
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper catalyzed alkaline aerobic lignin depolymerization: effect of botanical origin and industrial extraction process on reactivity supported through characterization†
Antonio
Hernández-Mañas
ab,
Alex
Martínez-Martin
a,
Johan
Madignier
a,
Pascal
Fongarland
b,
Frédérique
Bertaud
c,
Léa
Vilcocq
 *b and
Laurent
Djakovitch
*a
*b and
Laurent
Djakovitch
*a
aIRCELYON, UMR 5256, CNRS-Université de Lyon 1, 2 Avenue Albert Einstein, F-69626 Villeurbanne Cedex, Lyon, France. E-mail: laurent.djakovitch@ircelyon.univ-lyon1.fr
bCP2M, UMR 5128, CNRS-CPE-Université de Lyon 1, 43, bd du 11 novembre 1918, B.P. 82077, 69616 Villeurbanne Cedex, France. E-mail: lea.vilcocq@cnrs.fr
cCentre Technique du Papier, CTP, CS90251, 38044 Grenoble cedex 9, France
First published on 13th August 2024
Abstract
Different lignins, differing in their botanical origin or the extraction method, were subjected to aerobic catalytic depolymerisation in basic aqueous media using a copper-based catalyst (CuO/TiO2) towards aromatic compounds. Lignins were obtained from different kinds of biomass, including hardwood, softwood and wheat straw, and using different extraction methods such as kraft, soda or organosolv. Extensive characterization (elemental analysis; 1H, 13C, 31P and HSQC NMR; FTIR-ATR; DLS and SEC) revealed structural differences between lignin samples. For example, organosolv softwood lignin shows a higher degree of β-O-4 linkages, while organosolv wheat straw lignin presents higher structural degradation. These changes resulted in differences during the catalytic aerobic depolymerisation. The structural properties were correlated with the reaction results. Thus, the average molecular weight is directly related to the observed degree of conversion, and the β-O-4 content is correlated with the yields of aromatic compounds obtained. It was also observed that the catalytic effect of the CuO/TiO2 catalyst is more pronounced when the non-catalytic reaction shows low yields of aromatics. Finally, despite higher reactivity, hardwood lignin did not produce high yields of aromatic compounds due to the rapid degradation of the products.
Sustainability spotlightActually, lignin is the only biopolymer that does not compete with edible resources and that can be upgraded to deliver biosourced aromatic compounds. Its conversion is the center of numerous currently developing research toward efficient processes. This manuscript reports the catalytic aerobic depolymerisation under mild conditions of different lignins varying by their botanical origin or extraction method. Focus was laid on the production of vanillin. We showed that the structural differences and alterations arising from lignin extraction induced variations during the catalytic aerobic depolymerization. The structural behaviors were correlated with lignin conversion and the yields of aromatic compounds. Data demonstrated that the copper catalyst used is stable under the reaction conditions, suggesting reuse or implementation in a continuous process. These research studies align with the ethos of RSC Sustainability, providing comprehensive routes for assessing and optimizing lignin conversion to chemicals as an alternative to petroleum, also emphasizing UN's Sustainable Development Goals as “Responsible Consumption and Production” (SG12). |
Introduction
Lignin is one of the most complex and heterogeneous components of the lignocellulosic biomass and has been extensively studied in order to elucidate its structure and chemical composition to provide data prior to its chemical transformation. Lignin is the least abundant biopolymer in biomass, compared to cellulose and hemicellulose, and its abundance varies significantly with the type of biomass. The proportion of lignin increases from hardwood (i.e., acacia, eucalyptus or maple) to softwood (i.e., pine, cedar or cypress), passing through other types of biomass such as wheat straw or bagasse. Typically, softwoods contain around 21–29% by weight of lignin and hardwoods contain 18–25% by weight of lignin, while herbaceous plants contain 15–24% by weight of lignin.1,2It is an aromatic polymer made up of three different phenylpropane units that differ by the number of methoxy groups attached to the phenolic moiety. These units are abbreviated by the letters H (p-Hydroxyphenyl), G (Guaiacyl), and S (Syringyl). In lignin, these units are linked by different ether linkages, the most abundant being β-O-4 and α-O-4, and also by several C–C linkages, such as 5–5, β-5, and β-1. The proportion of these phenylpropane units and linkages is mainly influenced by the type of wood used to obtain lignin from the lignocellulosic biomass, with the extraction method also being the first source of modifications affecting the proportion of linkages in the extracted lignin. Thus, softwood lignin is composed exclusively of G units, hardwood lignin contains G and S units, while herbaceous lignin contains all three phenylpropane units.3 In general, the β-O-4 linkage (C–O–C) is the most common, accounting for 45–50% of the total number of linkages.4 In addition to the structure, the botanical origin and extraction method of lignin also influence its molecular weight.
In this study, six lignins from different botanical origins and extracted by different industrial processes were evaluated. The latter included kraft pulping under alkaline conditions (aqueous NaOH and Na2S), one of the main pulping methods in the paper industry, which induces several changes in the lignin (polymer degradation, condensation reactions, reduction of the β-O-4 occurrence and partial sulfonation).5 Two other processes were also included: soda pulping, which is similar to kraft pulping without the use of Na2S and reduces the sulfur content in the extracted lignin and organosolv pulping, which uses an organic solvent (e.g., a carboxylic acid) in combination with water and mineral acids to solubilize the lignin. In this case, the lignin structure is strongly affected by β-O-4 cleavages, lignin condensations or hydrolysis of ester groups.6
Lignin is produced in an amount of about 80 million tons per year (of which about 55 Mt per year is produced as kraft and 1 Mt per year as lignosulfonate) by the different paper industries worldwide,7,8 but most of it is burned for energy recovery, and therefore several technologies are being studied to achieve lignin depolymerization and subsequent valorization of low molecular weight compounds. Kraft lignin is therefore a particularly interesting material given its production growth of around 7% per year;9 however, the current production for commercial purposes remains low at around 250 kt per year.10
Among all the lignin depolymerization methods that were recently reviewed in detail,11–13 oxidation remains of particular interest due to the low energy requirements and the large number of valuable functional compounds produced, especially phenolic compounds, carboxylic acids or benzoquinones, which are of particular interest to the synthetic chemist and the pharmaceutical industry. Since this is the focus of this paper, our literature review will be limited to oxidative lignin upgradation in aqueous media to produce functional aromatic compounds such as aromatic aldehydes or ketones. Initially, most of the studies were carried out using model compounds to simulate the different monomers, dimers or oligomers present in lignin. These allowed the study of some reactivity trends, such as cleavage of interunit bonds, oxidative modification of the propanyl side-chain oxidative modification, and the oxidation of the aromatic ring and ring cleavage reactions. Both homogeneous and heterogeneous catalysts have been included in these studies. In general, in the field of heterogeneous catalysis high selectivity in aldehydes has been observed by using metal oxides and mixed metal oxides.14,15
In the field of lignin oxidative depolymerisation, as for model compounds, several catalyst-free and homogeneous catalyst-based processes have been used, and are the subject of several review articles.16–18 Next, numerous processes based on the use of heterogeneous catalysts have been reported and have been reviewed.19 Initially, perovskites20 and polyoxometalates21 were intensively evaluated, followed by metal supported catalysts based, for example, on Co,22–24 Mn,25 Fe26 or noble metals (i.e., Au,27 Pd28–30 or Pt31,32). As with homogeneous catalytic systems, the studies were often related to evaluation using model compounds. When applied to lignin, procedures obtained on model compounds could often not be transferred to real cases and yields of phenolic compounds remained low. These observations are often related to the fact that the model compounds used could not properly represent the lignin polymeric material and that condensation reaction, which often occurs with lignin, does not operate on such low molecular weight compounds. In this paper, we focus mainly on heterogeneous copper catalysts applied to the oxidative depolymerization of lignin. The situation contrasts with other catalytic materials, as it was found that copper catalysts have been intensively studied and applied to various lignin materials. H. Deng et al. showed that the use of a copper-doped perovskite catalyst significantly increased aldehyde production using oxygen as the oxidant, yielding 5.3% of vanillin.33 Kumar et al. evaluated the properties of Cu/γ-Al2O3 on grass soda lignin (Protobind™ 1000) and alkali lignin. For the latter, in water, they achieved up to 4.9% yield of vanillin.34,35 A Cu–Mn mixed oxide has been described for vanillin production by the alkaline wet oxidation of a commercial kraft lignin with H2O2. The activity of the catalyst increased with increase of the copper content; however, the authors observed that over-oxidation of vanillin to vanillic acid and other compounds occurred under the reaction conditions.36 Numerous studies on the catalytic oxidative depolymerization of native and organosolv lignin using Cu/TiO2 catalysts have been described by Kuznetsov et al.37–42 In general, low yields of aromatic compounds were observed despite varying the reaction conditions used (70–100 °C, and 300 °C under supercritical conditions). The authors showed that these low yields were not due to a lack of lignin depolymerization but are rather due to over-oxidation of aromatics. Ce–Cu/MFI nanosheet catalysts were applied to the aerobic depolymerization of organosolv lignin in ethanol under oxygen at 150 °C. Yields ranging from 10 to 58% of aromatics were obtained;43 however, these include alkylated derivatives due to the use of ethanol as the solvent, which has been reported elsewhere.44,45 In addition to supported copper metal catalysts, massive or supported CuO catalysts have also been investigated. One of the first reports concerned the use of an over-stoichiometric amount of CuO as the oxidant. In this work, Villar et al. reported that CuO used in a ratio of 160 wt% CuO/lignin can induce lignin depolymerisation under an inert atmosphere, giving 6 wt% yield of syringaldehyde and 1.3 wt% yield of vanillin.46,47 Using a similar approach, Ninomiya et al. reported the oxidative depolymerization of enzyme-pretreated eucalyptus lignin using a stoichiometric amount of CuO to give up to 39% yield of aromatic aldehydes.48 Recently, Qu et al. reported that CuO acts as a catalyst when used in the presence of hydrogen peroxide under microwave irradiation. Initially applied to the conversion of lignin β-O-4 dimeric model compounds to guaiacol, vanillin and vanillic acid via a dihydroxylation,49 the authors extended the scope to softwood and hardwood lignin with yields of ca. 4.4% of vanillin and about 11.4% of aldehydes (mainly syringaldehyde).50 Similarly, Ouyang et al. reported the use of CuO/Fe2(SO4)3 combined with hydrogen peroxide for the alkaline oxidative depolymerisation of wheat alkali lignin in different solvents. The best results were obtained in a mixture of MeOH/water, yielding 18% phenolic compounds. The authors suggested that the use of methanol co-solvent facilitates bond cleavage by improving lignin solubility and prevents recondensation reactions as it disfavors side demethoxylation.51 CuO NPs were found to be highly efficient for depolymerizing lignin into aldehydes and ketones, yielding up to 49% of aromatic aldehyde, when applied directly to biomass by conducting lignin fractionation and simultaneous depolymerisation in aqueous NaOH under oxygen at 160 °C.52 A copper–vanadium supported catalyst on zirconia was found to be efficient in alkaline water at 150 °C for the conversion of LignoBoost kraft lignin to aromatic monomers under oxygen with a yield of up to 9% by weight.53 Similarly, a VO(Acac)2/Cu(OAc)2 catalyst was investigated in the oxidative depolymerisation of industrial softwood kraft lignin giving a bio-oil in 50% wt yield with a selectivity to aromatic monomers reaching 14%.54 Recently, we reported that CuO/TiO2 catalyst prepared by incipient wet impregnation, was active in lignin oxidative depolymerization in basic aqueous medium in air as an oxidant at 150 °C, producing vanillin in up to 5.1% wt yield,55 work that was extended then to kraft black liquor.56,57
Mechanistically, it appears that catalytic aerobic lignin depolymerization, irrespective of the catalysts, proceeds via the formation of phenoxyl radicals that have evolved in reaction pathways such as retro-aldol reactions.58,59 However, recent work suggests that the pathways depend on the nature of the initial radical oxidation sources.60 Recently, we have reported that metal catalysts influence the selectivity of the reaction towards different families of aromatic compounds via different reaction pathways.30
In our research, and encouraged by this literature review, we are carrying out studies on aerobic oxidative lignin depolymerization under basic conditions. Various lignins, differing in their botanical origin and the pulping process were included in the study, after full characterization. In addition to kraft lignin, soda lignin and organosolv lignin were also evaluated. Despite its lower availability, olignin is of interest due to its currently growing market, estimated at +5%/5 years.7 Following our previous reports,55–57 a home-made CuO/TiO2 catalyst was used. In addition to catalytic tests and post-test fractionation followed by chemical analysis of fractions, some “structural behaviors” of the lignin potentially affecting the catalytic process were studied with the aim of maximizing vanillin production.
Materials and methods
Materials
The lignin studied in this work was extracted from different plants using different pulping processes. Some are commercially available and they were used without further purification (Table 1).| Name of the lignin employed, puritya | Botanical origin | Commercial name/provider | Industrial plant | Pulping process |
|---|---|---|---|---|
| a Purity was determined from Klason and ash content determination following Tappi T22om02 and Tappi UM250. Sodium content <0.1% in all samples. | ||||
| KraftSoft 1, 93% | Pine (softwood) | —/FCBA (France) | Smurfit Kappa, Facture (France) | Kraft, CO2 precipitation |
| KraftSoft 2, 96% | Pine (softwood) | —/Centre technique du papier (France) | Smurfit Kappa, Facture (France) | Kraft, CO2 precipitation up to pH 9 |
| KraftSoft 3, 93% | Resinous mix (softwood) | BIOPIVA™ 100/UPM Biochemicals, (Finlande) | DOMTAR-plymouth factory (US) | Kraft, CO2 precipitation and mineral acidification |
| KraftHard, 96% | KraftHard (hardwood) | Eucalyptus kraft lignin/Fibria (Brazil) | Jacarei factory (Brazil) | Kraft, no detail on lignin extraction |
| OrganoSoft, >94% | Pine (softwood) | — | Kindly supplied by LRGP laboratory | Organosolv, (EtOH/water), precipitation by water addition |
| OrganoHerb, 86% | Wheat straw (herbaceous) | BioLignin™/CIMV (France) | Pomacle (France) | Organosolv, (acetic or formic acid/water) |
| SodaHerb, 92% | Wheat straw (herbaceous) | Protobind 1000/green value (US) | India | Soda, no detail on lignin extraction |
All other chemicals are commercially available and were purchased from Sigma-Aldrich or Alfa Aesar. All solvents were used as supplied without further purification. TiO2 was kindly provided by Evonik (Aerolyst 7711 extrudates 1 × 4 mm).
Catalyst synthesis
The CuO/TiO2 (Cu 5% wt) catalyst was prepared by incipient wet impregnation using an aqueous solution of Cu(NO3)2·5H2O, 98% (6.9 g L−1) according to a previously reported procedure.55 In short, an aqueous copper nitrate solution was used to impregnate TiO2 extrudates. After 4 h of impregnation, the catalyst was dried overnight at 110 °C and calcined for 5 h at 550 °C (5 °C min−1) in air. After calcination, the catalyst pellets were crushed and sieved. The 90–200 μm fraction was used in all catalytic experiments.Typical catalytic oxidation reaction
The reaction was carried out in a 300 mL batch reactor (Vinci Technologies). 150 mL of an aqueous basic (NaOH, >98%, 10 g L−1) solution of lignin (5 g L−1 on the dry basis, the mass varies depending on the purity and moisture content) was added to the reactor together with the catalyst if present (5% wt Cu/lignin). Under these conditions, lignin is completely soluble.The reactor was first flushed with N2, and then heated up to 150 °C under stirring (1000 rpm). Once the temperature was reached, the reactor was pressurized with 20 bar of synthetic air and the mechanical stirring was adjusted at 1800 rpm. The completion of these operations defines the start time of the reaction. A control valve was used throughout the reaction time to keep the pressure constant.
