
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic synthesis of renewable 2-methylfuran from furfural†
Yuanyuan
Han
a,
Xing
Zhang
a,
Wei
Wang
 a,
Shaobo
Guo
a,
Xiaohui
Ji
*a and
Guangyi
Li
*b
a,
Shaobo
Guo
a,
Xiaohui
Ji
*a and
Guangyi
Li
*b
aShaanxi Key Laboratory of Catalysis, School of Chemical & Environment Science, Shaanxi University of Technology, Hanzhong, Shaanxi 723001, P. R. China. E-mail: jixiaohui@snut.edu.cn
bCAS Key Laboratory of Science and Technology on Applied Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: lgy2010@dicp.ac.cn
First published on 6th June 2024
Abstract
Biomass energy stands at the forefront of remedial strategies for the current energy deficit and has garnered considerable attention. The deployment of highly efficient catalysts is pivotal to the success of this energy form. Prevailing literature established that high-valent metal presence contributed significantly to the hydrogenation of furfural to prepare 2-methylfuran. In this study, we elucidated the efficacy of a monometallic catalyst consisting solely of cobalt and its oxides, denoted as Co/CoOx, in the catalytic transformation of furfural derived from lignocellulosic biomass into 2-methylfuran. Leveraging the economical Co/CoOx catalyst, in conjunction with minimal addition of hydroquinone, we accomplished the selective hydrodeoxygenation of furfural, culminating in an augmented yield of 2-MF up to 73%. This investigation is the inaugural confirmation that minuscule quantities of hydroquinone can efficaciously mitigate side reactions such as the polymerization of furfural within the selective hydrodeoxygenation process. The Co/CoOx catalyst was characterized through an array of analytical techniques, including XRD, H2-TPR and N2-adsorption. These characterization studies unveiled that the optimal selectivity for 2-methylfuran was achieved when the ratio of Co0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co2+ in the catalyst approached approximately 95%.
:Co2+ in the catalyst approached approximately 95%.
Sustainability spotlightThe fuels derived from non-edible lignocellulosic biomass were referred to as second-generation biomass hydrocarbon fuels, representing an optimal low-carbon alternative to conventional fossil fuels. A significant impediment to the widespread deployment of second-generation biomass hydrocarbon fuel is its elevated production expenses. Presently, furfural stands as one of the most extensively produced lignocellulosic derivatives on an annual basis, with 2-methylfuran playing a pivotal role in the synthesis of second-generation biomass fuels. This research employed an economical cobalt-based catalyst to enable the efficient catalytic transformation of furfural into 2-methylfuran, laying the foundation for the cost-effective production of 2-methylfuran in the future, thus contributing to a decrease in the manufacturing expenses of second-generation biofuels. |
1 Introduction
In the contemporary period, scholarly attention has increasingly concentrated on the technological progress in transforming biomass and its derivatives into liquid biofuels and bulk chemicals.1–4 Within this domain, the processes of biomass refining and upgrading have surfaced as pivotal fields of inquiry. A salient intermediate, furfural, originating from lignocellulosic biomass, has elicited significant interest from the research community.5,6 Furfural exhibited the potential to be efficiently converted into a multitude of high-value chemicals, such as 2-methylfuran (2-MF),7 2-methyltetrahydrofuran,8,9 and furfuryl alcohol,10–12 through the application of various catalysts under diverse reaction conditions. The production of 2-MF from furfural has attracted widespread attention because it can serve as a fuel in its own right13 and is also a key intermediate in the preparation of renewable aviation kerosene or diesel.142-MF can be synthesized through various chemical reactions, with the selective hydrodeoxygenation of furfural being the most extensively examined route for its production. Historically, the generation of 2-MF utilized the catalytic oxidation of 1,4-pentadiene.15 Nonetheless, the refinement of furfural production processes has led to the evolution of more efficient synthetic methodologies. In the year 1947, Bremenr et al.16 pioneered the development of CuAl and CuCr catalysts, with magnesium serving as a promoter. Employed at reaction temperatures of around 250 °C, these catalysts predominantly yielded 2-MF. Notably, CuCr catalysts demonstrated exceptional selectivity towards 2-MF, consistently delivering yields in excess of 85% within a temperature spectrum of 200 °C to 300 °C. Subsequent comparative studies of CuZnAl, CuMgAl, Cu2Cr2O5, and CuNiAl catalysts for the gas-phase selective hydrodeoxygenation of furfural were conducted by Ma et al.17 Their findings indicated that Cu2Cr2O5 catalysts afforded superior selectivity for 2-MF, with a yield of 73.5% being attained at 240 °C. Nevertheless, owing to the toxicological concerns associated with chromium, there was a concerted effort within the research community to identify environmentally benign alternatives to chromium-based catalysts in the pursuit of green chemical processes.18
In the domain of synthesizing 2-MF through the selective hydrodeoxygenation of furfural, scholarly investigations have revealed that noble metal catalysts exhibited superior selective hydrodeoxygenation performance. For example, Aldosari et al.19 synthesized a 4%Pd–1%Ru/TiO2 catalyst, achieving a 51.5% yield of 2-MF under ambient conditions. Vlachos et al.20 utilized a Ru/C catalyst to facilitate selective hydrodeoxygenation, culminating in a 76% yield of 2-MF after a reaction duration of 10 hours at a temperature of 180 °C, employing 2-pentanol as the reaction medium. In another notable study, Date et al.21 reported a 95% yield of 2-MF using an Ir/C catalyst at a reaction temperature of 220 °C, attributing this enhanced yield to the abundance of acidic sites on the catalyst provided by the presence of iridium oxide. Despite these advancements, the broader industrial application of such catalysts was constrained by factors including the availability and cost of noble metals.
