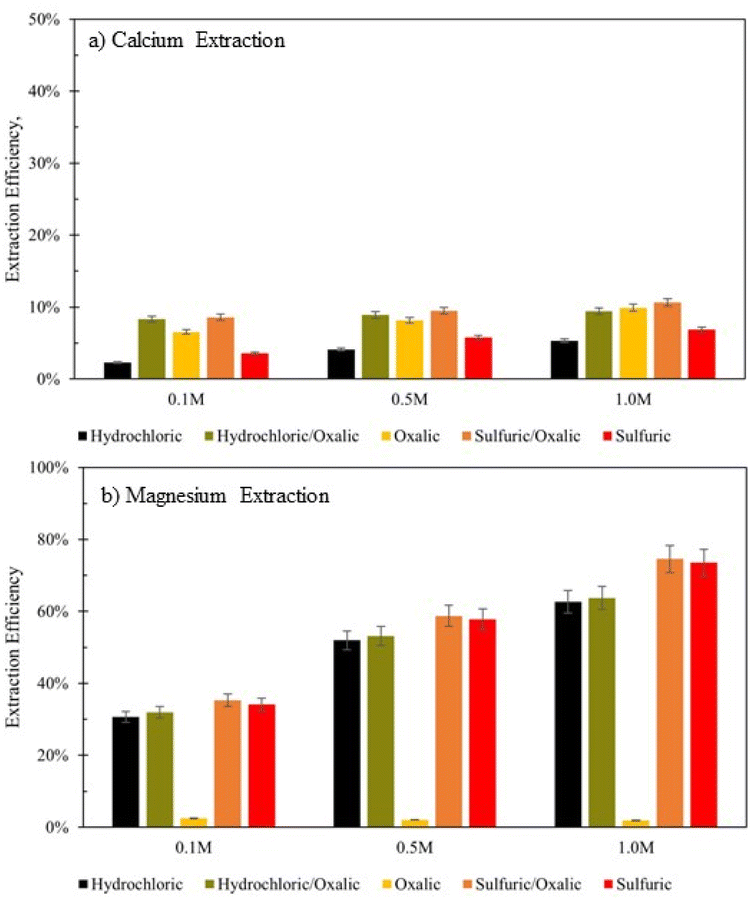 | ||
| Fig. 12 Extraction efficiency of (a) Ca2+ and (b) Mg2+ in mineral acid series dissolution, compared to single acid dissolution. Data organized by the solution concentration step of the series. Naming convention refers to the order of the acid(s) used. | ||
As expected from the poor Mg extraction of oxalic acid alone, the mineral/oxalic acid series dissolution had minimal effect on Mg2+ extraction. However, in all cases, the mineral/oxalic acid series dissolution extracted more Ca than any acid alone. In fact, the Ca extraction of mineral/oxalic series dissolution is nearly equal to the sum of the extraction from the two individual acids. The increase in Ca extraction might have been caused, in part, by the longer dissolution time of the oxalic acid step – which was 72 hours, compared to the 18 hour duration of the single acids. But another explanation could be that different mineral phases within Tail 2a are targeted by proton- and ligand-promoted dissolution. Perhaps oxalic acid effectively extracts Ca from anorthite (plagioclase), while the mineral acids extract Ca from diopside (pyroxene). Organic acids like oxalic acid have been shown to be more effective than mineral acids for dissolving plagioclase structures, while having a negligible effect on Mg extraction from pyroxene.14,24 Further, proton-promoted dissolution has been suggested to preferentially occur with pyroxenes over plagioclase.29
Again, the total calcium and magnesium extraction from the mineral acid series is displayed on a molar basis in Fig. 13. Both series dissolutions outperformed any acid alone, and the sulfuric/oxalic series consistently had the highest overall dissolution. As is the case with citric/oxalic dissolution, the increase of sulfuric/oxalic over sulfuric alone may not be worth the cost of additional acid consumption.
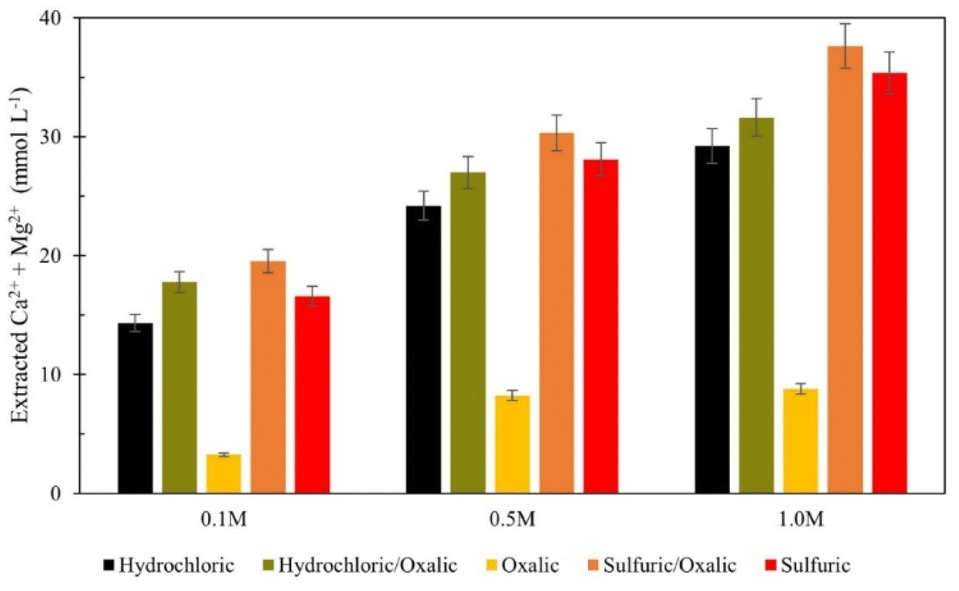 | ||
| Fig. 13 Combined calcium and magnesium extraction from mineral acid series dissolution, compared to single acid dissolution. | ||
Estimating Tail 2a dissolution rates
In this study, the parameter that was most influential on extraction efficiency was the time of dissolution. From a standpoint of additional emissions, elevation of time might be less impactful than parameters like energy and material usage, which can cause additional emissions and might defeat the purpose of carbon mineralization.30 Discussion of the dissolution rates observed in this study can give further insight into the extent the extraction efficiency can be improved by dissolution time. Using the data in Fig. 3 and the BET reactive surface