At the end of the reaction, the reactor was cooled to 15 °C using an ice bath and depressurized.
Fractionation of products
At the end of the reaction, the reaction mixture was filtered to recover the solid catalyst. Various post-reaction treatments were then carried out to separate the different reaction products (unreacted lignin, aromatic and aliphatic products) in order to develop qualitative and quantitative analyses. The fractionation protocol has been described in detail in a previous report (Fig. 1).55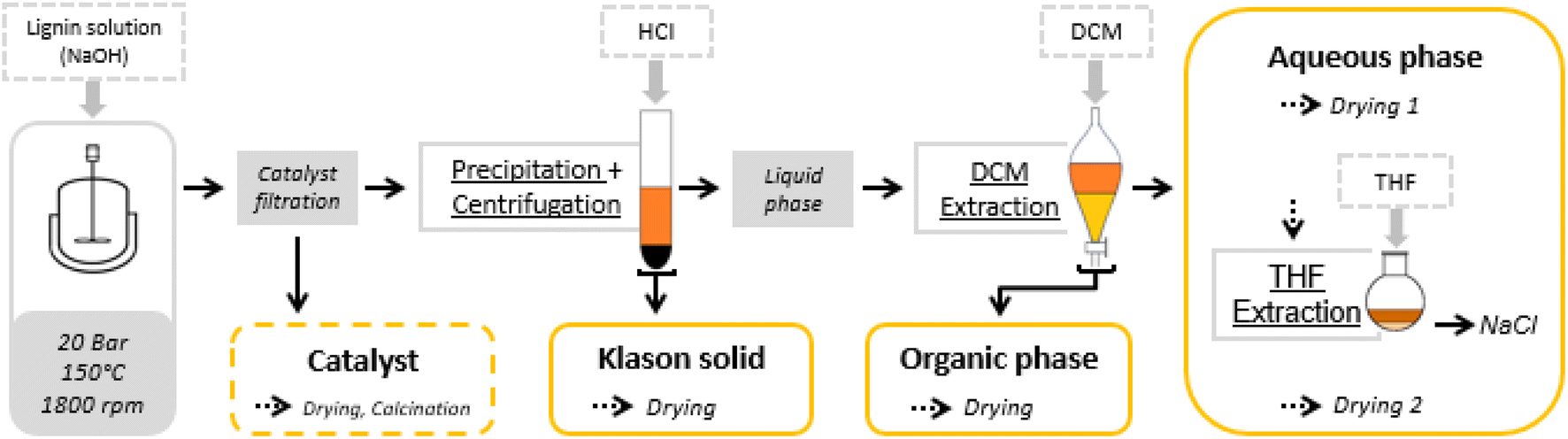 | ||
| Fig. 1 Oxidation test and fractionation protocol.55 | ||
Briefly, 100 mL of the reaction mixture was filtered to remove the solid catalyst. It was then acidified by adding a dilute solution of hydrochloric acid (10% wt/wt HCl) to pH 1 (∼15 mL) to cause the precipitation of the non-depolymerized lignin. The precipitate was separated by centrifugation at 4000 rpm for 20 min, washed with dilute hydrochloric acid and then with water. It was dried at 60 °C for 9 hours under vacuum (5 × 10−2 mmHg). The solid obtained was called Klason phase.
The supernatant liquid was extracted with dichloromethane (200 mL). The dichloromethane fraction was concentrated in a rotavapor and dried under vacuum (5 × 10−2 mmHg) at room temperature for 30 minutes. The residue obtained was called the organic phase, which consisted mainly of non-polar compounds (phenolic compounds and oligomers).
The remaining aqueous fraction was concentrated in the rotavapor and then dried at 60 °C under vacuum (5 × 10−2 mmHg) for 1 hour. The residue obtained contained the polar compounds (aliphatic acids), and NaCl formed during the acidification. It was taken up with THF and then filtered through a 0.4 μm filter. The THF fraction was then concentrated in the rotavapor and dried under vacuum (5 × 10−2 mmHg) at 60 °C for 1 hour giving a residue called the aqueous phase.
Characterization techniques
A calibration curve, obtained with polystyrene standards (Polymer Standards Service, Mainz, Germany) in the range of 500 to 4.2 × 106 g mol−1, was used to calculate the molecular weight distribution of the samples. The molecular weight was determined from the RID chromatogram and the PS calibration curve. Data were acquired and processed using OmniSEC 5.12 software. The lignins were dissolved in THF prior to analysis.
Results
Characterization of lignins
The lignins studied in this work were characterized using different complementary analytical techniques. The aim was to evaluate the influence of lignin structure and composition on the performance of their conversion into aromatic compounds produced upon catalytic aerobic depolymerization in alkaline media.Elemental analysis (Table 2) showed similar results for the different lignins, except for the Kraftsoft 2 which had a slightly higher carbon content than the others. The elemental compositions are close to those corresponding to the phenylpropane units (ca. C: 67.16%; H: 6.71%; O: 26.13%), which are known to form the main fraction of lignin, although some lignins have different proportions of other phenolic compounds, such as hydroxycinnamates or p-hydroxybenzoate3 and different proportions of condensate units.61
| % wt | C | H | O | N | S |
|---|---|---|---|---|---|
| KraftSoft 1 | 61.0 | 5.9 | 28.1 | 0.2 | 2.2 |
| KraftSoft 2 | 71.0 | 5.6 | 21.3 | <0.1 | 2.0 |
| KraftSoft 3 | 65.5 | 5.6 | 21.1 | <0.1 | 2.1 |
| KraftHard | 63.6 | 5.3 | 26.4 | 0.1 | 2.4 |
| OrganoSoft | 66.1 | 6.1 | 27.7 | 0.2 | — |
| OrganoHerb | 61.6 | 5.6 | 28.0 | 2.2 | 0.2 |
| SodaHerb | 64.6 | 5.3 | 25.5 | 0.5 | 0.7 |
Kraft lignin (KraftSoft 1, KraftSoft 2, KraftSoft 3 and KraftHard) has a low sulfur content not observed in other lignins, which is due to Na2S used in the process to increase the delignification efficiency.5
The TGA analysis (Fig. S1†) showed that the lignins burned before 500 °C with similar behaviors as a function of the temperature. The two exceptions were observed with the KraftSoft 3 lignin due to the higher moisture content, and the OrganoHerb lignin, which seems to burn at a lower temperature, around 440 °C which could be probably related to the higher residual saccharide content (i.e., 4.8% versus < 2% for the other lignins).
TGA also provides moisture and ash contents for each lignin (Table 3). The moisture contents were similar in all cases, around 5% wt, with the exception of KraftSoft 3, which has a significantly higher content of 19% w/w. Moisture was measured frequently for each lignin and showed constant values over time.
| % wt | Water | Ash |
|---|---|---|
| KraftSoft 1 (ref. 55) | 4.5 | 2.6 |
| KraftSoft 2 | 3.9 | 2.0 |
| KraftSoft 3 | 19.0 | 0.9 |
| KraftHard | 3.9 | 1.8 |
| OrganoSoft | 2.5 | 0.4 |
| OrganoHerb | 5.8 | 1.6 |
| SodaHerb | 4.3 | 2.1 |
FTIR-ATR was used to qualitatively analyze the structure of the different lignins (Fig. 2, S2 and Table S1†). The bands at 1420, 1504, and 1588 cm−1 are assigned to vibrations related to the aromatic backbone. The band at 1504 cm−1 is generally considered to be a “pure” band characteristic of the aromatic moieties in the biopolymer. According to the literature, for softwood lignin (i.e., Kraftsoft 1, Kraftsoft 2, Kraftsoft 3 and Organosoft) it is higher in intensity than the band at 1452 cm−1, which is not the case for the other lignins. Besides this difference related to the botanical origin,62 softwood lignins show the bands at 1264, 1206, and 1028 cm−1 (resp. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching; C–C and C–O stretching; aromatic C–H deformation) characteristic of guaiacyl units, while others show additional bands at 1329 and 1120 cm−1 (resp. aromatic C–H deformation S-band; CO stretching) corresponding to syringyl units. Bands characteristic of p-hydroxyl units, generally at ca. 1155 cm−1, are not clearly observed. Bands at 1694 cm−1 correspond to unconjugated carbonyl groups and those at 1452 are assigned to C–H stretching in CH2 and CH3 alkyl moieties. Bands at 852 and 814 cm−1 (G units) and 834 cm−1 (S units) are assigned to aromatic C–H out-of-plane deformations. Finally, all of them present bands corresponding to the different aliphatic C–H bonds at 2934 and 2840 cm−1 and the band at 3360 cm−1 is assigned to OH phenolic stretching.
O stretching; C–C and C–O stretching; aromatic C–H deformation) characteristic of guaiacyl units, while others show additional bands at 1329 and 1120 cm−1 (resp. aromatic C–H deformation S-band; CO stretching) corresponding to syringyl units. Bands characteristic of p-hydroxyl units, generally at ca. 1155 cm−1, are not clearly observed. Bands at 1694 cm−1 correspond to unconjugated carbonyl groups and those at 1452 are assigned to C–H stretching in CH2 and CH3 alkyl moieties. Bands at 852 and 814 cm−1 (G units) and 834 cm−1 (S units) are assigned to aromatic C–H out-of-plane deformations. Finally, all of them present bands corresponding to the different aliphatic C–H bonds at 2934 and 2840 cm−1 and the band at 3360 cm−1 is assigned to OH phenolic stretching.
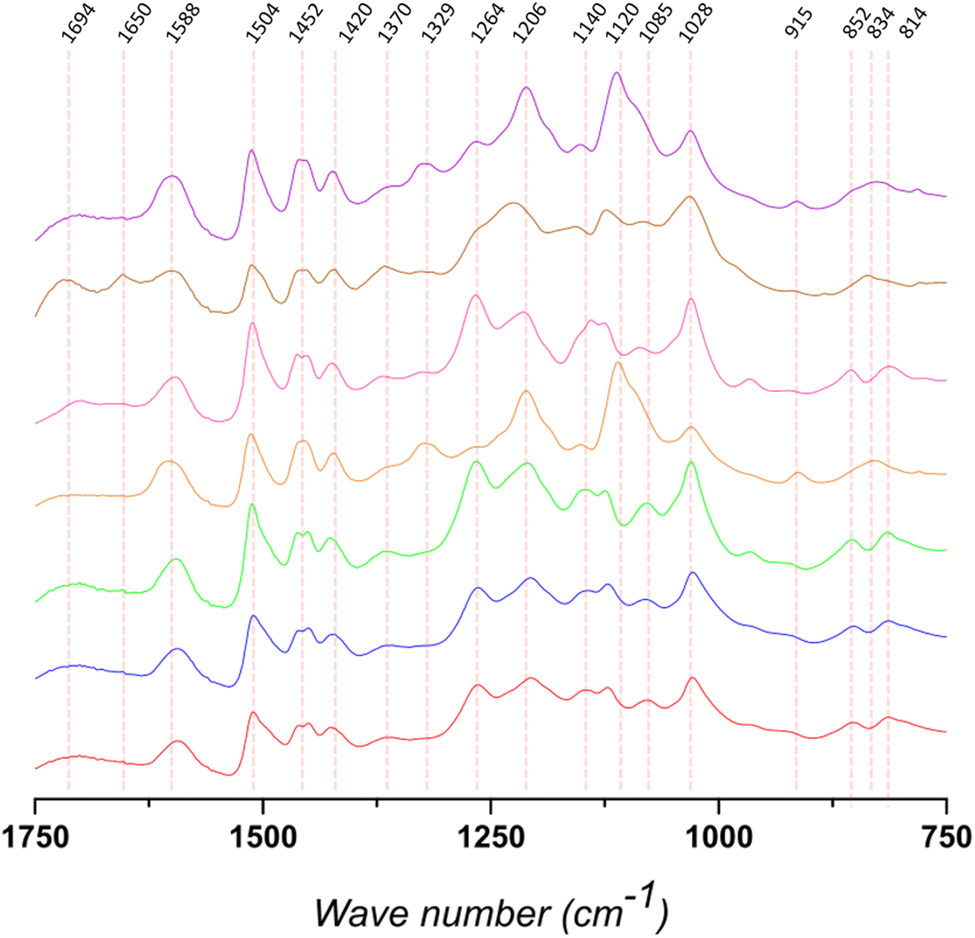 | ||
| Fig. 2 Extension (1800–750 cm−1) of FTIR-ATR spectra of the different lignins: KraftSoft 1 (red), KraftSoft 2 (blue), KraftSoft 3 (green), KraftHard (orange), OrganoSoft (pink), OrganoHerb (brown) and SodaHerb (violet). | ||
Remarkably, similar spectra are also observed for the organosolv pine lignin (OrganoSoft) and the kraft pine lignin (Kraftsoft 1 or 2), showing that the pulping method did not induce remarkable structural changes in the polymeric lignin structure. However, we cannot exclude that both pulping methods resulted in similar structural changes in the extracted lignin or that these differences cannot be observed by FTIR-ATR analysis.
KraftHard and SodaHerb, which have very similar FTIR spectra despite their different botanical origins (hardwood and wheat straw), show the presence of syringyl units (i.e., 1329 and 1120 cm−1). OrganoHerb, although it is also a wheat straw lignin, presents a very different FTIR spectra related to the modification carried out by the organosolv pulping method used,63 particularly due to the incorporation of alkyl moieties as esters (i.e., 1694, 1028 cm−1).64 These results are consistent with those obtained from the TGA analyses and show a significant degree of modification in OrganoHerb. Thus, the TGA data show that OrganoHerb could be fully oxidized under softer conditions, which could be related either to an originally highly oxidized structure, which is discarded in elemental analyses, or to a lower molecular weight. In fact, a lower molecular weight would not be expected from a lignin produced by an organosolv process, since it is generally carried out at lower temperatures and in the absence of or strong nucleophilic agents, which strongly limit modifications in the lignin structure.65 However, both TGA and FTIR results support chemical modifications produced during organosolv pulping. The various aromatic bands are of lower intensity than in the case of SodaHerb. On the other hand, the bands at 1694 and 1650 cm−1 are slightly higher, which is associated with a higher presence of esters (and/or carboxylic acids) and benzoquinones. Such modifications are not directly related to the organosolv method, since OrganoSoft did not show the same behavior, but rather to the conditions used by the supplier.
Semi-quantitative 1H and quantitative 13C NMR analyses (Tables S2, S3, Fig. S3 and S4†) were performed to obtain information on the composition of lignin in terms of aliphatic and aromatic content.
1H NMR analyses showed that most of the protons are present in the aliphatic structures (incl. oxygenated aliphatic moieties) (δppm: 0.5–5.8 ppm; 42.8–48.5%), the rest being mainly associated with aromatic and phenolic protons (δppm: 6.1–9.2 ppm; 6.8–14.3%). Low levels of protons corresponding to aldehydes and carboxylic acids were observed. Of note is the higher content of aliphatic protons in SodaHerb, OrganoHerb, and Organosoft lignin, especially in OrganoHerb lignin, which indicates condensation reactions with organic solvents during biomass fractionation. Consequently, the aromatic proton content is significantly lower, which is consistent with FTIR-ATR analyses. According to previous report,31,32 all protons are observed and quantified by 1H NMR and the data correlate with those obtained from elemental analyses.
13C NMR (Table S3†) allowed the identification of between 76% and 29% of the carbon content when compared to elemental analyses. This fraction is particularly low for wheat straw lignin. The difference in the identified carbon content of KraftSoft 1 and KraftSoft 2 lignin, although they are similar materials, is noteworthy. In addition, KraftSoft 2 has a higher aromatic and lower aliphatic carbon content. These differences could be related to the extraction conditions used by the supplier to produce the pulp, or to variations in the wood used.