Catalysts composed of non-precious metals were extensively employed in the selective hydrodeoxygenation of furfural, favored for their cost-effectiveness and reduced environmental footprint. Among them, copper-based catalysts were distinguished for their superior catalytic performance and were supported by relatively plentiful reserves. Xu et al.22 successfully synthesized a copper-based catalyst utilizing a CuMgAlNi hydrotalcite-like precursor and examined the implications of varied reduction temperatures on the selective hydrodeoxygenation process of furfural. The research findings indicated that at a reduction temperature of 300 °C, the gaseous-phase catalytic selective hydrodeoxygenation of furfural predominantly generated 2-MF, achieving a selectivity of 48%. An escalation in the reduction temperature was observed to inversely affect the selectivity towards 2-MF, while concurrently augmenting the selectivity for furfuryl alcohol. This phenomenon underscored the significant role of copper particle size in dictating the selectivity of the selective hydrodeoxygenation reaction. Moreover, the incorporation of cobalt has been shown to augment the catalytic efficacy of copper-based catalysts. Kalong et al.23 employed a wet impregnation technique to synthesize both monometallic Cu/γ-Al2O3 and bimetallic CuCo/γ-Al2O3 catalysts, subsequently evaluating their catalytic performance. The findings of the study suggested that the bimetallic catalysts exhibited an alloyed state, which served to bolster their capacity for C–O bond cleavage, thereby advancing the selectivity toward 2-MF. Despite these advancements, such catalysts were encumbered by limitations including elevated reaction temperatures, protracted durations of reaction, and suboptimal substrate-to-catalyst ratios. Consequently, there existed a critical imperative to devise a catalyst that was not only environmentally benign and cost-efficient but also exhibited superior performance in the conversion of furfural to 2-MF, with the aim of achieving high product yields.
Notably, in recent years, the Cu/AC catalyst disclosed by Gong et al.24 achieved nearly 100% yield of 2-MF. Likewise, Li et al.25 reported that the Ni–Cu/C catalyst attained a high yield of 98% of 2-MF. The Co–Cu/ZrO2 catalyst reported by Akmaz et al.26 achieved a 94.1% yield of 2-MF at 200 °C. Moreover, the Co-CoOx/AC catalyst, as reported by Zhang et al.,27 stood out for operating at the lowest reaction temperature, as low as 120 °C, yet yielding 87.4% of 2-MF (see Table 1). It was worth noting that the presence of a certain proportion of metal oxides in the catalyst contributed to achieving high yields of 2-MF. For example, in the Cu/AC catalyst, a desired ratio of Cu0:Cu+:Cu2+ efficiently catalyzed the conversion of furfural to 2-MF. An et al.28 confirmed that the addition of Mo in the Cu–Mo/CoOx catalyst promoted the formation of the CuCo alloy and Coδ+ species, which were beneficial for the adsorption of furfural and catalytic cleavage of C–O bonds. The Co-CoOx/AC catalyst has also been confirmed to simultaneously contain Co0 and Co2+. It can be observed that a certain proportion of high-valent metal presence contributed significantly to the hydrogenation of furfural to prepare 2-MF. However, due to the presence of carriers or the interaction of the second metal with metal oxides, it's challenging to quantitatively describe the content of high-valent metals in the catalysts mentioned above.
| Entry | Catalyst | T (°C) | 2-MF yield (%) | Time (h) | Furfural to catalyst ratio | Ref. |
|---|---|---|---|---|---|---|
| 1 | Cu/AC | 170 | 100 | 4 | 1:1 |
24 |
| 2 | NiCu/C | 220 | 98 | 5 | 10:1 |
25 |
| 3 | Co–Cu/ZrO2 | 200 | 94.1 | 6 | 5:2 |
26 |
| 4 | Cu–Co/γ-Al2O3 | 180 | 94 | 3.5 | 8:1 |
23 |
| 5 | NiCo–MgAlO | 220 | 92.1 | 6 | 6:1 |
42 |
| 6 | Cu3–Mo1/CoOx | 180 | 92 | 4 | 13:4 |
28 |
| 7 | Co–CoOx/AC | 120 | 87.4 | 5 | 10:1 |
27 |
| 8 | Cu–Co/Al2O3 | 220 | 87 | 5 | 7:3 |
43 |
| 9 | Cu2Cr2O5 | 240 | 73.5 | 5 | 1:1 |
15 |
| 10 | CuCo/γ-Al2O3 | 200 | 50 | 6 | 10:1 |
21 |
| 11 | CuMgAlNi | 300 | 48 | 2 | 5:1 |
20 |
| 12 | WxC/SiO2 | 200 | 45 | 24 | 7:1 |
44 |
| 13 | Co/CoOx | 170 | 73 | 2 | 10:1 |
This work |
Recently, Xiang et al.29 elucidated the efficient catalytic capability of the Co@CoO catalyst in the conversion of 5-hydroxymethylfurfural to 2,5-dimethylfuran. This monometallic catalyst, comprising solely a metal and its corresponding oxides, facilitated the precise determination of the metal-to-metal oxide ratio. The present investigation was centred on the catalytic transformation of furfural over a Co/CoOx catalyst, which has been shown to facilitate the efficient conversion of furfural to 2-MF. Through the modulation of the reduction temperature, it was possible to convert pristine Co3O4 into an active composite consisting of metallic Co and CoOx, collectively referred to as Co/CoOx, which was confirmed through characterization analysis of its composition and structural features. Reaction conditions of 170 °C, a reaction duration of 2 hours, and a pressure of 2 MPa, coupled with trace amounts of hydroquinone, culminated in a notable yield of 73% for 2-MF. It is obvious that the Co/CoOx catalyst used in this study is a non-precious metal catalyst that can efficiently achieve a high yield of 2-MF when the furfural/catalyst ratio was 10:1. This work posited a simplified mechanistic route for the conversion process of furfural to 2-MF, augmenting our understanding of the underlying chemical processes. Additionally, employing H2-TPR, the ratio of Co0:Co2+ in the catalyst was quantitatively described, observing that the optimal selectivity for 2-MF was achieved when this ratio approached approximately 95%.
