Complete depolymerization of poly(ester-alt-thioether)s under mild conditions into AB functional monomers†
Received
13th September 2023
, Accepted 4th January 2024
First published on 5th January 2024
Abstract
The chemical recyclability of poly(ester-alt-thioether) structures is investigated. Different polymers were synthesized by alternating anionic ring-opening copolymerization of thiolactone and epoxide monomers using benzyl alcohol – BEMP phosphazene base as initiating system. Controlled structures with number-average molar masses between 2000 and 5000 g mol−1 were obtained. In a second time, degradation studies were carried out by methanolysis in acidic and basic conditions. The influences of the base/acid concentration and of the epoxide unit lateral substituent on the degradation rate were also investigated. The structure of degradation product has been confirmed by 1H, 13C NMR, SEC and ESI mass spectrometry, and corresponds to α-hydroxy-ω-methyl ester repeating unit molecules. These molecules were subsequently used as A–B monomers in step-growth polymerization syntheses using either p-toluene sulfonic acid (PTSA) or titaniumbutoxide (Ti(OBu)4) as catalyst. The resulting condensation polymers were analyzed by NMR, SEC, MALDI ToF, TGA and DSC. Identical structures, comparable molar masses and slightly higher glass transition temperatures have been obtained compared to the ones of the native polymers synthesized by AROP.
Sustainability spotlight
Plastic wastes have dramatic consequences on the environment and thus, the question of plastic end-of-life becomes increasingly critical. The mechanical recycling of polymers is currently strongly promoted. However, recycled materials suffer from several drawbacks, such as inevitable loss of mechanical properties due to the partial degradation of the polymer chains and contamination by the accumulation of additives. Therefore, chemical recycling is a promising alternative to move towards a virtuous circular economy. In this work, we investigated for the first time the depolymerization by alcoholysis of an original family of functional polyester in order to prepare new functional AB monomers. The polycondensation reactions of these AB monomers were subsequently studied in order to regenerate the native macromolecular structure, thus demonstrating the possibility of sustainable life cycle for these materials. This work is therefore aligned with goal 12 of the UN SDG(s) in order to ensure sustainable consumption and production patterns.
|
Introduction
In 2021, 390.7 million tons of plastic were produced and it is estimated that the volume will continue to grow to an annual production of 500 million tons of plastic depending on the model used.1,2 One of the key challenges facing polymer science is the question of sourcing and the replacement of petro-resources with bio-resources as raw materials for the manufacture of thermoplastic materials. In addition, it is now well-recognized that plastic wastes have dramatic consequences on the environment and thus, the question of plastic end-of-life becomes increasingly critical.3 In recent years, a great deal of research work has been done on the synthesis of biodegradable and functional polymers.4–7 However, their degradation in landfills or directly in ecosystems leads to a loss of recoverable materials and is therefore not the most interesting strategy from an economic point of view. In addition, the accumulation of degradation products in the environment could eventually have harmful consequences. Three methods are concomitantly used to deal with these wastes: landfill, incineration/energy recovery and recycling. For obvious reasons and to avoid ecosystem pollutions, state regulations wish to reduce or even ban the landfill of plastics. In many countries, energy recovery is currently the main outlet for plastic waste. For example, in the EU, 42% of plastic waste is used for energy recovery, 23% goes to landfill, and 35% is recycled while only about 9% is aimed to be recycled worldwide.1,8 Nevertheless, incineration generates atmospheric pollution and the production of so-called ultimate waste. Alternatively, the physical (mechanical) recycling of polymers is strongly promoted. However, recycled materials suffer from several drawbacks, such as inevitable loss of mechanical properties due to the partial degradation of the polymer chains and contamination by the accumulation of additives.9–14 Given the difficulties in recycling the polymers currently in circulation, an alternative route is being explored: converting the use of the “classic” petro-based polymers with polymers originally designed to be recycled.15,16 A growing number of studies is interested in the development of chemically recyclable plastics that would allow to move towards a virtuous circular economy.9 Two main strategies have been developed: “repurposing” and depolymerization processes. “Repurposing” consists of breaking down polymer chains by adjusting the pH or in the presence of a chemical reagent in order to convert them into new “building blocks” that will be used to synthesize new virgin materials with high added value.9,16 For example, the glycolysis of PET by ethylene glycol in excess in the presence of an organic catalyst generates a new diol monomer that can be subsequently used in polycondensation.17 Depolymerization processes feature a polymerization–depolymerization cycle that regenerates the original pure monomer and thus re-synthesizes a virgin polymer with its native properties. Aliphatic polyesters, such as poly(lactic acid) (PLA), poly(caprolactone) (PCL) or poly(hydroxyalkanoate) (PHA) have become very popular in the chemical recyclable plastic field: efficient synthesis methods have been developed and degradation-recycling studies have been performed.16,18 In particular, promising results have been obtained for the polymerization–depolymerization cycle of PLA-lactic acid.19–21 Poly(γ-butyrolactone) (PγBL) is another very promising bio-based polymer that depolymerizes into butyrolactone by simple heating the bulk material at 260–300 °C for one hour or at room temperature in the presence of metal catalysts.22,23
Beyond classical aliphatic polyesters, the synthesis and depolymerization of new functional polyesters is of great interest to the field. In addition, the use of sulfur in materials and polymer chemistry is receiving growing interest from the scientific community. For example, the introduction of sulfur atoms in the polymer backbone enables to introduce additional properties in the polymer materials, such as high refractive index, ROS-responsiveness, ionic conductivity, metal-chelating properties, self-healing capacities, improved mechanical, electrical or thermal properties.24–29 For example, polythioesters are sulfur analogues of the aliphatic polyesters, that can be synthesized by AROP of thiolactones.30 Interestingly, thioester groups are more electrophilic than ester groups and thus, more susceptible to nucleophilic attacks and to degradation.31 In addition, thiolactones have lower ring strains than lactones, which thermodynamically favors the depolymerization of polythioesters compared to corresponding polyesters.32 For example, the homopolymer resulting from the ring-opening polymerization of the γ-thiolactone derived from 4-hydroxyproline is fully depolymerizable into its original monomer in the presence of DBU under mild conditions.33 Similarly, the poly(thioester) resulting from the ring-opening polymerization of bridged bicyclic [221] BTL can be depolymerized in the bulk or in solution in the presence of lanthanum catalyst or organic base catalyst at room temperature.34
Our group recently reported the synthesis of well-defined poly(ester-alt-thioether)s by organo-catalyzed anionic ring-opening copolymerization of thiolactones with various epoxides under mild conditions.35–37 This versatile technique enables the preparation of a large number of thioether-containing functional polyesters. In this paper, the efficiency of the degradation–depolymerization by methanolysis of four different poly(ester-alt-thioether)s is presented. The study focuses on the sensitivity of the degradation rate to the pH and to the structure of the epoxide unit lateral substituents. A particular attention will be given to the stability of thioether bonds during the depolymerization process. The molecules obtained after degradation were carefully characterized by NMR and mass spectrometry and subsequently used in step-growth (re)polymerizations. The structure, molecular characteristics and properties of the recycled polymers have been compared to the ones of the original polyesters.
Results and discussion
Poly(ester-alt-thioether) synthesis
N-Acetyl homocysteine thiolactone (NHTL) was copolymerized by AROP with butylene oxide (BO), tert-butyl glycidyl ethers (tBuGE), ethoxyethyl glycidyl ethers (EEGE) and eugenol glycidyl ether (EGE) according to previously published protocols (Tables 1 and S1†).35–37 The final polymer structures are shown Fig. 1. The poly(NHTL-alt-EGE) of the run 4, Table 1 was already synthesized and characterized in a previous publication and the full characterization of this polymer has therefore not been repeated here.36
Table 1 Experimental conditions and molecular characteristics of the polymers obtained by the copolymerization of NHTL with various epoxides in THF (1.8 mol L−1) at 50 °C using benzyl alcohol and BEMP base as initiating system and [OH]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[BEMP]:[NHTL]:[epoxide] ratio equal to 1:1:25:25
:[BEMP]:[NHTL]:[epoxide] ratio equal to 1:1:25:25
| Run |
Epoxide |
Time (h) |
Cva (%) |
M
n,th (g mol−1) |
M
n,RMN
(g mol−1) |
M
n,SEC (g mol−1) |
M
w,SEC (g mol−1) |
Đ
|
|
Monomer conversion calculated by 1H NMR spectroscopy of the reaction medium by comparison of the free monomer signal integration with the corresponding polymer chain signal integration.