In terms of quantification, most of the carbons are aromatics (δppm: 102–162 ppm), with a percentage of 60 to 72%. The remainder is mainly linked to aliphatic carbons (δppm: 10–90 ppm). OrganoHerb is an exception among the lignins studied, since it has a lower proportion of aromatic carbon (18.5 mmol C glignin−1), compared to the other lignins (30.5–42.7 mmol C glignin−1), which is in agreement with the previous analyses (1H NMR and FTIR).
Quantitative 31P NMR analyses were carried out after phosphorylation of the free hydroxyl groups, allowing the analysis of the different structural groups present in the studied lignin, especially the condensate units (Table S4 and Fig. S5†).
The data showed that 37–52% of the oxygen present in lignin could be quantified by 31P NMR as free hydroxyl groups, the rest being bound either to ether bonds or carbonyl moieties (i.e., CO in COR, CO–OH). OrganoHerb showed the lowest free hydroxyl content by this method, which is consistent with FTIR analyses showing the incorporation of alkyl moieties, probably as ethers or esters, due to the use of a mixture of acetic and formic acid during the extraction process.
In terms of quantification, as expected, the amount of hydroxyls attached to the guaiacyl unit, is higher in KraftSoft lignin (1.77 to 2.07 mmol O glignin−1) than in other lignins (0.74–0.93 mmol O glignin−1), in agreement with observations made in FTIR-ATR analyses. It is also noteworthy that the proportion of hydroxyls in guaiacyl units is similar in hardwood and herbaceous lignin (KraftHard, SodaHerb and OrganoHerb; resp. 0.89; 0.93 and 0.74 mmol O glignin−1, resp.).
Determination of the proportion of hydroxyls present in syringyl or condensate units is complicated by the overlap of the corresponding regions in this analysis (140.2–144.3 ppm). Therefore, the sum of both contributions was analysed. It is higher in KraftHard (3.44 mmol O glignin−1) than in all KraftSoft (1.80–2.10 mmol O glignin−1) lignin according to the generally accepted lignin structures in both woods. The proportion of free hydroxyl linked to syringyl units is lower in the case of the wheat straw OrganoHerb (1.29 mmol O glignin−1) and SodaHerb (2.38 mmol O glignin−1) lignin. These observations are consistent with the lower proportion of phenolic protons observed in the 1H NMR. However, in the FTIR analyses the conclusion is a little different as it showed that KraftHard and SodaHerb lignin have similar syringyl proportions, which could mean that KraftHard has an important fraction of condensed units. Similarly, KraftSoft 1, KraftSoft 2, and KraftSoft 3, with a free hydroxyl amount of 1.80–2.10 mmol O glignin−1 in this spectral region, showed an important amount of condensed unit associated with the kraft extraction process. OrganoSoft lignin shows a lower value of free hydroxyls associated with condensed units than that observed for the Kraftsoft 1 lignin despite the use of the same wood chips for lignin extraction, which may be related to a lower degree of degradation during the organosolv pulping process reducing the formation of condensate units. Alternatively, the formation of esters during the process, masking some of the OH groups initially present, could not be excluded.
Finally, all lignins were further characterized by recording HSQC NMR spectra that help to evaluate the different structures and inter-unit bonds present in lignin (Fig. S6 and S7†).66 This information is important, as the proportion of the β-O-4 bonds in the lignin matrix is strongly related to the yields of aromatics that could be obtained by its oxidative depolymerisation.67
The HSQC NMR spectra are generally decomposed in three regions: the aliphatic region (δH/δC: 0.5–5.0/5–50 ppm), the oxygenated aliphatic region (δH/δC: 2.5–6/50–105 ppm) and the aromatic region (δH/δC: 4.9–8.0/90–160 ppm). Only the last two regions are informative and will be analyzed. The different spectra of lignin are provided in the ESI (Fig. S6–S9),† and two examples are shown in Fig. 3.
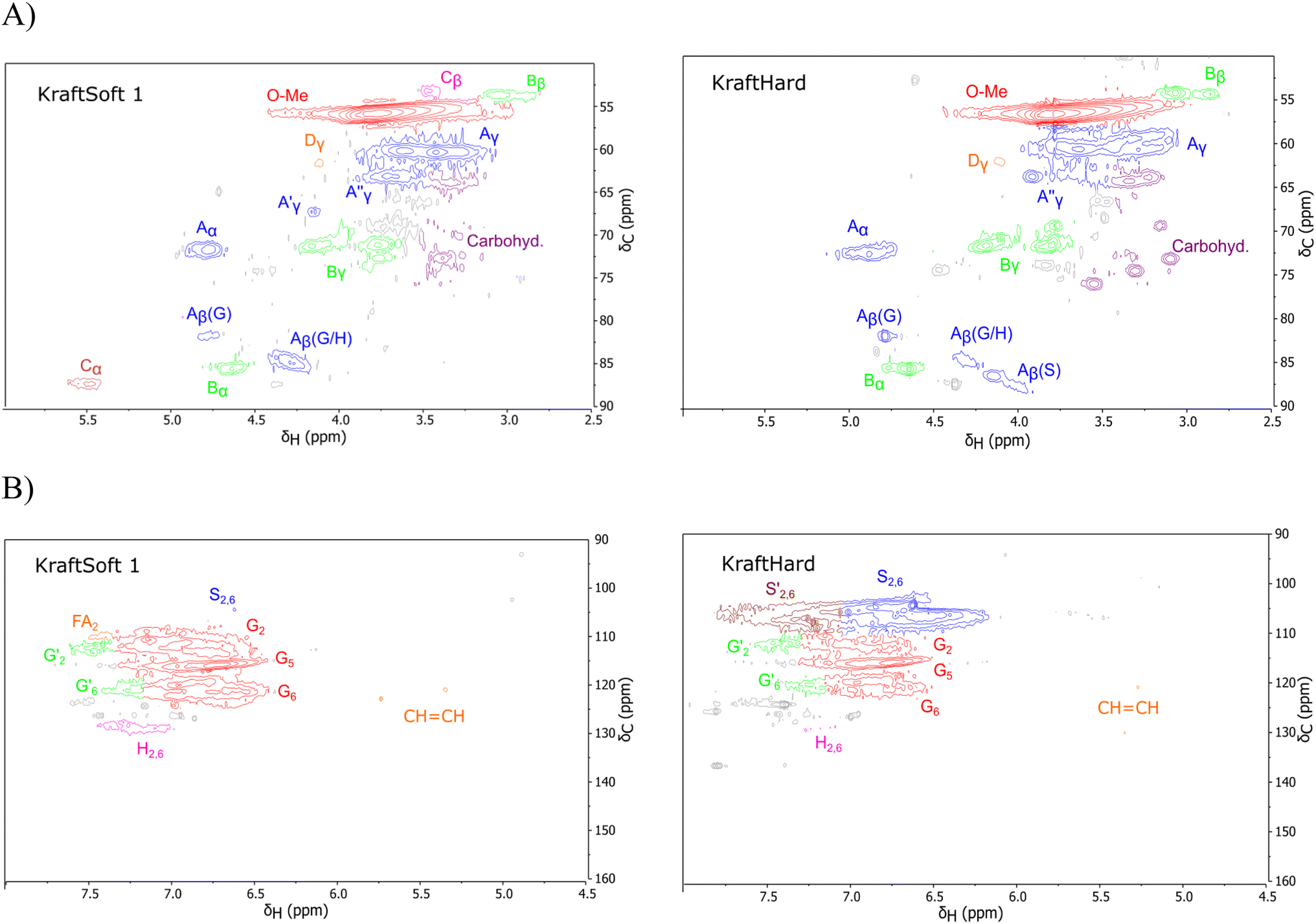 | ||
| Fig. 3 Example of spectra obtained by HSQC analysis of the initial lignin. (A) Oxygenated aliphatic region, (B) aromatic region. | ||
The oxygenated aliphatic region (Fig. S8†) of all lignins shows several similarities. Those of the kraft pine lignin, namely KraftSoft 1, KraftSoft 2 and KraftSoft 3 are very similar due to the botanical origin of the woods used for their production. The same conclusion applies to Organosoft lignin, which is produced from the same wood chips as Kraftsoft 1. KraftHard and SodaHerb show few differences related to the presence of syringyl moieties.
More specifically, all isolated lignins show the presence of carbohydrates (several correlations, depending on the lignin, in general, δH/δC: 3.09–3.81/63.7–79.4 ppm). The presence of carbohydrates may limit the valorization due to a higher stabilization of C–C bonds with carbohydrates in basic media.68 Next, all lignins show correlations corresponding to methoxy moieties (ca. δH/δC: 3.74/56.9 ppm) and the correlation corresponding to the inter-unit bonds, namely β-O-4 (A) (ca. δH/δC: Aα: 4.76/72.1 ppm; Aβ (G): 4.76/81.8 ppm; Aβ (G/H): 4.28/84.8 ppm; and Aβ (S): 4.13/86.6 ppm for SodaHerb and KraftHard lignin; Aγ: 3.44/60.4 ppm; 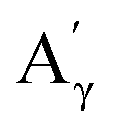 : 4.14/67.6 ppm;
: 4.14/67.6 ppm; 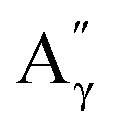 : 3.70/63.58 ppm), β–β (B) (ca. δH/δC: Bα: 4.64/85.6 ppm; Bβ: 3.02/54.4 ppm; Bγ: 3.78, 4.16/71.6 ppm) in resorcinol moieties and β-5 (C) (ca. δH/δC: Cα: 5.49/87.8 ppm; Cβ: 3.45/53.8 ppm) in phenylcoumaran moieties, mainly in softwood lignin. Correlations corresponding to cinnamic alcohol are also observed (D) (ca. δH/δC: Dγ: 4.10/62.1 ppm). Moieties containing β–β and β-5 linkages can be considered as condensate structures, possibly present in the native lignin or formed during the pulping process by the formation of C–C bonds after the degradation of C–O–C linkages, which have a lower bond energy. The formation of β-5 bonds as inter-unit linkages requires the presence of a free carbon at the position 5 in the aromatic ring, which limits the formation of these structures from lignin with syringyl units, such as from hardwood or herbaceous species. The chemical structure of all the types detected by the HSQC-NMR analysis is shown in Fig. S7.†
: 3.70/63.58 ppm), β–β (B) (ca. δH/δC: Bα: 4.64/85.6 ppm; Bβ: 3.02/54.4 ppm; Bγ: 3.78, 4.16/71.6 ppm) in resorcinol moieties and β-5 (C) (ca. δH/δC: Cα: 5.49/87.8 ppm; Cβ: 3.45/53.8 ppm) in phenylcoumaran moieties, mainly in softwood lignin. Correlations corresponding to cinnamic alcohol are also observed (D) (ca. δH/δC: Dγ: 4.10/62.1 ppm). Moieties containing β–β and β-5 linkages can be considered as condensate structures, possibly present in the native lignin or formed during the pulping process by the formation of C–C bonds after the degradation of C–O–C linkages, which have a lower bond energy. The formation of β-5 bonds as inter-unit linkages requires the presence of a free carbon at the position 5 in the aromatic ring, which limits the formation of these structures from lignin with syringyl units, such as from hardwood or herbaceous species. The chemical structure of all the types detected by the HSQC-NMR analysis is shown in Fig. S7.†
The aromatic region (Fig. S9†) shows the characteristic correlations corresponding to guaiacyl units (G) (ca. δH/δC: G2: 6.87/112.6 ppm, G5: 6.74/115.77 ppm, G6: 6.75/120.27 ppm) and those of p-hydroxyphenyl units (H) (ca. δH/δC: H2,6: 7.21/128.85 ppm), and syringyl units (S) (ca. δH/δC: S2,6: 6.68/105.8 ppm) and Cα-oxidized syringyl units (ca. δH/δC: 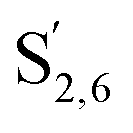 : 7.26/106.9 ppm) for KraftHard and SodaHerb lignins in particular, and p-hydroxyphenyl units (H) (ca. δH/δC: H2,6: 7.16/128.9 ppm). Correlations corresponding to ferulic derivatives were also observed (ca. δH/δC: FA2: 7.42/109.5 ppm, FA6: 7.17/124.1 ppm, FA7: ppm, 7.50/114.6 ppm, p-CE2.6: 7.49/130.4 ppm) and (CHCH) (δH/δC: 5.35/120.86, 5.73/122.88 ppm). Correlations corresponding to guaiacyl units substituted by carbonyl moieties (G′) are also observed (
: 7.26/106.9 ppm) for KraftHard and SodaHerb lignins in particular, and p-hydroxyphenyl units (H) (ca. δH/δC: H2,6: 7.16/128.9 ppm). Correlations corresponding to ferulic derivatives were also observed (ca. δH/δC: FA2: 7.42/109.5 ppm, FA6: 7.17/124.1 ppm, FA7: ppm, 7.50/114.6 ppm, p-CE2.6: 7.49/130.4 ppm) and (CHCH) (δH/δC: 5.35/120.86, 5.73/122.88 ppm). Correlations corresponding to guaiacyl units substituted by carbonyl moieties (G′) are also observed (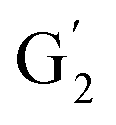 : δH/δC: 7.47/111.89 ppm,
: δH/δC: 7.47/111.89 ppm, 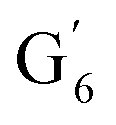 : δH/δC: 7.27/121.12 ppm).
: δH/δC: 7.27/121.12 ppm).
