
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and structural characterization of L-prolinol derived chiral eutectic mixtures as sustainable solvents in asymmetric organocatalysis†
Jose A.
Ñíguez
 a,
Sarah J.
Burlingham
a,
Rafael
Chinchilla
*a,
Juana M.
Pérez
*b,
Ignacio
Fernández
*b and
Diego A.
Alonso
*a
a,
Sarah J.
Burlingham
a,
Rafael
Chinchilla
*a,
Juana M.
Pérez
*b,
Ignacio
Fernández
*b and
Diego A.
Alonso
*a
aDepartamento de Química Orgánica, Facultad de Ciencias, and Instituto de Síntesis Orgánica (ISO), Universidad de Alicante, Apdo. 99, 03080, Alicante, Spain. E-mail: diego.alonso@ua.es
bDepartment of Chemistry and Physic, Research Centre CIAIMBITAL, University of Almería, Ctra. Sacramento s/n, 04120, Almería, Spain. E-mail: ifernan@ual.es
First published on 21st December 2023
Abstract
The present study focuses on the synthesis, characterization, and examination of the organocatalytic properties of a series of structurally novel L-prolinol-based chiral solvents. Different L-prolinol/TBAB and L-prolinol/glycolic acid (GA) mixtures have been characterized and evaluated for their structural characteristics using ATR-FTIR spectroscopy, differential scanning calorimetry (DSC), and NMR techniques including diffusion and NOESY NMR measurements. The structural characteristics of the L-prolinol-based chiral solvents enabled these novel materials to display organocatalytic activities in the asymmetric conjugate addition of ketones to nitroolefins, with the chiral liquid L-prolinol/GA 1/1 showing the highest yields and selectivities, which are similar or superior to those displayed by L-prolinol in VOC or under neat conditions. Moreover, the new chiral liquid was successfully recovered and reused with minimal loss of performance over several cycles. Regarding the reaction mechanism, a rapid formation of the oxazoline intermediate has been detected by NMR spectroscopy.
Sustainability spotlightAsymmetric organocatalysis is a valuable tool in green chemistry although it usually involves toxic volatile organic compounds (VOCs) as the reaction medium. Reducing chemicals and waste in organocatalysis is an important development in sustainable chemistry, especially using recyclable chiral liquids where the organocatalyst itself is part of the liquid through a challenging balance between its interactions with the components of the liquid and the reagents involved in the catalytic process. Herein, we present our latest studies on the use of small molecules as partners of chiral solvents in asymmetric organocatalyzed processes. Our work emphasizes the importance of the following UN sustainable development goals: industry, innovation, and infrastructure (SDG 9), responsible consumption and production (SDG 12), and climate action (SDG 13). |
Introduction
Sustainable chemistry1 is a concept that has garnered increasing interest in the scientific community, given the concerning environmental context in which the world currently exists. One of the primary goals of sustainable chemistry is to reduce the usage of traditional organic solvents and replace them with ecofriendly alternatives.2,3 In particular, a variety of solvents have been examined, including ionic liquids (IL), eutectic mixtures, deep eutectic solvents (DES), supercritical fluids, liquid polymers, and biodegradable solvents derived from renewable sources.4 Deep eutectic solvents (DES) are emerging as one of the most appropriate green solvents to replace volatile organic compounds (VOCs), due to the limitations of ionic liquids as a reaction medium.5,6 The biodegradability of DES depends on the components that make up them, resulting in exceptionally high biodegradability rates7 and no toxicity.8 Furthermore, the development of DES does not generate by-products and they do not require purification, resulting in a favourable ecological footprint.9 DESs are formed when two or more components interact, primarily through hydrogen bonding, to create a liquid mixture. When these interactions are established, a new structure, formed by an extensive network of hydrogen bonds and other weak interactions, is created. However, not all eutectic mixtures are DES. To be considered a DES, a eutectic mixture must meet two conditions: have a melting point and a significantly deviated eutectic point temperature that is lower than what is expected in respect of an ideal mixture of the components.10,11 If either of these conditions is not met, the most appropriate term to use is low transition temperature mixtures (LTTMs),12 which are still sustainable and alternative solvents. The characterization of eutectic mixtures typically involves performing an analysis of eutectic profiles, checking for the presence of a melting point, and verifying interactions between the components using techniques such as nuclear magnetic resonance (NMR) and infrared (IR) spectroscopies.Despite the many benefits offered by eutectic mixtures, their use as a reaction medium in asymmetric organocatalysis remains limited,13 with conjugate additions,14–19 α-amination reactions,20 aldol reactions21–25 and reduction reactions26 being the most extensively studied processes. The utilization of chiral DES (CDES), where at least one of the components is a chiral organocatalyst,13,27 as environmentally friendly organocatalytic liquids is a unique and noteworthy example of catalysis performed by this innovative category of liquids. Although promising, the utilization of CDES poses significant challenges and remains largely unexplored in the realm of green asymmetric catalysis. For example, (+)-camphorsulfonic acid has been recently used as a hydrogen bond donor to create liquids at room temperature when mixed with ammonium methanesulfonates as hydrogen bond acceptors.28 Despite exhibiting low enantioselectivity, the resulting liquids were able to work as chiral organocatalysts, green solvents, and acid catalysts in the asymmetric Michael-type Friedel–Crafts addition of indole to chalcone. On the other hand, our research group has recently shown the potential of L-proline to form chiral room temperature eutectic mixtures with glycolic acid, glycerol, 1,4-butanediol, p-toluenesulfonic acid, thymol, and diethylene glycol, materials that were successfully employed as green solvents in the addition of butanone to trans-β-nitrostyrene.29