Experimental molar mass determined by 1H NMR spectroscopy of the purified product by comparison of a initiator signal integration with a polymer chain signal integration.
|
| 1 |
BO |
7 |
100 |
5890 |
5890 |
3820 |
5230 |
1.37 |
| 2 |
tBuGE |
6 |
100 |
7360 |
7360 |
4500 |
5990 |
1.33 |
| 3 |
EEGE |
5 |
95 |
7350 |
6190 |
5200 |
6600 |
1.27 |
| 4 (ref. 36) |
EGE |
17 |
97 |
8500 |
7600 |
4450 |
7120 |
1.60 |
 |
| | Fig. 1 Structures of the poly(ester-alt-thioether)s synthesized by the alternating anionic ring-opening copolymerization of NHTL with BO, tBuGE, EEGE and EGE, respectively. | |
In all cases, the 1H NMR spectra of the polymers purified by dialysis are in good agreement with alternating structures (Fig. S2–S4†). Nevertheless, it can be noted that traces of BEMP phosphazene base are still present on the NMR spectra at 3.28, 3.18, 2.74 and 1.18 ppm. The perfectly alternating structures of poly(NHTL-alt-tBuGE) and poly(NHTL-alt-BO) are confirmed by MALDI-TOF analyses, carried out for the first time in the case of poly(NHTL-alt-epoxide) (Fig. 2 and S5†). In both cases, two populations of linear macromolecules are visible on the spectra: the first one corresponding to an initiation by benzyl alcohol and the second one to an initiation by moisture traces. The poly(NHTL-alt-EEGE) and poly(NHTL-alt-EGE) were not detected through mass spectrometry in reflectron mode. The molar mass differences between two consecutive peaks of a same population are always equal to the exact molar mass of the NHTL–epoxide repeating unit of a perfectly alternating structure (289 g mol−1 for NHTL–tBuGE or 231 g mol−1 for NHTL–BO). In order to avoid the use of THF, the alternating copolymerization can also be performed under solventless conditions as shown in run A9, Table S1.† It is worth noting that solventless conditions resulted in lower Mn and higher dispersity obtained for AROP carried out in solution (Fig. S1†).
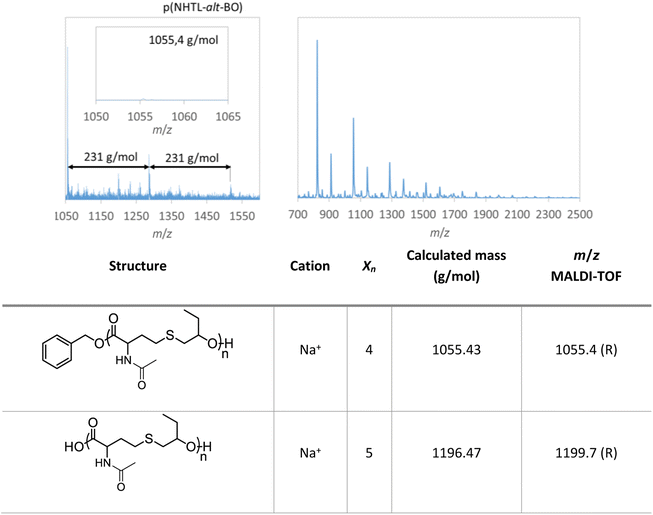 |
| | Fig. 2 MALDI-TOF analysis of poly(NTHL-alt-BO) (run 1, Table 1), reflectron mode on the left and linear mode on the right. | |
Methanolysis of the poly(ester-alt-thioether)s
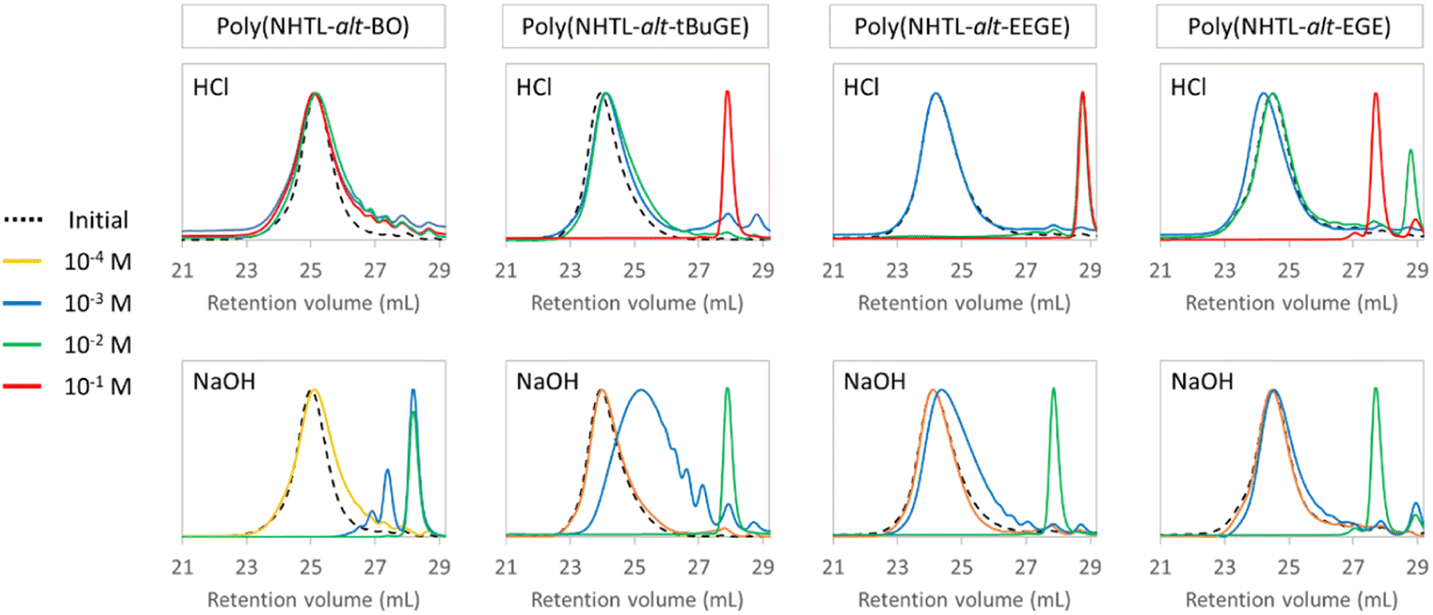
The previous poly(ester-alt-thioether)s have been subsequently used as model structures for chemical degradation studies by methanolysis. Indeed, transesterification or hydrolysis degradation reactions are widely studied for the chemical recycling of polyesters into repolymerizable units.38–42 In particular the methanolysis of PET resulting in dimethyl terephthalate and ethylene glycol is the most preferred alcoholysis method, which may proceed under three different forms of methanol namely liquid, vapor or supercritical methanol.43 In addition, previous works have shown that the addition of ethereal solvents, such as THF, during the methanolysis of PET under alkaline conditions resulted in speeding up the degradation rate.44,45 It should be noted that despite its toxicity, methanol is widely used industrially. In addition, because of its low environmental impact and its high exposure limits, the use of methanol is recommended by solvent selection guides.46,47 In our case, methanol was also preferred over ethanol because previous studies have demonstrated that the rate of a transesterification reaction dropped sharply between methoxy and ethoxy leaving group.48 The methanolysis of poly(ester-alt-thioether)s will not regenerate the initial monomers but produce new polymerizable units, namely the methoxy-ester of the repeating unit resulting from the polymerization by ring opening of epoxides and thiolactones. The methanolysis were carried out at various HCl or NaOH concentrations in 50/50 (v/v) THF/methanol mixture, allowing an easy solubilization of the polymers (Table 2). All the experiments were carried out overnight (during 16 h) at room temperature and at a polymer concentration of 50 mg mL−1. SEC analyses were performed in THF on the native polymers and on the crude products just after degradation (Fig. 3). The depolymerization rates of the poly(ester-alt-thioether) chains were calculated according to the formula given in Table 2. When a full depolymerization was demonstrated by SEC, the absence of the signal corresponding to –CHR– groups next to an ester function was also verified by 1H NMR. As expected, the degradation rate depends on the base or acid concentrations: the higher the concentration, the faster the degradation. In a general way, the methanolysis is more efficient in basic media and occurred under milder conditions. This results is in agreement with similar results already reported in the literature for poly(carbonate)s and poly(ester)s.49,50 Whatever the copolymer, a full degradation of the polymer chains is observed after one night for [NaOH] ≥ 10−2 mol L−1. Moreover, the degradation is rapid compared to the hundreds of hours in very concentrated acidic or basic aqueous solutions (C = 10−1 mol L−1),50 or to the several tens of hours in organic solvents in the presence of organic base required for the full degradation of poly(ε-caprolactone) (Mn = 10.7 kg mol−1, Đ = 1.59), a well-studied commercially available aliphatic polyester with a similar number of carbon atoms in the main chain repeating unit.51 It is worth noting that even for NaOH concentrations as low as 10−3 or 10−4 mol L−1, significant decrease of Mn can be observed overnight. The structure of the epoxide monomer unit has little influence on the efficiency of the degradation under basic conditions. The influence of the epoxide lateral substituent is more visible during the degradation in acidic conditions. Copolymers with hydrophobic side group exhibits superior resistance to acid methanolysis: poly(NHTL-alt-EEGE) is more sensitive to degradation under acidic conditions: a full depolymerization is observed after only 16 hours at a [HCl] = 10−2 mol L−1 whereas under similar conditions, the other structures are only slightly degraded. This result is explained by the hydrolysis of the acetal lateral groups and the generation of hydrophilic glycidol unit. The deprotection of the acetal groups is supported by the SEC results: the product obtained after degradation under acidic conditions has a lower molar mass than the one resulting from the depolymerization under basic conditions (Fig. 3). The most hydrophobic copolymers are more resistant to the degradation under acidic conditions: poly(NHTL-alt-EGE) required [HCl] = 10−1 mol L−1 for full degradation within 16 hours whereas only 5.6% degradation was observed at the same HCl concentration for poly(NHTL-alt-BO) for the same time.