In addition, HSQC-NMR spectra were used to quantify the different inter-unit linkages in a semi-quantitative manner by using the method developed by Wen et al.66 (Table 4). The data show that softwood lignins have a higher proportion of β-O-4 linkages compared to KraftHard or SodaHerb lignin, which makes them better candidates as raw materials for the production of aromatic aldehydes since these linkages can lead to the production of aromatic compounds.67 On the other hand, comparison of data obtained for OrganoSoft (i.e., 10.3%β-O-4) and Kraftsoft 2 (i.e., 9.8%β-O-4) lignin produced from the same wood chips suggests that the Organosoft process retains β-O-4 linkages to some extent compared to the kraft process. These results are consistent with the higher aromatic content observed by the other NMR analyses. With the exception of SodaHerb lignin, which has a low β–β content, little difference is observed for the other lignins. KraftHard and KraftSoft 1 have the highest β–β content, which is consistent with data obtained from 31P NMR analysis for the quantification of condensate units. Finally, hardwood and herbaceous lignin do not present a significant amount of β-5 linkages, which is generally low except for the OrganoSoft lignin.
| %Linkage/C9 unit | KraftSoft 1 | KraftSoft 2 | KraftSoft 3 | KraftHard | OrganoSoft | SodaHerb |
|---|---|---|---|---|---|---|
| β-O-4 | 6.7% | 9.8% | 6.7% | 5.8% | 10.3% | 3.6% |
| β–β | 5.8% | 4.1% | 3.2% | 5.0% | 4.0% | 2.7% |
| β-5 | 2.4% | 3.5% | 2.0% | 0.5% | 13.1% | 0.7% |
Table 5 shows the quantification of the different phenylpropane units carried out using the aromatic region of the HSQC NMR analyses. According to the FTIR analyses, softwood lignin (KraftSoft 1, KraftSoft 2, KraftSoft 3 and OrganoSoft) has a very low proportion of syringyl units (1–6%), in contrast to KraftHard and SodaHerb, where this is the main phenylpropane unit with, respectively, 77% and 55%. The content of p-hydroxyphenyl units is higher in the case of the softwood lignin and SodaHerb (i.e., 4–8%), whereas this unit is almost absent in KraftHard (i.e., 2%).
| % Of the total | KraftSoft 1 | KraftSoft 2 | KraftSoft 3 | KraftHard | OrganoSoft | SodaHerb |
|---|---|---|---|---|---|---|
| Guaiacyl units (IG2) | 89% | 87% | 90% | 21% | 91% | 35% |
| Syringyl units (IS2,6) | 3% | 6% | 1% | 77% | 5% | 59% |
| p-Hydroxyphenyl units (IH2,6) | 7% | 7% | 8% | 2% | 4% | 5% |
The data for the SEC analyses are given in Table 6. The results of average molecular weight and the degree of polymerization are significantly higher for softwood lignin (i.e., KraftSoft 2, KraftSoft 3, OrganoSoft) (except for KraftSoft 1, which does not correlate) than for KraftHard and SodaHerb lignin, a situation already reported.69 These results may be related to condensation reactions during the delignification process of softwood lignin, which increase the molecular weight. Condensation reactions generally produce a significant proportion of 5–5 or β-5 linkages from guaiacyl units, and are therefore predominant with softwood lignin. Thus, SEC analyses correlate with HSQC NMR analyses and semi-quantitative determination of the proportion of linkages between units (Table 4). It appears from the SEC analyses that the botanical origin of wood used to produce lignin determines its molecular weight, being less affected by the pulping method used.
| Lignin | M n (g mol−1) | M w (g mol−1) | M w/Mn | Polymerization degrees aver. (Mw/Mmonomer) |
|---|---|---|---|---|
| KraftSoft 1 | 745 | 1505 | 2.0 | 9.1 |
| KraftSoft 2 | 1133 | 2964 | 2.6 | 17.9 |
| KraftSoft 3 | 1262 | 2925 | 2.3 | 17.6 |
| KraftHard | 609 | 1456 | 2.4 | 8.8 |
| OrganoSoft | 1064 | 2405 | 2.3 | 14.5 |
| SodaHerb | 697 | 1503 | 2.2 | 9.1 |
In a previous study55 we showed that the formation of lignin agglomerates in alkali solution could be a limiting factor for the aerobic depolymerisation. Table 7 shows the results of DLS analyses of the different lignins studied in this work. Agglomerates of different sizes are observed depending on the type of lignin, with softwood lignin providing the smallest agglomerates, which could reduce the mass transfer limitations.
| Lignin | Z average (nm) |
|---|---|
| KraftSoft 1 | 71.7 |
| KraftSoft 2 | 115.1 |
| KraftSoft 3 | 153.5 |
| KraftHard | 267.3 |
| OrganoSoft | 70.0 |
| OrganoHerb | 637.1 |
| SodaHerb | 341.6 |
For lignins, agglomeration and solubility under basic conditions are closely related, the smaller the agglomerate the higher the solubility. Zulfiqar et al.70,71 showed that, for polyamides, the aryl ether content can produce a significant increase in solubility. Following these studies, Fig. 4 shows the correlation between the β-O-4 linkage content and the size of the lignin agglomerates under basic conditions, with the highest β-O-4 linkage content resulting in the smallest agglomerates (and thus the highest solubility). With the exception of KraftSoft 1 for which there may be an experimental variation (which may be due to its lower average molecular weight), all lignins showed a strong correlation between the two parameters. Therefore, these results show that the observations made for polyamides also exist well for lignin.
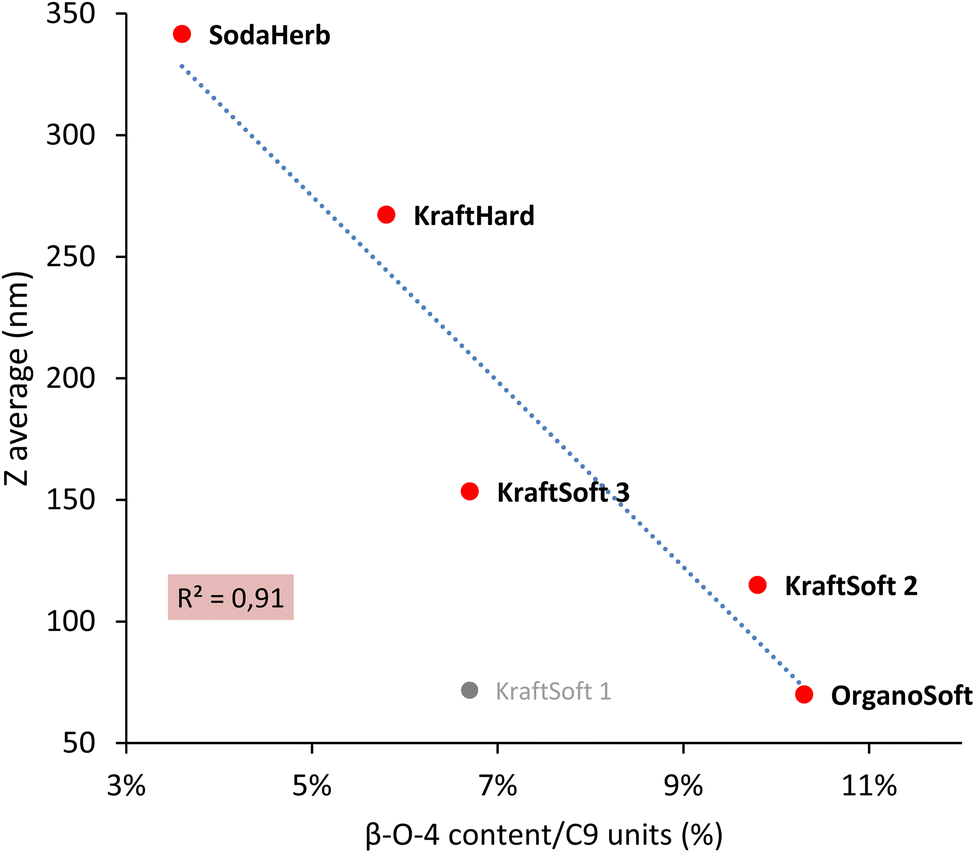 | ||
| Fig. 4 Correlation between the β-O-4 content obtained by HSQC-NMR analysis, and the floc size (Z) obtained by DLS analysis. | ||
Table 8 summarizes some of the analyses carried out on lignin. Herbaceous lignins, i.e., OrganoHerb and SodaHerb lignins, have the lowest content of phenolic hydroxyls and β-O-4 linkages, which resulted in the formation of the largest flakes under basic aqueous conditions, resp. 637 and 342 nm, versus < 270 nm for the other lignins. The molecular weight seems to be mainly influenced by the origin of the wood, since softwood lignin has generally a higher molecular weight value, and thus a higher average degree of polymerization.
| Analysis | 31P-NMR | HSQC-NMR | SEC | DLS |
|---|---|---|---|---|
| Parameter | Phenolic OH (mmol g−1 lignin) | β-O-4 (% linkage/C9 units) | M n (g mol−1) | Z average (nm) |
| KraftSoft 1 | 4.53 | 6.7 | 745 | 71.7 |
| KraftSoft 2 | 3.83 | 9.8 | 1133 | 115.1 |
| KraftSoft 3 | 4.21 | 6.7 | 1262 | 153.5 |
| KraftHard | 4.6 | 5.8 | 609 | 267.3 |
| OrganoSoft | 4.04 | 10.3 | 1064 | 70 |
| OrganoHerb | 2.47 | — | — | 637.1 |
| SodaHerb | 3.66 | 3.6 | 697 | 341.6 |
While our analyses to determine the β-O-4 content of the KraftSoft, OrganoSoft and SodaHerb lignins show a similar value to those reported in the literature,72,73 the obtained phenolic hydroxyl content is significantly higher. Here, the experimental method and standard used can significantly influence the results.
Similarly, the data obtained from SEC analyses differ from those reported in the literature, a situation already pointed out by Constant et al.73 as the experimental method used for the SEC analysis has an important influence on the molecular weight results, limiting the extrapolation of the results to other studies. Nevertheless, the trends observed follow those already reported, i.e., in general softwood lignins have higher molecular weights than hardwood or wheat straw lignins, with the exception of Kraftsoft 1, which differs from the other softwood lignins. A chemical interaction between lignin and SEC column cannot be excluded in some cases.
Non-catalytic oxidation experiments
All reaction experiments were carried out using the reaction conditions previously optimized for softwood kraft lignin (NaOH 10 g L−1, 150 °C, 20 bar of air and 1800 rpm).31,32Fig. 5 shows the mass balance obtained for the reaction with the different lignins. Most of the experiments give a mass balance in the range of ca. 80%, varying from 70 to 93%. As previously reported,55 the mass loss can be explained by the different losses observed during the fractionation process and the gases formed during the oxidation. The main gas formed during aerobic depolymerisation of lignin under basic conditions is carbon dioxide, with small amounts of carbon monoxide and methanol.74 However gas analysis was not performed during our study.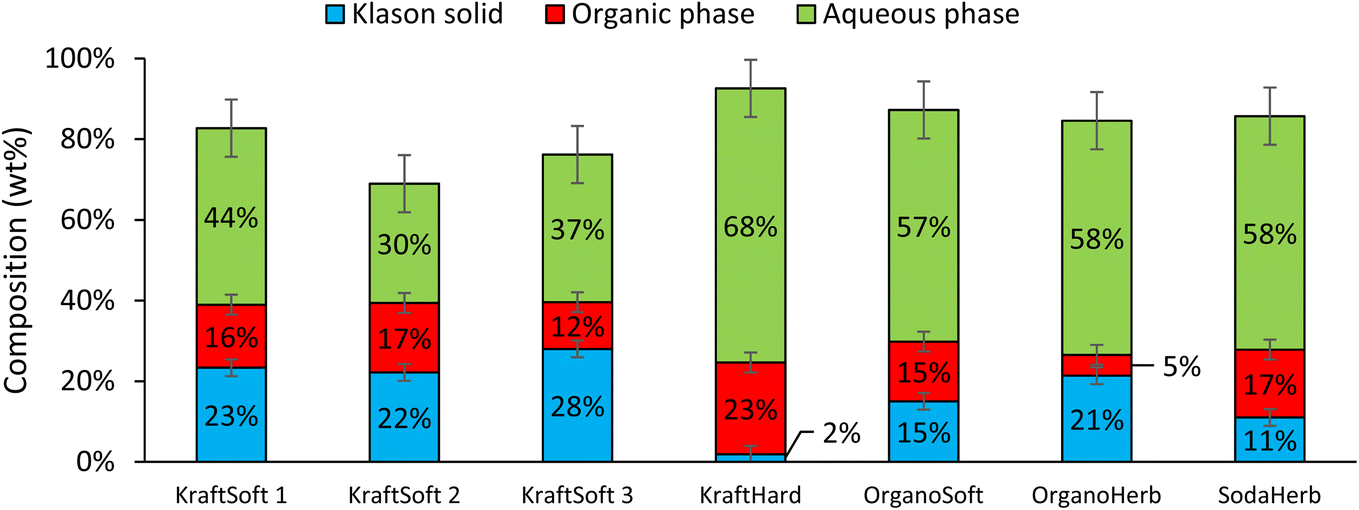 | ||
| Fig. 5 Mass balance of the different fractions (% wt on lignin basis) after non catalytic oxidative depolymerization tests, 150 °C, 1800 rpm, 1 hour, 20 bar of air. | ||
In addition to the mass balance, these experiments also provide the degree of lignin conversion. Lignin conversion was calculated as the percentage of lignin that was not recovered upon acidification of the reaction mixture in the form of a Klason phase, representing the lignin converted to aromatics (organic phase), linear (di)acids (aqueous phase) and gases.
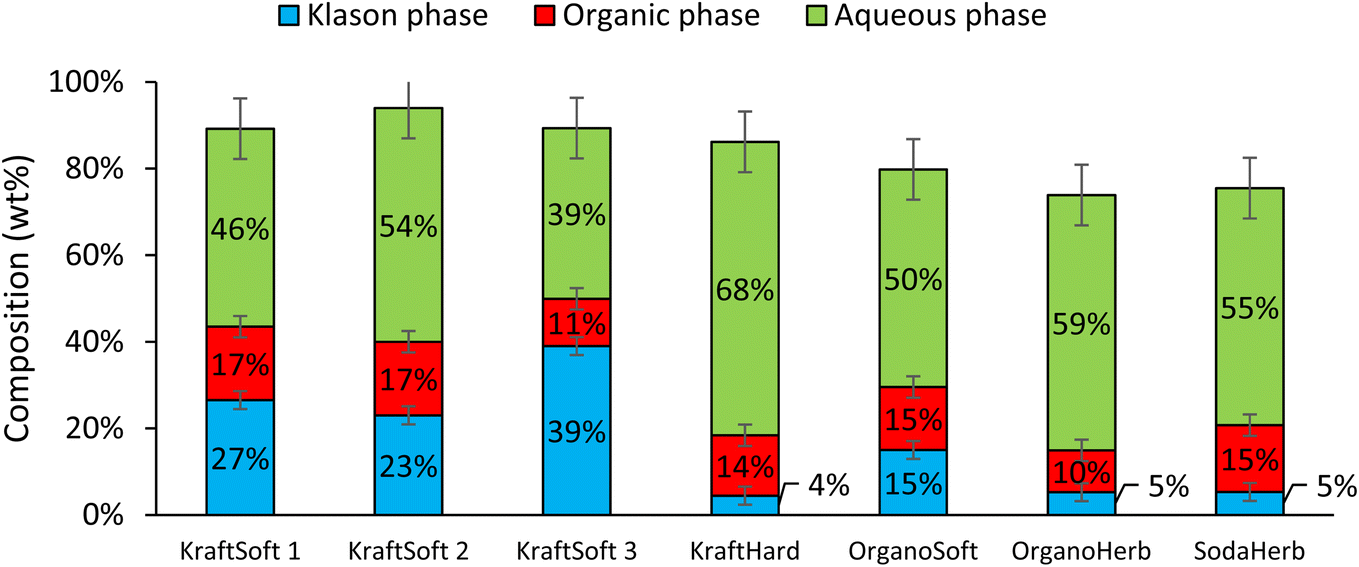
Fig. 5 shows remarkable differences in terms of reactivity for the different lignins. KraftSoft lignin has almost the same amount of Klason phase, corresponding to a very close reactivity, whereas OrganoSoft lignin, despite the same type of wood chips used, has a slightly higher reactivity as the amount of Klason phase is lower. This can be directly related to the lignin extraction process used, as the organosolv process is known to provide lignin with less condensed units despite few structural modifications,31,32 as observed from 31P NMR. Surprisingly, SodaHerb and OrganoHerb lignin give very different results in terms of reactivity, which may be related to the incorporation of alkyl moieties in the former during the extraction process as suggested by FTIR analyses and correlated by 31P NMR studies. KraftHard lignin shows the highest reactivity, which is directly related to the high proportion of syringyl units, which prevents condensation reactions during the aerobic oxidation process. In addition, since this lignin also has the lowest molecular weight, it was expected from the SEC analyses that it would lead to the highest monomer and oligomer production, and more generally, the lignin conversion can be correlated with the molecular weight calculated from the SEC analyses (Fig. 6). As the molecular weight is proportional to the degree of polymerization in polymers, the results show that a higher degree of polymerization implies a longer reaction time to produce the monomeric units.