area of Tail 2a, dissolution rates can be calculated in mol m−2 s−1.where M represents the metal cation being dissolved, nM,t is the number of moles of M released at a given time, t (seconds), and ABET is the BET reactive surface area of Tail 2a (m2 g−1). Measured dissolution rates are plotted in Fig. 13 with the dissolution rates of other minerals published by Palandri and Kharaka.31 Additionally, the release of Ca in a flow-through dissolution experiment at near-ambient conditions (i.e., 22 °C, 1.013 bar, dissolved CO2 at pH 4.4) measured previously is plotted for comparison (the measured release of Mg at near-ambient conditions is omitted because of data below instrument detection limit).10
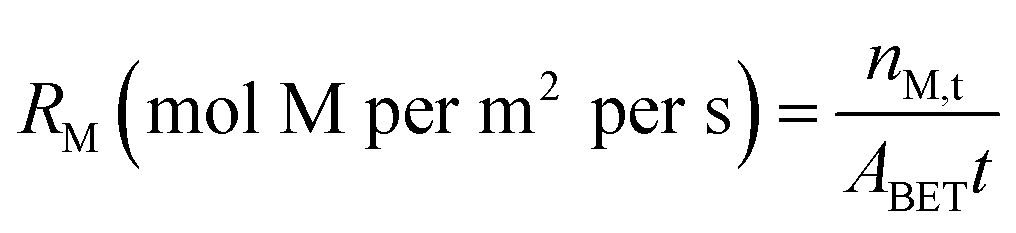
In both acid solutions, Ca release rates exceed the rate measured at ambient conditions. Based on previous findings, it is expected that Ca2+ is being leached from anorthite.9 As shown in Fig. 14, the steady state release rates of Ca2+ in both acid solutions converge upon the steady state rate of anorthite dissolution at ambient conditions. The release rate of Mg in citric acid solution converges upon the steady state dissolution rate of lizardite, suggesting that citric acid is leaching Mg from the phyllosilicate content in Tail 2a.
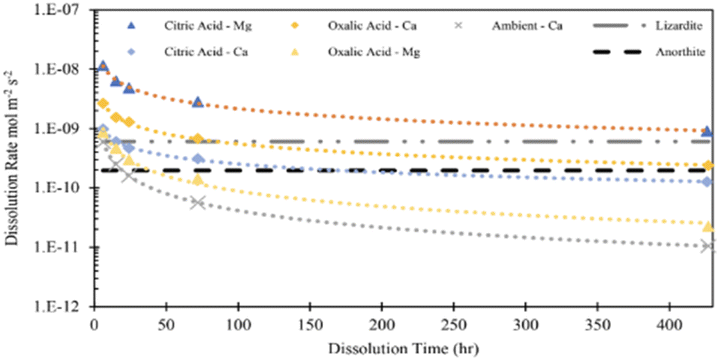 | ||
| Fig. 14 Measured dissolution rates based on release of Mg and Ca in 0.5 M citric and oxalic acid solutions. Compared to measured Ca release rate and steady state anorthite dissolution, both at ambient temperature/pressure and pH = 4.19.10,31 | ||
Relating dissolution mechanisms and mineral phases
In this study, the ability of various mineral and organic acids to extract calcium and magnesium from the Stillwater tailings was tested. The comparison of dissolution in single acid solutions with “parallel” and “series” dissolution configurations gives insight into how the acids attack the varying mineral surfaces within heterogeneous PGM tailings.In 0.5 M citric acid, about 44% of Mg was extracted in 72 hours. Notably, 43.7% of the Mg in the Stillwater tailings is found in phyllosilicate minerals. A majority of the Mg extraction occurred before steady state dissolution, indicating that the phyllosilicates dissolved quickly, relative to the other silicate phases present in the tailings (Fig. 3). As dissolution extends from 3–18 days, the Mg extraction in 0.5 M citric acid nearly doubles. This evidence suggests that citric acid extracts Mg from phyllosilicates in Tail 2a (about 44% of the Mg in Tail 2a) at a relatively fast rate and continues to extract Mg from inosilicates over extended time periods.