Table 2 Experimental conditions of poly(ester-alt-thioether) degradations in acidic or basic conditions overnight. Degradation rate is equal to the difference between the number of ester bond before and after the degradation, relative to the number
| Polymer |
Acid/base nature |
Acid/base (mol L−1) |
M
n,SEC before depolymerization (M0, g mol−1) |
M
n,SEC after depolymerization (M(t), g mol−1) |
Degradation (%) |
| Poly(NHTL-alt-BO) |
HCl |
10−4 |
2900 |
2550 |
1.6 |
| 10−3 |
2300 |
3.0 |
| 10−2 |
2250 |
3.3 |
| 10−1 |
1950 |
5.6 |
| NaOH |
10−4 |
2250 |
3.3 |
| 10−3 |
400 |
72.1 |
| 10−2 |
300 |
100.0 |
| 10−1 |
300 |
100.0 |
| Poly(NHTL-alt-tBuGE) |
HCl |
10−4 |
5400 |
5400 |
0.0 |
| 10−3 |
5300 |
0.1 |
| 10−2 |
4800 |
0.9 |
| 10−1 |
380 |
100.0 |
| NaOH |
10−4 |
5180 |
0.3 |
| 10−3 |
1885 |
14.1 |
| 10−2 |
380 |
100.0 |
| 10−1 |
380 |
100.0 |
| Poly(NHTL-alt-EEGE) |
HCl |
10−4 |
5200 |
5200 |
0.0 |
| 10−3 |
5200 |
0.0 |
| 10−2 |
205 |
100.0 |
| 10−1 |
205 |
100.0 |
| NaOH |
10−4 |
5200 |
0.0 |
| 10−3 |
3640 |
3.5 |
| 10−2 |
390 |
100.0 |
| 10−1 |
390 |
100.0 |
| Poly(NHTL-alt-EGE) |
HCl |
10−4 |
4450 |
4400 |
0.1 |
| 10−3 |
4450 |
0.0 |
| 10−2 |
4400 |
0.1 |
| 10−1 |
460 |
100.0 |
| NaOH |
10−4 |
3290 |
4.1 |
| 10−3 |
3070 |
5.2 |
| 10−2 |
460 |
100.0 |
| 10−1 |
460 |
100.0 |
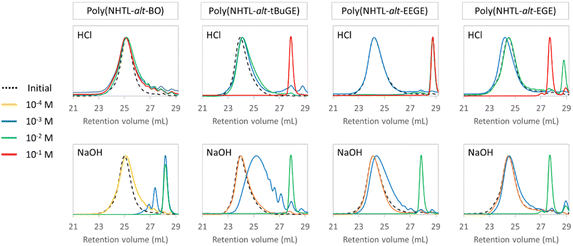 |
| | Fig. 3 SEC analysis of alternating copolymer degradation through acidic and basic methanolysis in presence of different concentrations of NaOH and HCl during 16 h. | |
AB monomer characterization
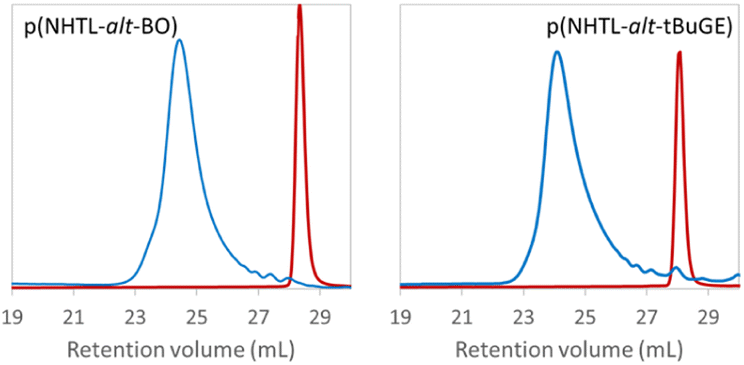
Poly(NHTL-alt-BO) derived from an alkyl epoxide and poly(NHTL-alt-tBuGE) derived from a glycidyl ether have been selected for the rest of the work as model structure for our study. One poly(ester) bear a simple aliphatic side chain and the other one a more hindered tert-butyl ether group. The full polymer degradation were performed overnight under basic conditions at [NaOH] = 10−1 mol L−1 for runs 1 and 2 of Table 1 (Fig. 4).