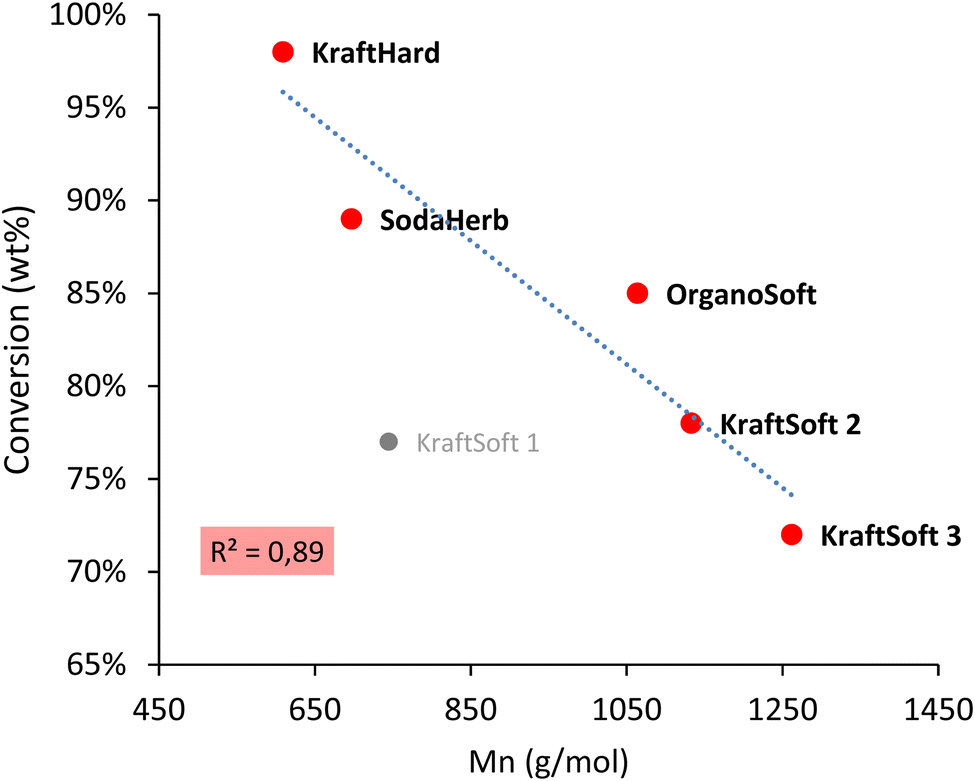 | ||
| Fig. 6 Correlation between the molecular weight (Mn) obtained by the SEC analysis, and conversion obtained by the non-catalytic oxidation reaction. | ||
In all cases, the aqueous phase is the major fraction produced during the reaction. KraftSoft lignins (KraftSoft 1, KraftSoft 2 and KraftSoft 3) provide similar proportions, between 30 and 44%. OrganoSoft lignin, which has a slightly higher reactivity, provides 57% of the aqueous phase, similar to SodaHerb and OrganoHerb lignins; KraftHard lignin, which has the highest reactivity, is also the lignin that resulted in the highest production of the aqueous phase with 68% wt.
In this project, the product of interest, i.e., the aromatic compounds are found in the organic phase. From Fig. 5, it can be seen that KraftHard lignin produces the highest fraction of interest with the highest proportion of the organic phase at 23%, followed by the softwood lignin (ca. 15%). A detailed analysis of this phase is given below.
In addition to the determination of lignin conversions and of the mass balance, we were particularly interested in a detailed analysis of the organic phase containing the aromatic compounds of interest. The nature and the proportion of the different aromatics obtained during aerobic depolymerization of lignins in basic media depend not only on the efficiency of the reaction but also on the botanical origin of the wood used to produce the lignin. The aromatics, at least the major ones, have been quantified by GC analysis of the organic phase and the results are summarized in Fig. 7.
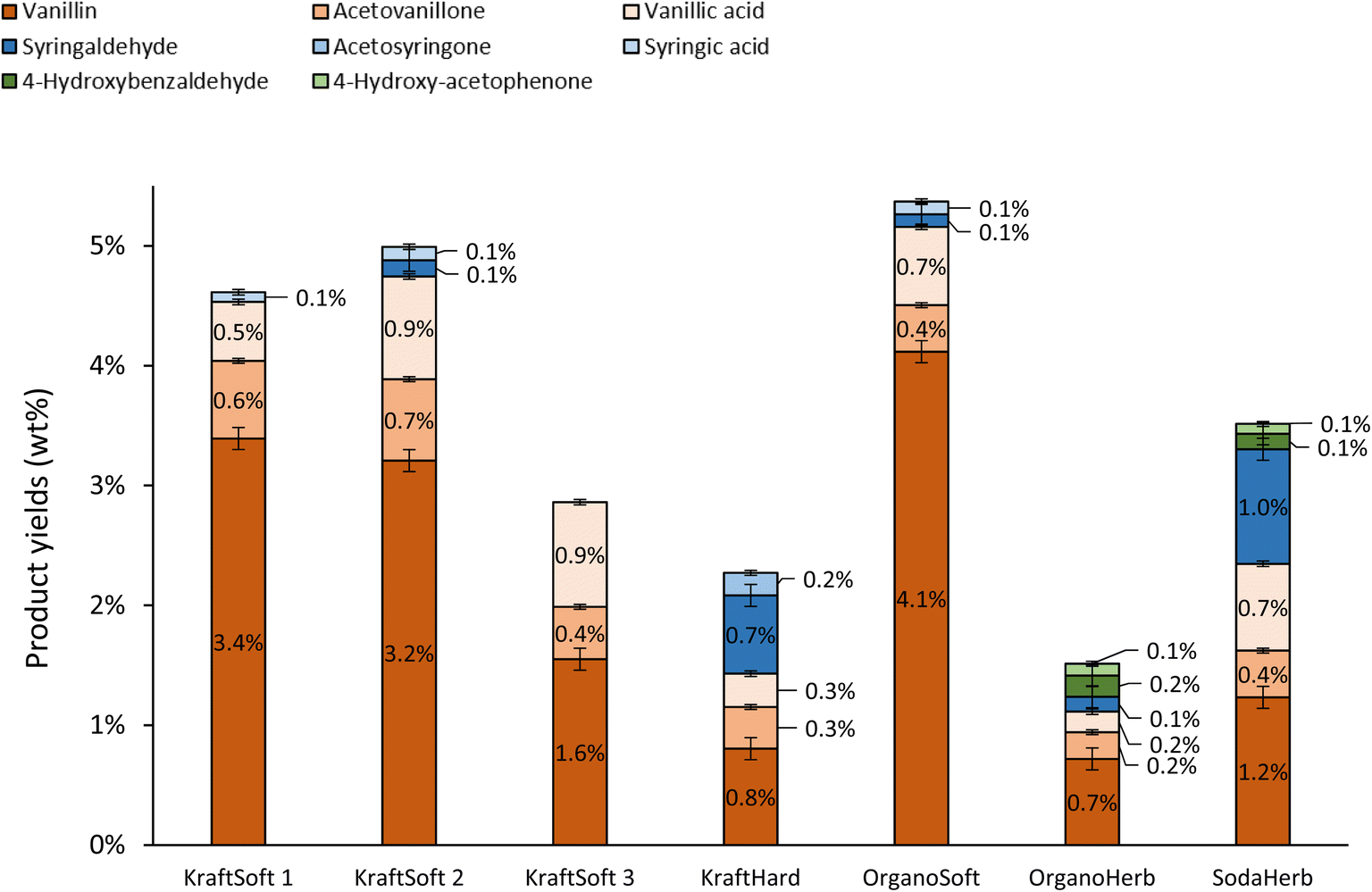 | ||
| Fig. 7 Yields of aromatics in the organic phases, calculated on lignin basis, obtained from the non-catalytic oxidation of the different lignins (150 °C, 20 bar air, 60 minutes). | ||
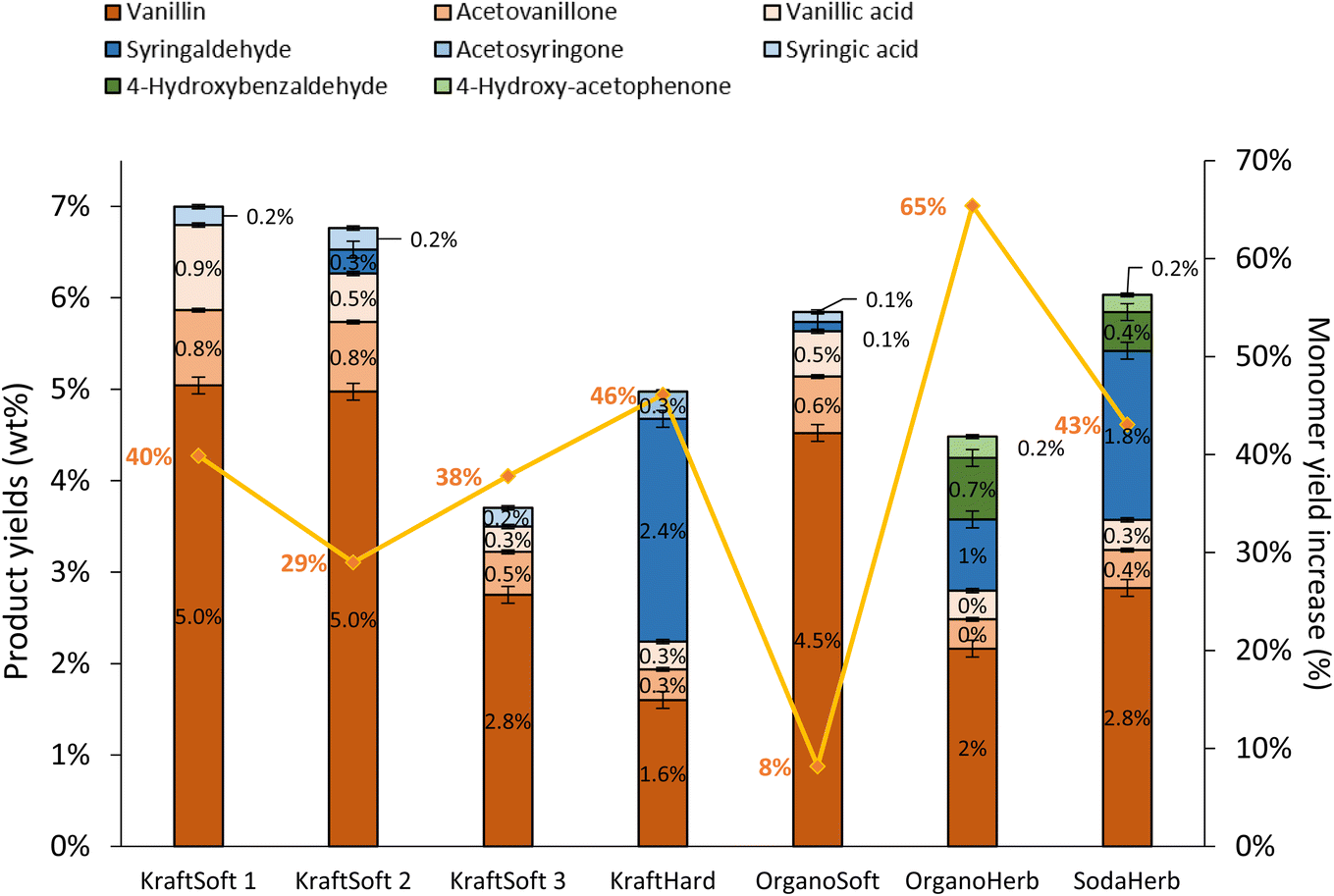
Softwood lignins (i.e., KraftSoft 1, KraftSoft 2, KraftSoft 3 and OrganoSoft), which are mainly composed of guaiacyl units, yield mainly vanillin, acetovanillone, vanillic acid and homovanillic acid as a result of the aerobic oxidation of the guaiacyl derivatives. On the other hand, KraftHard, SodaHerb and OrganoHerb lignins, in addition to products derived from guaiacyl units, also led to a significant amount of syringyl derivatives such as syringaldehyde, syringic acid and acetosyringone. Finally, herbaceous lignin (i.e., OrganoHerb and SodaHerb) also produce compounds derived from the p-hydroxyphenyl units, such as 4-hydroxybenzaldehyde and 4-hydroxyacetophenone.
Irrespective of the lignin studied, aerobic depolymerisation in alkaline media produced aromatic aldehydes as the main products (i.e., vanillin, syringaldehyde and 4-hydroxybenzaldehyde). Several authors have shown that acetovanillone is formed as a by-product of the vanillin formation75 while vanillic acid is formed by a parallel guaiacyl oxidation mechanism.59 We have recently reported routes to explain the formation of these products from kraft lignin, and believe that vanillin, acetovanillone and vanillic acid are derived from guaiacyl units via three different routes.30
The highest yields of aromatic compounds are obtained from softwood lignin, i.e., KraftSoft 1, KraftSoft 2 and OrganoSoft lignin, and are in the range of 4.9 and 5.5% wt. It is noteworthy that OrganoSoft lignin gives interesting yields of aromatic compounds despite the low organic phase content. On the other hand, KraftSoft 3 lignin resulted in lower yields of aromatic compounds, which can be related to the lower amount of organic phase. However, this may also be due to either an unknown proportion of “impurities”, or to the process used to separate it from the black liquor during acidification with sulphuric acid, which is known to induce structural changes and sulfation. Inorganic sulfates are commonly found as mineral impurities.76 KraftHard and SodaHerb produced aromatic compound yields of 2.8 and 3.6% wt resp., as a mixture of syringaldehyde and vanillin approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, corresponding to the initial proportion of guaiacyl and syringyl units. OrganoHerb produced the lowest aromatic yield of 1.6% wt, probably due to the very low amount of the organic phase obtained after the reaction.
:1, corresponding to the initial proportion of guaiacyl and syringyl units. OrganoHerb produced the lowest aromatic yield of 1.6% wt, probably due to the very low amount of the organic phase obtained after the reaction.
Wang et al.67 reported a correlation between the production of aromatic from lignin oxidation and the content of β-O-4 linkages. The main known mechanism to produce these aromatic compounds starts with the oxidation of these β-O-4 linkages, therefore it is logical to correlate the yield of the aromatic compounds with the content of β-O-4 linkages in lignin (Fig. 8). Most lignins show a significant direct correlation between yields of aromatic compounds and β-O-4 content. KraftHard lignin does not follow the trend, which can be related to its significantly higher conversion resulting in a high amount of aqueous phase where the aromatic overoxidation compounds are found. KraftSoft 3 did not fire as well, for the reason explained above. Finally, the β-O-4 content of the OrganoHerb lignin could not be quantified due to the insolubility in the solvent used for the NMR experiments.