2 Materials and methods
2.1 Materials
Furfural (99.0%), furfuryl alcohol (99.0%), tetrahydrofurfuryl alcohol (THFA, 99.0%), 2-methylfuran (2-MF, 99.0%), 2,5-dimethylfuran (99.0%), and hydroquinone (99.0%) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. Isopropanol (99.0%) was purchased from Guangdong Guanghua Sci-Tech Co., Ltd. Analytical grade cobalt nitrate hexahydrate and ammonium carbonate were purchased from Xilong Scientific Co., Ltd. Except for furfural and furfuryl alcohol, which were pretreated by vacuum distillation before use, no other reagents were treated.2.2 Catalyst synthesis
A precipitation method was used to synthesize Co/CoOx, in which cobalt nitrate was used as a precursor for synthesis.29 In a typical procedure, 60 mmol of cobalt nitrate and 69 mmol of (NH4)2CO3 were dissolved separately in 200 mL of distilled water, and the (NH4)2CO3 solution was added dropwise into the cobalt nitrate solution with vigorous stirring. The resulting suspension was aged with stirring at 60 °C for 1 h and allowed to stand at room temperature for 12 h. After filtration and washing with distilled water, the solid product was dried at 100 °C for 12 h and finally calcined in air at 450 °C for 4 h to obtain the desired Co/CoOx sample precursor.The prepared Co/CoOx precursor was further reduced in a tube furnace under H2 (10% H2–Ar mixed gas) at the desired temperature (200–350 °C) with a heating rate of 2 °C min−1 for 2 h. The resulting reduced cobalt oxide was labeled as Co/CoOx-m, where m represents the reduction temperature.
2.3 Catalytic activity testing in a batch reactor
The selective hydrodeoxygenation of furfural was conducted in a 100 mL stainless steel high-pressure reactor. The required amount of furfural, catalyst, and solvent was sealed in the reactor, which was then purged three times with N2 and H2 respectively to remove air and fill in the desired H2 pressure. The reactor was rapidly heated to the predetermined temperature, and after the reaction, it was cooled in an ice bath. The reaction liquid was filtered through a 13 mm × 0.22 μm organic membrane filter for product analysis. Qualitative analysis was performed using a gas chromatography-mass spectrometry system (QP2020 NX), and quantitative analysis was conducted using a gas chromatography system (Agilent 7890B) equipped with an HP-5 column and an FID detector. The carrier gas type of gas chromatography is helium.The conversion of furfural and the yield of products were calculated using 2,5-dimethylfuran as an internal standard, based on the product yield.
| Conversion (%) = [(Moles of reacted furfural)/(Moles of initial furfural)] × 100 | (1) |
| Yield (%) = [(Moles of product)/(Moles of initial furfural)] × 100 | (2) |
2.4 Catalyst characterization
The crystal structure information of the catalyst was determined using an Empyrean-100 X-ray diffractometer from the Dutch company PANalytical. The instrument was operated at a scanning speed of 10° min−1 in the range of 10–80°, with a Cu Kα radiation source (λ = 0.15432 nm), a tube voltage of 40 kV, and a tube current of 40 mA.The specific surface area, average pore size, and pore volume of the catalyst were determined using a Micromeritics ASAP 2460 physical adsorption analyzer. Prior to the analysis, the sample was pretreated at 110 °C under vacuum for one hour, followed by a 6 h degassing at 350 °C to remove various gases and impurities adsorbed on the catalyst surface. The N2 adsorption–desorption measurement was conducted at 77 K liquid nitrogen temperature under a vacuum pressure of 10−6 torr. The specific surface area was calculated using the BET equation after completion of the analysis. The method adopted for calculating the aperture distribution is the Barrett–Joyner–Halenda method.
The H2-TPR (hydrogen temperature programmed reduction) technique on an Autochem II 2920 chemisorption analyzer from Micromeritics was used to characterize the differences in reduction temperatures of various catalysts. First, the samples were pretreated at 400 °C in an Ar atmosphere for 30 minutes to remove surface-adsorbed impurities. The samples were then cooled to 50 °C and the carrier gas was switched to a 10 vol% H2/Ar mixed gas. After the baseline had stabilized, the samples were heated from 50 °C to 800 °C at a rate of 10 °C min−1. A thermal conductivity detector (TCD) was used to record the test signal throughout the process, and a cold trap was installed before the TCD to remove water produced during the reduction process.
3 Results and discussion
3.1 Catalyst characterization
To investigate the influence of the reduction process on the catalyst structure, the specific surface area and pore structure of the catalyst were characterized. As shown in Fig. S1,† the N2 adsorption–desorption isotherms for Co/CoOx at varying temperatures of reduction displayed characteristics of type IV isotherms, indicative of the presence of mesoporous structures within the materials.30Table 2 presents the BET specific surface area and pore volume data. Specifically, the Co/CoOx-200 sample exhibited a specific surface area of 25.7 m2 g−1 and a pore volume of 0.01 cm3 g−1. A discernible trend was observed wherein an elevation in the reduction temperature resulted in a concomitant decrease in the catalyst's specific surface area, which diminished to 3.4 m2 g−1, and the pore volume concurrently contracted to 0.0012 cm3 g−1. This reduction in surface area and pore volume may be attributed to the collapse of pores during the reduction process.31| Reduction temperature (°C) | BET surface area (m2 g−1) | Total pore volume (cm3 g−1) |
|---|---|---|
| a Note: 200 °C represents Co/CoOx reduced at 200 °C; 250 °C represents Co/CoOx reduced at 250 °C; 300 °C represents Co/CoOx reduced at 300 °C; 350 °C represents Co/CoOx reduced at 350 °C. | ||
| 200 °C | 25.7 | 0.010 |
| 250 °C | 6.1 | 0.0024 |
| 300 °C | 5.8 | 0.0021 |
| 350 °C | 3.4 | 0.0012 |
XRD analysis was also used to characterize the phases of these catalysts (Fig. 1). For unreduced Co/CoOx, only the spinel phase Co3O4 was observed. The main spinel phases of Co/CoOx-200 were Co3O4 and a small amount of cubic phase CoO. The XRD pattern of Co/CoOx-250 showed a mixture of cubic phases of Co, CoO and Co3O4. Co/CoOx-300 and Co/CoOx-350 mainly exhibited a metallic Co structure. Five significant diffraction peaks were observed in the XRD pattern for Co3O4 at 2θ = 31.2°, 36.8°, 44.8°, 59.3°, and 65.2°, corresponding to the (220), (311), (400), (511), and (440) planes,32 respectively. With the increase in reduction temperature, the peak belonging to Co3O4 became weaker, and diffraction relative to CoO and metallic Co was detected. The diffraction at 2θ = 42.4° was assigned to the (200) plane of cubic phase CoO.