The present study focuses on the synthesis and examination of the organocatalytic properties of novel L-prolinol-based chiral liquids in the asymmetric Michael addition of ketones to nitroolefins as well as in the unravelling of the real structure in solution of these new mixtures with the help of advanced NMR methods.
Results and discussion
Preparation and characterization of novel L-prolinol-based eutectic mixtures
Initially, a set of compounds which are known to form eutectic mixtures as hydrogen bond acceptors (HBA) were evaluated as counterparts of L-prolinol to prepare the corresponding chiral mixtures at room temperature. Choline chloride (ChCl), acetylated choline chloride (Ac-ChCl), and glycerol were examined in different molar ratios, with the formation of a homogeneous liquid phase not detected in any case after heating the mixture up to 80 °C. In contrast, n-tetrabutylammonium bromide (TBAB) and glycolic acid (GA) gave stable liquids at room temperature (Table 1, entries 8–10). As depicted in entries 5–7, L-prolinol/TBAB mixtures with lower molar ratios than 2/1 did not afford homogeneous liquid phases at room temperature.| Entry | HBA | L-Prolinol/HBAa | T formation (°C) |
|---|---|---|---|
| a Molar ratio. b Determined by heating mixtures and visually checking the formation of a stable homogeneous liquid phase. c Solid phase mixtures at room temperature. ND: not determined. | |||
| 1 | TBAB | 4/1 | 30 |
| 2 | TBAB | 3/1 | 30 |
| 3 | TBAB | 2.5/1 | 35 |
| 4 | TBAB | 2/1 | 40 |
| 5 | TBAB | 1.5/1 | 50c |
| 6 | TBAB | 1/1 | 60c |
| 7 | TBAB | 1/2 | ND |
| 8 | GA | 2/1 | 30 |
| 9 | GA | 1/1 | 25 |
| 10 | GA | 1/2 | 25 |
A characterization study based on DSC, ATR-FTIR and NMR was carried out for the lowest formation temperature chiral mixtures: L-prolinol/TBAB 2/1 and 4/1 and L-prolinol/GA 1/1 and 2/1. Regarding the thermal analysis, the four chiral materials showed, in the second heating cycle, low temperature (between −76.89 and −55.87) glass transitions (Tg) while only the L-prolinol/TBAB 2/1 mixture presented a melting point at 43.05 °C in the second heating run (Fig. 1). These results led to the conclusion that, except for the L-prolinol/TBAB 2/1 mixture, the other three chiral mixtures seemed to form low transition temperature mixtures.12,36
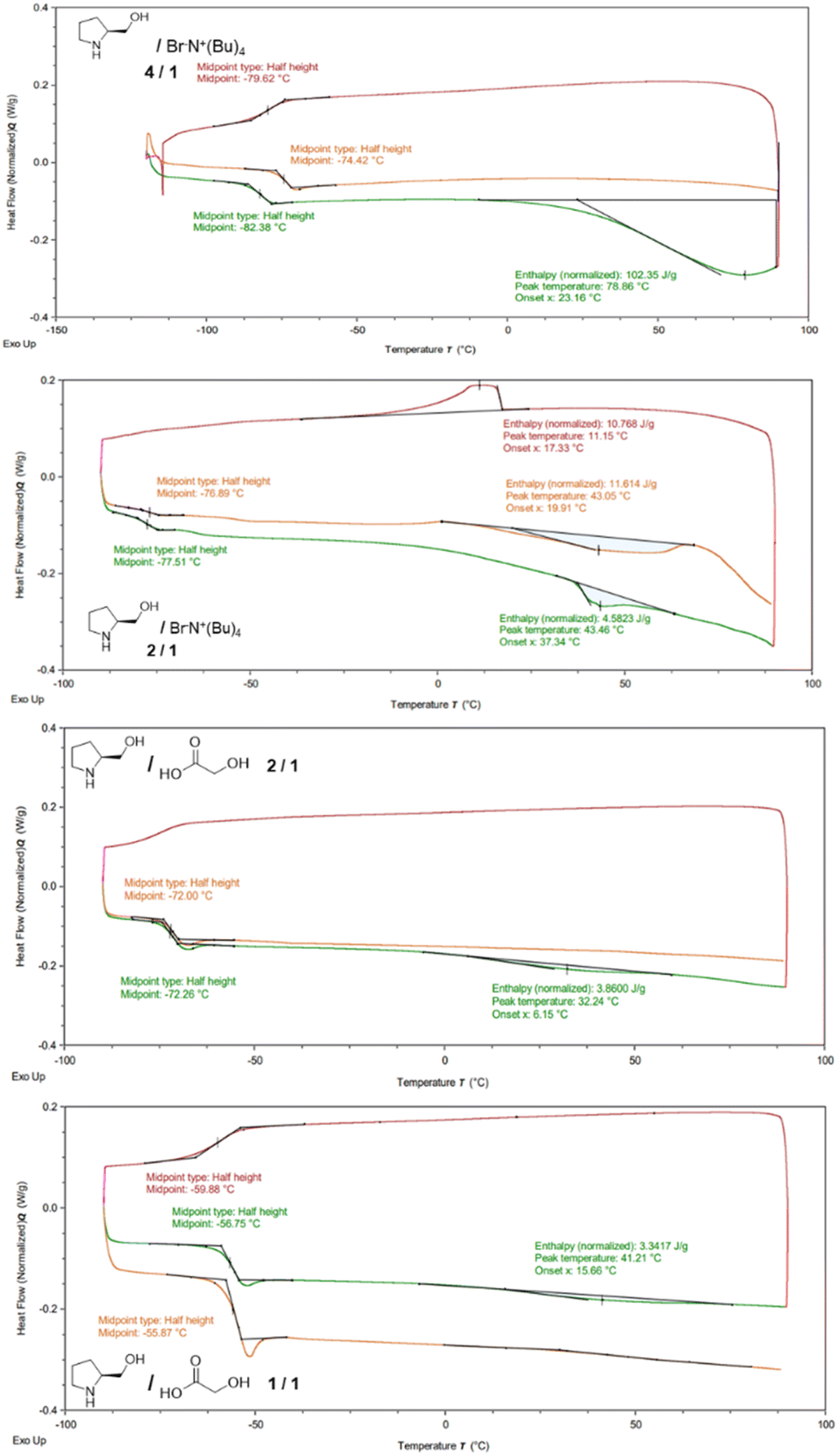 | ||
| Fig. 1 DSC thermograms of L-prolinol/TBAB 2/1 and 4/1 and L-prolinol/GA 1/1 and 2/1 mixtures. | ||
The chemical structures of the L-prolinol/TBAB 2/1 and 4/1 mixtures were initially studied by ATR-FTIR. The spectra of these two mixtures compared with the spectra of pure L-prolinol and TBAB are presented in Fig. 2 and Table 2. As shown, the two chiral mixtures have very similar FTIR spectra with no significant variations between them. However, a change in shape and wavenumber was observed for the νOH stretching vibration of L-prolinol in the chiral mixtures when compared to pure L-prolinol. The broad character of this absorption and the overlap presence of the νNH stretching vibration in the same range hid the shift due to the interaction of the O–H⋯Br− hydrogen bond-type as it has been previously detected in other eutectic mixtures.37 On the other hand, a blue-shift wavenumber variation (from 602 to 584–585 cm−1) was observed for the γOH out of the plane bending vibration of L-prolinol in chiral mixtures when compared to the pure sample, while no changes were detected for the νC–O absorption band (Fig. 2 and Table 2). Finally, changes in the absorption bands of the asymmetrical and symmetrical stretching vibrations of C–N present in both components of the mixture could not clearly confirm the association between L-prolinol and TBAB, since the differences between the IR spectra of each component and mixtures were insignificant and difficult to identify. Based on the obtained spectra, it can be proposed that a weak interaction (probably by hydrogen bonding) is formed in the studied mixtures between the hydroxyl group of L-prolinol and the bromine atom from TBAB.
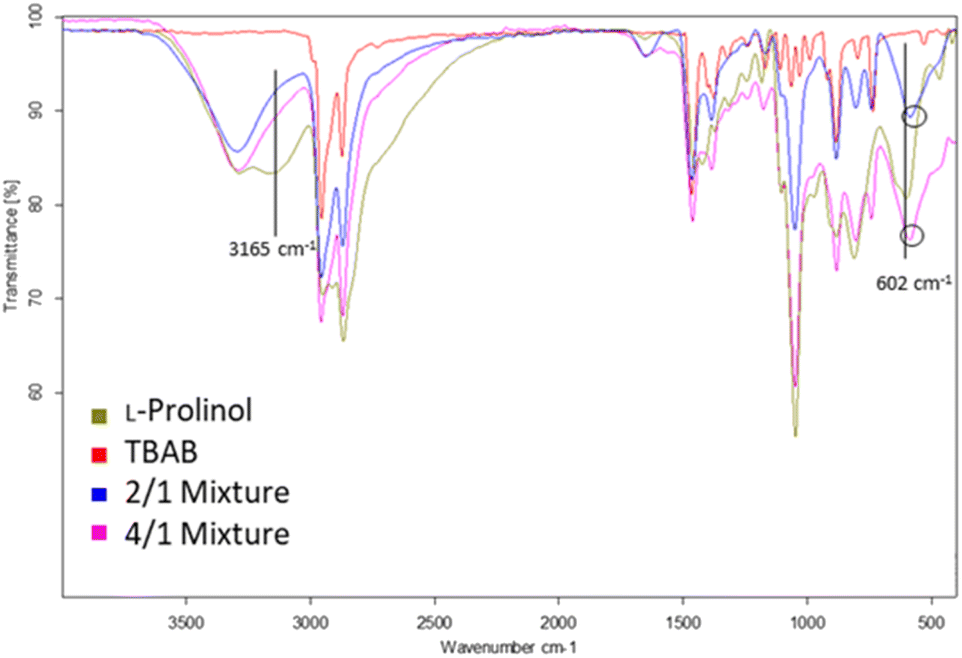 | ||
| Fig. 2 FTIR spectra of L-prolinol (green), TBAB (red), L-prolinol/TBAB 2/1 (blue) and L-prolinol/TBAB 4/1 (pink) mixtures. | ||
| Entry | Mixture | ν OH (cm−1) | γ OH (cm−1) | ν C–O (cm−1) | ν NH (cm−1) |
|---|---|---|---|---|---|
| 1 | L-Prolinol | 3165 | 602 | 1047 | 3294 |
| 2 | 2/1 | ND | 584 | 1049 | 3293 |
| 3 | 4/1 | ND | 585 | 1048 | 3290 |
ATR-FTIR studies on the two L-prolinol/GA mixtures showed a change in the carbonyl νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of GA from 1700.9 cm−1 (pure GA) to 1581.3 (2/1 mixture) and to 1579.5 cm−1 (1/1 mixture) (Fig. 3). These displacements clearly indicated the formation of the carboxylate and the corresponding ammonium salt of L-prolinol, a fact that was corroborated when preparing the sodium salt of GA which showed a νCO stretching vibration frequency at 1589 cm−1.
O stretching vibration of GA from 1700.9 cm−1 (pure GA) to 1581.3 (2/1 mixture) and to 1579.5 cm−1 (1/1 mixture) (Fig. 3). These displacements clearly indicated the formation of the carboxylate and the corresponding ammonium salt of L-prolinol, a fact that was corroborated when preparing the sodium salt of GA which showed a νCO stretching vibration frequency at 1589 cm−1.