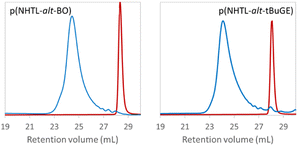 |
| | Fig. 4 Crude product SEC analysis of poly(NHTL-alt-BO) (run 1, Table 1 on the left) and of poly(NHTL-alt-tBuGE) (run 2, Table 1 on the right) before and after degradation through basic methanolysis in presence of 10−1 mol L−1 of NaOH. | |
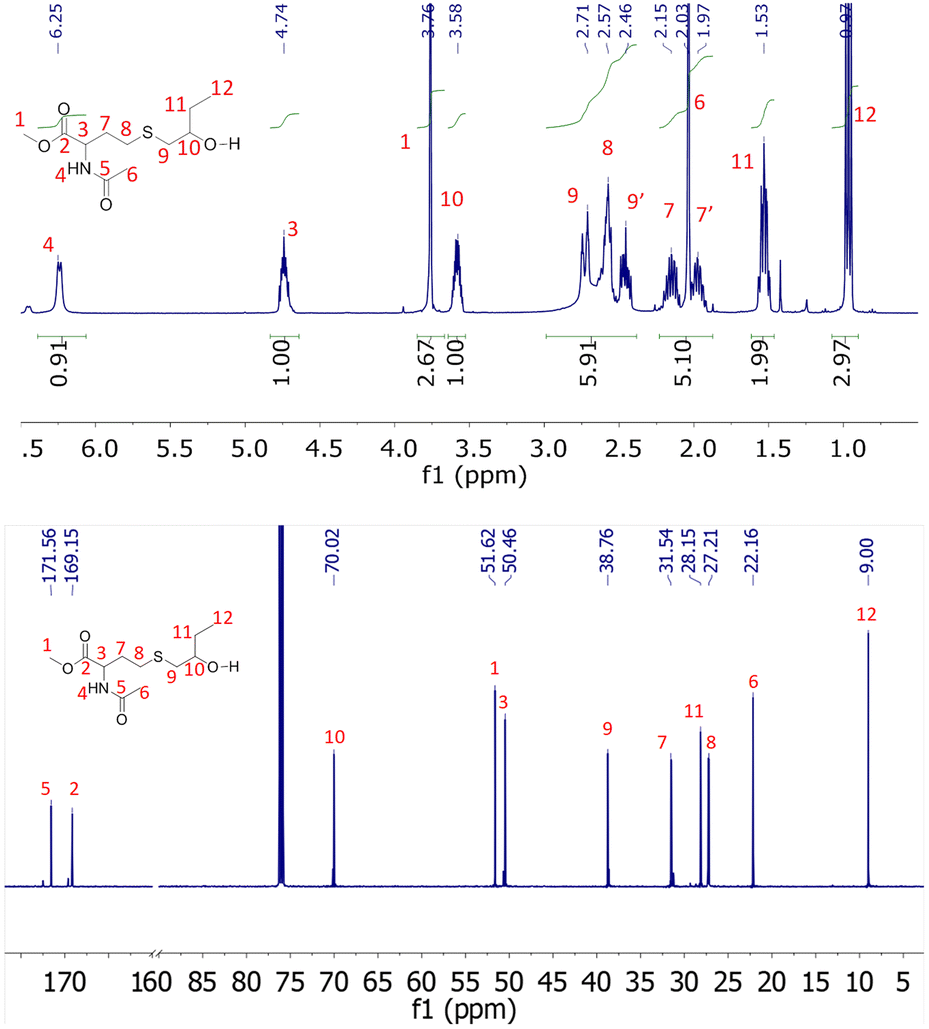
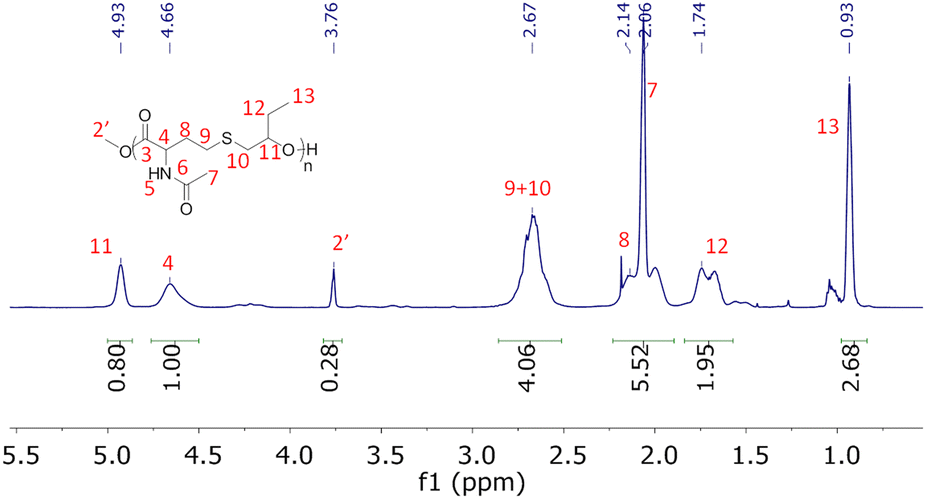
The 1H NMR spectra of the products obtained after degradation are shown in ESI (Fig. S6 and S7†). They were subsequently purified by flash silica column chromatography and colourless oils were obtained with yields of approximately 70%. We assume that this non-quantitative yield can be explained by the elimination during the purification of carboxyl containing molecules resulting from the partial hydrolysis of methyl ester groups. The purified products were analyzed by 1H, 13C NMR and ESI high-resolution mass spectrometry. For both copolymer degradation products, the molar masses obtained by HRMS-ESI are compatible with the α-methyl ester-ω-hydroxy repeating units (Fig. S12†). In addition, all 1H and 13C NMR signals could be attributed to the expected structures (Fig. 5 and S8–S11†). The presence at 3.76 ppm (Fig. 5) and 3.73 ppm (Fig. S10†) of signals corresponding to methyl esters confirm the transesterification depolymerization mechanism. For NHTL–tBuGE monomer, no 1H or 13C NMR signals could be attributed to the presence of carboxylic acids and the integration values of the methyl ester group is compatible with a fully esterified product (Fig. S10†). For NHTL–BO product, the integral ratio of the methoxy group (signal 1 in Fig. 5) is slightly smaller than 3.0, which is probably due to partial hydrolysis into a carboxyl group (about 5%). The presence of a small percentage of carboxyl group is confirmed by the small signals at 6.48 ppm on the 1H NMR spectrum and at 172.50 and 169.57 ppm on the 13C NMR spectra (Fig. 5). It should be stressed that both the amide and thioether groups are stable under the depolymerization conditions. In particular, no oxidation of the thioether could be detected. Only the ester groups of the macromolecules were affected by the pH-catalyzed methanolysis.
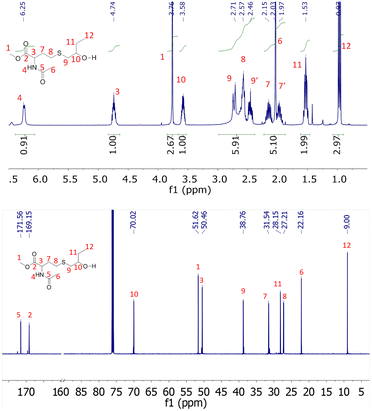 |
| | Fig. 5
1H (up) and 13C (bottom) NMR spectrum of degraded poly(NHTL-alt-BO) (run 1, Table 1) in CDCl3 at 25 °C. | |
Polycondensation of AB monomers
After purification and drying, the α-methyl ester-ω-hydroxy repeating units have been used as AB monomers in step-growth polymerizations according to the conditions summarized in Table 3. Polymerization of secondary alcohols is known to be more challenging than the ones of primary alcohols owing to steric issues. When secondary hydroxyl groups are used as sole alcohol groups, the corresponding polyesters present a low molar masses.52–55 For example, the copolymerization of 2,3-butanediol with various diacids yielded polyesters with Mn in the range 2–10 kg mol−1.56–58 The repolymerizations have been carried out in a Gram-scale glass reactor equipped with mechanical stirring and connected to vacuum (Fig. S13†). p-Toluenesulfonic acid (PTSA) and titanium(IV) butoxide (Ti(OBu)4) were used as catalysts. They are both commonly encountered in step-growth polymerizations.59–64 PTSA does not present any significant toxicity.65 Ti(OBu)4 is commonly considered as less toxic than conventional transesterification antimony catalysts.66 Preliminary thermogravimetric analysis (TGA) were performed on both AB monomers to determine the maximum polymerization temperature (Fig. S14†). No degradation is observed for temperatures below 200 °C. Therefore, a maximum temperature of 160 °C was set for the polycondensation in order to prevent any degradation of the structures during time.
Table 3 Experimental conditions and molecular characteristics of the polycondensation experiments. Catalyst was added with an amount of 1% in mass
| Run |
Structure type (precursor) |
Catalyst |
Pressure |
T (°C) |
Time (day) |
M
n,SEC (g mol−1) |
M
w,SEC (g mol−1) |
Đ
|
|
Polymerization without monomer purification.