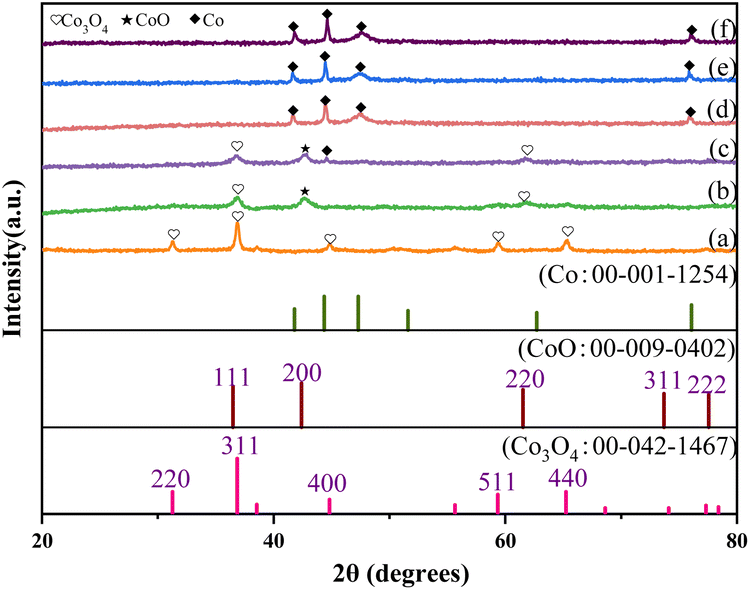 | ||
| Fig. 1 XRD patterns of (a) unreduced Co/CoOx, (b) 200 °C reduced Co/CoOx, (c) 250 °C reduced Co/CoOx, (d) 300 °C reduced Co/CoOx and (e) 350 °C reduced Co/CoOx. | ||
The Co/CoOx samples at different reduction temperatures were tested using H2-TPR. Prior to the TPR measurements, the catalysts were pre-treated, ensuring the absence of any organic or other residues. Clear H2 consumption peaks were observed in the temperature ranges of 250–320 °C and 320–450 °C (Fig. S2†). The low-temperature peak corresponded to the reduction of Co3+ to Co2+ and the formation of CoO,33 while the high-temperature peak corresponded to the reduction of Co2+ to metallic Co.34 Previous studies reported the sequential reduction process that transitions from Co3O4 to CoO and subsequently to metallic Co.35,36 The data delineated in Table 3 revealed that, among the catalyst specimens evaluated, Co/CoOx-200 manifested the most substantial hydrogen uptake. As the reduction temperature increased, the amount of hydrogen consumption by the sample significantly decreased, confirming that a large amount of CoOx in Co/CoOx catalysts had been reduced. The amount of hydrogen consumption through the Co/CoOx catalysts could be used to approximately calculate the proportion of Co in different valence states in the catalysts. The detailed results are shown in Table 3. The H2-TPR outcomes substantiated the coexistence of CoOx and metallic Co within the synthesized catalysts. The H2-TPR outcomes were in agreement with those of XRD.
| Reduction temperaturea (°C) | Quantityb (mmol g−1) | Atomic ratioc |
|---|---|---|
| a 200 °C represents Co/CoOx reduced at 200 °C; 250 °C represents Co/CoOx reduced at 250 °C; 300 °C represents Co/CoOx reduced at 300 °C; 350 °C represents Co/CoOx reduced at 350 °C. b Hydrogen consumption in the H2-TPR test. c The atomic ratio of Co atoms with different valence states calculated from the hydrogen consumption in H2-TPR. | ||
| 200 °C | 16.06 | Co2+:Co3+ = 69.7%:30.3% |
| 250 °C | 1.872 | Co0:Co2+ = 84.9%:15.1% |
| 300 °C | 0.875 | Co0:Co2+ = 94.8%:5.2% |
| 350 °C | 0.801 | Co0:Co2+ = 95.2%:4.8% |
3.2 Synthesis of 2-MF by furfural selective hydrodeoxygenation