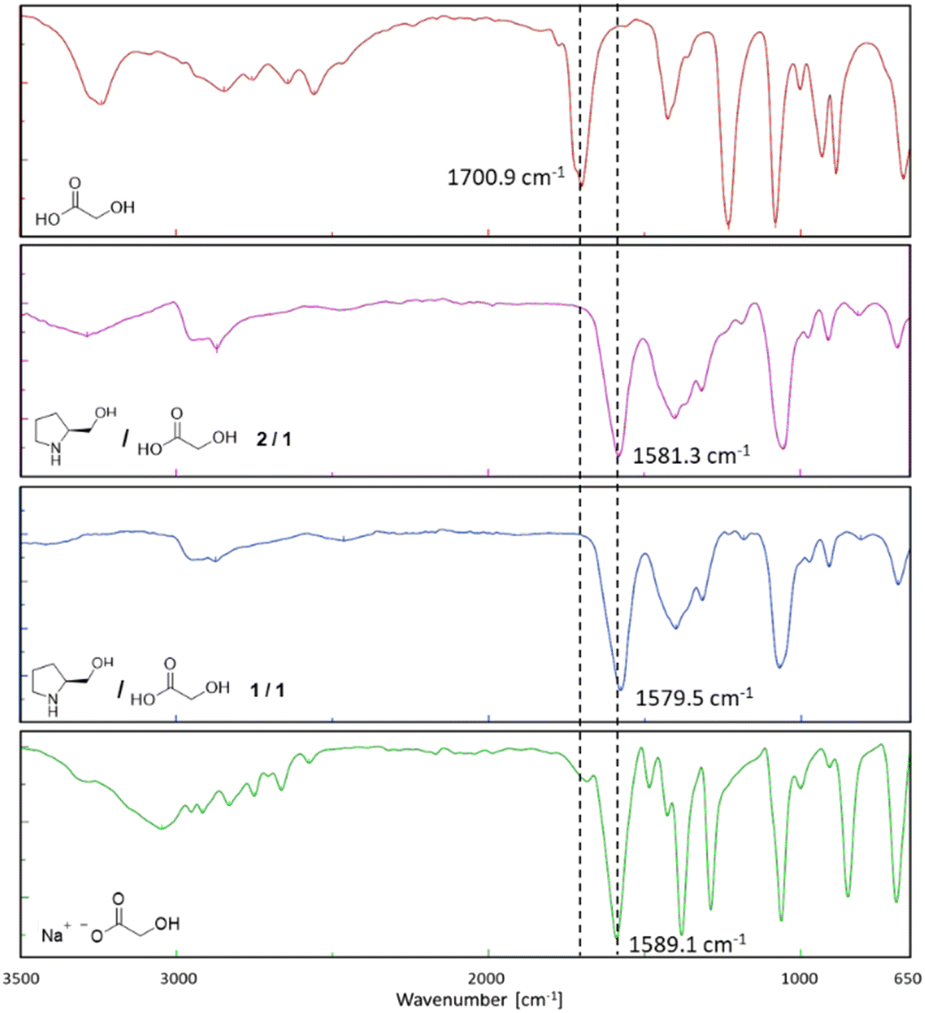 | ||
| Fig. 3 FTIR spectra of glycolic acid (red), L-prolinol/GA 2/1 (violet), L-prolinol/GA 1/1 (blue), and L-prolinol sodium salt (green). | ||
Next, 1H and 14N NMR studies of the 4/1 and 2/1 L-prolinol/TBAB mixtures were performed in neat and CD3CN solutions (Table 3). In the tests carried out in CD3CN, a deshielding of 0.53 and 0.65 ppm was obtained for the nitrogen-14 of TBAB in 4/1 and 2/1 mixtures (entries 1 and 2). In both tests a slight shielding of 1/1′P, 2P and 5P protons was observed, and an important shielding of the proton of L-prolinol's hydroxyl (entries 5–14), facts that seemed to indicate the presence of weak interactions between the two entities probably through the bromide.
|
|
||||
|---|---|---|---|---|
| Entry | Mixture | Position | Δδ (ppm, CD3CN) | Δδ (ppm, neat) |
| a Coordination shifts defined as δmixture − δligand. | ||||
| 1 | 4/1 | N | −0.53 | ND |
| 2 | 2/1 | −0.65 | ||
| 3 | 4/1 | 1T | +0.01 | ND |
| 4 | 2/1 | +0.01 | ||
| 5 | 4/1 | 1P | +0.02 | +0.02 |
| 6 | 2/1 | +0.03 | +0.01 | |
| 7 | 4/1 | 1P′ | +0.01 | +0.04 |
| 8 | 2/1 | +0.01 | +0.04 | |
| 9 | 4/1 | 5P | +0.04 | +0.06 |
| 10 | 2/1 | +0.03 | +0.05 | |
| 11 | 4/1 | 2P | +0.04 | +0.04 |
| 12 | 2/1 | +0.03 | +0.04 | |
| 13 | 4/1 | –OH | ND | +0.38 |
| 14 | 2/1 | ND | +0.55 | |
1H NMR diffusion measurements in CD3CN were conducted on both samples 4/1 and 2/1, monitoring signals from both entities L-prolinol and TBAB. We were interested in the study of their diffusion properties through PGSE NMR, an important tool used nowadays in the analysis of ion-pairing and molecular weight estimations, among many other applications.38,39 In both mixtures, diffusion values of 1.49228 and 1.48103 × 10−10 m2 s−1 were obtained for TBAB and values of 2.64001 and 2.71421 × 10−10 m2 s−1 for L-prolinol. The Stokes–Einstein relation allowed us to transform the D-values into the corresponding hydrodynamic radii (Table 4), making evident the lack of any kind of interaction in solution.
| Component | Mixture | D × 10−10a (m2 s−1) | r H (Å) |
|---|---|---|---|
| a The experimental error in the D-values is ±2%. b The viscosities used in the Stokes–Einstein equation were taken from Perry's Chemical Engineers' handbook 8th edition and the value was 0.36023 × 10−3 kg s−1 m−1 for acetonitrile. The signals at δH 3.12, 3.35 and 1.97 ppm were monitored for TBAB, L-prolinol and CD3CN, respectively. | |||
| L-Prolinol | 4/1 | 2.61001 | 2.3 |
| 2/1 | 2.71421 | 2.2 | |
| Pure | 2.68786 | 2.3 | |
| TBAB | 4/1 | 1.49228 | 3.9 |
| 2/1 | 1.48103 | 4.0 | |
| Pure | 1.43697 | 4.1 | |
PGSE signal attenuations are shown in Fig. 4 for both mixtures in CD3CN at 294 K, together with both in independent samples. The hydrodynamic radii obtained for free L-prolinol and free TBAB of 2.3 and 4.1 Å, which are almost equal those obtained in the mixtures, validated the previous statement. However, that a small proportion of bromide interacting species is in fast exchange with the main species cannot be rejected and could be this minor percentage of the coordinated assembly, the one catalytically active.
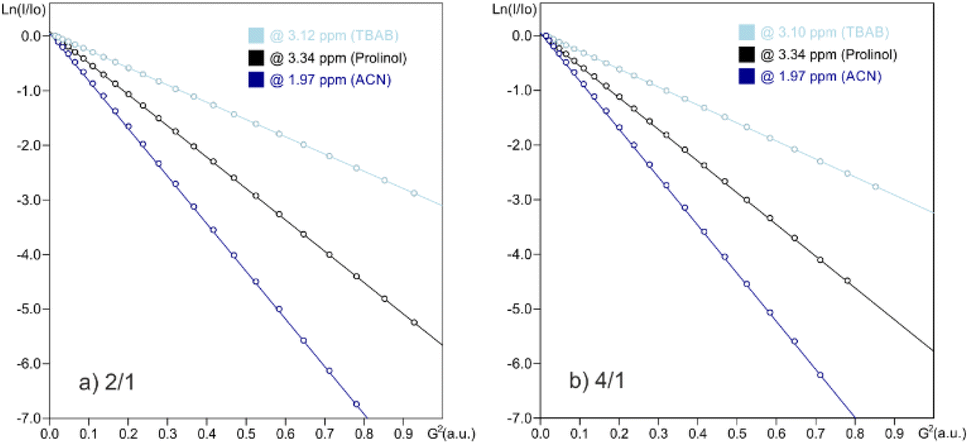 | ||
| Fig. 4 Stejskal–Tanner plots from 1H PGSE NMR diffusion experiments in CD3CN at 292 K using the stimulated echo with the bipolar pair pulse (stebpgp1s1d) sequence for 2/1 and 4/1 (L-prolinol/TBAB) mixtures. | ||
Driven by the IR and NMR results, it was tentatively proposed an equilibrium between a minor species in where an ionic–hydrogen bond-like interaction between the hydroxyl group of L-prolinol and TBAB was responsible for the formation of the eutectic mixture (Fig. 5).
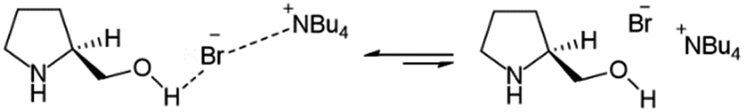 | ||
| Fig. 5 Proposed structure in L-prolinol/TBAB chiral mixtures. | ||
1H and 13C NMR studies for the 2/1 and 1/1 L-prolinol/GA mixtures showed different chemical shifts compared to the free compounds (Table 5). In 1H NMR, the most significant changes occurred in the 2G protons, with a shielding of 0.32 and 0.36 ppm in 2/1 and 1/1 mixtures (entries 1 and 2), and in the 1P, 1P′, 2P and 5P protons in the 2/1 mixtures, with a deshielding of 0.20, 0.15, 0.22 and 0.16 ppm respectively (entries 7, 9, 11, and 13), suggesting the presence of a possible hydrogen bonding interaction between these positions. In 13C NMR, the most relevant changes occurred in 1G, observing a deshielding of 3.81 and 3.93 ppm for the 2/1 and 1/1 mixtures, respectively (entries 3 and 4), suggesting the presence of an ionic-type interaction between the carboxylate and the ammonium that form the liquid salt. Other important displacement changes occurred in 2G, 1P, 2P and 5P (entries 6, 15, 17, 18 and 19, Table 5) reinforcing the idea of hydrogen bond interactions between those positions.