Ligand-promoted dissolution of Mg-bearing minerals in Tail 2a might also be expected to occur in oxalic acid solution, especially because oxalate makes strong complexes with Mg (Table 4). However, Mg extraction efficiency in oxalic acid was extremely limited, only reaching about 2% after 72 hours without improving over 18 days. Oxalate might have irreversibly adhered to the mineral surface of the phyllo- and inosilicates within Tail 2a, as suggested by multiple experiments in this study. When oxalic and citric acids were combined in solution, Mg extraction matched that of oxalic acid alone, rather than the much higher Mg extraction of citric acid (Fig. 8). Further, Mg extraction efficiency in the oxalic/citric series dissolution decreased with increasing oxalic acid dissolution time (Fig. 9B). These results indicate that oxalate strongly interacts with Mg atoms at the phyllo- and inosilicate surface, irreversibly binding to the surface and rendering it inert. This finding is different than what has been previously reported with oxalate and serpentine, potentially due to such high oxalate concentrations and low pH used in these experiments.32
The mineral acids (sulfuric, hydrochloric) readily released Mg with much higher extraction efficiencies in 18 hours than citric acid did in 72 hours, extracting up to about 74% with 1.0 M sulfuric acid solution. This amount exceeds the Mg present in any one mineral phase, suggesting that both ino- and phyllosilicates containing Mg in Tail 2a can quickly undergo proton-promoted dissolution.
In addition to Mg release, inosilicate dissolution would result in Ca release (diopside, tremolite). This might explain the limited Ca extraction exhibited by the mineral acids, which are known to be less effective in dissolving tectosilicates such as plagioclase.14,15,22,24,32–34 This hypothesis is reinforced by the mineral/oxalic acid series dissolution, where the mineral acids appeared to have extracted Ca from different mineral sources than oxalic acid (Fig. 10a).
The Ca extraction of citric and oxalic acid are controlled by ligand-promoted dissolution of anorthite, where the ligands primarily attack the Al–O sites to liberate Ca by forming Ca–ligand complexes. Based on the comparative XRD diffractograms of the solid residue left behind after dissolution, both organic acids effectively dissolved anorthite over the 72 hour period, especially oxalic acid. However, despite anorthite holding about 77% of the Ca in Tail 2a, dissolution in 0.5 M oxalic and citric acid solutions resulted in Ca extraction efficiencies of only about 20% and 10%, respectively.
For oxalic acid, the low Ca extraction efficiency can be explained by precipitation of a Ca-oxalate salt, whewellite. When solid whewellite precipitates, calcium is removed from solution and is not measured in the extraction solution. However, this does not explain the low Ca extraction efficiency of citric acid. Another possibility is that some stable Ca complexes with citrate and oxalate are undetected by ion chromatography, but evidence of this in the literature is lacking.
The behavior of mineral phases within Tail 2a undergoing dissolution with the acids tested in this study is summarized in Table 5, corroborated with other published literature results. This table can be used to develop strategies on how to approach acid-promoted carbon mineralization with heterogeneous tailings like those of Stillwater. Tailings rich in a particular silicate group may have higher extraction in a corresponding acid with darker shading in Table 5.
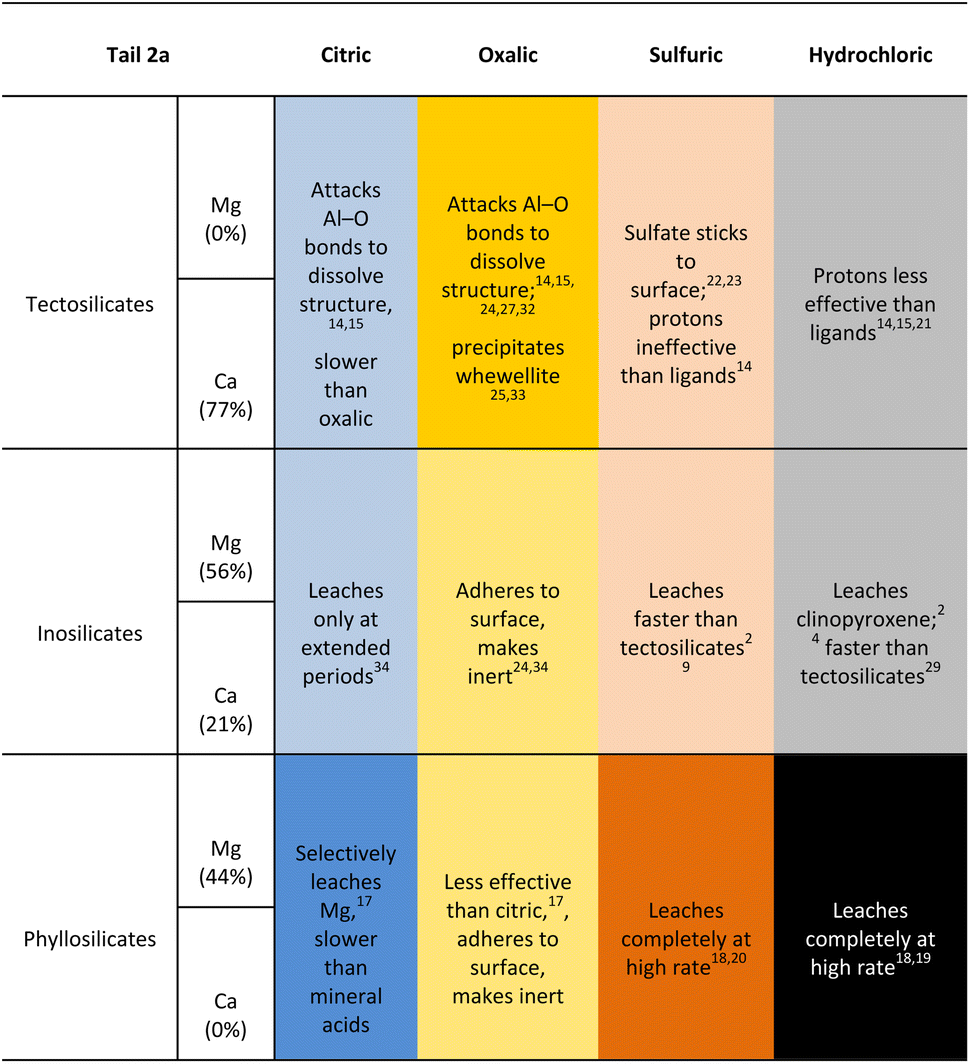
|
Effect of pressure on mineral carbonation
To complete the pH swing process from extraction to carbonation depicted in Fig. 1, synthetic solutions mimicking the calcium levels from the extraction trials were used as starting materials for the carbonation trials. These trials aimed to assess the impact of varying pressure on the amount of CO2 consumed by the mineral phase, which would affect the cost at scale due to its relation to compression costs and the potential need for a recycle loop. Additionally, the pH swing was examined by comparing the efficiency of adding the base “in situ” and “ex situ,” methods explored in literature but not in the exact same proportions for a direct comparison.35,36An investigation of the effects of PCO2 on the carbonation reaction, using synthetic solutions made from calcium hydroxide, demonstrated the critical relationship between the transport phenomena of CO2 in water and the reaction of CO32− and Ca2+ in aqueous media.