The synthesis of 2MF was achieved through the selective hydrodeoxygenation of furfural using a high-pressure Parr reactor system. The selective hydrodeoxygenation procedure was executed under a hydrogen atmosphere for a duration of 2 h, utilizing the Co/CoOx catalyst at its specified reduction temperature. This catalyst was synthesized by a straightforward precipitation technique, followed by a reduction process under 10% H2 for 2 h. The literature review suggested that the synthesis of 2-MF via catalytic selective hydrodeoxygenation of furfural was initiated through the hydrogenation of its aldehyde functional group, yielding furfuryl alcohol. This was followed by the selective hydrodeoxygenation of the hydroxyl moiety in furfuryl alcohol to produce 2-MF. Nevertheless, during this latter stage of selective hydrodeoxygenation, there existed a proclivity for concurrent hydrogenation of the furan ring, culminating in the incidental generation of tetrahydrofurfuryl alcohol as an undesired byproduct.37,38 Moreover, an elevated hydrogen pressure or a prolonged reaction duration could lead to the additional hydrogenation of the furan ring present in 2-MF, resulting in the formation of 2-methyltetrahydrofuran and 1,4-pentanediol. Simultaneously, the polymerization of furfural and furfuryl alcohol constituted prevalent presumable side reactions within this experimental framework. The aforementioned reaction sequence is delineated in Scheme 1.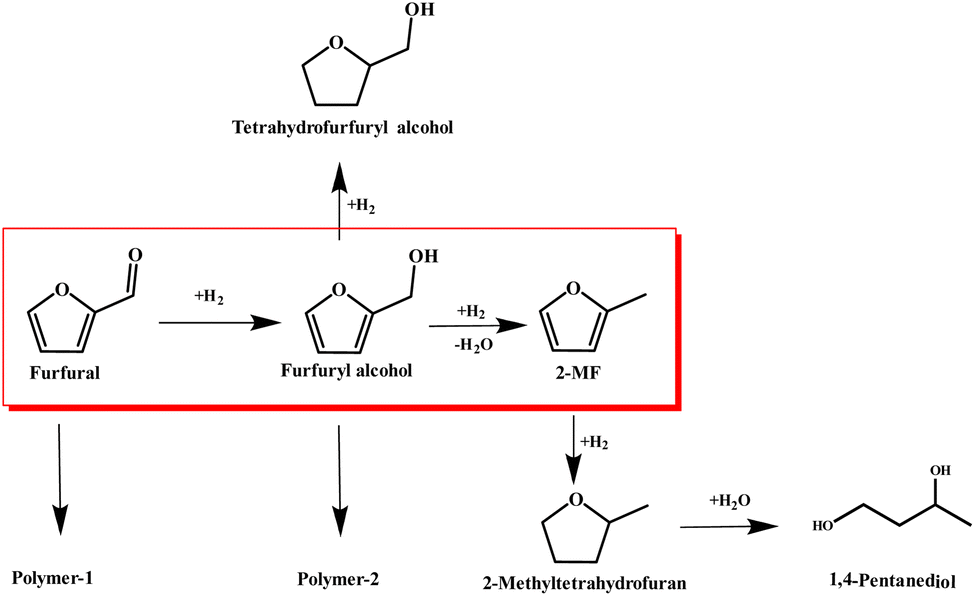 | ||
| Scheme 1 The pathway for the selective hydrodeoxygenation of furfural. | ||
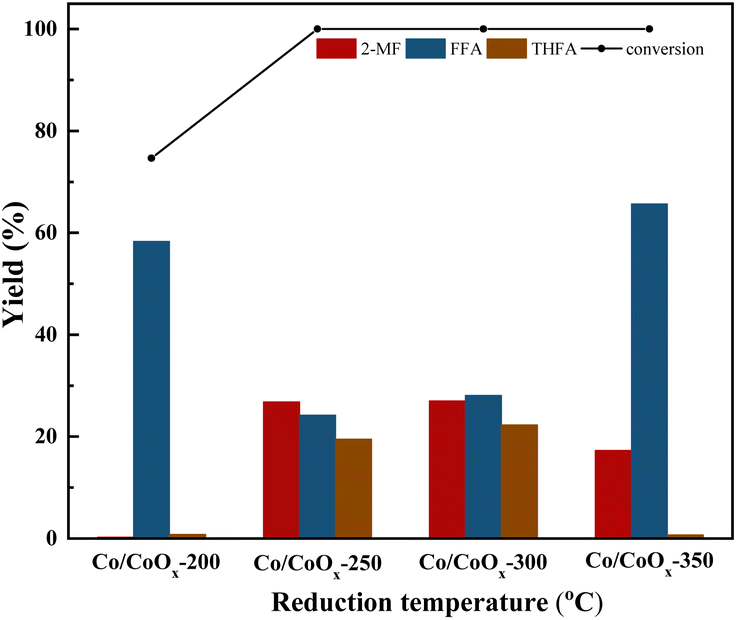 | ||
| Fig. 2 Effect of reduction temperature on furfural selective hydrodeoxygenation conversion and yield using Co/CoOx catalysts. Reaction conditions: furfural 2 g, isopropyl 40 mL, catalyst 200 mg, agitation speed 700 rpm, H2 pressure 2 MPa, temperature 130 °C, time 2 h. 2-MF: 2-methylfuran, FFA: furfuryl alcohol, THFA: tetrahydrofurfuryl alcohol. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O bonds, accelerating the reaction rate and promoting the generation of 2-MF.
O and C–O bonds, accelerating the reaction rate and promoting the generation of 2-MF.