|
|
||||
|---|---|---|---|---|
| Entry | Nucleus | Mixture | Position | Δδ (ppm, CD3CN) |
| a Coordination shifts defined as δmixture − δligand. | ||||
| 1 | 1H | 2/1 | 2G | +0.32 |
| 2 | 1/1 | +0.36 | ||
| 3 | 13C | 2/1 | 1G | −3.81 |
| 4 | 1/1 | −3.93 | ||
| 5 | 13C | 2/1 | 2G | +0.06 |
| 6 | 1/1 | −0.70 | ||
| 7 | 1H | 2/1 | 1P | −0.20 |
| 8 | 1/1 | −0.19 | ||
| 9 | 1H | 2/1 | 1P′ | −0.15 |
| 10 | 1/1 | −0.13 | ||
| 11 | 1H | 2/1 | 2P | −0.22 |
| 12 | 1/1 | −0.21 | ||
| 13 | 1H | 2/1 | 5P | −0.16 |
| 14 | 1/1 | −0.14 | ||
| 15 | 13C | 2/1 | 1P | −1.62 |
| 16 | 1/1 | −0.10 | ||
| 17 | 13C | 2/1 | 2P | −0.86 |
| 18 | 1/1 | −0.67 | ||
| 19 | 13C | 2/1 | 5P | −0.45 |
| 20 | 1/1 | −0.10 | ||
Diffusion studies were also carried out for these L-prolinol/GA mixtures. PGSE signal attenuations are shown in Fig. 6 for the mixture of reagents L-prolinol (P) + glycolic acid (GA) in CD3CN at 294 K, together with both in independent samples. Often the Stokes–Einstein equation40 and its modifications41,42 are useful and enable molecular size estimation of particles larger than the solvent. These calculated hydrodynamic radii, rH, assume spherical shapes; hence, they do not represent the real shape of the molecules. Nevertheless, their use is well established for comparison, since they offer a rapid and easy method to recognize ion pairing and/or aggregation. As is shown in Table 6, diffusion experiments showed decreased D-values and increased rH values of both components when they are forming the mixture with respect to their free states (Table 2). This fact suggested and confirmed the formation of a new entity, where both components diffused together due to the established interactions.
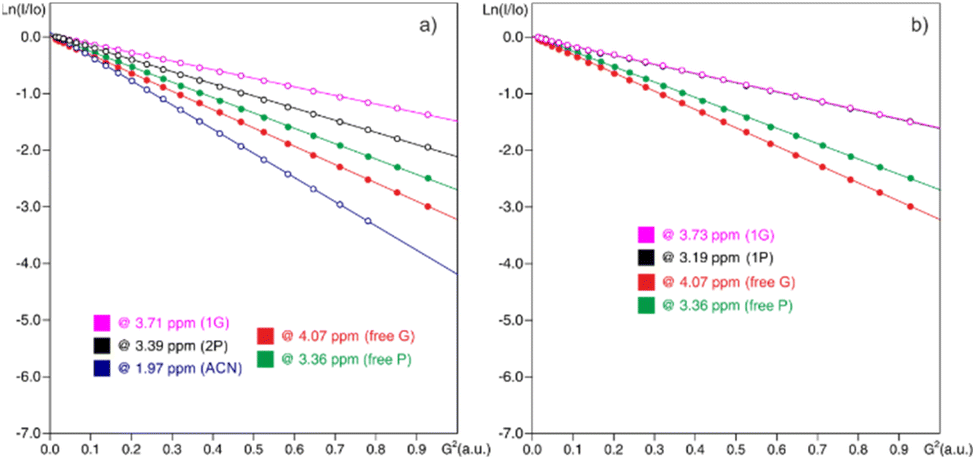 | ||
| Fig. 6 Stejskal–Tanner plots from 1H PGSE NMR diffusion experiments in CD3CN at 294 K using the stimulated echo with the bipolar pair pulse (stebpgp1s1d) sequence for (a) free L-prolinol (P), free glycolic acid (G) and DES (2P + 1G); (b) free L-prolinol (P), free glycolic acid (G) and DES (1P + 1G). | ||
| Component | Mixture | D × 10−10a (m2 s−1) | r H (Å) |
|---|---|---|---|
| a The experimental error in the D-values is ±2%. b The viscosities used in the Stokes–Einstein equation were taken from Perry's Chemical Engineers' handbook 8th edition and the value was 0.36023 × 10−3 kg s−1 m−1 for acetonitrile. The signals at δH 3.12, 3.35 and 1.97 ppm were monitored for TBAB, L-prolinol and CD3CN, respectively. | |||
| L-Prolinol | 2/1 | 2.09974 | 2.9 |
| 1/1 | 1.57077 | 3.9 | |
| Pure | 2.68786 | 2.3 | |
| GA | 2/1 | 1.48276 | 4.1 |
| 1/1 | 1.57049 | 3.9 | |
| Pure | 3.12463 | 2.0 | |
Interestingly, in this polar and non-protic solvent media, the hydrodynamic radius slightly increased with concentration (ca. 0.3–0.5 Å) probably due to the extent of aggregation.
To shed more light on the existing interactions that discriminate between the real conformation of the chiral mixture we measured one-dimensional build-up NOESY curves to estimate inter-moiety distances between each of the two constitutional partners. As seen in Fig. 7, NOE experiments showed proximity between the 2G and 1P, 1P′, 2P and 5P protons. One of the 2G protons was selectively excited, giving an NOE effect with the 1P, 1P′, 2P and 5P protons (Fig. 7c); on the other hand, the same test was carried out by exciting the 3P diastereotopic proton up the plane, giving an NOE effect with the 1P, 1P′, 2P, 4P and 5P protons (Fig. 7b). The information extracted from these tests was the distance of the 3P protons from 2G, and the proximity of the 2G with 1P, 1P′, 2P and 5P, these latter being the positions at which the interactions occurred.
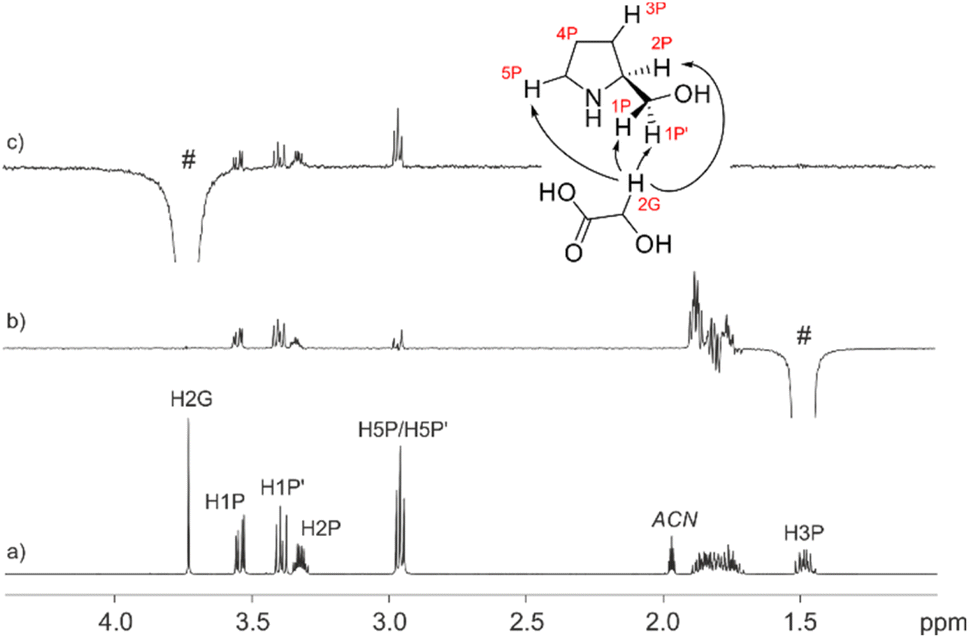 | ||
| Fig. 7 (a) 1H NMR (500 MHz, ACN-d3) spectrum of the 1/1 mixture; (b) 1D 1H, 1H DPFGSE NOESY spectrum (tm 1.0 s) with selective excitation of the up-field shifted diastereotopic proton H3P, and (c) 1D 1H, 1H DPFGSE NOESY spectrum (tm 1.0 s) with selective excitation of the methylenic proton of glycolic acid H2G. | ||
Inter-protonic distances between GA and L-prolinol have been calculated by the −1/6 power of the cross-relaxation rates ratio between the unknown distance and that for a reference fixed distance times the reference distance. The respective cross-relaxation rates, σ, were obtained by linear fitting of the normalized growing curves, using the excited peak intensity at the same mixing time to minimize relaxation bias.43 The selective 1D DPFGSE-NOESY spectrum of H2G and differentiated signals of L-prolinol showed clear and well-resolved NOE enhancements with very flat baselines, which are representative of all the spectra obtained in this study. The analysis to determine inter-proton distances on both samples is illustrated in Fig. 8. Selective 1D DPFGSE-NOESY was obtained over a range of mixing times, i.e., 0.1, 0.2, 0.3, 0.4, 0.5, 0.7 and 1.0 s.34,35,44,45
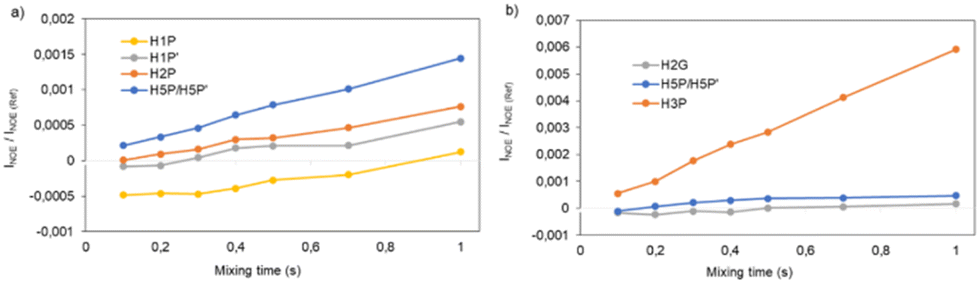 | ||
| Fig. 8 1D DPFGSE-NOESY intensities (INOE) normalized with respect to the inverted peak intensity and plotted against mixing time (tm). (a) Selective excitation of H2G; (b) selective excitation of H1P. | ||
The NOE intensities, normalized with respect to the inverted peak intensity, were plotted against the mixing time to obtain build-up rates that, to a large extent, canceled the effect of external relaxation at moderate mixing times.46 The average interproton distances were then calculated from the obtained cross-relaxation rates (σ values) and the intramolecular reference distance, assuming that the tumbling of the system can be described by a single rotational correlation time. The corresponding reference distance of 2.6 Å between protons H5 and H3 in L-prolinol was derived from experimental data available from X-ray diffraction. For example, the cross-relaxation rate constants for the intramolecular H2G–H2P and H2G–H5P were σ = 0.0008207 s−1 and σ = 0.0006866 s−1, corresponding to estimated distances of 2.9 and 3.0 Å, respectively (Table 7). As expected, smaller distances are found within L-prolinol, whereas weaker contacts and larger distances are obtained between glycolic acid and L-prolinol. The estimations of the average cross relaxation rate constants and the distances derived from the whole analysis are shown in Table 7.