Second depolymerization–repolymerization cycle.
|
| 5 |
NHTL–BO (1) |
PTSA |
Atm |
120–160 |
5 |
560 |
740 |
1.34 |
| 6 |
NHTL–BO (A1) |
PTSA |
Under low pressure |
150 |
7 |
640 |
1100 |
1.73 |
| 7 |
NHTL–BO (A2) |
Ti(OBu)4 |
Under low pressure |
150 |
5 |
2440 |
12500 |
5.1 |
| 8 |
NHTL–BO (A4) |
Ti(OBu)4 |
Under low pressure |
150 |
5 |
3560 |
9340 |
2.62 |
| 9a |
NHTL–BO (A5) |
Ti(OBu)4 |
Under low pressure |
150 |
3 |
1850 |
5130 |
2.8 |
| 10b |
NHTL–BO (9) |
Ti(OBu)4 |
Under low pressure |
150 |
5 |
2350 |
6830 |
2.9 |
| 11 |
NHTL–tBuGE (2) |
PTSA |
Atm |
160 |
5 |
— |
— |
— |
| 12 |
NHTL–tBuGE (A6) |
PTSA |
Under low pressure |
150 |
5 |
640 |
1060 |
1.7 |
| 13 |
NHTL–tBuGE (A7) |
Ti(OBu)4 |
Under low pressure |
150 |
6 |
2780 |
6900 |
2.5 |
| 14 |
NHTL–tBuGE (A9) |
Ti(OBu)4 |
Under low pressure |
150 |
5 |
4370 |
18300 |
4.2 |
| 15b |
NHTL–tBuGE (13) |
Ti(OBu)4 |
Under low pressure |
150 |
5 |
1500 |
5950 |
3.3 |
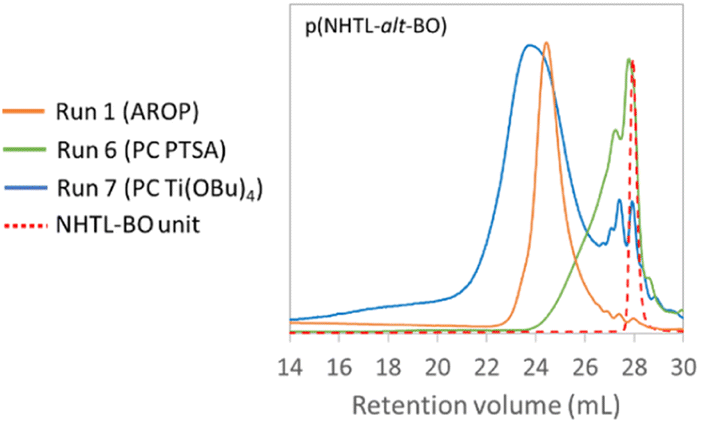
In a first part, the polymerisation of the NHTL–BO monomer was studied. A first polymerization reaction was performed using PTSA as catalyst under atmospheric pressure (Table 3, run 5). The temperature was gradually increased from 120 °C to 160 °C over five days. Very limited polymerization was observed over time and very low molar masses corresponding to dimers were obtained. Run 6 was performed under vacuum after applying a 24-hour prepolymerization step at 150 °C without the presence of catalyst, in order to eliminate the remaining traces of water or solvent and in order to enable the first transesterifications reactions with an efficient elimination of the MeOH co-product. The molar masses were only poorly improved. The SEC trace shows very limited conversion and very low molar mass (Fig. 6). Higher Mn were obtained when PTSA was replaced by titanium(IV) butoxide (Ti(OBu)4) as catalyst (Table 3, runs 7–8) (Fig. 6). The mass-average molar masses Mw are higher than the ones previously achieved by AROP (Table 1), nevertheless the Mn remain relatively low (2.5 and 3.5 kg mol−1). These Mn values are in the same range as those obtained in the literature when secondary hydroxyl groups are used as sole alcohol groups for the synthesis of polyesters.52–55 In addition, the presence of low molar mass macromolecules (monomer, dimers, trimers…) is evidenced by the SEC analyses (Fig. 6, run 7). The dispersity of the polycondensation polymer is higher than the one of the AROP copolymer. The long polymerization time can be essentially explained by a low polymerization temperature compared with temperatures conventionally used for polycondensation reactions and by the lower reactivity of secondary alcohols compared with primary alcohols.
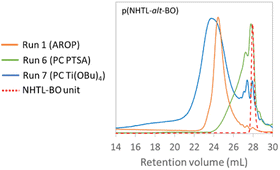 |
| | Fig. 6 SEC analysis of poly(NTHL-alt-BO) synthesized by AROP (run 1, Table 1 in orange), by PC with PTSA (run 6, Table 3 in green) and by PC with Ti(OBu)4 (run 7, Table 3 in blue) in THF at 40 °C. | |
With the exception of the signals corresponding to the chain ends, the 1H and 13C NMR spectra of run 7 polymer are identical to the NMR spectra of the poly(NHTL-alt-BO) synthesized by AROP, confirming identical structures. All the signals could be attributed to the expected structure of thiolactone–epoxide alternating copolymer (Fig. 7). The amide and thioether groups remained stable during the polycondensation. The 13C NMR spectrum also confirmed the expected structure (Fig. S15†).
 |
| | Fig. 7
1H NMR analysis in CDCl3 at 25 °C of poly(NHTL-alt-BO) synthesized by polycondensation (run 7, Table 3). | |
As previously observed for the MALDI-TOF analysis, the desorption of poly(NHTL-alt-BO) is difficult (Fig. 8). One major and four minor populations could be detected, each of them corresponds to macromolecules constituted of the expected NHTL–BO repeating unit. Four populations have been identified as follows in order of importance. The main distribution corresponds to perfectly alternating macrocycles cationized by sodium ion. The macrocyclization can be explained by an intramolecular transesterification reaction occurring during the polymerization process. The minor distributions can be attributed to the expected alternating polymer cationized by a sodium ion and end-terminated by a methoxy group at one end and by a hydroxy group at the other end and to linear alternating macromolecules end-terminated by an ethoxy group at one end and by a hydroxy group or an acetyl group at the other end. We assume that these population can be explained by the presence of a small amount ethyl acetate used as eluent during the column chromatography in the AB monomer. In conclusion, all the characterization methods suggests the reformation of the poly(NHTL-alt-BO) structure.
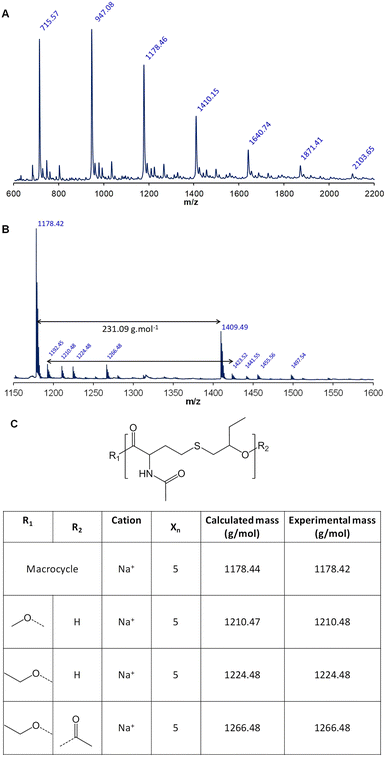 |
| | Fig. 8 MALDI-TOF analysis of poly(NTHL-alt-BO) (run 7, Table 3): (A) linear mode; (B) reflectron mode and (C) postulated structures. | |
The purification of the AB monomer by flash chromatography required significant volumes of organic solvents and cannot be easily transfer to an industrial process. However, the result of run 9 (Table 3) demonstrates that the polymerization is taking place even if the reaction is carried out on the AB monomer unit directly after methanolysis without any purification step other than a thorough drying. In the longer term, a greener process is therefore conceivable even if currently the Mn obtained remains low.