Analysing the yield of carbonates (Fig. 15), increasing pressure leads to the formation of more calcite within the residence time. At 9 bar CO2, 95% of the theoretical yield of calcite was formed within an hour. At 9 bar, 35% of the CO2 provided to the reactor was consumed, either by the solid phase or dissolving into the aqueous phase. In terms of calcite production, increasing the pressure shows a clear positive trend in the product yield. However, in terms of converting CO2 into carbonate minerals, at 3 bar half of the gas that was provided was converted into a mineral phase.
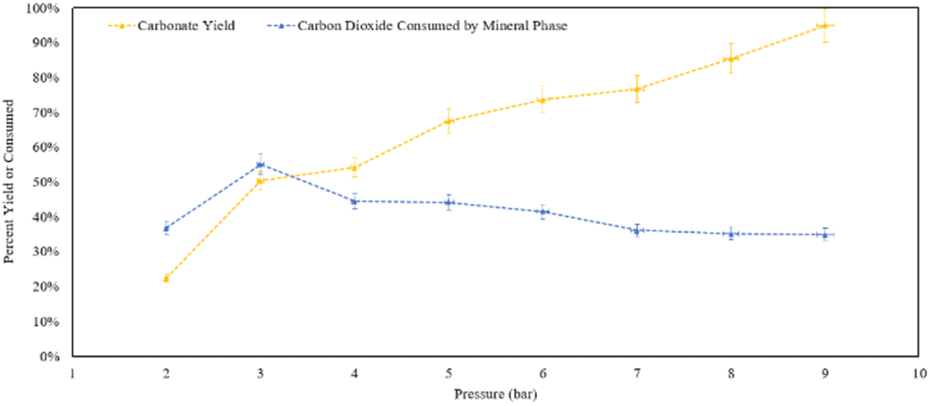 | ||
| Fig. 15 Percent yield of the residues and percent of CO2 mineralized from what was provided to the reaction in the pressure varied calcite formation trials. | ||
X-ray diffraction confirmed that the solid reaction product was calcium carbonate, in the form of calcite. At 9 bar, nearly all of the Ca provided was converted into calcium carbonate: consistent with the overall trend that higher pressure increased the carbonate yield. However, as the yield of calcium carbonate product increased, the relative amount of CO2 consumed by the mineral phase decreased. Providing excess CO2, propagated the conversion of additional carbonate products from the reaction. It should be noted that based on the reactor dimensions and amount of calcium hydroxide provided, the stoichiometric amount of CO2 would have been achieved at approximately ∼2.5 bar, indicating that CO2 was present in excess of the stoichiometric amount at pressures of 3–9 bar.
The reaction products were also analysed for particle size and shape to assess any potential compatibility with carbonates used in industrial applications. The particle size distribution of the reaction products by reaction pressure is displayed in Fig. 16, measured via laser diffractometry to quantify the amount of particles at varying diameters. The particle size distribution show a fairly similar distribution of d10, d50, and d90 metrics across all the reaction pressures, with the exception being the products created at 5 bar CO2 which are approximately twice the size of the products for the other reaction pressures. Notably, the 5 bar products saw the beginning of a shift in the physical appearance of the reaction products after filtration, as the products created at pressures less than 5 bar had a finer grain size, while above 5 bar there were visible pores and a more variable grain size (Fig. 17).
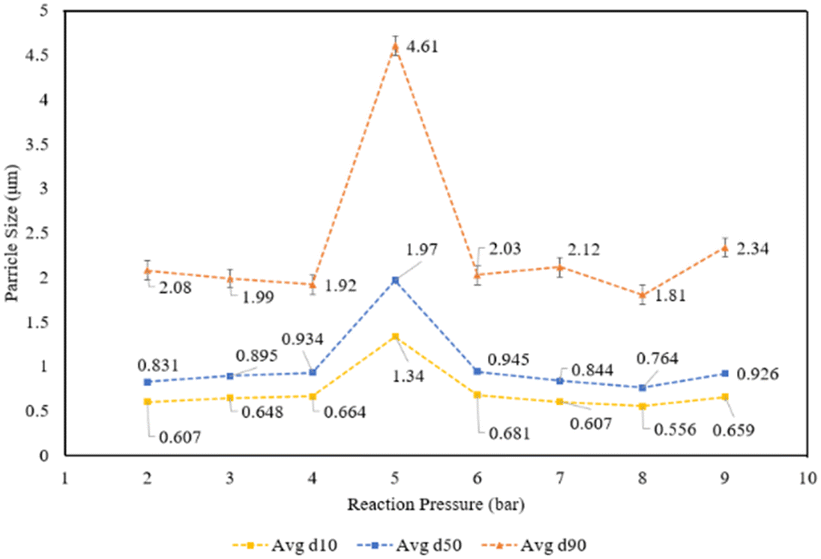 | ||
| Fig. 16 Particle size distribution (d10, d50, d90) of reaction products at varying pressures. | ||
 | ||
| Fig. 17 Carbonate filter cakes produced from calcite hydroxide experiments at 3 bar (left) and 9 bar (Right). | ||
Outside of outliers at 5 bar, the particle size distribution of the reaction products demonstrate that nearly all of the particles are distinctly less then 3 μm in size. When combined with the yield and mineralization metric data, the higher pressures produced more carbonate products but not larger carbonate products. This phenomenon suggests that carbonate products were undergoing nucleation, but not propagation into larger calcite crystals.