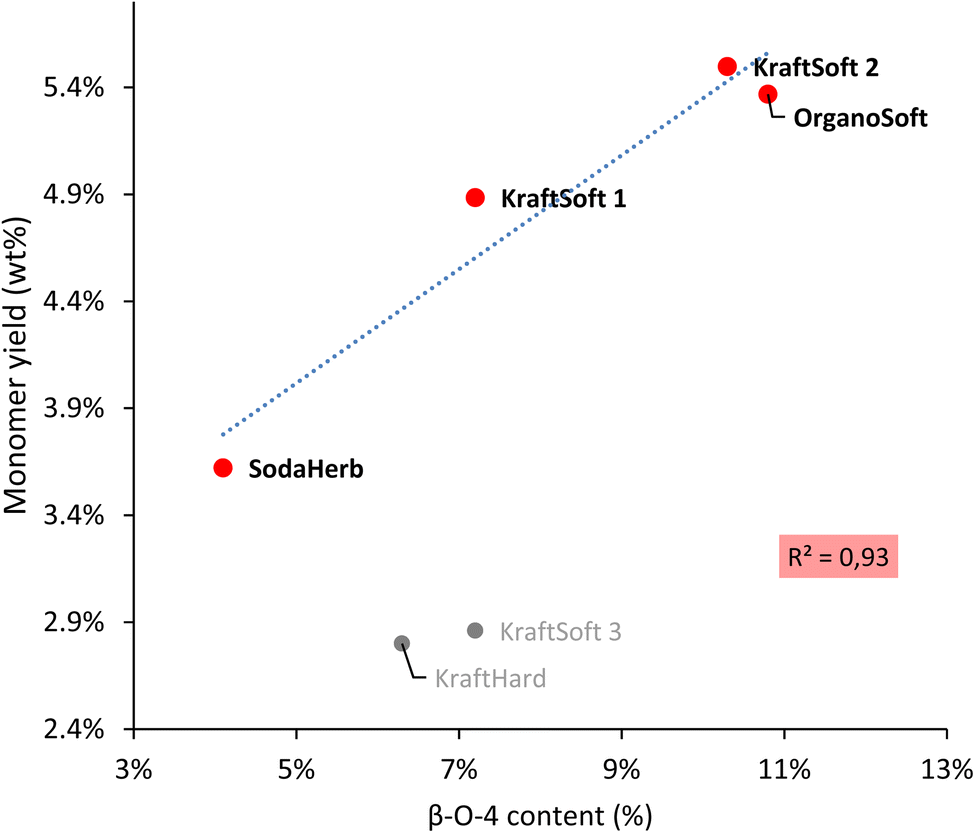 | ||
| Fig. 8 Correlation between the β-O-4 content obtained by HSQC-NMR analysis, and the aromatic monomeric yield obtained from the non-catalytic oxidation reaction. | ||
Characterization of the products of reaction
Klason phases obtained from the non-catalytic oxidation of the different lignins were analyzed by elemental analysis (Table 9). While no strong differences were observed for the initial lignin, the Klason phase obtained from OrganoHerb lignin shows a significantly higher carbon content, a lower oxygen content and a significant nitrogen content. This may be related to alkylation of the lignin structure during the extraction process, according to analyses (i.e., TGA, FTIR and NMR) of the initial lignin.| % wt | C | H | O | N | S |
|---|---|---|---|---|---|
| KraftSoft 1 | 59.4 | 4.5 | 35.3 | 0.2 | 0.6 |
| KraftSoft 2 | 57.3 | 4.1 | 37.8 | 0.2 | 0.6 |
| KraftSoft 3 | 50.0 | 3.7 | 45.1 | 0.2 | 1.0 |
| KraftHard | 57.2 | 4.2 | 36.4 | 0.2 | 2.1 |
| OrganoHerb | 62.0 | 6.8 | 29.6 | 1.5 | 0.0 |
| SodaHerb | 59.7 | 4.8 | 34.3 | 0.7 | 0.5 |
FTIR-ATR analyses of the Klason phases are shown in Fig. S10 and S11.† Comparison of these FTIR spectra with those of the initial lignin (Fig. 2) shows a marked decrease in the band at 1504 cm−1, characteristic of the aromatic backbone, while the band at 1694 cm−1, corresponding to the non-conjugated carbonyl groups, increases. These results confirm an oxidation of this material during the reaction. The Klason of OrganoHerb lignin presents a higher quantity of aliphatic (2934 and 2840 cm−1) and carboxylic acids (1694 cm−1) due to the initial alkylation during the lignin extraction process and the Klason phase of SodaHerb lignin shows a lower level of structural modifications, when referring to the relative intensity of the different IR bands, than KraftHard lignin mainly due to a lower conversion.
HSQC spectra of the Klason phases obtained after non-catalytic oxidation experiments for the aliphatic and aromatic regions are shown in Fig. S12 and S13,† respectively.
Irrespective of whether the oxygenated aliphatic region or the aromatic region is considered, all correlations corresponding to the main structural units of lignin are still visible. However, when compared to HSQC NMR of the starting lignin, noticeable differences are observed in the aromatic region: correlations corresponding to guaiacyl units (ca. δH/δC: G2: 6.87/112.6 ppm, G5: 6.74/115.77 ppm, G6: 6.75/120.27 ppm) or syringyl units (ca. δH/δC: S2,6: 6.68/105.8 ppm) show reduced intensity. Correlations corresponding to H-units are not observed. The decreased intensity of these correlations is associated with a clear increase of the intensity of correlations associated with G′ units (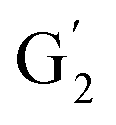 : δH/δC: 7.47/111.89 ppm,
: δH/δC: 7.47/111.89 ppm, 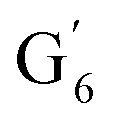 : δH/δC: 7.27/121.12 ppm) and S′ units (
: δH/δC: 7.27/121.12 ppm) and S′ units (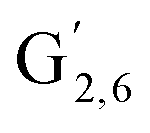 : 7.26/106.9 ppm) due to a partial oxidation of the guaiacyl and syringyl units present in the lignin. KraftHard and SodaHerb lignins are particularly affected by this reaction, which is related to the higher conversion of these lignins. Considering the oxygenated aliphatic region, a significant decrease of intensity for each signal is observed, especially for KraftHard lignin. The application of the semi-quantitative method allowed us to express this decrease as shown in Table 10. Thus, in most cases, the content of β-O-4 linkages in Klason phases decreased by 47% to 78%, except for SodaHerb probably due to its lower initial content. The β-5 linkages showed a similar trend. The decrease for the β–β linkages is significantly lower (13% to 56%), which can be attributed to the higher resilience of C–C linkages compared to ether linkages under the reaction conditions.
: 7.26/106.9 ppm) due to a partial oxidation of the guaiacyl and syringyl units present in the lignin. KraftHard and SodaHerb lignins are particularly affected by this reaction, which is related to the higher conversion of these lignins. Considering the oxygenated aliphatic region, a significant decrease of intensity for each signal is observed, especially for KraftHard lignin. The application of the semi-quantitative method allowed us to express this decrease as shown in Table 10. Thus, in most cases, the content of β-O-4 linkages in Klason phases decreased by 47% to 78%, except for SodaHerb probably due to its lower initial content. The β-5 linkages showed a similar trend. The decrease for the β–β linkages is significantly lower (13% to 56%), which can be attributed to the higher resilience of C–C linkages compared to ether linkages under the reaction conditions.
| % Linkage/C9 | KraftSoft 1 | KraftSoft 2 | KraftSoft 3 | KraftHard | OrganoSoft | SodaHerb |
|---|---|---|---|---|---|---|
| β-O-4 | 2.6% | 2.2% | 2.6% | 7.6% | 2.6% | 1.9% |
| β–β | 3.5% | 1.8% | 2.8% | 3.9% | 3.5% | 2.3% |
| β-5 | 0.8% | 1.5% | 0.8% | 0.5% | 0.8% | 0.7% |
|
||||||
| Decrease produced by the non-catalytic oxidation (%) | ||||||
| β-O-4 | 61% | 78% | 61% | — | 75% | 47% |
| β–β | 40% | 56% | 13% | 22% | 13% | 15% |
| β-5 | 67% | 57% | 60% | — | 94% | 0% |
We were particularly interested in correlating the decrease in β-O-4 linkage content in Klason phases with monomeric aromatic production (Fig. 9) as β-O-4 linkage is a key point to obtaining significant yields of aromatic compounds during lignin oxidative depolymerization. Most of the lignins show linear correlation between the aromatic yield and the decrease of β-O-4 content, with the exception of KraftSoft 3, which resulted in a significantly lower yield of monomeric aromatic compounds compared to what could be expected from its initial β-O-4 content.
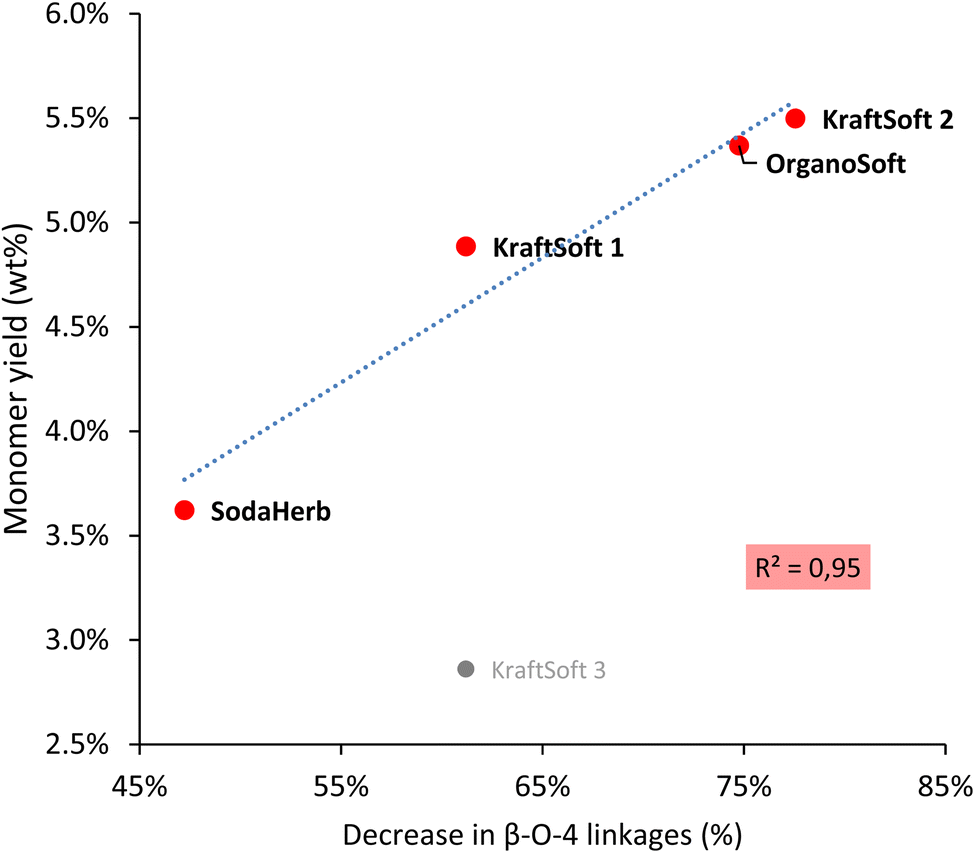 | ||
| Fig. 9 Correlation between the decrease in the β-O-4 content obtained by HSQC-NMR analysis of the Klason phases, and the monomeric yield obtained from the non-catalytic oxidation reaction of the different lignins under standard conditions. | ||
The molecular weights of the Klason Phases measured by SEC chromatography are given in Table 11. The molecular weight was significantly reduced during the oxidation reaction due to the depolymerization process. The rate of reduction seems to be related to the initial molecular weight of the lignin: thus KraftSoft 2 and KraftSoft 3, which have the highest molecular weight (ca. 2900 g mol−1) show the highest rate of reduction up to 86%, whereas SodaHerb and KraftSoft 1 with low molecular weight show a lower rate of reduction. Although these data are informative, they suffer from some limitations: during acidification if the molecular weight of the lignins is too low, they will not precipitate, so it is agreed that the molecular weight of the Klason phase could present a minimum value of about 200–400 g mol−1. On the other hand, the SEC analysis is also limited for molecular weights close to or below 400 g mol−1.
| Lignin | M n (g mol−1) | M w (g mol−1) | M w/Mn | Polymerization degree aver. (Mw/Mmon.) | Decrease in Mn from the initial lignin |
|---|---|---|---|---|---|
| KraftSoft 1 | 426 | 813 | 1.9 | 4.9 | 46.0% |
| KraftSoft 2 | 226 | 425 | 1.9 | 2.6 | 85.7% |
| KraftSoft 3 | 351 | 606 | 1.7 | 3.7 | 79.3% |
| KraftHard | 271 | 433 | 1.6 | 2.6 | 70.3% |
| OrganoSoft | 445 | 767 | 1.7 | 4.7 | 68.1% |
| SodaHerb | 306 | 711 | 2.4 | 4.3 | 52.7% |
The molecular weight distributions for the different lignin and Klason phases are shown in the ESI (Fig. S13).† KraftSoft 1 shows an asymmetric distribution with a secondary peak in the low molecular weight region, a trend that is followed by KraftSoft 2 and KraftSoft 3. These peaks are associated with the presence of low molecular weight condensates. According to the report by Akim et al., under oxidation conditions, stable 5–5 linkages can be formed, which limit the depolymerization rate due to their significantly unreactive character.61 Other lignins show a more symmetrical contribution, which is explained by the higher proportion of syringyl units. Such units have a methoxy substituent at the carbon 5 position, which prevents the formation of 5–5 linkages and reduces the formation of condensates. Accordingly, it was expected that hardwood and herbaceous lignin (i.e., KraftHard, SodaHerb) could produce higher yields of aromatic compounds due to the lower amount of unreactive condensates formed during the oxidation reaction, which is however limited by side degradation reactions.
Catalytic oxidation experiments
Catalytic oxidation experiments were carried out using CuO/TiO2 (5% wt Cu, 5% Cu/lignin, 90–200 μm) under previously optimized reaction conditions.55 Under these conditions, like before, the catalyst was found to be stable by the absence of leaching (<5 ppm).Fig. 10 shows the proportions of the different phases obtained from these experiments for each lignin evaluated. The results can be compared with those shown in Fig. 5 obtained in the absence of a catalyst. The depolymerization of softwood lignin (KraftSoft 1, KraftSoft 2 and OrganoSoft) and KraftHard, are not affected by the presence of the catalyst under our reaction conditions. Similar values for lignin conversion and similar proportions of the different phases are observed. KraftSoft 3, however, shows a higher amount of Klason phase, which is associated with a lower conversion (i.e., ca. 60% versus 70%). SodaHerb and OrganoHerb show strong differences with the non-catalytic experiments, especially in terms of conversion, which increases to about 95% versus 85–89%.
 | ||
| Fig. 10 Mass balance obtained from the catalytic oxidation of the different lignins, calculated in lignin basis (150 °C, 20 bar of air, 60 minutes, CuO/TiO2 5% wt Cu, 5% Cu/lignin). | ||
We have already addressed the issue of internal mass transfer limitation in such reactions.55 Thus, limited diffusion of lignin into the catalyst limits its effect on the conversion of lignin into oligomeric intermediates towards aromatic compounds. It is particularly pronounced for lignin with a number average molecular weight above 1000 g mol−1, such as softwood lignin (i.e., KraftSoft 2 and KraftSoft 3). Therefore, the initial steps of the depolymerization reaction are carried out by non-catalytic oxidation, mainly under the action of the base, and oligomers with lower diffusion limits are formed, which undergo depolymerization towards the formation of aromatic compounds under the control of the catalytic system. The lignin with a lower molecular weight, such as SodaHerb (697 g mol−1), shows a significantly higher conversion driven by the catalyst related to its lower number average molecular weight and the probable presence of oligomers.