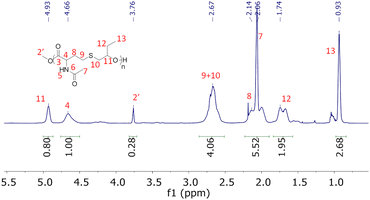
The first reaction with the NHTL–tBuGE AB monomer (run 11, Table 3) was performed in presence of PTSA at 160 °C under atmospheric pressure resulted in the formation of a brittle solid, insoluble in all the tested organic solvents. Thus, no SEC analysis could be performed. Nevertheless, the product could be dissolved in deuterated water and analyzed by NMR. The 1H NMR spectrum shows the complete disappearance of the signal corresponding to the tert-butyl group, which might result from the deprotection of the hydroxy groups, resulting in the presence of one primary and one secondary hydroxy group on each epoxide unit of the repeating unit (Fig. 9). In addition, the signal of the methine group at 4.17 ppm in the 1H NMR spectrum suggests a preferential polycondensation by the primary hydroxy groups. The presence of the second hydroxy group increased the polymer hydrophilicity and could also lead to the formation of hyperbranched structures, explaining the lack of solubility in organic solvent. The presence of cross-linking reactions is also to be considered, although the latter could not be identified. When the polymerization reactions are performed at 150 °C using Ti(OBu)4 as catalyst (Table 3, runs 13–14), the tert-butoxy groups remain stable, no deprotection of the hydroxy group is observed and the polymer are soluble in THF, DMSO and CDCl3. The best results were obtained for Table 3 run 14 when the polycondensation was carried out at 150 °C using Ti(OBu)4 as catalyst. A Mn above 4000 g mol−1 was determined by SEC (Fig. S16†). A signal corresponding to high molar masses can be observed on the SEC trace (Fig. S19†). This signal is even more visible with a UV detection at 254 nm. After a 30-minute acidic treatment with a 10−1 mol L−1 solution of HCl in THF, the high molar mass signal is strongly reduced while the polymer signal remains intact, suggesting the presence of metal catalyst aggregates. All the signals of the 1H NMR spectrum could be attributed to the expected poly(NTHL-alt-tBuGE) structure (Fig. S17†). The MALDI-TOF spectrum is shown in Fig. S18,† it is worth noting that the molar mass difference between two consecutive peaks of a same population is always equal to the exact molar mass of the NHTL/tBuGE repeating unit of a perfectly alternating structure (289 g mol−1). Two major and three minor populations could be attributed. The main distribution corresponds to the expected linear polymer cationized by a sodium ion and end-terminated by a methoxy group at one end and by a hydroxy group at the other end. The second most important distribution can be attributed to macrocycles cationized by sodium ion. The minor distributions can be attributed to the expected linear polymer cationized by a sodium ion and end-terminated by a methoxy or an ethoxy group at one end and by a hydroxy or an acetyl group at the other end.
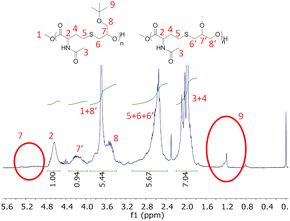 |
| | Fig. 9
1H NMR spectrum of poly(NHTL-alt-tBuGE) synthesized by polycondensation in presence of PTSA (run 11, Table 3) in CDCl3 at 25 °C with hypothetical attributions of the branched moiety. | |
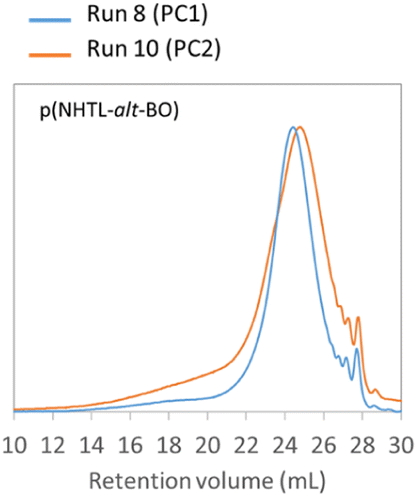
The poly(ester-alt-thioether)s structures obtained by AROP can be depolymerized and subsequently repolymerized by polycondensation. Finally, the possibility of repeating the protocol and performing several cycles was evaluated. As a proof of concept, a second depolymerization–repolymerization experiment has been performed on the poly(NHTL-alt-BO) and the poly(NHTL-alt-tBuGE) synthesized by polycondensation in the run 8 and run 14, Table 3 respectively. The repolymerization of the purified AB monomer obtained after the basic methanolysis was possible. The second repolymerization was carried in the exact same condition than the run 8 or 14 that is with a pre-polymerization step of one day, using 1% in mass of the titanium butoxide as the catalyst and carrying out the polycondensation during four more days at 150 °C. As shown by the SEC analysis, the second repolymerization of the poly(NHTL-alt-BO) gave very consistent results and the polymer obtained through the run 19 shows very similar molar mass distribution as that of the polymer of the run 8 (Fig. 10). Concerning the poly(NHTL-alt-tBuGE) structure, the second cycle was also possible and a polymer chain was obtained. However the Mn and Mw of the second repolymerization (run 15, Table 3) are lower than those obtained in the previous experiments (run 14, Table 3) (Fig. S21†).
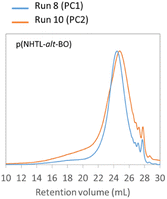 |
| | Fig. 10 SEC analysis in THF at 40 °C of poly(NTHL-alt-BO) synthesized by a first (run 8, Table 3 in blue) and a second repolymerization (run 10, Table 3 in orange). | |
In fact, the polymer obtained was only partially soluble in organic solvent meaning that some branching may occur due to the deprotection of the tBuGE unit. 1H NMR spectrum shows that if the poly(NTHL-alt-tBuGE) is the major structure of the compound, undesired signals also indicate a partial deprotection of the lateral tert-butyl group and confirming the probable occurrence of branching (Fig. S22†).
Thermal characterization
The poly(NHTL-alt-BO)s and poly(NHTL-alt-tBuGE)s synthesized by AROP and by step-growth polymerization were characterized by TGA and DSC. The polymers prepared by both polymerization methods have similar degradation temperatures that is, around 270 °C and 275 °C for BO and tBuGE respectively (Fig. S20†). This temperature is in agreement with the degradation temperature that have been observed for poly(thioether)s in the literature, suggesting that carbon–sulfur bond is the weakest linkage in the polymer structure.67 All copolymers are amorphous, only displaying glass transition temperatures (Tg) (Fig. 11). Interestingly, the polymer synthesized by polycondensation have higher Tg than those of the corresponding polymers obtained by AROP: 39.4 vs. 17.4 °C for the poly(NHTL-alt-BO) and 35.1 vs. 18.5 for the poly(NHTL-alt-tBuGE). These increases can be explained by the higher Mw of the repolymerized compounds. The high dispersity values also suggest that cross-linking might occurred, which could hinder the chain mobility and increases glass transition temperature.
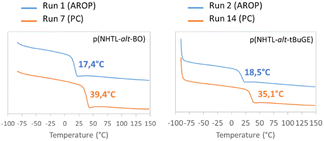 |
| | Fig. 11 DSC curves of second heating scans at 10 °C min−1 of poly(ester-alt-thioether)s. Superposition of the thermal response of poly(NHTL-alt-BO) on the left and poly(NHTL-alt-tBuGE) on the right with polymers obtained by AROP (runs 1 and 2, Table 1 in blue) and by polycondensation (runs 7 and 14, Table 3 in orange). | |
Conclusion
The chemical depolymerization of poly(ester-alt-thioether) by alcoholysis is reported for the first time. The methanolysis in THF/MeOH of poly(NHTL-alt-BO), poly(NHTL-alt-tBuGE), poly(NHTL-alt-EEGE) and poly(NHTL-alt-EGE) copolymers was carried out under acidic and basic conditions. As expected, the degradation is easier in basic medium and the presence of hydrophobic lateral substituents slightly increases the copolymer stability. For all the copolymer structures, a full degradation is achieved after one night at room temperature under relatively mild basic conditions by the cleavage of the ester groups without degrading the thioether groups and thus generating α-hydroxy-ω-methyl ester repeating units.
In a second step, the α-hydroxy-ω-methyl ester molecules were isolated for the first time and successfully used as AB monomers in step-growth homopolymerizations leading to the regeneration of the original poly(ester-thioether) structures. The polymer molar masses remained relatively low but the use of the AB building blocks as low percentage comonomer is an opportunity to investigate.
Experimental section
Materials
2-Tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine solution (BEMP, 1.0 mol L−1 in hexane, Sigma-Aldrich), benzyl alcohol (99.8% anhydrous, Sigma-Aldrich), N-acetyl-DL-homocysteine thiolactone (NHTL, >99%, Sigma-Aldrich), p-toluenesulfonic acid monohydrate (PTSA, 98%, Sigma-Aldrich) and titanium(IV) n-butoxide (99+%, Thermo Scientific Chemicals) were used as received. Ethoxyethyl glycidyl ether (EEGE) was synthesize in the laboratory from glycidol (96%, Sigma-Aldrich) and ethyl vinyl ether (99%, Sigma-Aldrich). Eugenol glycidyl ether (EGE) was synthesized according to the methods already published.34 Butylene oxide (BO, 99%, Sigma-Aldrich), EEGE and tert-butyl glycidyl ether (tBuGE, 99%, Sigma-Aldrich) were cryo-distilled over CaH2 twice prior use. THF was dried with an MBRAUN MB SPS-800 solvent purification system under nitrogen.