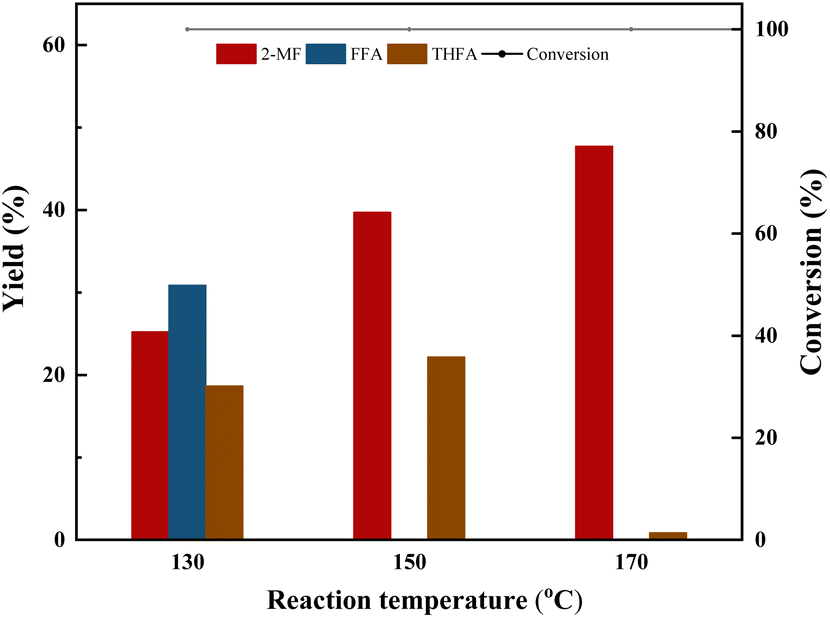 | ||
| Fig. 3 Effect of reaction temperature on furfural selective hydrodeoxygenation conversion and yield using Co/CoOx-300 catalysts. Reaction conditions: furfural 2 g, isopropyl 40 mL, catalyst 200 mg, agitation speed 700 rpm, H2 pressure 2 MPa, time 2 h. 2-MF: 2-methylfuran, FFA: furfuryl alcohol, THFA: tetrahydrofurfuryl alcohol. | ||
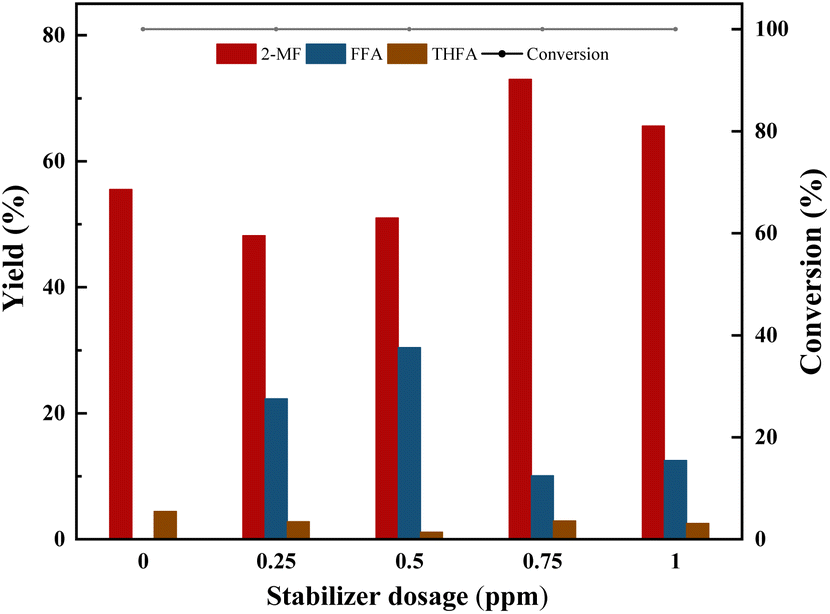 | ||
| Fig. 4 Effect of stabilizer dosage on furfural selective hydrodeoxygenation conversion and yield using Co/CoOx-300 catalysts. Reaction conditions: furfural 2 g, isopropyl 40 mL, catalyst 200 mg, temperature 170 °C, agitation speed 700 rpm, H2 pressure 2 MPa, time 2 h. 2-MF: 2-methylfuran, FFA: furfuryl alcohol, THFA: tetrahydrofurfuryl alcohol. | ||
To investigate the role of hydroquinone, three sets of comparative experiments were conducted. From Table 4, it can be observed that furfural underwent only minor polymerization reactions (6–7%) in the absence of a catalyst under the reaction conditions, and the addition of hydroquinone didnt inhibit this slight polymerization reaction. However, upon the addition of a Co-based catalyst, furfural underwent significant polymerization (37%). By adding hydroquinone, the polymerization reaction was significantly reduced to 16%, indicating that, in this process, the polymerization reaction was mainly due to the polymerization of furfural catalyzed by the Co-based catalyst.
| Catalysts (g) | Reactant | Stabilizer dosage (ppm) | Conversion (%) | 2-MF yield (%) | Furfuryl alcohol yield (%) | Polymer yieldb (%) |
|---|---|---|---|---|---|---|
| a Reaction conditions: furfural 2 g, furfural alcohol 2 g isopropyl 40 mL, catalyst 200 mg, temperature 170 °C, agitation speed 700 rpm, H2 pressure 2 MPa, time 2 h. 2-MF: 2-methylfuran. b The polymer yield was calculated by using the carbon balance, which was determined by subtracting the quantifiable amounts of reactants and products from 100%. | ||||||
| 0 | Furfural | 0 | 6 | 0 | 0 | 6 |
| 0 | Furfuryl alcohol | 0 | 11.4 | 0 | 0 | 11.4 |
| 0.2 | Furfural | 0 | 100 | 58 | 5 | 37 |
| 0.2 | Furfuryl alcohol | 0 | 15.1 | 10.3 | 0 | 4.8 |
| 0 | Furfural | 0.75 | 7 | 0 | 0 | 7 |
| 0.2 | Furfural | 0.75 | 100 | 73 | 11 | 16 |
Another particularly interesting phenomenon was observed with furfuryl alcohol, which behaved oppositely to furfural. In the absence of a catalyst, the polymerization was 11.4%. However, upon the addition of the catalyst, the polymerization decreased to 4.8%, indicating that the Co-based catalyst inhibited the polymerization of furfuryl alcohol. Consequently, it was feasible to delineate the reaction pathway for the selective hydrodeoxygenation of furfural into 2-MF by modulating the reaction selectivity. This involved consideration of side reactions, such as the polymerization of furfural and furfuryl alcohol, the reduction of furfuryl alcohol to tetrahydrofurfuryl alcohol, and the excessive hydrogenation of 2-MF resulting in the production of 2-methyltetrahydrofuran and 1,5-pentanediol. These processes are comprehensively illustrated in Scheme 1. Moreover, the observations from this study were in alignment with findings put forth by other researchers in this field.41
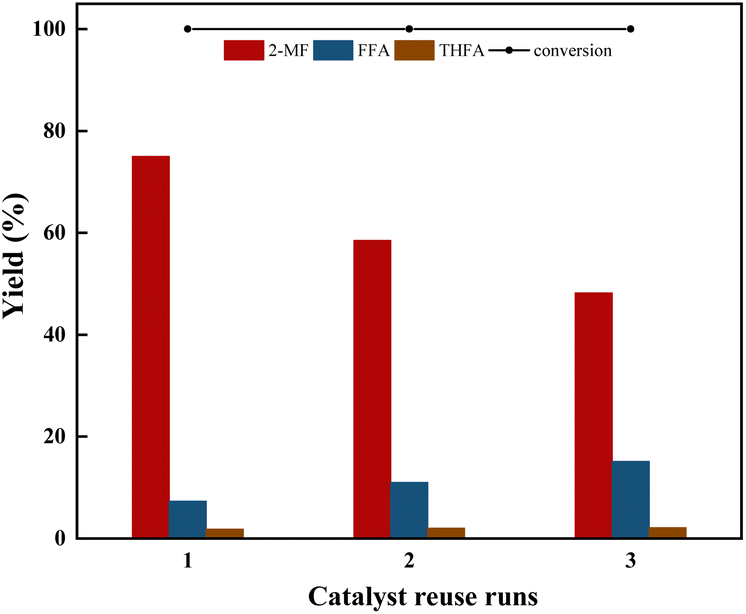 | ||
| Fig. 5 Reusability of the catalyst for furfural selective hydrodeoxygenation conversion and yield using Co/CoOx-300 catalysts. Reaction conditions: furfural 2 g, isopropyl 40 mL, catalyst 200 mg, stabilizer dosage 0.75 ppm, temperature 170 °C, agitation speed 700 rpm, H2 pressure 2 MPa, time 2 h. 2-MF: 2-methylfuran, FFA: furfuryl alcohol, THFA: tetrahydrofurfuryl alcohol. | ||