Fig. 11 shows the GC analyses of the organic phases. The data show that the catalyst has a significant activity in the production of aromatic compounds from lignin regardless of the lignin source, but with a limited catalytic effect depending on the lignin source. Most aromatic compound yields were increased by 29% to 65%, except for the OrganoSoft lignin where no significant improvement was observed. KraftSoft 1 and KraftSoft 2 show an increase in vanillin yield up to 5% wt, while the yields of other aromatics remain unchanged. KraftSoft 3 shows an increase in vanillin production up to 2.8% wt, and KraftHard and SodaHerb show a similar aromatic aldehyde production when vanillin and syringaldehyde yields are added (resp. 4% and 4.6%). The yields of aromatics obtained with OrganoHerb are particularly improved in the presence of catalysts with an increase of 65%, reaching a total aromatic aldehyde yield of 3.7% wt.
 | ||
| Fig. 11 Yields of aromatics in the organic phase, calculated on lignin basis, obtained from the catalytic oxidation of the different lignin (bars) and total yield increase attributed to the copper catalyst (yellow points) (150 °C, 20 bar air, 60 minutes, CuO/TiO2 5% wt Cu, 5% Cu/lignin). | ||
Catalytic effect on the products
As before, the Klason phases obtained from the catalytic oxidation experiments of the different lignins were analyzed. The FTIR-ATR analyses showed no significant changes due to the presence of the catalyst (Fig. S14 and S15†).The HSQC NMR spectra (Fig. S16 and S17†) of the Klason phases show few differences compared to those obtained from the non-catalytic runs. The oxygenated aliphatic region shows a higher degradation of the polymeric structure, which is supported by the much lower intensities of the correlations assigned to the inter-unit bonds as the β-O-4 (A) (ca. δH/δC: Aα: 4.76/72.1 ppm; Aβ (G): 4.76/81.8 ppm; Aβ (G/H): 4.28/84.8 ppm; Aγ: 3.44/60.4 ppm; 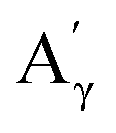 : 4.14/67.6 ppm;
: 4.14/67.6 ppm; 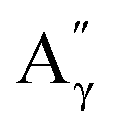 : 3.70/63.58 ppm), β-β (B) (ca. δH/δC: Bα: 4.64/85.6 ppm; Bγ: 3.78, 4.16/71.6 ppm; the Bβ: 3.02/54.4 ppm completely disappeared) in resorcinol moieties and β-5 (C) (ca. δH/δC: Cα: 5.49/87.8 ppm; the Cβ: 3.45/53.8 ppm is not always observed) in phenylcoumaran moieties. Correlations corresponding to cinnamic alcohol (D) (ca. δH/δC: Dγ: 4.10/62.1 ppm) are not observed. The aromatic region shows almost all the correlations expected for the lignin structure, with increased intensity of correlations related to oxidized guaiacyl and syringyl moieties (i.e.,
: 3.70/63.58 ppm), β-β (B) (ca. δH/δC: Bα: 4.64/85.6 ppm; Bγ: 3.78, 4.16/71.6 ppm; the Bβ: 3.02/54.4 ppm completely disappeared) in resorcinol moieties and β-5 (C) (ca. δH/δC: Cα: 5.49/87.8 ppm; the Cβ: 3.45/53.8 ppm is not always observed) in phenylcoumaran moieties. Correlations corresponding to cinnamic alcohol (D) (ca. δH/δC: Dγ: 4.10/62.1 ppm) are not observed. The aromatic region shows almost all the correlations expected for the lignin structure, with increased intensity of correlations related to oxidized guaiacyl and syringyl moieties (i.e., 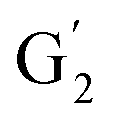 : δH/δC: 7.47/111.89 ppm,
: δH/δC: 7.47/111.89 ppm, 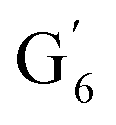 : δH/δC: 7.27/121.12 ppm;
: δH/δC: 7.27/121.12 ppm; 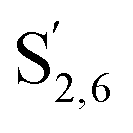 : 7.26/106.9 ppm).
: 7.26/106.9 ppm).
Table 12 shows the semi-quantitative analysis of the HSQC NMR spectra. The data show that β-O-4 are not significantly affected by the presence of the catalyst. The result obtained for the KraftHard lignin can be attributed to an experimental variation. However, the β-β bonds are, nevertheless, strongly affected by the presence of the catalyst, but this effect is not reflected in the yield obtained for the aromatic compounds, since the transformation of these key bonds could not lead to a depolymerization of the oligomeric chains, but rather to a recondensation reaction by the formation of new C–C bonds. The evaluation of the variations in the content of β-5 bonds is rather difficult due to their low concentration.
| %Linkage/C9 | KraftSoft 1 | KraftSoft 2 | KraftSoft 3 | KraftHard | OrganoSoft | SodaHerb |
|---|---|---|---|---|---|---|
| β-O-4 | 2.3% | 3.1% | 2.9% | 2.3% | 2.9% | 2.9% |
| β–β | 0.7% | 1.8% | 2.2% | 2.1% | 0.4% | 0.9% |
| β-5 | 0.6% | 0.6% | 0.8% | 0.6% | 1.6% | 0.5% |
|
||||||
| Decrease produced by the catalyst (%) | ||||||
| β-O-4 | 10% | — | — | 69% | — | — |
| β–β | 81% | — | 22% | 45% | 88% | 59% |
| β-5 | 34% | 58% | — | — | — | 23% |
SEC analysis shows no significant catalyst effect on molecular weight, as can be observed in the ESI (Table S5 and Fig. S18).† This is more likely related to the diffusion limitations of lignin into the catalyst, and to competitive recondensation reactions. For KraftSoft 1 and KraftSoft 3 lignins, an increase in the occurrence of low molecular weight condensates is observed, confirming a catalytic activity in the formation of condensate structures such as 5–5 biphenyl. For KraftSoft 2 lignin a similar distribution is observed for both runs in the presence or absence of the catalyst, probably due to the significant proportion of condensate units in both cases. KraftHard and SodaHerb lignins resulted in a similar molecular weight distribution in all cases, related to the presence of syringyl units that prevent the formation of the 5–5 biphenyl condensate.
Catalytic reaction kinetics
Catalytic oxidation experiments were carried out with KraftSoft1 and KraftHard lignin at different reaction times under the standard reaction conditions. The initial reaction time (i.e., 0 min) was fixed after the reactor was heated to the desired temperature under an inert atmosphere and suddenly pressurized under an air atmosphere. Fig. 12 shows the concentration of the different phases at each reaction time (0, 15, 30, 60, 120 minutes) for KraftHard (solid line) and KraftSoft 1 (dashed line). After 30 minutes of reaction, the conversion of KraftHard lignin was higher than that of KraftSoft lignin, due to its lower initial molecular weight.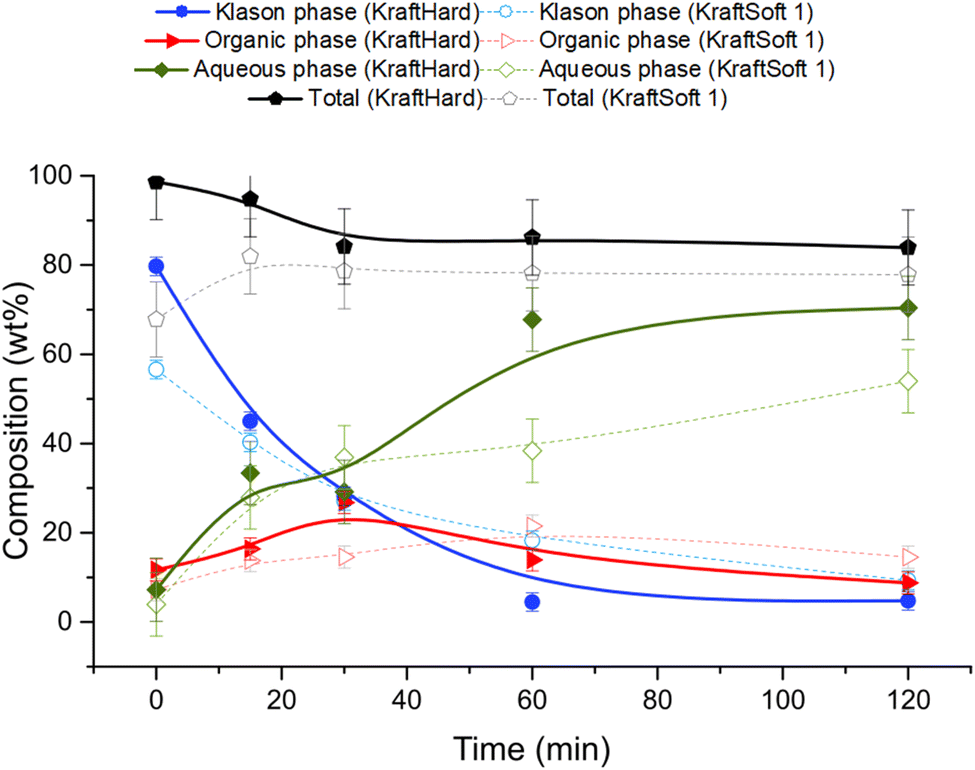 | ||
| Fig. 12 Mass balance of the different fractions, obtained from the catalytic oxidation (5% CuO/TiO2, 5% Cu/lignin, 90–200 μm, 150 °C, 20 bar of air and 1800 rpm) of KraftHard (continuous line), and KraftSoft 1 (dotted line). Each set of points corresponds to one experiment in the bath reactor. Lines are guide for the eyes. | ||
The organic phase yield of KraftHard, after increasing slowly during the first 30 minutes achieving to a maximum production of 26.8% wt, starts to decrease slightly over the reaction time. KraftSoft 1 shows this decrease after 60 minutes of reaction time. The shorter time observed for KraftHard lignin is due to the lower stability of the syringyl derivatives as reported by Casimiro et al.77 and the lower initial molecular weight. In addition, for the same reasons, KraftHard showed a higher production of aqueous phase as observed after 2 hours of reaction time reaching 70% wt.
Fig. 13A and B show the results of GC analyses of the organic phases. Fig. 13A shows the evolution of the aromatic aldehyde yields over time. Syringaldehyde is the main product obtained from the deoxidative depolymerisation of KraftHard lignin and shows a maximum yield (3.8% wt) at a reaction time of 30 minutes. Once this maximum was reached, the syringaldehyde yield decreased rapidly to 1.6% wt after 2 hours reaction time. It is noteworthy that the 30 minute reaction time also corresponds to the maximum yield of vanillin (1.7% wt), which is maintained for 2 hours. These data support the lower stability of syringaldehyde derivatives under oxidative conditions. Similar observations have been reported by Rodrigues Pinto et al.69
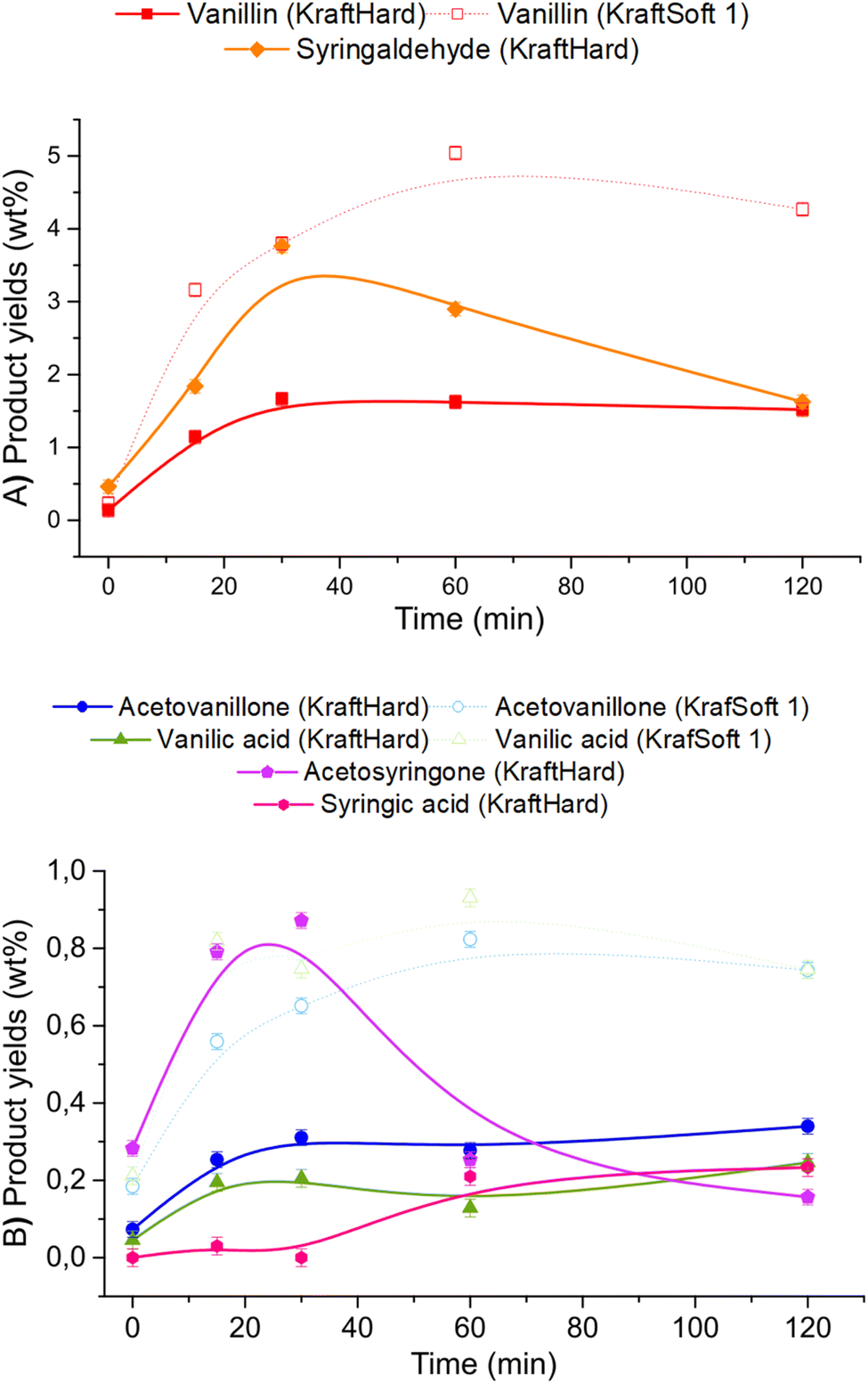 | ||
| Fig. 13 Product yields obtained by GC analyses of the organic phase, obtained from the catalytic oxidation (5% CuO/TiO2, 5% Cu/lignin, 90–200 μm, 150 °C, 20 bar of air and 1800 rpm) of KraftHard (continuous line), and KraftSoft 1 (dotted line). | ||
Acetosyringone and acetovanillone (Fig. 13B) follow the same trend as syringaldehyde and vanillin, which means that their formation mechanism may follow the same reaction pathway as those of aromatic aldehydes. The yields of syringic acid and vanillic acid increased over the first 60 minutes of reaction time and then remained constant over the entire reaction time. According to our own work, and as reported by Schutyser et al.,78 no traces of vanillic acid or syringic acid were obtained from oxidation experiments of vanillin or syringaldehyde. Therefore, these acids are not formed by vanillin or syringaldehyde oxidation, but by alternative pathways during aerobic depolymerisation of lignin as previously reported.30
Conclusion
In this work, seven lignins from different botanical origins and different pulping and extraction processes were subjected to oxidative depolymerization in an alkaline aqueous medium, with or without the presence of a solid copper catalyst. Before and after reaction, the lignins and oxidation products were characterized in detail by a combination of analytical techniques.In line with the literature, it has been observed that the pulping method used to extract lignin from biomass can have an important effect on its structures. This was particularly highlighted by comparing OrganoSoft and KraftSoft lignins extracted from the same woody biomass. OrganoSoft had a higher content of β-O-4 linkages than KraftSoft lignin. Similarly, SodaHerb showed a less degraded structure compared to OrganoHerb lignin, although these were not extracted from exactly the same biomass, as the way in which wheat straw is cultivated can affect the biomass. For all lignins, apart from the well-known distribution of phenylpropane units, it was observed that softwood lignins appeared to have a lower average molecular weight and a lower β-O-4 content than the others. In conclusion, the way in which lignins are produced from biomass affects not only their structures but also their physicochemical properties.
When analyzing the way in which the physicochemical properties affect lignin conversion, we observed that the conversion was significantly affected by the average molecular weight of the lignin used. Conversions were higher for lignin with lower average molecular weights. The benefit of using a catalyst is not always fully reflected in the lignin conversion. For example, SodaHerb (low average molecular weight lignin) and OrganoHerb were the only lignins to show increased conversion in the oxidative depolymerization reactions when using the CuO/TiO2 catalyst (89 to 95% wt and 79 to 95% wt, respectively). These results confirm that longer polymer chains present diffusion limitations in the catalyst, which affect its activity in lignin depolymerization. The role of the catalyst remains unclear to some extent. Under catalytic conditions, an increase in vanillin yield of up to 30% was obtained while no effect on lignin conversion was observed. This suggests that diffusion limitation exists, and that the heterogeneous catalyst acts mainly on low molecular weight lignin chains, thus increasing oxidative depolymerization towards expected aromatic compounds. However, related to this mode of action, under catalytic conditions, over-oxidation of the product could be observed, therefore limiting the yields of the products of interest.