In addition to the particle size distribution, the particle shape of the reaction products was also measured by way of sphericity for all reaction products, the sphericity was 0.87 which indicates largely spherical particles with some defects.
The size and shape of the carbonation reaction products suggests a possible synergy with the pulp & paper industry, where the 50th percentile of particle size, by count, is approximately 1 μm.37 The average sphericity of the products are compatible with precipitated calcium carbonate used as filler for printer quality paper (a $15 B annual market which produces 300 M tonnes each year).38,39
While particle size and shape were consistent across variable pressures, the quality of the precipitated carbonated became more porous as reaction pressure increased. As depicted in Fig. 17 carbonates produced at pressures of 4 bar and under displayed smooth, planar surfaces where carbonates produced above 5 bar showed highly porous surfaces. These pores are possibly caused by CO2 degassing from the liquid media during filtration. As predicted by Henry's law of solubility, the higher-pressure experiments are expected to see higher concentrations of CO2 solvate into the liquid phase: potentially contributing to the inverse relationship between the increase in residue yield and decrease in the percent of CO2 consumed by the mineralization reaction. The concentration of CO2 in the aqueous phase may influence the physical properties of the resultant carbonate cakes, as previously noted, which can affect the suitability of the carbonates for specific applications, thus impacting the economics of an industrial scale mineral carbonation process.
In situ and ex situ base addition for carbonation
The base addition experiments demonstrated that comparable product yields can be achieved using both ex situ and in situ base addition methods under constant volumes of base and pressures (Fig. 18). This indicates that maintaining high pH levels during the reaction under pressure may not be necessary at an industrial scale.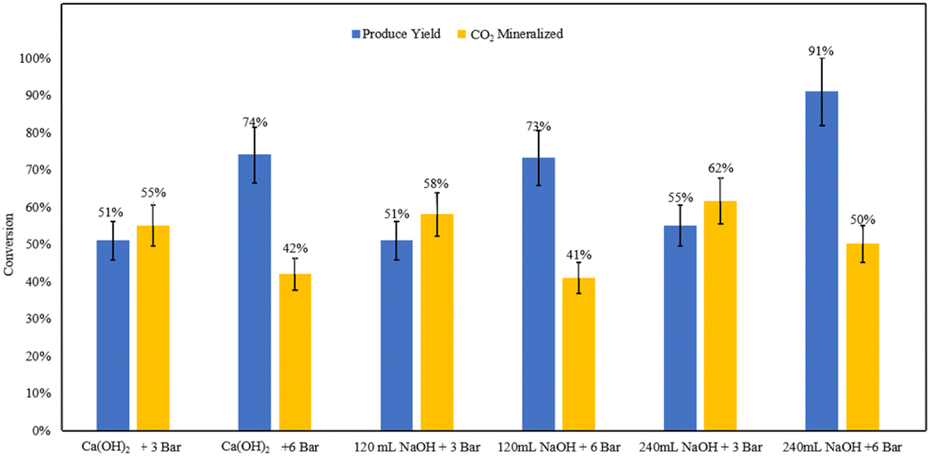 | ||
| Fig. 18 Measured product yields and CO2 mineralization quantities based on mineral carbonation experiments. | ||
The pH measurements of the effluent from the ex situ trials provided further clarity. For the 120 mL NaOH ex situ trial at 6 bar, the pH was recorded at 6.53. In contrast, the 240 mL NaOH ex situ trial at the same pressure resulted in a significantly higher pH of 10.21. These results suggest that an excess amount of base can sustain sufficiently high pH levels for effective carbonation.
The findings imply that in situ base addition may be prone to pH gradients at an industrial scale, potentially leading to inconsistent reaction conditions. On the other hand, ex situ base addition produces a more homogeneous mixture with a uniformly high pH, which can endure the residence time required for the reaction. This homogeneity could be particularly advantageous for scaling up the process, ensuring consistent and efficient carbonation across the entire reactor volume.