Instruments
1H and 13C NMR spectra were recorded in CDCl3 using a Bruker 400 MHz NMR spectrometer. Size Exclusion Chromatography Experiment (SEC) were carried out on three PL gel Mixed-C 5 μm columns (7.5 × 300 mm; separation limits: 0.2 to 2000 kg mol−1) maintained at 40 °C and sample Viscotek GPCmax delivery module and 2 modular detectors: a Viscotek 3580 differential refractive index (RI) detector and a Shimadzu SPD20-AV diode array UV detector. THF was used as the mobile phase at a flow rate of 1 mL min−1, toluene was used as a flow rate marker. All polymers were injected (50 μL) at a concentration of 5 mg mL−1 after filtration through a 0.45 μm pore-size membrane. The OmniSEC 4.6.2 software was used for data acquisition and analysis. Number-average molar masses (Mn), weight-average molar masses (Mw) and dispersities were determined by SEC with a calibration curve based on poly(methyl methacrylate) standard (calibration range 0.2–1000 kg mol−1), using the RI detector. The thermal decomposition was evaluated by thermogravimetric analysis (TGA) using a TA Q50 apparatus. Data were collected using a 20 °C min−1 ramp to 600 °C. The experiment was run under a stream of nitrogen. DSC measurements were carried out using a TA DSC Q2000 calibrated with indium standard. Sample (ca. 15 mg) was placed in an aluminum cell and initially cooled at −70 °C. Two heating (10 K min−1)–cooling (10 K min−1) cycles were then applied in the −70–150 °C temperature range.
Matrix-assisted laser desorption and ionization time-of-flight mass spectrometry (MALDI-TOF MS)
Mass spectra were recorded by MALDI-TOF MS using dithranol as a matrix and NaI as cationizing agent using a Bruker Autoflex Speed mass spectrometer, equipped with a laser that produces pulses at 337 nm. Spectra were recorded in reflectron mode at an accelerating potential of 20 kV. Samples were prepared by dissolving the polymer in THF at a concentration of 5 mg mL−1. A 10 μL aliquot of this solution was mixed with 20 μL of matrix solution and 10 μL of NaI solution (both at 20 mg mL−1 in THF). Poly(ethylene oxide) standards (polymer standards service) of known structures, Mn = 1500 g mol−1 and Mn = 4000 g mol−1 were used to calibrate the mass scale. In all cases, to determine m/z, the molar mass of the sodium cation was added.
General AROP procedure
The anionic alternating copolymerization was carried out according to the following typical procedure. In a glove-box, 4 mL of THF, 60 μL of benzyl alcohol (0.58 mmol), 2.1 mL of tert-butyl glycidyl ether (14.5 mmol), and 2.3 g of N-acetylhomocysteine thiolactone (14.5 mmol) were introduced under nitrogen into a polymerization tube. Then, 580 μL of BEMP solution (0.58 mmol) were added using a microsyringe. After closure of the flask, the reaction mixture was stirred at 50 °C and left to react for the required period of time (6–7 h). After removing residual monomers and solvent by rotary evaporation, the polymer was dissolved in THF and dialyzed in cellulose ester membrane (Repligen Spectra/Por 6 dialysis tubing, flat width = 45 mm, molecular cutoff = 1 kDa) against THF for 24 h. After removing the solvent by rotatory evaporation and dried under vacuum at 80 °C a white solid was obtained. Yield: 87%. Deviations from this general procedure are summarized in Table 1.
Methanolysis conditions used for the preparation of AB monomers
In a 100 mL round bottom flask, 3.0 g of the poly(ester-alt-thioether) are dissolved in a mixture of methanol and THF (50:50 volume ratio) at a concentration of 0.1 g mL−1 of polymer and 0.1 mol L−1 of NaOH (0.12 g). The mixture is placed under stirring in an oil bath at 50 °C overnight. The reaction medium is then neutralized with HCl. After removing the solvent under rotary evaporation, 3.20 g of product were obtained (yield = 100%, AB monomer + NaCl salt). The product is subsequently purified through a flash silica column using 100% of ethyl acetate as eluent. The fraction of the repeating unit is collected and after removing the solvent under rotary evaporation, the product is dried under vacuum at 50 °C overnight. A colorless oil is obtained (yield = 70%) and analyzed by 1H NMR.
General polycondensation procedure
In a polycondensation reactor, 4 g of the AB monomer is added. The reactor is placed in a oil bath, mechanical stirring is started and temperature is increased at 150 °C. Then, vacuum is slowly applied to the system. After 24 h of pre-polymerization, the reactor is opened and 1% in mass of Ti(OBu)4 is added (40 mg). The polymerization is then let under mechanical stirring during four days. The polymer is then dissolved in THF of CHCl3 for reconditioning. After removing the solvent by rotatory evaporation and dried under vacuum at 80 °C a yellow to brown brittle solid is obtained.
Author contributions
Simon Le Luyer – investigations and writing, original draft; Philippe Guégan – resources, supervision, review and editing; Nicolas Illy – conceptualization, resources, supervision, review and editing.
Conflicts of interest
There are no conflicts to declare.
References
-
Plastics Europe, Plastics - The Facts 2022, Brussells, 2022 Search PubMed.
- H. Sardon and A. P. Dove, Science, 2018, 360, 380–381 CrossRef CAS PubMed.
- C. Q. Y. Yong, S. Valiyaveettil and B. L. Tang, Int. J. Environ. Res. Public Health, 2020, 17, 1509 CrossRef CAS PubMed.
- S. Ramesh Kumar, P. Shaiju, K. E. O’Connor and R. Ramesh Babu, Curr. Opin. Green Sustainable Chem., 2020, 21, 75–81 CrossRef CAS.
- P. Rai, S. Mehrotra, S. Priya, E. Gnansounou and S. K. Sharma, Bioresour. Technol., 2021, 325, 124739 CrossRef CAS PubMed.
- S. Rodríguez-Fabià and G. Chinga-Carrasco, J. Bioresour. Bioprod., 2022, 7, 161–172 CrossRef.
- D. Wei, H. Wang, Z. Ziaee, F. Chibante, A. Zheg and H. Xiao, Mater. Sci. Eng., C, 2016, 58, 986–991 CrossRef CAS PubMed.
- D. Kawecki, P. R. W. Scheeder and B. Nowack, Environ. Sci. Technol., 2018, 52, 9874–9888 CrossRef CAS PubMed.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- S. Yao, A. Tominaga, Y. Fujikawa, H. Sekiguchi and E. Takatori, Nihon Reoroji Gakkaishi, 2013, 41, 173–178 CrossRef CAS.
- I. A. Ignatyev, W. Thielemans and B. Vander Beke, ChemSusChem, 2014, 7, 1579–1593 CrossRef CAS PubMed.
- J. N. Hahladakis, C. A. Velis, R. Weber, E. Iacovidou and P. Purnell, J. Hazard. Mater., 2018, 344, 179–199 CrossRef CAS PubMed.
- A. Jansson, K. Möller and T. Gevert, Polym. Degrad. Stab., 2003, 82, 37–46 CrossRef CAS.
- N. George and T. Kurian, Ind. Eng. Chem. Res., 2014, 53, 14185–14198 CrossRef CAS.
- M. Hong and E. Y. X. Chen, Trends Chem., 2019, 1, 148–151 CrossRef CAS.
- X. Tang and E. Y. X. Chen, Chem, 2019, 5, 284–312 CAS.
- K. Fukushima, O. Coulembier, J. M. Lecuyer, H. A. Almegren, A. M. Alabdulrahman, F. D. Alsewailem, M. A. Mcneil, P. Dubois, R. M. Waymouth, H. W. Horn, J. E. Rice and J. L. Hedrick, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1273–1281 CrossRef CAS.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070 CrossRef CAS PubMed.