| Entry | Inter-moieties | σ IS (s−1) | r IS (Å) |
|---|---|---|---|
| a All the fits have R2 values higher than 0.99 and the estimated error for a single σIS value is about 10%. b Reference distance of 2.6 Å between protons H5 and H3 in L-prolinol. | |||
| 1 | H2G–H1P | 0.0006881 | 2.96 |
| 2 | H2G–H1P′ | 0.000674 | 2.97 |
| 3 | H2G–H2P | 0.0008207 | 2.87 |
| 4 | H2G–H5P/H5P′ | 0.0006866 | 2.96 |
| 5 | H1P–H2G | 0.0004315 | 3.20 |
| 6 | H1P′–H2G | 0.0003826 | 3.26 |
| 7 | H2P–H2G | 0.0004399 | 3.19 |
| Entry | Intra-moieties | σ IS (s−1) | r IS (Å) |
|---|---|---|---|
| 8 | H5P/H5P–H2G | 0.0004238 | 3.21 |
| 9 | H1P–H5P/H5P | 0.0002933 | 3.41 |
| 10 | H1P–H3P | 0.0060052 | 2.06 |
| 11 | H1P′–H5P/H5P | 0.0012299 | 2.69 |
| 12 | H1P′–H3P | 0.0078786 | 1.97 |
| 13 | H5P/H5P–H3P | 0.0016386 | 2.56 |
| 14 | H3P–H1P | 0.0048353 | 2.14 |
| 15 | H3P–H1P′ | 0.0067618 | 2.02 |
| 16 | H2P–H5P/H5P | 0.0019036 | 2.50 |
With all the information obtained, a structure was proposed for L-prolinol/GA mixtures where there were present ionic and hydrogen bond interactions (Fig. 9).
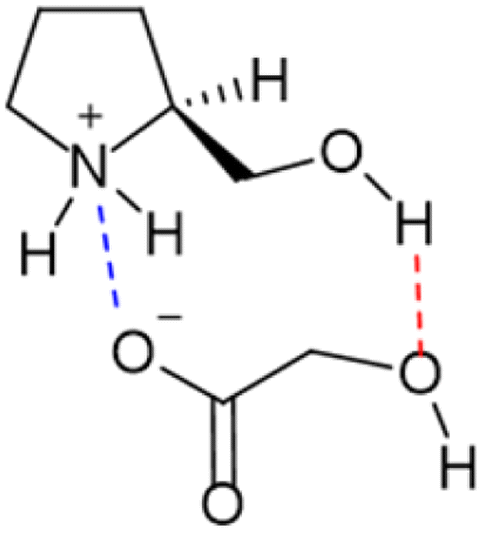 | ||
| Fig. 9 Proposed structure in L-prolinol/GA chiral mixtures. | ||
Application of L-prolinol-based chiral liquids in asymmetric organocatalysis
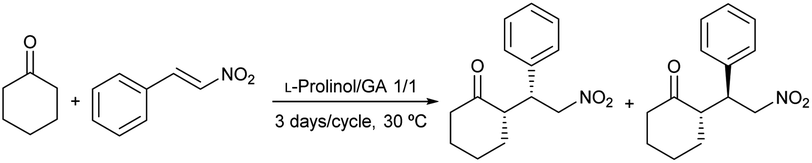
The organocatalytic activity of the synthesized chiral mixtures was studied next. Thus, L-prolinol/TBAB 2/1 (61 ± 7 cP at 50 °C; 0.9% H2O by Karl-Fischer) and 4/1 (27 ± 15 cP, at 50 °C; 0.7% H2O by Karl-Fischer) as well as L-prolinol/GA 1/1 (931 ± 2 cP, at 50 °C; 0.9% H2O by Karl-Fischer) and 2/1 (197 ± 2 cP, at 50 °C; 1.4% H2O by Karl-Fischer) were employed as a reaction medium in the model conjugate addition of cyclohexanone to β-nitrostyrene. The reactions were carried out at 30 °C using 5/1 and 3/1 chiral solvent/limiting reagent mass ratios (Table 8). As shown, when using L-prolinol/GA chiral liquids, full conversions were obtained with good diastereo- and enantioselectivities (Table 8, entries 1–4). The best results were observed when using L-prolinol/GA 1/1 (mass ratio 5/1) with a dr (syn/anti) of 83/17 and a 74% ee for the major syn diastereomer (entry 1). These results demonstrated similar or superior performance compared to the use of L-prolinol as a catalyst (10 mol%) in both neat conditions (Table 8, entry 5) and in dichloromethane (entry 6) after 3 days. In the case of dichloromethane, the enantioselectivity for the major diastereomer increased to 81% ee, however, the reaction conversion was only 61%. Much lower enantioselectivity was obtained when using L-prolinol/GA 1/1 as catalyst (10 mol%) under neat conditions (57% ee for the syn diastereoisomer), as depicted in entry 7. On the other hand, lower conversions and selectivities were generally obtained for the chiral liquids L-prolinol/TBAB which again led to poorer results when decreasing the chiral liquid/limiting reagent mass ratio (Table 8, entries 8–11). Subsequently, the influence of water on the selectivity of the model conjugate addition of the most selective chiral liquid (L-prolinol/GA 1/1) was evaluated. As demonstrated in entry 12 of Table 8, the mixture L-prolinol/GA/H2O 1/1/0.4 afforded the conjugate addition product with better syn diastereoselectivity (90 versus 66%), but lower enantioselectivities for both diastereomers (Table 8, compare entries 1 and 12). Finally, model conjugate addition was scaled up to 10 g of chiral liquid obtaining an 82/2 (syn/anti) diastereomeric ratio and a 77% ee for the major diastereomer (Table 8, entry 13).|
|
||||
|---|---|---|---|---|
| Entry | Medium | Conv.f (%) | drg (syn/anti) | eeg (syn/anti) |
| a Chiral liquid/limiting reagent mass ratio of 5/1. b Chiral liquid/limiting reagent mass ratio of 3/1. c L-Prolinol (10 mol%) was used as catalyst. d L-Prolinol/GA 1/1 mixture (10 mol%) was used as catalyst. e Reaction performed employing 10 g of chiral solvent and 8 mmol of reagents. f Determined by 1H NMR analysis of the crude reaction mixture. g Determined by chiral HPLC analysis of the crude reaction mixture. Major diastereomer syn (S,R).47 | ||||
| 1 | L-Prolinol/GA 1/1a | >99 | 83/17 | 74/11 |
| 2 | L-Prolinol/GA 1/1b | >99 | 80/20 | 52/48 |
| 3 | L-Prolinol/GA 2/1a | >99 | 73/27 | 44/37 |
| 4 | L-Prolinol/GA 2/1b | >99 | 76/24 | 56/56 |
| 5 | Neatc | >99 | 72/28 | 67/61 |
| 6 | CH2Cl2c | 61 | 88/12 | 81/34 |
| 7 | Neatd | >99 | 94/6 | 57/14 |
| 8 | L-Prolinol/TBAB 2/1a | >99 | 83/17 | 64/6 |
| 9 | L-Prolinol/TBAB 2/1b | 73 | 62/38 | 41/12 |
| 10 | L-Prolinol/TBAB 4/1a | 89 | 66/34 | 51/4 |
| 11 | L-Prolinol/TBAB 4/1b | 51 | 55/45 | 19/23 |
| 12 | L-Prolinol/GA/H2O 1/1/0.4a | >99 | 95/5 | 63/5 |
| 13 | L-Prolinol/GA 1/1a,e | >99 | 85/15 | 74/12 |
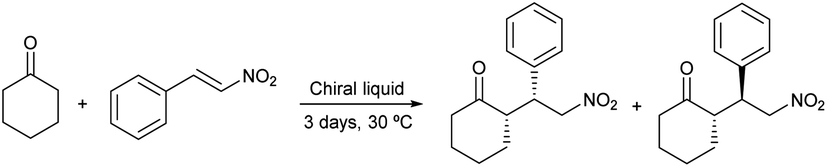
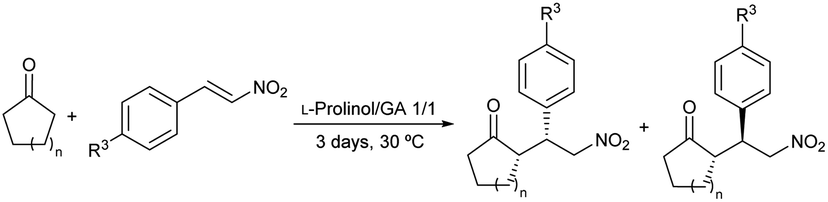
After establishing the optimized reaction conditions (Table 1, entry 1), the conjugate addition of ketones to nitrostyrenes was further investigated to determine the reaction scope (Table 9). Various nitroolefins were reacted with cyclohexanone and cyclopentanone, which yielded the corresponding adducts with moderate to high yields and moderate to good diastereo- (38–66%) and enantioselectivities (59–74% for the syn isomer) (Table 9, entries 1–4).