Conclusions
The highest carbonation efficiencies measured in this study are tabulated in Table 6, coupled with estimations of the CO2 that could potentially be stored in all of the tailings at Stillwater (∼1 Mt tailings per year) and in all of the tailings projected to be produced at nickel and PGM mining operations (3.5 Gt per year).40 These data demonstrate the maximum amount of CO2 that could be stored in PGM tailings as indicated by the experimental results above.:1 ratio of cation to CO2
| Acid | Acid conc. (mol L−1) | Time (h) | S/L ratio (g L−1) | Extraction efficiency | Maximum CO2 capacity kg CO2 per t tailings | Potential CO2 storage (t CO2) | ||
|---|---|---|---|---|---|---|---|---|
| Ca2+ | Mg2+ | Sibanye-Stillwater (1 Mt per year) | World (3.5 Gt per year) | |||||
| Citric | 0.5 | 72 | 40 | 4.28% | 41.09% | 4.3 | 4300 | 15000000 |
| Oxalic | 0.5 | 72 | 40 | 10.03% | 2.36% | 1.3 | 1300 | 5000000 |
| Citric | 0.5 | 426 | 30 | 10.29% | 81.73% | 8.7 | 8800 | 31000000 |
| Oxalic | 0.5 | 427 | 30 | 19.82% | 2.10% | 2.3 | 2300 | 8000000 |
| Hydrochloric | 1.0 | 18 | 20 | 5.34% | 62.63% | 6.4 | 6500 | 23000000 |
| Sulfuric | 1.0 | 18 | 20 | 6.88% | 73.55% | 7.6 | 7600 | 27000000 |
| Citric/oxalic | 0.5/0.5 | 72 + 16 | 30/20 | 6.79% | 45.08% | 4.9 | 5000 | 18000000 |
| Sulfuric/oxalic | 1.0/0.5 | 18 + 72 | 20/20 | 10.65% | 74.53% | 8.1 | 8100 | 29000000 |
Overall, the highest Mg extraction efficiencies were found with elevated conditions of time (citric acid, 18 days) and pH (1.0 M sulfuric acid solutions). These Mg extraction efficiencies far exceed those achieved previously at near-ambient conditions (<1% Mg was labile) and by the alternative ÅA route (∼38% Mg extracted at 450 °C using ammonium sulfate).9,11
While the Ca extraction in this study also exceeded that of near-ambient conditions (<3% Ca was labile), it only exceeded the level achieved by the alternative ÅA route (13%) when the tailings dissolved in 0.5 M oxalic acid solution for 18 days. The structure of anorthite which binds more than 75% of the Ca in Tail 2a has been proven too restrictive for proton-promoted dissolution at the conditions tested in this study.41,42
In this study, for the first time, the effect of different acid solutions, solution concentration, dissolution time, and solid/liquid ratio on the calcium and magnesium extraction of plagioclase-rich PGM mine tailings was assessed for the purpose of carbon mineralization. While results give insight into how to best approach pH swing carbon mineralization for PGM mine tailings, further work is necessary to develop an efficient and economic process that can store CO2 in the billions of tonnes of ultramafic mine tailings that will be generated in the coming century.40
Additionally, future work could include optimizing the solid/liquid ratio to minimize acid consumption or undertaking molecular studies to increase understanding of surface chemistry on cation extraction in heterogeneous tailing sources. Future studies may also consider measuring the pH of the resultant leachate from the extraction process to analyze the amount of acid being consumed during dissolution. In addition, monitoring the amount of Al and Si leaching from the mineral structure to compare the effects of insoluble complexes limiting dissolution to the actual amount of mineral dissolving in the acid.
If the use of acids to leach divalent cations from the tailings is to be continued, another consideration to be made is the emissions associated with the acids used. For instance, sulfuric acid production has associated SO2 emissions of 2–48 kg per t H2SO4 (depending on process efficiency) and CO2 emissions of 4.05 kg per t H2SO4.43 These potential impacts might warrant a study on new potential sources of acids to use in a mineralization process like the one used in this study. Acid might be able to be sourced from acid mine drainage, but this would vary at each mine, as it is unlikely to be available at Stillwater.44,45 Alternatively, there are some studies of using CO2 to produce organic acids like acetic acid and oxalic acid.45–47 The potential to use a carbon-negative reagent would further increase the environmental beneficiation of the process.48
To store CO2 in mine tailings with the calcium and magnesium extraction methods used in this study, it is necessary to optimize the carbonation step. Future efforts should work to tune the reaction conditions to be mindful of the life cycle and techno economic implications of engineering decision on an industrial scale pH-swing process for mineral carbonation. In particular, life cycle analysis can provide insights into environmental impacts other than the carbon balance of the process at scale, including water use, ecotoxicity, and other key factors that are of concern for the mining industry. Given that the tailings did not completely dissolve under any of the tested experimental conditions, there will likely be a solid residue leftover. If this solid residue cannot be valorized at scale, then it would likely be returned to the waste impoundments. Although this process generates a residue waste stream, it would significantly reduce the amount of waste being sent to the impoundments. Economic efforts should include considering the energy requirements at scale, the impacts of continuous and batch operations on economics, and the choice of base as a reagent. Similar to considering the emissions associated with the acids used for leaching, the upstream emissions of the producing base should also be considered. Sodium hydroxide was used in these experiments, which has an emissions factor of 0.46 kg CO2eq per kg NaOH.49 The implicit emissions in the choice of base should be weighed with the co-location to the Stillwater Mine in Nye, Montana.
Another way to do this would be to use an acid that was not tested in this study, carbonic acid (H2CO3). Using carbonic acid has the potential to allow the dissolution and carbonation to occur in one step. Further, acid consumption would not be a problem like it is with the acids tested in this study, because carbonic acid consumption would indicate the tailings are being carbonated. Hence, process economics could be improved by requiring less process equipment and consuming less raw material throughout the process. This was tested previously with pH of 4.4 with very limited cation release.9 However, by using a more concentrated acid solution (with lower pH), stirring, and perhaps pressure, Ca and Mg extraction could be increased.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge Martin Uxa for his help with providing data acquisition in the lab. The authors would like to thank the Max Planck Institute, Grantham Foundation, and Climeworks Foundation for supporting the work.References
- Intergovernmental Panel on Climate Change (IPCC), Climate Change 2021: The Physical Science Basis, 2021 Search PubMed.