4 Conclusions
A collection of Co/CoOx catalysts was synthesized via a simple precipitation technique, and their efficacy in catalyzing the selective hydrodeoxygenation of furfural to 2-MF was substantiated. In the series, Co/CoOx-300 demonstrated superior selectivity towards the formation of 2-MF. The introduction of a small amount of hydroquinone, serving as a polymerization inhibitor, facilitated the attainment of 2-MF yields as high as 73% under mild reaction parameters (170 °C, 2 MPa H2, for a duration of 2 hours) despite employing a substantial furfural/catalyst mass ratio of 10:1. Analytical techniques, including XRD and H2-TPR ascertained that Co/CoOx-300 predominantly consisted of metallic Co and CoO when the ratio of Co0 to Co2+ within the catalyst was around 95% leading to optimal selectivity towards 2-MF. Conclusively, a mechanistic schematic delineating the selective hydrodeoxygenation of furfural to 2-MF was constructed, predicated on the observations gleaned from the experimental procedures (see Scheme 1).
Author contributions
Y. Han: conceptualization, methodology, investigation, writing – original draft, writing – review and editing. X. Zhang: methodology, investigation. W. Wang: data curation, methodology, investigation. S. Guo: methodology, investigation, writing – original draft, writing – review and editing. X. Ji: writing – review and editing, formal analysis. G. Li: writing – review and editing, formal analysis, project administration, funding acquisition.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful for the support from the National Natural Science Foundation of China (No. 22078318), and the Youth Innovation Promotion Association CAS (grant No. 2020186).References
- X. Wu, R. Daniel, G. Tian, H. Xu, Z. Huang and D. Richardson, Appl. Energy, 2011, 88(7), 2305–2314 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41(24), 8075–8098 RSC.
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2016, 117(10), 6834–6880 CrossRef PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41(4), 1538–1558 RSC.
- A. S. Mamman, J. M. Lee, Y. C. Kim, I. T. Hwang, N. J. Park, Y. K. Hwang, J. S. Chang and J. S. Hwang, Biofuels, Bioprod. Biorefin., 2008, 2(5), 438–454 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12(4), 539–554 RSC.
- B. Wang, C. Li, B. He, J. Qi and C. Liang, Energy Chem., 2017, 26(4), 799–807 CrossRef.
- P. Biswas, J. H. Lin, J. Kang and V. V. Guliants, Appl. Catal., A, 2014, 475, 379–385 CrossRef CAS.
- X. Chang, A. F. Liu, B. Cai, J. Y. Luo, H. Pan and Y. B. Huang, ChemSusChem, 2016, 9(23), 3330–3337 CrossRef CAS PubMed.
- B. J. Liu, L. H. Lu, B. C. Wang, T. X. Cai and I. Katsuyoshi, Appl. Catal., A, 1998, 171(1), 117–122 CrossRef CAS.
- J. J. Musci, A. B. Merlo and M. L. Casella, Catal. Today, 2017, 296, 43–50 CrossRef CAS.
- V. Vetere, A. B. Merlo, J. F. Ruggera and M. L. Casella, J. Braz. Chem. Soc., 2010, 21, 914–920 CrossRef CAS.
- Y. Román-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447(7147), 982–985 CrossRef PubMed.
- G. Z. Ren, G. Y. Li, Y. Zhang, A. Q. Wang, X. D. Wang, Y. Cong, T. Zhang and N. Li, Sustainable Energy Fuels, 2022, 6(4), 1156–1163 RSC.
- M. Wu, T. Wang, W. H. Li, Q. Zhang, B. Zhang, K. Q. Chen, S. Y. Peng, G. J. Li, J. N. Huang, Q. Wang and C. Wang, Chem. Eng. J., 2023, 461, 141944 CrossRef CAS.
- J. G. M. Bremner and R. K. F. Keeys, J. Chem. Soc., 1947, 1068–1080 RSC.