|
|
|||||
|---|---|---|---|---|---|
| Entry | n | R3 | Conv.a (%) | drb (syn/anti) | eeb (syn/anti) |
| a Determined by 1H NMR analysis of the crude reaction mixture. b Determined by chiral HPLC analysis of the crude reaction mixture. Major diastereomer syn (S,R).47 | |||||
| 1 | 2 | –H | >99 | 83/17 | 74/11 |
| 2 | 2 | –OMe | 90 | 82/18 | 60/69 |
| 3 | 2 | –Cl | 37 | 69/31 | 59/62 |
| 4 | 1 | –H | >99 | 72/28 | 61/55 |
The importance of studying the new chiral liquids capacity from an economic perspective stems from the concept of using the solvent multiple times. To optimize the regeneration method, recyclability of chiral liquids L-prolinol/TBAB 2/1 and L-prolinol/GA 1/1 was investigated. Initially, the solubility of the two liquids was examined in various solvents including ethyl acetate, methyl tert-butyl ether (MTBE), cyclopentyl methyl ether (CPME), toluene, ethanol, and cyclohexane. The chiral mixture L-prolinol/TBAB 2/1 was found to be completely soluble in all the solvents evaluated and was therefore not included in further studies. In contrast, the L-prolinol/GA 1/1 liquid showed low solubilities (1.6–8 wt%) in all tested solvents except for ethanol (soluble up to 40% by weight according to 1H NMR). Ethyl acetate was chosen for further investigation due to the low (1–2 wt%) and complete solubility showed by the chiral mixture and the conjugate addition products in this solvent, respectively. During five reaction cycles of the model addition of cyclohexanone to β-nitrostyrene, no decrease in conversion or diastereoselectivity was observed.48 However, the enantioselectivity of the major syn diastereomer decreased progressively from 74% to 46% ee probably due to the slow but progressive slight loss of organocatalyst after each wash (Table 10). To give some clarity to this last result, the EtOAc washing regeneration method was analyzed. Thus, two freshly prepared batches of the L-prolinol/GA 1/1 chiral liquid were washed with two different batches of EtOAc (0.27 and 0.16 wt% H2O content, respectively) and then dried under vacuum to completely remove the organic solvent. In both experiments, insignificant extraction of the chiral liquid to the EtOAc phase was observed (1.6 and 1.3 wt%, respectively), although the L-prolinol/GA molar ratio in the extracted material (determined by 1H NMR) was 1/1.7 and 1/0.7, respectively. This probably indicated the presence of small amounts of free L-prolinol in the freshly prepared chiral liquid, which was extracted in the washing process, especially when using EtOAc with a higher water content.
|
|
||||
|---|---|---|---|---|
| Entry | Cycle | Conv.a (%) | dr (syn/anti)b | ee (syn/anti)b |
| a Determined by 1H NMR analysis of the crude reaction mixture. b Determined by chiral HPLC analysis of the crude reaction mixture. Major diastereomer syn (S,R).47 | ||||
| 1 | 1 | >99 | 83/17 | 74/11 |
| 2 | 2 | >99 | 87/13 | 72/15 |
| 3 | 3 | >99 | 85/15 | 65/18 |
| 4 | 4 | >99 | 85/15 | 65/15 |
| 5 | 5 | >99 | 78/22 | 46/19 |
The reaction mechanism and intermediates involved in the L-prolinol derived organocatalysts reaction with aldehydes have been previously studied by in situ NMR spectroscopy in DMSO-d6.49 Using this dipolar aprotic solvent Gschwind and colleagues have been able to detect the enamine and its rapid cyclization to the corresponding oxazolidine (unreactive hemiaminal) in the reaction between L-prolinol and L-prolinol ethers with aldehydes. In the case of the stoichiometric reaction between cyclohexanone and the chiral liquid L-prolinol/GA 1/1 we have been able to detect and characterize, for the first time in a chiral liquid, the corresponding spirooxazolidine as a function of time (Fig. 10), not being able to detect under these conditions neither the iminium salt nor the enamine intermediates. As depicted in Fig. 10, after 1 h the conversion into the oxazolidine is 37%, and only after 20 hours the conversion is above 87%.
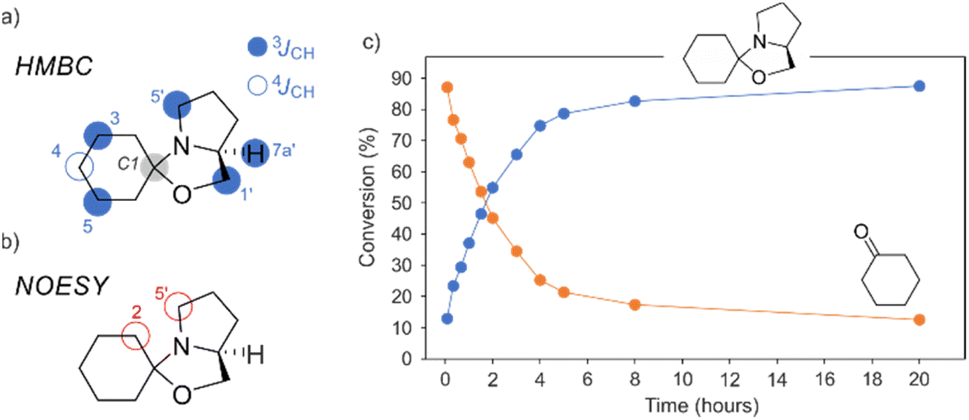 | ||
| Fig. 10 (a) 1H, 13C gHMBC and (b) 1H, 1H gNOESY key interactions; (c) evolution of the amount of cyclohexanone and spirooxazolidine as a function of time as monitored by 1D 1H NMR spectroscopy. | ||
In the 1H NMR spectrum of the oxazolidine intermediate, it is worth mentioning the signal at 3.75 ppm which was assigned to the bridgehead 7a′. In the 13C NMR spectrum, the signal at 97.9 ppm, corresponding to the spirocyclic carbon C1, also indicated the formation of an oxazolidine compound. The HMBC spectrum provided definitive proof by observing key cross-peaks, especially those involving the spirocyclic carbon C1. Fig. 10a illustrates the interactions found, where three-bond couplings (to H3, H5, H1′, H5′, and H7a′) as well as four-bond couplings (to H4) were all observed in the two-dimensional map (see Fig. 10a). In addition to conducting long-range NMR experiments, we performed a 2D NOESY experiment, revealing intense cross-peaks between the methylenic protons of C2 (located at 1.51 ppm) and the two diastereotopic protons of C5′ (located at 2.69 and 2.54 ppm). These moieties are highlighted in Fig. 10b for clarity. Fig. 10c displays the kinetic profile, demonstrating the concurrent formation of the oxazolidine product and the simultaneous decrease in the conversion of cyclohexanone over time.