- National Academies of Sciences, Engineering, and Medicine, Negative Emissions Technologies and Reliable Sequestration: A Research Agenda, The National Academies Press, Washington, DC, 2019 Search PubMed.
- J. Wilcox, B. Kolosz and J. Freeman, Carbon Dioxide Removal Primer, 2021 Search PubMed.
- C. M. Woodall, I. Piccione, M. Benazzi and J. Wilcox, Capturing and Reusing CO2 by Converting it to Rocks, Front. Young Minds, 2021, 8, 592018 CrossRef.
- Climeworks, Mammoth construction update, 2023, available from: https://climeworks.com/news/climeworks-mammoth-construction-update-jul23, accessed 2024 Aug 5 Search PubMed.
- Heirloom, Two direct air capture facilities in northwest Louisiana, 2024, available from: https://www.heirloomcarbon.com/news/two-direct-air-capture-facilities-in-northwest-louisiana, accessed 2024 Aug 5 Search PubMed.
- CarbonCure Technologies, A year after the Carbon XPRIZE: how CarbonCure accelerated innovation and growth, 2023, available from, https://www.carboncure.com/concrete-corner/a-year-after-the-carbon-xprize-how-carboncure-accelerated-innovation-and-growth/, accessed 2024 Aug 5 Search PubMed.
- Montana Department of Environmental Quality and US Department of Agriculture, Stillwater Mining Company's Revised Water Management Plans and Boe Ranch LAD, Montana Department of Environmental Quality, Helena, MT, 2012 Search PubMed.
- T. Watari, B. C. McLellan, D. Giurco, E. Dominish, E. Yamasue and K. Nansai, Total material requirement for the global energy transition to 2050: a focus on transport and electricity, Resour., Conserv. Recycl., 2019, 148, 91–103 CrossRef.
- C. M. Woodall, X. Lu, G. Dipple and J. Wilcox, Carbon Mineralization with PGM Mine Tailings – Characterization and Reactivity Analysis, Minerals, 2021, 11, 844 CrossRef CAS.
- R. Zevenhoven, M. Slotte, E. Koivisto and R. Erlund, Serpentinite Carbonation Process Routes Using Ammonium Sulfate and Integration in Industry, Energy Technol., 2017, 5(6), 945–954 CrossRef CAS.
- C. M. Woodall, N. McQueen and J. Wilcox, Carbon mineralization using plagioclase feldspar-rich mine tailings: Characterization and cation extraction, in GHGT-15, 2021 Search PubMed.
- A. Azdarpour, M. Asadullah, E. Mohammadian, H. Hamidi, R. Junin and M. A. Karaei, A review on carbon dioxide mineral carbonation through pH-swing process, Chem. Eng. J., 2015, 279, 615–630 CrossRef CAS.
- S. A. Welch and W. J. Ullman, The effect of organic acids on plagioclase dissolution rates and stoichiometry, Geochim. Cosmochim. Acta, 1993, 57, 2725–2736 CrossRef CAS.
- W. Shotyk and H. W. Nesbitt, Ligand-Promoted Dissolution of Plagioclase Feldspar: A Comparison of the Surface Chemistry of Dissolving Labradorite and Bytownite Using SIMS, in Geochemistry of the Earth's Surface and of Mineral Formation, 1990, pp. 320–321 Search PubMed.
- E. R. Bobicki, Q. Liu and Z. Xu, Ligand-promoted dissolution of serpentine in ultramafic nickel ores, Miner. Eng., 2014, 64, 109–119 CrossRef CAS.
- S. C. M. Krevor and K. S. Lackner, Enhancing serpentine dissolution kinetics for mineral carbon dioxide sequestration, Int. J. Greenhouse Gas Control, 2011, 5(4), 1073–1080 CrossRef CAS.
- S. Teir, H. Revitzer, S. Eloneva, C. J. Fogelholm and R. Zevenhoven, Dissolution of natural serpentinite in mineral and organic acids, Int. J. Miner. Process., 2007, 83(1–2), 36–46 CrossRef CAS.
- S. Teir, S. Eloneva, C. J. Fogelholm and R. Zevenhoven, Dissolution of steelmaking slags in acetic acid for precipitated calcium carbonate production, Energy, 2007, 32(4), 528–539 CrossRef CAS.
- K. Yoo, B. S. Kim, M. S. Kim, J. C. Lee and J. Jeong, Dissolution of magnesium from serpentine mineral in sulfuric acid solution, Mater. Trans., 2009, 50(5), 1225–1230 CrossRef CAS.
- L. Wei, H. Hu, Q. Chen and J. Tan, Effects of mechanical activation on the HCl leaching behavior of plagioclase, ilmenite and their mixtures, Hydrometallurgy, 2009, 99(1–2), 39–44 CrossRef CAS.
- J. Ma, Y. Zhang, Y. Qin, Z. Wu, T. Wang and C. Wang, The leaching kinetics of K-feldspar in sulfuric acid with the aid of ultrasound, Ultrason. Sonochem., 2017, 35, 304–312 CrossRef CAS PubMed.
- W. Stumm, Reactivity at the mineral-water interface: dissolution and inhibition, Colloids Surf., A, 1997, 120(1–3), 143–166 CrossRef CAS.
- N. A. Meyer, J. U. Vögeli, M. Becker, J. L. Broadhurst, D. L. Reid and J.-P. Franzidis, Mineral carbonation of PGM mine tailings for CO2 storage in South Africa: a case study, Miner. Eng., 2014, 59, 45–51 CrossRef CAS.