- F. Ma, H. Li and J. Jiang, Chem. Res. Chin. Univ., 2019, 35(3), 498–503 CrossRef CAS.
- A. O'Driscoll, T. Curtin, W. Y. Hernández, P. Van Der Voort and J. J. Leahy, Org. Process Res. Dev., 2016, 20(11), 1917–1929 CrossRef.
- O. F. Aldosari, S. Iqbal, P. J. Miedziak, G. L. Brett, D. R. Jones, X. Liu, J. K. Edwards, D. J. Morgan, D. K. Knight and G. J. Hutchings, Catal. Sci. Technol., 2016, 6(1), 234–242 RSC.
- P. Panagiotopoulou, N. Martin and D. G. Vlachos, J. Mol. Catal. A: Chem., 2014, 392, 223–228 CrossRef CAS.
- N. S. Date, A. M. Hengne, K. W. Huang, R. C. Chikate and C. V. Rode, Green Chem., 2018, 20(9), 2027–2037 RSC.
- C. Xu, L. Zheng, D. Deng, J. Liu and S. Liu, Catal. Commun., 2011, 12(11), 996–999 CrossRef CAS.
- M. Kalong, P. Hongmanorom, S. Ratchahat, W. Koo-amornpattana, K. Faungnawakij, S. Assabumrungrat, A. Srifa and S. Kawi, Fuel Process. Technol., 2021, 214, 106721 CrossRef CAS.
- W. b. Gong, C. Chen, H. Zhang, G. Wang and H. Zhao, ChemistrySelect, 2017, 2(31), 9984–9991 CrossRef CAS.
- H. Li, H. Liu, C. Cai, H. Wang, Y. Huang, S. Li, B. Yang, C. Wang, Y. Liao and L. Ma, Catal. Commun., 2023, 175, 106625 CrossRef CAS.
- S. Akmaz, S. Algorabi and S. N. Koc, Can. J. Chem. Eng., 2021, 99, S562–S574 CrossRef CAS.
- Z. Zhang, Z. Zhang, X. Zhang, F. Wang, Z. Wang, Y. Li, X. Wang, R. Ahishakiye and X. Zhang, Appl. Surf. Sci., 2023, 612, 155871 CrossRef CAS.
- Y. An, Q. Wu, L. Niu, C. Zhang, Q. Liu, G. Bian and G. Bai, J. Catal., 2024, 429, 115271 CrossRef CAS.
- S. Xiang, L. Dong, Z. Q. Wang, X. Han, L. L. Daemen, J. Li, Y. Q. Cheng, Y. Guo, X. H. Liu, Y. F. Hu, A. J. Ramirez-Cuesta, S. H. Yang, X. Q. Gong and Y. Q. Wang, Nat. Commun., 2022, 13(1), 3657 CrossRef CAS PubMed.
- J. Zhu, F. Q. Chen, Z. G. Zhang, M. Li, Q. W. Yang, Y. W. Yang, Z. B. Bao and Q. L. Ren, ACS Sustain. Chem. Eng., 2019, 7(15), 12955–12963 CrossRef CAS.
- A. Takagaki, M. Ohara, S. Nishimura and K. Ebitani, Chem. Commun., 2009, 41, 6276–6278 RSC.
- H. Wu, G. Pantaleo, G. Di Carlo, S. Guo, G. Marcì, P. Concepción, A. M. Venezia and L. F. Liotta, Catal. Sci. Technol., 2015, 5(3), 1888–1901 RSC.
- W. P. Ma, Y. J. Ding and L. W. Lin, Ind. Eng. Chem. Res., 2004, 43(10), 2391–2398 CrossRef CAS.
- S. R. De Miguel, M. C. Rom n-Martınez, E. L. Jablonski, J. L. G. Fierro, D. Cazorla-Amorόs and O. A. Scelza, J. Catal., 1999, 184(2), 514–525 CrossRef CAS.
- W. Chu, P. Chernavskii, L. Gengembre, G. Pankina, P. Fongarland and A. Khodakov, J. Catal., 2007, 252(2), 215–230 CrossRef CAS.
- B. Solsona, T. E. Davies, T. Garcia, I. Vázquez, A. Dejoz and S. H. Taylor, Appl. Catal., B, 2008, 84(1–2), 176–184 CrossRef CAS.
- P. Koley, S. Chandra Shit, B. Joseph, S. Pollastri, Y. M. Sabri, E. L. H. Mayes, L. Nakka, J. Tardio and J. Mondal, ACS Appl. Mater. Interfaces, 2020, 12(19), 21682–21700 CrossRef CAS PubMed.
- T. Varila, E. Mäkelä, R. Kupila, H. Romar, T. Hu, R. Karinen, L. R. Puurunen and U. Lassi, Catal. Today, 2021, 367, 16–27 CrossRef CAS.
- Z. L. Zhang, Z. W. Zhang, X. B. Zhang, F. M. Wang, Z. Wang, Y. W. Li, X. T. Wang, R. Ahishakiye and X. Zhang, Appl. Surf. Sci., 2023, 612, 155871 CrossRef CAS.
- K. Yan, G. Wu, T. Lafleur and C. Jarvis, Renewable Sustainable Energy Rev., 2014, 38, 663–676 CrossRef CAS.
- S. Liu, Y. Amada, M. Tamura, Y. Nakagawa and K. Tomishige, Green Chem., 2014, 16(2), 617–626 RSC.
- X. Gong, N. Li, Y. Li and R. Hu, Mol. Catal., 2022, 531, 112651 CrossRef CAS.
- S. Srivastava, G. C. Jadeja and J. Parikh, Chem. Eng. Res. Des., 2018, 132, 313–324 CrossRef CAS.
- P. Bretzler, M. Huber, S. Nickl and K. Köhler, RSC Adv., 2020, 10(46), 27323–27330 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00229f |
| This journal is © The Royal Society of Chemistry 2024 |