Experimental
General
Unless otherwise noted, all commercial reagents and solvents were employed without further purification. Reactions under an argon atmosphere were carried out in oven-dried glassware sealed with a rubber septum. 1H NMR (300 MHz) spectra for catalysis experiments were recorded on a Bruker AC-300, employing CDCl3 as solvent and TMS (0.003%) as reference; 1H NMR (500.13 MHz), 13C NMR (125.75 MHz) and 14N NMR (36.14 MHz) spectra for characterization studies were recorded on a Bruker Avance III 500 employing a 5 mm BBFO 1H/BB(19F) probe; unless otherwise stated, standard Bruker software routines (TOPSPIN) were used for the 1D and 2D NMR measurements. 1H, 13C and 14N chemical shifts (δ) are reported in ppm values relative to TMS and liquid NH3, respectively, and coupling constants (J) in Hz. ATR-IR spectra were recorder on a JASCO FT/IR 4100 and on an FT-IR Bruker Alpha spectrophotometer (Alpha II, Bruker Optik, Ettlingen, Germany) equipped with an attenuated total reflection module (ATR) containing a diamond crystal. Chiral HPLC analyses were performed on an Agilent 1100 Series (Quat Pump G1311A, DAD G1315B detector and automatic injector) equipped with Chiralpack (AS-H, AD-H and OD-H) chiral columns using mixtures of hexane/isopropanol as mobile phase, at 25 °C. Differential Scanning Calorimetry (DSC) studies were carried out on DSC heat flow equipment with MDSC temperature from TA Instruments, model Q250. Karl-Fischer water content analyses were performed using a Mettler Toledo KF V20 Compact Volumetric Titrator. Viscosity analyses were performed on a Brookfield high torque viscosimeter CAP 1000+, the measurements were performed at 50 °C, 750 rpm with a 10 s hold time and 25 s run time using spindle 5.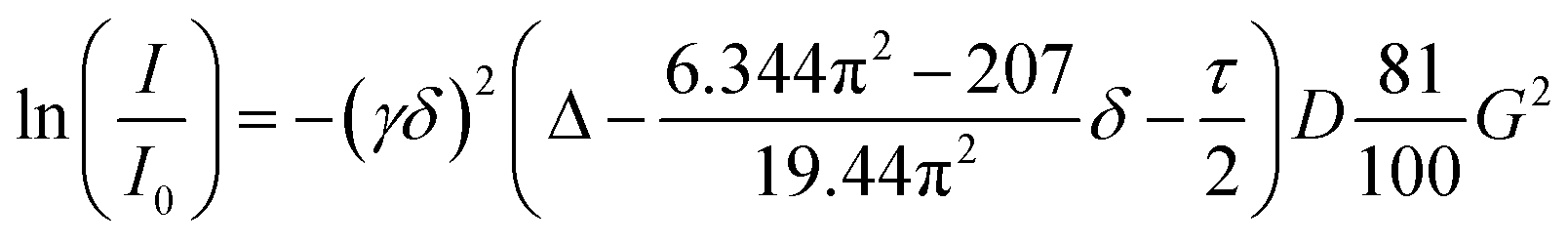 | (1) |
Chiral eutectic mixtures preparation
Choline chloride, acetyl-choline chloride, glycine, n-tetrabutylammonium bromide (TBAB), glycolic acid (GA) and L-prolinol were purchased from Merck, Sigma-Aldrich, BLDpharm or Alfa Aesar (purities >98%). To achieve the desired molar ratio, the respective amounts of each component were added to a 5 mL glass vial, resulting in a final mass of 0.1 g. The resulting mixture was introduced into a sand bath under stirring at 25 °C and kept under an argon atmosphere. Subsequently, the mixtures were heated gradually at a rate of 4 °C every three minutes until the temperature reached 80 °C over the course of an hour, while monitoring the formation of a single liquid phase. After the liquid phase was observed, the vials were taken out of the sand bath and allowed to settle at room temperature to ensure that a uniform and liquid phase was sustained. Once confirmed, the mixtures were stored under an argon atmosphere at room temperature for further characterization and subsequent utilization in catalytic experiments.Diffusion experiments
PGSE NMR diffusion measurements were carried out using the stimulated echo sequence containing bipolar pair pulses.30 A smoothed rectangular shape was used for the gradient pulses and their strength varied automatically during the experiments. The D values were determined from the slope of the regression line ln(I/I0) versus G2, according to the Stejskal–Tanner equation for smoothed rectangular shaped gradient pulses (eqn (1))31 and employing the DiffAtOnce package.32I/I0 = observed spin echo intensity/intensity without gradients, G = gradient strength, Δ = delay between the midpoints of the gradients, D = diffusion coefficient, δ = gradient length, τ = duration of the gradient recovery delay and the 180° pulse.The measurements were carried out without spinning. The delay after the radiofrequency pulses was set to 200 μs, and the delay after the gradient pulses was set to 1 ms. Gradient calibration was carried out by means of a diffusion measurement of HDO in D2O (D(HDO) = 1.902 × 10−9 m2 s−1).33 To check reproducibility and lack of convection, three different measurements with different diffusion parameters (Δ and/or δ) were always carried out. The experimental error in D values was estimated to be smaller than ±2%. All the data leading to the reported D values afforded lines whose correlation coefficients were above 0.999. The gradient strength was incremented in 4% steps from 8% to 96% so that 23 points could be used for regression analysis. The recycle delay was set to 5 s. Experiments were carried out without active temperature regulation, at the probe ambient temperature of 294 ± 0.1 K.
1D NOESY experiments
Selective 1D DPFGSE NOESY experiments were carried out by using the double pulse field gradient echo sequence34,35 with different mixing times varying from 0.1 to 1.0 s. The data were collected using a spectral width of 6849 Hz and 32 K complex data points for a 2.39 s acquisition time with a 2 s recycle delay. Selective Gaussian pulses of 61.5 ms were used to invert the target resonances. Processing of the spectra was accomplished by zero filling to 64 K followed by an exponential multiplication using a line width of 1 Hz. All NOESY peaks areas were subsequently divided by the area of the inverted signal at the same mixing time. This ensures that the enhancements are corrected for relaxation effects. Inter-proton distances were obtained from the slopes of the normalized growing rates versus mixing time obtained by linear regression and applying the isolated spin pair approach (ISPA) using the H4–H5 distance of 2.8 Å in L-prolinol as reference and the peak at the same mixing time to account for relaxation effects. All the distances were calculated from σ ratios by including the appropriate correction factors to account for the number of protons involved, i.e., those involving H5/H5′ of L-prolinol, the σIS was calculated dividing the slope of the normalized growing rate by a factor of two. All the fits have R2 values higher than 0.99 and the estimated error for a single σIS value was about 10%. 2D NOESY spectra were collected with 1.0 second of mixing time using a spectral width of 3894 Hz in F2 and 1 K complex data points for an acquisition time of 0.132 s with a 2 s recycle delay. 256 points were collected in the indirect F1 dimension for a 0.033 s acquisition time. 16 scans were collected per F1 increment and F1 quadrature detection was achieved using the States-TPPI method.General procedure of conjugate additions. Cyclohexanones to β-nitrostyrene
Initially, 0.08 mmol of β-nitrostyrene was added to a glass vial containing 100 mg of the appropriate chiral solvent, and the mixture was stirred for 5 minutes at room temperature. The corresponding ketone (0.08 mmol) was then added to the reaction mixture, which was stirred for 3 days at 30 °C. In the case of using L-prolinol/GA mixtures as the reaction medium, after this time, 0.5 mL of EtOAc was added to the mixture and vigorously stirred. The organic phase was collected, and this procedure was repeated twice. The combined organic phases were then evaporated under vacuum to give the crude reaction product, which was analyzed by 1H NMR and chiral HPLC. On the other hand, when L-prolinol/TBAB mixtures were used as the solvent, after 3 days, 3 mL of EtOAc were added to the mixture until it was completely dissolved. Then, this organic mixture was washed with 5 mL of water, which was decanted and extracted with 3 mL of EtOAc twice. The organic phases were collected and evaporated under vacuum to give the crude reaction product, which was analyzed by 1H NMR and chiral HPLC.Experimental procedure for scaling up the conjugate addition of cyclohexanone to β-nitrostyrene using the L-prolinol/GA 1/1 mixture
β-nitrostyrene (8 mmol) was added to the L-prolinol/GA 1/1 mixture (10 g) and the obtained mixture was stirred for 5 min at RT. Then, cyclohexanone (8 mmol) was added to the reaction mixture, which was stirred for 3 days at 30 °C. Then, EtOAc (10 mL) was added to the mixture, which was vigorously stirred, and the organic phase was collected. This extraction procedure was repeated twice, and the combined organic phases were evaporated under vacuum to give the crude reaction product which was analyzed by 1H NMR and chiral HPLC.Study of the recyclability of the L-prolinol/GA 1/1 mixture in the addition of cyclohexanone to β-nitrostyrene
0.08 mmol of β-nitrostyrene was added to a glass vial containing 100 mg of the L-prolinol/GA 1/1 mixture, and the mixture was stirred for 5 minutes at room temperature. Cyclohexanone (0.08 mmol) was then added to the reaction mixture, which was stirred for 3 days at 30 °C. After this time, 0.5 mL of EtOAc was added to the mixture and vigorously stirred. The organic phase was collected, and this procedure was repeated twice. The combined organic phases were then evaporated under vacuum to give the crude reaction product, which was analyzed by 1H NMR and chiral HPLC. The remaining chiral mixture was used directly in a new reaction cycle following the above-described procedure, and this was repeated for a total of five reaction cycles.Conclusions
In this paper, L-prolinol-based chiral mixtures were prepared and used as green and organocatalytic reaction media in the asymmetric conjugate addition of ketones to nitroolefins. L-Prolinol/TBAB 4/1 and 2/1, as well as L-prolinol/glycolic acid 2/1 and 1/1 chiral liquids, were structurally characterized using ATR-FTIR spectroscopy, differential scanning calorimetry (DSC), and NMR techniques. In particular, the chiral solvent L-prolinol/glycolic acid 1/1 showed very good activity and moderate to good diastereo- and enantioselectivities in the organocatalyzed conjugate addition of ketones to β-nitrostyrenes. A protocol for conjugate addition at a gram-scale was developed, and the recovered chiral liquid was found to be reusable for up to five reaction cycles. However, the enantioselectivity of the resulting product decreased with each cycle, suggesting that further optimization of the recovery protocol is necessary. Also, by in situ NMR studies, we have been able to detect the oxazolidine intermediate generally observed in enamine activated organocatalyzed asymmetric reactions involving L-prolinol derived organocatalysts.Author contributions
All the authors equally contributed to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research has been funded by Generalitat Valenciana (Project AICO 2021/013), Junta de Andalucía (projects 102C2000004, UAL2020-AGR-B1781 and P20_01041), the State Research Agency of the Spanish Ministry of Science, Innovation and Universities (projects PID2021-126445OB-I00, PDC2021-121248-I00, PID2020-119116RA-I00 and PID2021-127332NB-I00) together with EU FEDER funds, and the University of Alicante (Project VIGROB-173 and grants UAUSTI 2022). J. M. P. acknowledges the University of Almeria (grant no. HIPATIA2021_04) for her fellowship.Notes and references
- I. T. Horváth, Chem. Rev., 2018, 118, 369–371 CrossRef PubMed.