- C. Alberti and S. Enthaler, ChemistrySelect, 2020, 5, 14759–14763 CrossRef CAS.
- P. Majgaonkar, R. Hanich, F. Malz and R. Brüll, Chem. Eng. J., 2021, 423, 129952 CrossRef CAS.
- L. Cederholm, J. Wohlert, P. Olsén, M. Hakkarainen and K. Odelius, Angew. Chem., Int. Ed., 2022, 61, e202204531 CrossRef CAS PubMed.
- M. Hong and E. Y. X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS PubMed.
- M. Hong and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2016, 55, 4188–4193 CrossRef CAS PubMed.
- A. Kausar, S. Zulfiqar and M. I. Sarwar, Polym. Rev., 2014, 54, 185–267 CrossRef CAS.
-
X. H. Zhang and P. Theato, Sulfur-Containing Polymers: From Synthesis to Functional Materials, Wiley, 2021 Search PubMed.
- H. Li, S. M. Guillaume and J.-F. Carpentier, Chem.–Asian J., 2022, 17, e202200641 CrossRef CAS PubMed.
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC.
- H. Mutlu, E. B. Ceper, X. Li, J. Yang, W. Dong, M. M. Ozmen and P. Theato, Macromol. Rapid Commun., 2019, 40, 1800650 CrossRef PubMed.
- P. Carampin, E. Lallana, J. Laliturai, S. C. Carroccio, C. Puglisi and N. Tirelli, Macromol. Chem. Phys., 2012, 213, 2052–2061 CrossRef CAS.
- N. Illy and E. Mongkhoun, Polym. Chem., 2022, 13, 4592–4614 RSC.
- N. M. Bingham, Q. u. Nisa, S. H. L. Chua, L. Fontugne, M. P. Spick and P. J. Roth, ACS Appl. Polym. Mater., 2020, 2, 3440–3449 CrossRef CAS.
- A. S. Narmon, C. A. M. R. van Slagmaat, S. M. A. De Wildeman and M. Dusselier, ChemSusChem, 2023, 16, e202202276 CrossRef CAS PubMed.
- J. Yuan, W. Xiong, X. Zhou, Y. Zhang, D. Shi, Z. Li and H. Lu, J. Am. Chem. Soc., 2019, 141, 4928–4935 CrossRef CAS PubMed.
- C. Shi, M. L. McGraw, Z.-C. Li, L. Cavallo, L. Falivene and E. Y. X. Chen, Sci. Adv., 2020, 6, eabc0495 CrossRef CAS PubMed.
- N. Illy, V. Puchelle, S. Le Luyer and P. Guégan, Eur. Polym. J., 2021, 153, 110490 CrossRef CAS.
- S. Le Luyer, B. Quienne, M. Bouzaid, P. Guégan, S. Caillol and N. Illy, Green Chem., 2021, 23, 7743–7750 RSC.
- V. Puchelle, Y. Latreyte, M. Girardot, L. Garnotel, L. Levesque, O. Coutelier, M. Destarac, P. Guégan and N. Illy, Macromolecules, 2020, 53, 5188–5198 CrossRef CAS.
- H. Tsuji, T. Ono, T. Saeki, H. Daimon and K. Fujie, Polym. Degrad. Stab., 2005, 89, 336–343 CrossRef CAS.
- H. Tsuji, Y. Yamamura, T. Ono, T. Saeki, H. Daimon and K. Fujie, Macromol. React. Eng., 2008, 2, 522–528 CrossRef CAS.
- S. Thiyagarajan, E. Maaskant-Reilink, T. A. Ewing, M. K. Julsing and J. van Haveren, RSC Adv., 2022, 12, 947–970 RSC.
- J. Jiang, K. Shi, X. Zhang, K. Yu, H. Zhang, J. He, Y. Ju and J. Liu, J. Environ. Chem. Eng., 2022, 10, 106867 CrossRef CAS.
- H. Chen, K. Wan, Y. Zhang and Y. Wang, ChemSusChem, 2021, 14, 4123–4136 CrossRef CAS PubMed.
- B. Shojaei, M. Abtahi and M. Najafi, Polym. Adv. Technol., 2020, 31, 2912–2938 CrossRef CAS.
- A. Oku, L.-C. Hu and E. Yamada, J. Appl. Polym. Sci., 1997, 63, 595–601 CrossRef CAS.
- L.-C. Hu, A. Oku, E. Yamada and K. Tomari, Polym. J., 1997, 29, 708–712 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
- G. Pomalaza, R. De Clercq, M. Dusselier and B. Sels, Appl. Catal., B, 2022, 300, 120747 CrossRef CAS.
- J. H. Jung, M. Ree and H. Kim, Catal. Today, 2006, 115, 283–287 CrossRef CAS.
- G. P. Sailema-Palate, A. Vidaurre, A. J. Campillo-Fernández and I. Castilla-Cortázar, Polym. Degrad. Stab., 2016, 130, 118–125 CrossRef CAS.
- B. Dong, G. Xu, R. Yang and Q. Wang, Chem.–Asian J., 2022, 17, e202200667 CrossRef CAS PubMed.
- ü. Kanca, J. Van Buijtenen, B. A. C. Van As, P. A. Korevaar, J. A. J. M. Vekemans, A. R. A. Palmans and E. W. Meijer, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2721–2733 CrossRef.
- K. R. Yoon, S.-P. Hong, B. Kong and I. S. Choi, Synth. Commun., 2012, 42, 3504–3512 CrossRef CAS.
- B. A. C. van As, J. van Buijtenen, T. Mes, A. R. A. Palmans and E. W. Meijer, Chem.–Eur. J., 2007, 13, 8325–8332 CrossRef CAS PubMed.
- F. van der Klis, R. J. I. Knoop, J. H. Bitter and L. A. M. van den Broek, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1903–1906 CrossRef CAS PubMed.
- E. Gubbels, L. Jasinska-Walc and C. E. Koning, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 890–898 CrossRef CAS.
- S. Thiyagarajan, W. Vogelzang, R. J. I. Knoop, A. E. Frissen, J. van Haveren and D. S. van Es, Green Chem., 2014, 16, 1957–1966 RSC.
- T. Debuissy, E. Pollet and L. Avérous, Polymer, 2016, 99, 204–213 CrossRef CAS.
-
K. Fukushima and T. Fujiwara, in Polymers for Biomedicine, 2017, pp. 149–189, DOI:10.1002/9781118967904.ch6.
- A. Bandzerewicz, M. Cegłowski, K. Korytkowska and A. Gadomska-Gajadhur, Appl. Sci., 2022, 12, 12445 CrossRef CAS.
- I. Shigemoto, T. Kawakami, H. Taiko and M. Okumura, Polymer, 2011, 52, 3443–3450 CrossRef CAS.
- Z. Terzopoulou, E. Karakatsianopoulou, N. Kasmi, V. Tsanaktsis, N. Nikolaidis, M. Kostoglou, G. Z. Papageorgiou, D. A. Lambropoulou and D. N. Bikiaris, Polym. Chem., 2017, 8, 6895–6908 RSC.
- P.-J. Roumanet, N. Jarroux, L. Goujard, J. Le Petit, Y. Raoul, V. Bennevault and P. Guégan, ACS Sustain. Chem. Eng., 2020, 8, 16853–16860 CrossRef CAS.
- T. H. N. Nguyen, F. Balligand, A. Bormann, V. Bennevault and P. Guégan, Eur. Polym. J., 2019, 121, 109314 CrossRef CAS.
-
Screening assessment of benzenesulfonic acid, 4-methyl-(p-toluenesulfonic acid), Government of Canada, 2022, ISBN 978-0-660-43596-1 Search PubMed.
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737–4753 RSC.
- F. Goethals, S. Martens, P. Espeel, O. van den Berg and F. E. Du Prez, Macromolecules, 2014, 47, 61–69 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of reaction mixtures and polymers, MALDI-TOF spectra and SEC traces. See DOI: https://doi.org/10.1039/d3su00320e |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Nicolas
Illy
* and
Nicolas
Illy