- G. Gadikota, C. Natali, C. Boschi and A. H. A. Park, Morphological changes during enhanced carbonation of asbestos-containing material and its comparison to magnesium silicate minerals, J. Hazard. Mater., 2014, 264, 42–52 CrossRef CAS PubMed.
- M. J. Eick, P. R. Grossl, D. C. Golden, D. L. Sparks and D. W. Ming, Dissolution of a lunar basalt simulant as affected by pH and organic anions, Geoderma, 1996, 74(1–2), 139–160 CrossRef CAS.
- S. A. Welch and W. J. Ullman, Feldspar dissolution in acidic and organic solutions: compositional and pH dependence of dissolution rate, Geochim. Cosmochim. Acta, 1996, 60(16), 2939–2948 CrossRef CAS.
- W. C. Graustein, K. Cromack and P. Sollins, Calcium oxalate: occurrence in soils and effect on nutrient and geochemical cycles, Science, 1977, 198(4323), 1252–1254 CrossRef CAS PubMed.
- A. C. McAdam, M. Y. Zolotov, T. G. Sharp and L. A. Leshin, Preferential low-pH dissolution of pyroxene in plagioclase-pyroxene mixtures: implications for martian surface materials, Icarus, 2008, 196(1), 90–96 CrossRef.
- M. S. Ncongwane, J. L. Broadhurst and J. Petersen, Assessment of the potential carbon footprint of engineered processes for the mineral carbonation of PGM tailings, Int. J. Greenhouse Gas Control, 2018, 77, 70–81 CrossRef CAS.
- J. L. Palandri and Y. K. Kharaka, A Compilation of Rate Parameters of Water-Mineral Interaction Kinetics for Application to Geochemical Modeling, 2004 Search PubMed.
- W. Shotyk and H. W. Nesbitt, Incongruent and congruent dissolution of plagioclase feldspar: effect of feldspar composition and ligand complexation, Geoderma, 1992, 55(1–2), 55–78 CrossRef CAS.
- L. L. Stillings, J. I. Drever and S. R. Poulson, Oxalate adsorption at a plagioclase (An47) surface and models for ligand-promoted dissolution, Environ. Sci. Technol., 1998, 32(19), 2856–2864 CrossRef CAS.
- S. V. Golubev and O. S. Pokrovsky, Experimental study of the effect of organic ligands on diopside dissolution kinetics, Chem. Geol., 2006, 235(3–4), 377–389 CrossRef CAS.
- R. Erlund, E. Koivisto, M. Fagerholm and R. Zevenhoven, Extraction of magnesium from four Finnish magnesium silicate rocks for CO2 mineralisation—part 2: aqueous solution extraction, Hydrometallurgy, 2016, 166, 229–236 CrossRef CAS.
- A. H. Park and L.-S. Fan, CO2 mineral sequestration: physically activated dissolution of serpentine and pH swing process, Chem. Eng. Sci., 2004, 59(22–23), 5241–5247 CrossRef CAS.
- M. A. Hubbe and R. A. Gill, Fillers for Papermaking: A Review of Their Properties, Usage Practices, and Their Mechanistic Role, Bioresources, 2016, 11(1), 2886–2963 CrossRef CAS.
- L. McGuire, The Problem with Paper, 2022 Search PubMed.
- Globe Newswire, Global Printing Paper Industry, 2020 Search PubMed.
- P. Renforth, The negative emission potential of alkaline materials, Nat. Commun., 2019, 10, 1401 CrossRef PubMed.
- R. M. Smith and A. E. Martell, Critical Stability Constants Volume 4: Inorganic Complexes, Springer, 1976 Search PubMed.
- J. M. Riley, H. Kim, T. D. Averch and H. J. Kim, Effect of magnesium on calcium and oxalate ion binding, J. Endourol., 2013, 27(12), 1487–1492 CrossRef PubMed.
- United States Environmental Protection Agency (EPA), Sulfuric Acid Emission Final Report (AP 42, Fifth Edition, Volume I, Chapter 8), 2022 Search PubMed.
- A. D. Paktunc, Characterization of mine wastes for prediction of acid mine drainage, in Environmental Impacts of Mining Activities, 1999, pp. 19–40 Search PubMed.
- Montana Department of Environmental Quality and United States Department of Agriculture, Final Environmental Impact Statement Appendices – Stillwater Mining Company's Revised Water Management Plans and Boe Ranch LAD, 2012 Search PubMed.
- Michigan Technological University, Captured carbon dioxide converts into oxalic acid to process rare earth elements, ScienceDaily, 2019 Search PubMed.
- M. A. Murcia Valderrama, R. J. van Putten and G. J. M. Gruter, The potential of oxalic – and glycolic acid-based polyesters: towards CO2 as a feedstock (carbon capture and utilization – CCU), Eur. Polym. J., 2019, 119(August), 445–468 CrossRef CAS.
- A. Wilk, L. Wieclaw-Solny, T. Spietz and A. Tatarczuk, CO2-to-methanol conversion – an alternative energy storage solution, Chemik, 2016, 70(10), 630–633 Search PubMed.
- L. Thannimalay, S. Yusoff and N. Zawawi, Life cycle assessment of sodium hydroxide. Aust. J. Basic Appl. Sci., 2013, 7(2), 421–431, available from: https://www.ajbasweb.com/old/ajbas/2013/February/421-431.pdf Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00443d |
| This journal is © The Royal Society of Chemistry 2024 |