The production of aromatic monomers, especially the phenolic aldehydes vanillin and syringaldehyde, was found to be governed by the β-O-4 content in lignin. The yields of monomers are significantly increased for most of the lignins when the CuO/TiO2 catalyst is used. The production of aromatic monomers seemed to reach a maximum that could not be exceeded regardless of the conditions used. This can be explained by the over-oxidation reactions and condensation reactions, which degrade the aromatic monomers, or by the low β-O-4 bond content that limits the potential for aromatic production during depolymerization. It may also be related to a lack of reactivity for some lignins, such as OrganoSoft, despite the use of catalysts. Finally, although hardwood lignin has a lower molecular weight than softwood lignin, the yields of aromatics obtained from hardwood lignin did not reflect the higher conversions, mainly due to faster degradation reactions. Such limitation is difficult to overcome in a batch reactor as implemented in this study. It could probably be improved in a continuous flow reactor, which is the subject of ongoing studies.
Finally, regarding the CuO/TiO2 catalyst, we demonstrated that it is stable under the reaction conditions by the absence of copper leaching (<5 ppm), indicating that it could be reused, or implemented in a continuous process to perform similar lignin conversion.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the National Agency of Research (Phenoliq No. ANR-16-CE06-0007-03) for funding. IRCELYON scientific and technical services are kindly acknowledged. Chantal Lorentz (IRCELYON) is warmly thanked for her help in NMR experiments. Evonik is thanked for providing TiO2 catalyst support. Dr Yann Le Brech (LRGP) is acknowledged for providing organosolv softwood lignin.References
- S. Malherbe and T. E. Cloete, Rev. Environ. Sci. Biotechnol., 2002, 1, 105–114 CrossRef CAS.
- H. M. N. Iqbal, G. Kyazze and T. Keshavarz, BioResources, 2013, 8, 3157–3176 CrossRef.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- J. M. Pepper, Nature, 1972, 237, 54 CrossRef.
- J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, in Top Value-Added Chemicals from Biomass Volume II—Results of Screening for Potential Candidates from Biorefinery Lignin, Pacific Northwest National Laboratory, U.S. Department of Energy, Richland, WA United States, 2007, pp. 1–79 Search PubMed.
- R. M. Ede and G. Brunow, Holzforschung, 1989, 317–322 CrossRef CAS.
- D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crop. Prod., 2019, 111526 CrossRef CAS.
- M. Kienberger, S. Maitz, T. Pichler and P. Demmelmayer, Processes, 2021, 9, 804 CrossRef.
- L. Dessbesell, M. Paleologou, M. Leitch, R. Pulkki and C. C. Xu, Renew. Sustain. Energy Rev., 2020, 109768 CrossRef CAS.
- S. Tapin-Lingua and L. Djakovitch, unpublished work.
- D. Bourbiaux, J. Pu, F. Rataboul, L. Djakovitch, C. Geantet and D. Laurenti, Catal. Today, 2021, 373, 24–37 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- T. Ren, W. Qi, R. Su and Z. He, ChemCatChem, 2019, 11, 639–654 CrossRef CAS.
- R. Ma, Y. Xu and X. Zhang, ChemSusChem, 2015, 8, 24–51 CrossRef CAS PubMed.
- T. Vangeel, W. Schutyser, T. Renders and B. F. Sels, Top. Curr. Chem., 2018, 376, 1–16 CrossRef CAS PubMed.
- F. S. Chakar and A. J. Ragauskas, Ind. Crop. Prod., 2004, 20, 131–141 CrossRef CAS.
- R. Abejón, H. Pérez-Acebo and L. Clavijo, Processes, 2018, 6, 98 CrossRef.
- O. Y. Abdelaziz, I. Clemmensen, S. Meier, C. A. E. Costa, A. E. Rodrigues, C. P. Hulteberg and A. Riisager, ChemSusChem, 2022, 15, e202201232 CrossRef CAS PubMed.
- R. Behling, S. Valange and G. Chatel, Green Chem., 2016, 18, 1839–1854 RSC.
- P. Gao, C. Li, H. Wang, X. Wang and A. Wang, Chin. J. Catal., 2013, 34, 1811–1815 CrossRef CAS.
- H. Zhang and S. Fu, ChemCatChem, 2024, 16, e202301204 CrossRef CAS.
- J. Zakzeski, A. D. Ebczak, P. C. A. Bruijnincx and B. M. Weckhuysen, Appl. Catal. A: Gen., 2011, 394, 79–85 CrossRef CAS.
- A. Jha, K. R. Patil and C. V. Rode, ChemPlusChem, 2013, 78, 1384–1392 CrossRef CAS PubMed.
- K. Sun, S. Chen, J. Zhang, G. P. Lu and C. Cai, ChemCatChem, 2019, 11, 1264–1271 CrossRef CAS.
- B. Kurek and F. Gaudard, J. Agric. Food Chem., 2000, 48, 3058–3062 CrossRef CAS PubMed.
- L. Hdidou, K. Khallouk, A. Solhy, B. Manoun, A. Oukarroum and A. Barakat, Catal. Sci. Technol., 2018, 8, 5445–5453 RSC.
- Y. Song, J. K. Mobley, A. H. Motagamwala, M. Isaacs, J. A. Dumesic, J. Ralph, A. F. Lee, K. Wilson and M. Crocker, Chem. Sci., 2018, 9, 8127–8133 RSC.
- W. Deng, H. Zhang, X. Wu, R. Li, Q. Zhang and Y. Wang, Green Chem., 2015, 17, 5009–5018 RSC.
- F. G. Sales, L. C. A. Maranhão, N. M. Lima Filho and C. A. M. Abreu, Ind. Eng. Chem. Res., 2006, 45, 6627–6631 CrossRef CAS.
- D. Bourbiaux, Y. Xu, L. Burel, F. Goc, P. Fongarland, R. Philippe, G. Aubert, C. Aymonier, F. Rataboul and L. Djakovitch, Catalysts, 2021, 11, 1311 CrossRef CAS.
- C. Cabral Almada, A. Kazachenko, P. Fongarland, D. Da Silva Perez, B. N. Kuznetsov and L. Djakovitch, Biomass Convers. Biorefin., 2020, 12, 3795–3808 CrossRef.
- C. Cabral Almada, A. Kazachenko, P. Fongarland, D. Da Silva Perez, B. N. Kuznetsov and L. Djakovitch, Catalysts, 2021, 11, 467 CrossRef.
- H. Deng, L. Lin and S. Liu, Energy Fuels, 2010, 24, 4797–4802 CrossRef CAS.
- A. Kumar, B. Biswas and T. Bhaskar, Bioresour. Technol., 2020, 299, 1–7 Search PubMed.
- A. Kumar, B. Biswas, K. Saini, A. Kumar, J. Kumar, B. B. Krishna and T. Bhaskar, Biomass Convers. Biorefin., 2021, 12, 115–128 CrossRef.
- W. Jeon, I.-H. Choi, J.-Y. Park, J.-S. Lee and K.-R. Hwang, Catal. Today, 2020, 352, 95–103 CrossRef CAS.
- B. N. Kuznetsov, N. V. Garyntseva, A. V. Levdansky, L. Djakovitch and C. Pinel, J. Sib. Fed. Univ., Chem., 2014, 7, 316–325 CAS.
- B. N. Kuznetsov, I. G. Sudakova, N. V. Garyntseva, L. Djakovitch and C. Pinel, React. Kinet., Mech. Catal., 2016, 1–14 Search PubMed.
- B. N. Kuznetsov, N. V. Chesnokov, N. V. Garyntseva, I. G. Sudakova, A. V. Pestunov, L. Djakovitch and C. Pinel, Kinet. Catal., 2018, 59, 48–57 CrossRef CAS.
- B. N. Kuznetsov, I. G. Sudakova, O. V. Yatsenkova, N. V. Garyntseva, F. Rataboul and L. Djakovitch, Ind. Catal., 2018, 10, 360–367 CrossRef.
- B. N. Kuznetsov, N. V. Chesnokov, I. G. Sudakova, N. V. Garyntseva, S. A. Kuznetsova, Y. N. Malyar, V. A. Yakovlev and L. Djakovitch, Catal. Today, 2018, 309, 18–30 CrossRef CAS.
- B. N. Kuznetsov, I. G. Sudakova, N. V. Garyntseva, V. E. Tarabanko, O. V. Yatsenkova, L. Djakovitch and F. Rataboul, Catal. Today, 2021, 375, 132–144 CrossRef CAS.
- L. Li, J. Kong, H. Zhang, S. Liu, Q. Zeng, Y. Zhang, H. Ma, H. He, J. Long and X. Li, Appl. Catal., B, 2020, 279, 119343 CrossRef CAS.
- W. Sebhat, A. El-Roz, A. Crepet, C. Ladavière, D. D. S. Perez, S. Mangematin, C. C. Almada, L. Vilcocq, L. Djakovitch and P. Fongarland, Biomass Convers. Biorefin., 2020, 10, 351–366 CrossRef CAS.
- W. Sebhat, A. El Roz, P. Fongarland, L. Vilcocq and L. Djakovitch, Catalysts, 2021, 11, 875 CrossRef CAS.
- J. C. Villar, A. Caperos, F. García-Ochoa and J. Wood, Chem. Technol., 1997, 17, 259–285 CAS.
- J. C. Villar, A. Caperos and F. García-Ochoa, Wood Sci. Technol., 2001, 35, 245–255 CrossRef CAS.
- K. Ninomiya, K. Ochiai, M. Eguchi, K. Kuroda, Y. Tsuge, C. Ogino, T. Taima and K. Takahashi, Ind. Crop. Prod., 2018, 111, 457–461 CrossRef CAS.
- C. Qu, K. Ito, I. Katsuyama, T. Mitani, K. Kashimura and T. Watanabe, ChemSusChem, 2020, 13, 4510–4518 CrossRef CAS PubMed.
- C. Qu, M. Kaneko, K. Kashimura, K. Tanaka, S. Ozawa and T. Watanabe, ACS Sustain. Chem. Eng., 2017, 5, 11551–11557 CrossRef CAS.
- X. Ouyang, T. Ruan and X. Qiu, Fuel Process. Technol., 2016, 144, 181–185 CrossRef CAS.
- Y. Zhu, Y. Liao, L. Lu, W. Lv, J. Liu, X. Song, J. Wu, L. Li, C. Wang, L. Ma and B. F. Sels, ACS Catal., 2023, 13, 7929–7941 CrossRef CAS.
- O. Y. Abdelaziz, I. Clemmensen, S. Meier, S. Bjelić, C. P. Hulteberg and A. Riisager, Top. Catal., 2023, 66, 1369–1380 CrossRef CAS.
- F. Walch, O. Y. Abdelaziz, S. Meier, S. Bjelić, C. P. Hulteberg and A. Riisager, Catal. Sci. Technol., 2021, 11, 1843–1853 RSC.
- A. Hernández Mañas, L. Vilcocq, P. Fongarland and L. Djakovitch, Waste Biomass Valori., 2023, 14, 3789–3809 CrossRef.
- L. Vilcocq, N. Chaussard, A. Hernández Mañas, O. Boyron, M. Taam, F. Bertaud, P. Fongarland and L. Djakovitch, Green Chem., 2023, 25, 4793–4807 RSC.
- A. Hernández Mañas, N. Chaussard, F. Bertaud, L. Vilcocq, P. Fongarland and L. Djakovitch, Ind. Eng. Chem. Res., 2022, 61, 7430–7437 CrossRef.
- V. E. Tarabanko, N. A. Fomova, B. N. Kuznetsov, N. M. Ivanchenko and A. V. Kudryashev, React. Kinet. Catal. Lett., 1995, 55, 161–170 CrossRef CAS.
- V. E. Tarabanko, D. V. Petukhov and G. E. Selyutin, Kinet. Catal., 2004, 45, 569–577 CrossRef CAS.
- M. Davaritouchaee, W. C. Hiscox, E. Terrell, R. J. Mancini and S. Chen, Green Chem., 2020, 22, 1182–1197 RSC.
- L. G. Akim, J. L. Colodette and D. S. Argyropoulos, Can. J. Chem., 2001, 79, 201–210 CAS.
- R. Vanholme, K. Morreel, C. Darrah, P. Oyarce, J. H. Grabber, J. Ralph and W. Boerjan, New Phytol., 2012, 196, 978–1000 CrossRef CAS PubMed.
- J. Snelders, E. Dornez, B. Benjelloun-Mlayah, W. J. Huijgen, P. J. de Wild, R. J. Gosselink, J. Gerritsma and C. M. Courtin, Bioresour. Technol., 2014, 156, 275–282 CrossRef CAS PubMed.
- H. Labauze, N. Cachet and B. Benjelloun-Mlayah, Ind. Crop. Prod., 2022, 187, 115328 CrossRef CAS.
- C. Heitner, D. Dimmel and J. Schmidt, Lignin and Lignans: Advances in Chemistry, CRC, 2016 Search PubMed.
- J.-L. Wen, S.-L. Sun, B.-L. Xue and R.-C. Sun, Materials, 2013, 6, 359–391 CrossRef PubMed.
- Y. Wang, S. Sun, F. Li, X. Cao and R. Sun, Ind. Crop. Prod., 2018, 116, 116–121 CrossRef CAS.
- J. Gierer and S. Wännström, Holzforschung, 1984, 38, 181–184 CrossRef CAS.
- P. C. Rodrigues Pinto, E. A. Borges da Silva and A. r. E. d. Rodrigues, Ind. Eng. Chem. Res., 2011, 50, 741–748 CrossRef CAS.
- S. Zulfiqar, Z. Ahmad and M. I. Sarwar, Colloid Polym. Sci., 2007, 285, 1749–1754 CrossRef CAS.
- S. Zulfiqar, I. Lieberwirth and M. I. Sarwar, Chem. Phys., 2008, 344, 202–208 CrossRef CAS.
- F. P. Bouxin, A. McVeigh, F. Tran, N. J. Westwood, M. C. Jarvis and S. D. Jackson, Green Chem., 2015, 17, 1235–1242 RSC.
- S. Constant, H. L. J. Wienk, A. E. Frissen, P. de Peinder, R. Boelens, D. S. van Es, R. J. H. Grisel, B. M. Weckhuysen, W. J. J. Huijgen, R. J. A. Gosselink and P. C. A. Bruijnincx, Green Chem., 2016, 18, 2651–2665 RSC.
- H. Paananen, E. Eronen, M. Mäkinen, J. Jänis, M. Suvanto and T. T. Pakkanen, Ind. Crop. Prod., 2020, 152, 1–9 CrossRef.
- S. M. Shevchenko and A. G. Apushkinskii, Russ. Chem. Rev., 1992, 61, 105–130 CrossRef.
- Valmet, in The next generation LignoBoost – tailor-maid lignin production for different lignin bioproduct markets, Valmet, 2017, May 26, pp. 1–8 Search PubMed.
- F. M. Casimiro, C. A. E. Costa, C. M. Botelho, M. F. Barreiro and A. E. Rodrigues, Ind. Eng. Chem. Res., 2019, 58, 16442–16449 CrossRef CAS.
- W. Schutyser, J. S. Kruger, A. M. Robinson, R. Katahira, D. G. Brandner, N. S. Cleveland, A. Mittal, D. J. Peterson, R. Meilan, Y. Román-Leshkov and G. T. Beckham, Green Chem., 2018, 20, 3828–3844 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00144c |
| This journal is © The Royal Society of Chemistry 2024 |