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- V. Hessel, N. N. Tran, M. R. Asrami, Q. D. Tran, N. Van Duc Long, M. Escribà-Gelonch, J. O. Tejada, S. Linke and K. Sundmacher, Green Chem., 2022, 24, 410–437 RSC.
- J. García-Álvarez, Handbook of Solvents, ChemTec. Publishing, Toronto, Canada, 2019, vol. 2 Search PubMed.
- Q. Zhang, K. de Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- D. A. Alonso, A. Baeza, R. Chinchilla, G. Guillena and D. J. Ramón, Eur. J. Org Chem., 2016, 612–632 CrossRef CAS.
- K. Radošević, M. Cvjetko Bubalo, V. Gaurina Srček, D. Grgas, T. Landeka Dragičević and R. I. Redovniković, Ecotoxicol. Environ. Saf., 2015, 112, 46–53 CrossRef PubMed.
- M. Hayyan, M. A. Hashim, A. Hayyan, M. A. Al-Saadi, I. M. AlNashef, M. E. S. Mirghani and O. K. Saheed, Chemosphere, 2013, 90, 2193–2195 CrossRef CAS PubMed.
- Y. T. Liu, Y. A. Chen and Y. J. Xing, Chin. Chem. Lett., 2014, 25, 104–106 CrossRef CAS.
- B. Tang and K. H. Row, Monatsh. Chem., 2013, 144, 1427–1454 CrossRef CAS.
- M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, J. Solution Chem., 2019, 48, 962–982 CrossRef CAS.
- M. Francisco, A. van den Bruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef CAS PubMed.
- D. A. Alonso, S. J. Burlingham, R. Chinchilla, G. Guillena, D. J. Ramón and M. Tiecco, Eur. J. Org Chem., 2021, 4065–4071 CrossRef CAS.
- E. Massolo, S. Palmieri, M. Benaglia, V. Capriati and F. M. Perna, Green Chem., 2016, 18, 792–797 RSC.
- D. R. Ñíguez, G. Guillena and D. A. Alonso, ACS Sustain. Chem. Eng., 2017, 5, 10649–10656 CrossRef.
- J. Flores-Ferrándiz and R. Chinchilla, Tetrahedron: Asymmetry, 2017, 28, 302–306 CrossRef.
- J. Flores-Ferrándiz and R. Chinchilla, Tetrahedron: Asymmetry, 2014, 25, 1091–1094 CrossRef.
- J. Flores-Ferrándiz, B. Fiser, E. Gómez-Bengoa and R. Chinchilla, Eur. J. Org Chem., 2015, 1218–1225 CrossRef.
- A. Torregrosa-Chinillach, A. Sánchez-Laó, E. Santagostino and R. Chinchilla, Molecules, 2019, 24, 4058–4070 CrossRef CAS PubMed.
- D. Ros Ñíguez, P. Khazaeli, D. A. Alonso and G. Guillena, Catalysts, 2018, 8, 217/1–217/11 CrossRef.
- C. R. Müller, I. Meiners and P. Domínguez de María, RSC Adv., 2014, 4, 46097–46101 RSC.
- C. R. Müller, A. Rosen and P. Domínguez de María, Sustainable Chem. Processes, 2015, 3, 12 CrossRef.
- R. Martínez, L. Berbegal, G. Guillena and D. J. Ramón, Green Chem., 2016, 18, 1724–1730 RSC.
- D. Brenna, E. Massolo, A. Puglisi, S. Rossi, G. Celentano, M. Benaglia and V. Capriati, J. Org. Chem., 2016, 12, 2620–2626 CAS.
- N. Fanjul-Mosteirín, C. Concellón and V. Del Amo, Org. Lett., 2016, 18, 4266–4269 CrossRef PubMed.
- C. Faverio, M. F. Boselli, T. Ruggiero, L. Raimondi and M. Benaglia, Tetrahedron Chem, 2023, 6, 100038–100041 CrossRef.
- T. A. Hopkins, L. VandenElzen, B. P. Nelson, V. Vaid, J. Brickley, P. Ariza, G. Whitacre, I. Patel, O. Gooch, M. Bechman and C. Jordan, Ind. Eng. Chem. Res., 2023, 62, 1606–1613 CrossRef CAS.
- T. Palomba, G. Ciancaleoni, T. del Giacco, R. Germani, F. Ianni and M. Tiecco, J. Mol. Liq., 2018, 262, 285–294 CrossRef CAS.
- M. Tiecco, D. A. Alonso, D. R. Ñíguez, G. Ciancaleoni, G. Guillena, D. J. Ramón, A. Apio Bonillo and R. Germani, J. Mol. Liq., 2020, 313, 113573 CrossRef CAS.
- D. H. Wu, A. D. Chen and C. S. Johnson, J. Magn. Reson., Ser. A, 1995, 115, 260–264 CrossRef CAS.
- D. Sinnaeve, Concepts Magn. Reson., Part A, 2012, 40, 39–65 CrossRef.
- DiffAtOnce® is a registered program developed by I. Fernández and F. M. Arrabal-Campos in the University of Almería. 2013.
- H. J. V. Tyrrell and K. R. Harris, Diffusion in Liquids, Butterworths, London, 1984 Search PubMed.
- K. Stott, J. Stonehouse, J. Keeler, T.-L. Hwang and J. Shaka, J. Am. Chem. Soc., 1995, 117, 4199 CrossRef CAS.
- K. Stott, J. Keeler, Q. N. Van and A. J. Shaka, J. Magn. Reson., 1997, 125, 302–324 CrossRef CAS.
- Glass-transition temperatures do not denote a specific temperature for a first-order transition. It is plausible that our eutectic systems face kinetic barriers preventing crystallization, thereby only manifesting the Tg while evading the melting peak. All our efforts to prompt crystallization overriding kinetic limitations by low temperature annealing or aging processes have been unsuccessful so far with only low-temperature endothermic processes resembling Tg or Tg with enthalpic relaxation being observed, as depicted in Fig. 1.
- R. Chromá, M. Vilková, I. Shepa, P. Makoś-Chełstowska and V. Andruch, J. Mol. Liq., 2021, 330, 115617 CrossRef.
- E. O. Stejskal and J. E. Tanner, J. Chem. Phys., 1965, 42, 288–292 CrossRef CAS.
- B. Antalek, Magn. Reson., 2002, 14, 225–258 CAS.
- D = kT/(6πηrH), where rH is the hydrodynamic radius, D the diffusion coefficient, k the Boltzmann constant, T the absolute temperature and η the solvent viscosity.
- A. Macchioni, G. Ciancaleoni, C. Zuccaccia and D. Zuccaccia, Chem. Soc. Rev., 2008, 37, 479 RSC.
- R. Evans, Z. Deng, A. K. Rogerson, A. S. McLachlan, J. J. Richards, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed., 2013, 52, 3199 CrossRef CAS PubMed.
- S. Macura, B. T. Farmer and L. R. Brown, J. Magn. Reson., 1986, 70, 493–499 CAS.
- J. Stonehouse, P. Adel, J. Keeler and J. Shaka, J. Am. Chem. Soc., 1994, 116, 6037 CrossRef CAS.
- D. J. McCord, J. H. Small, J. Greaves, Q. N. Van, A. J. Shaka, E. B. Fleischer and K. J. Shea, J. Am. Chem. Soc., 1998, 120, 9763 CrossRef CAS.
- H. Hu and K. Krishnamurthy, J. Magn. Reson., 2006, 182, 173–177 CrossRef CAS PubMed.
- A. Martinez-Cuezva, M. Marin-Luna, D. A. Alonso, D. Ros-Niguez, M. Alajarin and J. Berna, Org. Lett., 2019, 21, 5192–5196 CrossRef CAS PubMed.
- Parallel experiments were conducted to analyze the chiral liquid using 1H NMR after each of the recycling cycles, not detecting either the starting material or the reaction product in the chiral phase.
- M. B. Schmid, K. Zeitler and M. Gschwind, J. Am. Chem. Soc., 2011, 133, 7065–7074 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3su00349c |
| This journal is © The Royal Society of Chemistry 2024 |